

Abstract
The Saccharomyces cerevisiae transporter Arn1p takes up the ferric-siderophore ferrichrome, and extracellular ferrichrome dramatically influences the intracellular trafficking of Arn1p. In the absence of ferrichrome, Arn1p sorts directly to the endosomal compartment. At low concentrations of ferrichrome, Arn1p stably relocalizes to the plasma membrane, yet little to no uptake of ferrichrome occurs at these low concentrations. At higher concentrations of ferrichrome, Arn1p cycles between the plasma membrane and endosome. Arn1p contains two binding sites for ferrichrome: one site has an affinity similar to the KT for transport, but the second site has a much higher affinity. Here we report that this high-affinity binding site lies within a unique extracytosolic, carboxyl-terminal domain. Mutations within this domain lead to loss of ferrichrome binding and uptake activities and missorting of Arn1p, including a failure to relocalize to the plasma membrane in the presence of ferrichrome. Mutation of phenylalanine residues in the cytosolic tail of Arn1p also lead to missorting, but without defects in ferrichrome binding. We propose that the carboxyl terminus of Arn1p contains a receptor domain that controls the intracellular trafficking of the transporter.
Keywords: iron, major facilitator, siderophore, trafficking, ubiquitin
Introduction
Iron is an essential nutrient for almost every organism on earth, as it is required for the formation of essential prosthetic groups, such as heme and iron–sulfur clusters. Iron exists stably in multiple oxidation states, and this property has proved both useful and problematic for cells. The bioavailability of iron is low, as most iron in an aerobic environment is present in the oxidized, ferric form, which is only sparingly soluble at neutral pH and forms precipitates of oxyhydroxides. Microorganisms and plants have solved this bioavailability problem through a variety of strategies, most notably through the synthesis and secretion of different types of small molecules that can bind ferric iron with remarkably high affinity and specificity, thereby solubilizing the iron. These small molecules, termed siderophores, can be captured in their ferric form by specific cellular transport systems, and the iron can be extracted and used by the metabolic machinery of the cell. The budding yeast Saccharomyces cerevisiae does not synthesize siderophores, but it can take up iron bound to siderophores secreted by other microbes (Neilands, 1995; Byers and Arceneaux, 1998).
S. cerevisiae expresses two genetically separate systems of siderophore iron uptake. One system relies on the extracellular reduction and release of ferrous iron from the siderophore ligand, followed by uptake through the high-affinity transport complex specific for ferrous iron (Lesuisse et al, 1987; Yun et al, 2000a). This system employs the ferric-chelate reductase activities of Fre1p, Fre2p, Fre3p, or Fre4p (Dancis et al, 1990; Georgatsou and Alexandraki, 1994; Yun et al, 2001), and the transport activities of Fet3p and Ftr1p (Askwith et al, 1994; Stearman et al, 1996). The second system consists of four homologous transporters that are members of the SIT subfamily (TC 2.A.1.16) of the major facilitator superfamily (MFS) of secondary transporters. These transporters are termed Arn1p, Arn2p/Taf1p, Arn3p/Sit1p, and Arn4p/Enb1p, and they exhibit specificity for intact siderophore–iron chelates of the hydroxamate and catecholate classes (Lesuisse et al, 1998; Heymann et al, 1999, 2000a, 2000b; Yun et al, 2000a, 2000b; Moore et al, 2003).
Arn1p specifically transports ferrichrome (FC)-type siderophores, and the intracellular trafficking of Arn1p is dramatically affected by the presence of the siderophore substrate (Heymann et al, 2000b; Yun et al, 2000b; Kim et al, 2002). In the absence of FC, newly synthesized Arn1p is sorted directly from the Golgi to the late endosomal compartment. In the presence of low concentrations of FC (10–100 nM), Arn1p stably relocalizes to the plasma membrane, yet little to no uptake of FC occurs at this low concentration. At higher FC concentrations (1 μM and higher), Arn1p cycles between the plasma membrane and endosomal compartments, where uptake of FC occurs. This pattern of Arn1p trafficking occurs only in the presence of FC and not in the presence of desferrioxamine B, a hydroxamate siderophore that is a transport substrate for Arn3p, but not for Arn1p. The FC transporter of Candida albicans, CaArn1p, also exhibits an altered pattern of intracellular localization in the presence of FC (Hu et al, 2002). The relocalization of Arn1p in response to low concentrations of FC suggests the presence of a specific, high-affinity receptor that can signal the presence of FC to Arn1p.
A clue to the identity of the FC receptor comes from kinetic studies of FC binding to Arn1p. These studies indicate that Arn1p contains two binding sites for FC, one site with an affinity similar to the KT for transport and a second site of much higher affinity (Moore et al, 2003). As no high-affinity binding sites for FC are detected in cells that do not express Arn transporters, these results suggest that the FC receptor is part of the Arn1p transporter itself. FC likely gains access to endosomal Arn1p through the highly dynamic process of fluid-phase endocytosis, as blocks in endocytosis inhibit the capacity of FC to induce plasma membrane relocalization of Arn1p (Moore et al, 2003). We have performed a structure–function analysis of Arn1p and here we report that mutations in predicted extracytosolic loops of Arn1p resulted in loss of FC uptake activity, loss of FC binding activity, and defects in the trafficking of Arn1p. These data indicated that Arn1p contains an FC receptor domain that influences the intracellular trafficking of the transporter. FC also enhanced the ubiquitination of Arn1p, a process that required the Rsp5p ubiquitin ligase.
Results
Mutagenesis of an extracytosolic domain unique to ARN transporters
Members of the ARN family of siderophore transporters have a predicted topology that consists of 14 transmembrane domains (TMDs), with the amino and carboxyl termini oriented toward the cytoplasm (Figure 1) (Goffeau et al, 1997). This predicted topology is similar to that derived from the crystal structures of GlpT and LacY (Abramson et al, 2003b; Huang et al, 2003), two bacterial members of the MFS that contain 12 TMDs. The region spanning the first 12 TMDs of Arn1p exhibited homolgy to other bacterial members of the MFS (data not shown), but the region spanning the 13th and 14th TMDs and the carboxyl terminus was unique to the ARN family of transporters. The extracytosolic loops of the MFS transporters are very short, both in the crystal structures of GlpT and LacY and in the predicted topology of the first 12 TMDs of Arn1p. The extracytosolic loop between the 13th and 14th TMDs of Arn1p was predicted to be larger, containing 44 amino-acid residues and a potential α-helical region, and we questioned whether this region constituted an FC binding domain. We tested the function of this and other domains within Arn1p by site-directed mutagenesis, followed by measurement of the capacity of the mutant alleles to transport FC into the cell, to bind FC, and to relocalize to the plasma membrane in the presence of FC.
Figure 1.
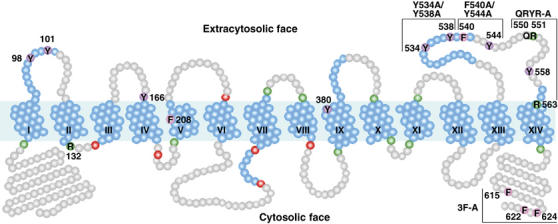
Predicted topology and mutagenesis of Arn1p. TMDs predicted from TMHMM and TMpred are numbered and shown coiled in blue, with additional potential helical regions predicted from HMMTOP and the structure of GlpT also in blue. Basic residues anchoring TMDs are shown in green, and acidic residues anchoring TMDs are shown in red. The region spanning the first 12 TMDs exhibits homology to other MFS family members. Mutated residues are numbered.
We explored the role of the last extracytosolic domain of Arn1p by making alanine substitutions for the tyrosine, phenylalanine, arginine, and glutamine residues of this domain. For comparison purposes, alanine substitutions were also made in tyrosine residues of predicted extracytosolic loops extending from the first 12 TMDs and in a cluster of three phenylalanine residues in the carboxyl-terminal tail of Arn1p. Amino-acid residues targeted for site-directed mutagenesis are shown numbered in Figure 1.
Effects of mutations in Arn1p on the uptake of FC
The capacity of Arn1p to move extracellular FC into the cytosol is dependent on its proper intracellular trafficking (Kim et al, 2002). We tested whether mutations in Arn1p that might inhibit FC binding and/or intracellular trafficking would also prevent cells from growing on or taking up FC. A strain from which all endogenous siderophore transport systems have been deleted was transformed with low-copy-number plasmids containing the wild-type and mutant alleles of ARN1. Three Arn1p mutants, Y380A, Y534A/Y538A, and QRYR-A, completely failed to grow on medium that contained FC as the sole source of iron, and two mutants, Y101A and R132A, exhibited reduced growth (Figure 2A). Quantitative measurements of FC transport activity were performed on the transformants (Figure 2B), and each of the Arn1p mutants, with the exception of F208A, exhibited defects in transport activity, with levels of less than 11% of wild-type Arn1p.
Figure 2.
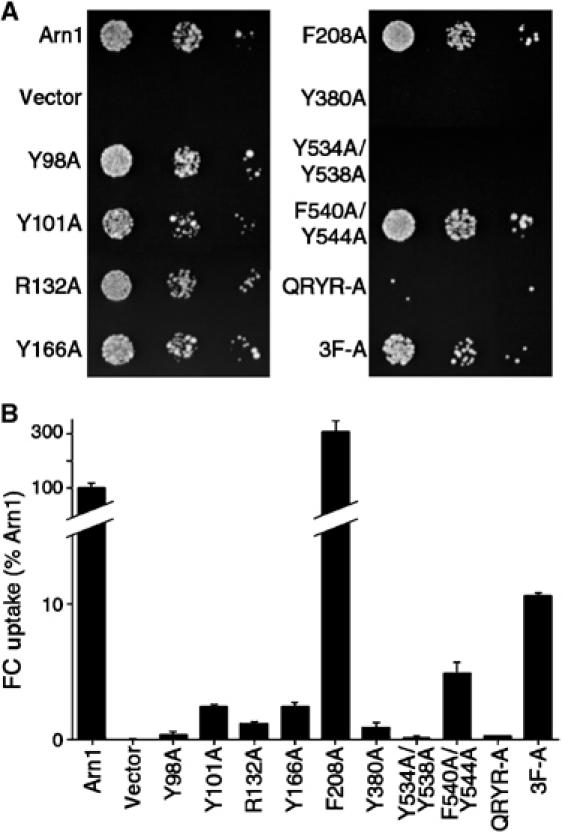
Impaired growth on FC and defects in uptake of FC due to mutations in Arn1p. A strain (CWY121) bearing deletions of all four Arn transporters and defective in ferrous iron uptake (fet3Δ) was transformed with low-copy-number plasmids containing the wild-type ARN1 allele, the indicated mutant arn1 alleles, or the empty vector. (A) Impaired growth on FC. Transformed strains were plated in serial 10-fold dilutions on medium containing 10 μM FC and incubated for 3 days at 30°C prior to imaging. (B) Defective uptake of FC. Transformed strains were grown in iron-poor medium to a density of 1 × 107 cells/ml, cells were harvested, and the uptake of [55Fe]FC was measured. FC uptake activity was normalized to the strain transformed with the wild-type allele. Assays were performed in duplicate, experiments were replicated three times, and a representative experiment is shown.
Many types of mutations, including amino-acid substitutions, can lead to misfolding of integral membrane proteins. Misfolded integral membrane proteins are typically retained in the endoplasmic reticulum (ER) and then degraded (Kostova and Wolf, 2003). We evaluated the mutant Arn1p transporters for proper folding by looking for retention in the ER. Only one mutant, R132A, exhibited the characteristic ‘signet ring' pattern typical of the ER by indirect immunofluorescence (data not shown). This arginine residue is highly conserved within the MFS (Goffeau et al, 1997) and may be involved in anchoring and orienting TMD II at the cytosolic face. The other mutant alleles of ARN1 that gave rise to properly folded proteins were further analyzed.
Amino-acid substitutions may also give rise to mutant proteins that are unstable and undergo rapid degradation. We evaluated the expression level of each of the mutant alleles of Arn1p by performing Western blot analysis on membranes collected from transformed cells (Figure 3). Each of the mutant proteins was readily detected and most were expressed at levels similar to that of wild-type Arn1p. Exceptions were mutants Y98A and Y166A, which were expressed at significantly lower levels than wild type. This lower level of expression could account for the reduced FC transport activity demonstrated for these mutants in Figure 2B. However, the observed level of expression was sufficient to allow for further analysis of FC binding and subcellular localization in these mutants, as these analyses are relatively unaffected by reduced levels of protein expression.
Figure 3.

Expression levels of wild-type and mutant alleles of Arn1p. The strain YPH499 was transformed with the wild-type and the mutant alleles of ARN1 in the low-copy number plasmid pRS316 and grown in iron-poor medium to induce expression of Arn1p. Cells were subjected to glass bead lysis and membranes were isolated. Identical quantities of membrane protein were analyzed by SDS–PAGE and Western blotting using antibodies against the HA epitope to detect alleles of Arn1p-HA. Molecular weight standards are indicated in kDa.
Loss of FC binding sites in Arn1p mutants
Although other MFS transporters, such as LacY and GlpT, contain a single binding site for their transport substrate (Abramson et al, 2003a), Arn1p contains two FC binding sites of differing affinities (Moore et al, 2003). Overexpression of Arn1p from the high-copy-number plasmid results in mislocalization of a portion of Arn1p to the plasma membrane, where it is available for FC binding. Thus, the number of FC binding sites on the surface of Arn1p and the dissociation constant for each of these sites can be determined. We have performed this analysis on each of the Arn1p mutants (Table I). Wild-type and mutant alleles of ARN1 were subcloned into a high-copy-number plasmid and used to transform a strain from which all four Arn transporters had been deleted. Transformed strains were grown in iron-poor medium to induce Arn1p expression, cells were harvested, and washed cells were incubated with [55Fe]FC at concentrations from 1.8 nM to 50 μM. Specifically bound [55Fe]FC was measured by scintillation counting and the data were examined by nonlinear regression analysis. Each mutant was analyzed to determine whether the data best fit a one- or two-site model, and the dissociation constant was determined for each site.
Table 1.
FC binding to wild-type and mutant Arn1p
| Mutation | Kd1 (nM) | 95% CI | Kd2 (μM) | 95% CI |
|---|---|---|---|---|
| Arn1 wild type | 7.5 | 5.9–9.2 | 1.2 | 0.8–1.5 |
| Y98A | 6.5 | 3.1–9.9 | 3.6 | 2.7–4.5 |
| Y101A | 11.5 | 7.0–16 | 3.4 | 2.7–4.1 |
| Y166A | 8.8 | 5.8–12 | 5.7 | 2.9–8.6 |
| Y380A | 82.4 | 51–113 | 6.1 | 5.4–6.8 |
| Y534A, Y538A (Y534A/Y538A) | ND | ND | 7.5 | 4.3–11 |
| F540A, Y544A (F540A/Y544A) | 2.5 | 0.5–4.5 | ND | ND |
| Q550A, R551A, Y558A, R563A (RQYR-A) | 53.8 | 38–69 | 7.4 | 2.2–13 |
| F615A, F622A, F624A (3F-A) | 3.5 | 2.6–4.3 | 1.5 | 0.2–2.8 |
| Kd1, dissociation constant, high-affinity site; Kd2, dissociation constant, low-affinity site; CI, confidence interval; ND, not detectable. | ||||
Alanine substitution of tyrosines 98, 101, and 166 was associated with little to no loss of FC binding affinity at the high-affinity site (KD1; Table I), and a three- to five-fold loss of affinity at the low-affinity site (KD2; Table I). These tyrosine residues are predicted to be located in the first and second extracytosolic loops linking TMDs I to II and III to IV, respectively. Substitution of tyrosine 380 to alanine was associated with an approximately 10-fold loss of FC binding affinity at the high-affinity site and five-fold loss at the low-affinity site. This residue is predicted to be located at the extracytosolic terminus of TMD IX. Alanine substitutions within the last extracytosolic loop between TMDs XIII and XIV were all associated with significant changes in the binding of FC. Mutation of both tyrosines 534 and 538 to alanines resulted in a complete loss of the high-affinity binding site with a greater than six-fold loss of affinity at the low-affinity site. In contrast, mutation of both phenylalanine 540 and tyrosine 544 to alanines resulted in no decrement in high-affinity FC binding, but a complete loss of the low-affinity binding site. Mutation of a cluster of conserved residues in the last extracytosolic domain, glutamine 550, arginine 551, tyrosine 558, and arginine 563, resulted in both a seven-fold loss of affinity at the high-affinity site and a six-fold loss at the low-affinity site. Finally, mutation of phenylalanine residues 615, 622, and 624, which are clustered in the cytosolic tail, resulted in no loss of affinity at either the high- or low-affinity binding sites. These data suggest that mutations within the last predicted extracytosolic loop of Arn1p affect both the high- and low-affinity FC binding sites, and that mutations in this loop have a greater effect on FC binding than do mutations in the first and second extracytosolic loops.
Plasma membrane relocalization of Arn1p with mutations in the first and second extracellular loops
We have suggested that Arn1p has a receptor-like domain that contains a high-affinity binding site for FC, and that the binding of FC to this domain affects the intracellular trafficking of the transporter. A prediction based on this hypothesis is that mutations in the receptor domain would not only affect FC binding, but also affect the intracellular trafficking of the mutant transporter. We tested this prediction by examining the intracellular localization of wild-type and mutant Arn1p, both before and after treatment with FC, using indirect immunofluorescence and fractionation and Western blotting of cellular membranes.
Cells transformed with low-copy-number plasmids containing the wild-type and mutant alleles of ARN1-HA were grown in iron-poor medium to mid-log phase, and then grown for an additional hour in the absence or presence of a low concentration of FC. Cells were then fixed and prepared for fluorescence microscopy (Figure 4A). In the absence of FC, wild-type Arn1p exhibited the expected punctate intracellular pattern of signal by fluorescence microscopy (Figure 4A, Arn1, −FC). When the cells expressing wild-type Arn1p were exposed to FC, the typical pattern of plasma membrane relocalization was observed: fluorescent signal was detected at both the periphery of the cells and in intracellular structures (Figure 4A, Arn1, +FC). Cells expressing the Y98A, Y101A, and Y166A mutant alleles also exhibited a predominately punctate, intracellular pattern of fluorescence in the absence of FC, and both a peripheral and intracellular pattern in the presence of FC. These results suggested that, similar to wild type, these mutant alleles of ARN1 were expressed in endosomes in the absence of FC, and that FC triggered their relocalization to the plasma membrane.
Figure 4.
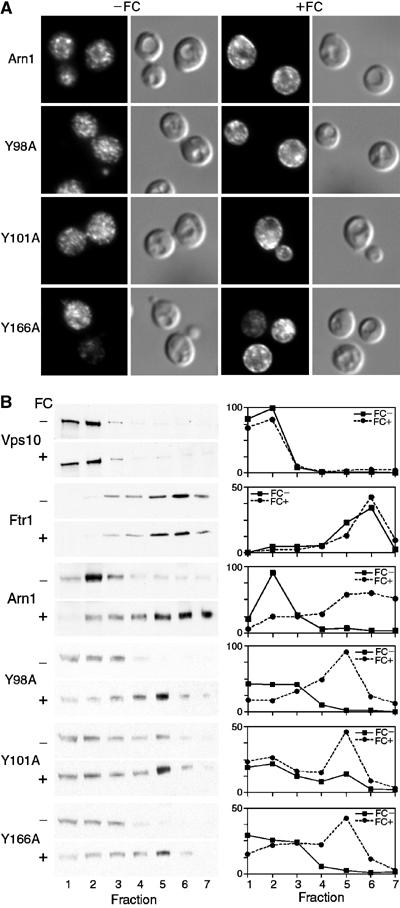
Relocalization to the plasma membrane of wild-type and mutant Arn1p after exposure to FC. (A) Plasma membrane relocalization by indirect immunofluorescence. The strain YPH499 was transformed with the wild-type and the indicated mutant alleles of Arn1p in the low-copy number plasmid pRS316 and grown in iron-poor medium to induce expression of Arn1p. Cells were divided and grown for an additional hour in the absence (−FC) or presence (+FC) of 100 nM FC prior to fixation and preparation for indirect immunofluorescence microscopy. Mouse monoclonal HA.11 was the primary antibody and Cy-3-conjugated donkey anti-mouse was the secondary antibody. Images are in pairs with fluorescence on the left and DIC on the right. (B) Redistribution of wild-type and mutant Arn1p to fractions containing plasma membranes after exposure to FC. The strain YPH499 FET3-HA FTR1-myc was transformed with the wild-type and the indicated mutant alleles of Arn1p in the low-copy number plasmid pRS316 and grown in the absence (FC−) or presence (FC+) of 100 nM FC as described above. Cells were harvested, membranes were collected and separated on sucrose step gradients, and the fractions were subjected to SDS–PAGE and Western blotting. Late Golgi vesicles were detected using an antibody directed against Vps10p, plasma membranes were detected using an anti-myc antibody directed against Ftr1p-myc, and HA-tagged Arn1p alleles were detected using anti-HA antibody. HA-tagged Fet3p (not shown) served as an internal loading control. Western blots are on the left, in pairs, and densitometric quantitation is shown on the right. Densitometry is reported in arbitrary units, which are proportional to the intensity of the bands on Western blots.
To confirm these observations, we analyzed the distribution of the Arn1p-containing membranes by equilibrium sedimentation in sucrose step gradients designed to separate intracellular vesicles from plasma membranes (Bagnat et al, 2001). Lysates were prepared from cells after growth in the absence or presence of FC. The cellular membranes were centrifuged on gradients, the gradients were fractionated, and Western blots were performed on the fractions (Figure 4B). Western blots using antibodies directed against resident proteins of the late Golgi (Vps10) and plasma membrane (Ftr1) demonstrated that intracellular vesicles were primarily detected in fractions 1–3, plasma membranes were primarily detected in fractions 5–7, and that FC had no effect on the distribution of the marker proteins. Wild-type Arn1p was detected primarily in fractions 1–3 in the absence of FC, but in the presence of FC, significant amounts of Arn1p were also detected in fractions 5–7. Membranes from cells expressing the Y98A, Y101A, and Y166A mutant alleles in the absence of FC were fractionated, and the mutant Arn1p was detected primarily in fractions containing intracellular vesicles. After cells were exposed to FC, mutant Arn1p transporters were also detected in fractions containing plasma membranes. An exception to this was mutant Y101A, in which a small amount of mutant protein was detected in plasma membrane fractions in the absence of FC, suggesting that this mutation was ‘leaky'. However, even Y101A demonstrated an increase in the amount of mutant Arn1p in plasma membrane fractions after treatment with FC. Taken together, these results indicated that mutations in tyrosine residues of the first and second extracytosolic loops did not impair either the high-affinity FC binding site or the capacity to relocalize to the plasma membrane in the presence of FC.
Impaired plasma membrane relocalization due to mutations within the last extracytosolic loop of Arn1p
Alanine substitutions of amino-acid residues in the last extracytosolic loop were associated with significant loss of FC binding at both the high- and low-affinity sites. We next examined the effects of these substitutions on the intracellular localization of mutant Arn1p in the absence and presence of FC (Figure 5). By indirect immunofluorescence, substitution of tyrosines 534 and 538 to alanine led to the detection of mutant Arn1p in both intracellular vesicles and at the periphery of the cell when FC was not present in the medium (Figure 5A). Addition of FC to the medium did not appreciably change the distribution of the Y534A/Y538A mutant. The F540A/Y544A mutant and the QRYR-A mutant exhibited similar patterns of localization by immunofluorescence. Both were detected in intracellular vesicles in the absence of FC and did not appreciably change localization with the addition of FC. We confirmed these observations using membrane fractionation and determined that although small amounts of Y534A/Y538A were located in plasma membrane fractions, the amount did not increase in the presence of FC (Figure 5B). Similarly, the F540A/Y544A mutant and the QRYR-A mutant were predominantly located in intracellular vesicle fractions in the absence of FC, and the amount of mutant protein in plasma membrane fractions did not change with the addition of FC. Together, these data indicated that mutations in the last predicted extracytosolic loop were associated with defects in FC binding and with impaired relocalization to the plasma membrane in the presence of FC.
Figure 5.
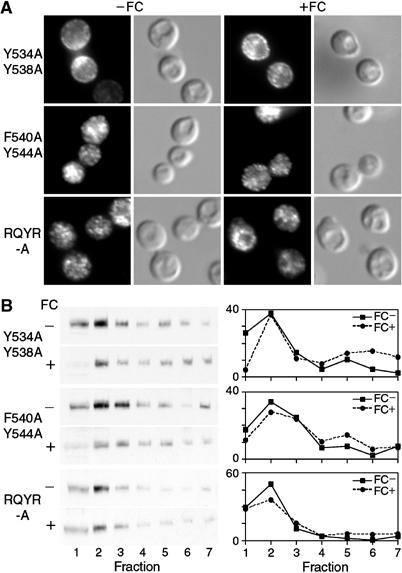
Loss of FC-induced plasma membrane relocalization in Arn1p mutants with substitutions in the last extracytosolic loop domain. Indirect immunofluorescence (A) and membrane fractionation (B) were performed as in Figure 4 using cells transformed with the indicated Arn1p alleles containing mutations in the last predicted extracytosolic loop between TMDs XIII and XIV.
Effects of alanine substitutions in TMD IX and the carboxyl terminus
To examine the effects of a mutation within a TMD, we made a single alanine substitution within predicted TMD IX. Tyrosine 380 is located near the predicted extracytosolic terminus of TMD IX and is highly conserved among all fungal siderophore transporters (Yun et al, 2000a). Substitution of tyrosine 380 to alanine led to defects in both the high- and low-affinity FC binding sites (see Table II) and we examined Y380A for defects in trafficking. By indirect immunofluorescence, Y380A was located both at the periphery of the cell and in intracellular vesicles in the absence of FC, and the intensity of the peripheral signal did not increase with the addition of FC (Figure 6A). Similar results were obtained using membrane fractionation, as Y380A was detected in both vesicular and plasma membrane fractions in the absence of FC, with no appreciable change in the presence of FC (Figure 6B). These results indicated that mutation of a conserved tyrosine within a TMD was associated with defects in FC binding and with trafficking to the plasma membrane in the absence of the transport substrate.
Table 2.
Oligonucleotides used in the site-directed mutagenesis of Arn1p
| Mutation | Oligonucleotide sequence |
|---|---|
| Y98A | 5′-CGGTAACATCCGTTATATTGCCACTGGTTACG CCACATCATCCTATAGTGAGC-3′ |
| Y101A | 5′-CGGTAACATCCGTTATATTTACACTGGTGCCG CCACATCATCCTATAGTGAGC-3′ |
| R132A | 5′-GCAAATCATATACGCTGCGTTGTCTGATGTGT TTGG-3′ |
| Y166A | 5′-CACAAGCTTATGATGTTCAAAGAGCCGCTGCA GGTGCCATTTTCTACAATGC-3′ |
| F208A | 5′-GTCCCGACTTGGCCTGCTATCATCAACACATG GATTGC-3′ |
| Y380A | 5′-CTATATGGCCGCTGATGCTTTGTACACGGTTA TGATTGTTGCCGTC-3′ |
| Y534A/Y538A | 5′-GGTGATGTTGCTCTAGCCACAACTGCCGCCGA ATCTCCAGCTACTTTCATTGAAACCTATACTTGG- 3′ |
| F540A/Y544A | 5′-GCCTACGAATCTCCATATACTGCCATTGAAAC CGCTACTTGGGGTACCCCACAAAGGAATGCC-3′ |
| QRYR-A | 5′-CTTGGGGTACCCCAGCAGCGAATGCCTAATGA ACGCTGCCAAATACGTTCAAGCATTGGAAACTATC G-3′ |
| 3F-A | 5′-CCAATTTTGGATTGGGCTGAAAAACTTCCATC AAAAGCCACTGCTAAACGGGAAGAACAAAAGCTGG -3′ |
| Nucleotide substitutions resulting in alanine mutations are underlined. | |
Figure 6.
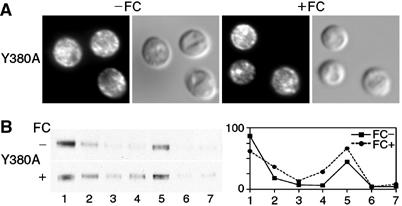
Localization of mutant allele Y380A to the plasma membrane in the absence of FC. Indirect immunofluorescence (A) and membrane fractionation (B) were performed as in Figure 4 using cells transformed with the Arn1p mutant Y380A, which contained a mutation in the predicted extracytosolic terminus of TMD IX.
To test the role of the cytosolic terminus in the trafficking of Arn1p, we examined mutant 3F-A, in which a cluster of three phenylalanines in the carboxyl terminus was substituted with alanines, for its localization in the presence and absence of FC. By both indirect immunofluorescence and membrane fractionation, 3F-A exhibited a predominantly intracellular location in both the presence and absence of FC (Figure 7A and B). Given that FC binding was normal in this mutant (see Table II), these results suggested that binding of FC at the high-affinity site is not sufficient to induce relocalization to the plasma membrane, but that structural elements in the cytosolic terminus were also involved in the relocalization process.
Figure 7.
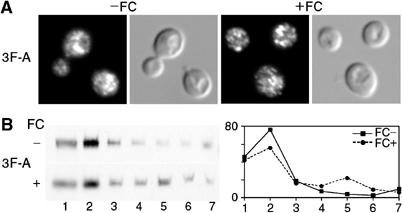
Impaired FC-induced relocalization to the plasma membrane in an Arn1p mutant with substitutions in the carboxyl-terminal cytosolic tail. Indirect immunofluorescence (A) and membrane fractionation (B) were performed as in Figure 4 using cells transformed with the Arn1p mutant 3F-A containing substitutions of three phenylalanine residues in the predicted carboxyl-terminal cytosolic tail.
FC-induced ubiquitination of Arn1p
FC binding was associated with alterations in the intracellular trafficking of Arn1p and we questioned whether FC could also affect the ubiquitination of Arn1p. Ubiquitination of integral membrane proteins has been demonstrated to direct their intracellular sorting to multivesicular bodies destined for degradation in the vacuole, and ubiquitination is especially important at the internalization step in the endocytosis of plasma membrane proteins (Dupre et al, 2001; Hicke, 2001). We examined the ubiquitination of wild-type Arn1p by expressing Arn1-HA in cells that also expressed a myc-epitope-tagged version of ubiquitin (Ellison and Hochstrasser, 1991), by immunoprecipitating Arn1p, and by probing the immunoprecipitated Arn1p for myc-tagged ubiquitin. We found that in the absence of FC, higher molecular weight forms of ubiquitinated Arn1p were readily detectable (Figure 8F). The addition of a low concentration (20 nM) of FC did not appreciably change the amount of ubiquitinated Arn1p, although the total amount of Arn1p did increase (Figure 8A and B). However, addition of FC at a higher concentration (10 μM) led to a large increase in the amount of ubiquitinated Arn1p (Figure 8F) without an appreciable change in the total amount of Arn1p (Figure 8A and B). The ubiquitinated proteins were not detected in immunoprecipitates from cells that did not express tagged Arn1p, thereby confirming that the ubiquitinated species represented modified Arn1p. These data indicated that a high concentration of FC, which bound to the Arn1p transport site and led to internalization of the transporter and uptake of FC, also led to an increase in ubquitinated forms of Arn1p. We tested whether the ubiquitin ligase encoded by Rsp5p was required for ubiquitination of Arn1p, as Rsp5p is required for the ubiquitination of other integral membrane proteins (Dupre et al, 2001). Ubiquitinated forms of Arn1p were greatly reduced in a strain carrying a temperature-sensitive mutation in RSP5 (Figure 8I), although total levels of Arn1p were substantially increased in the rsp5-1 mutant strain. These results indicated that Rsp5p was required for the ubiquitination of Arn1p, and that ubiquitination of Arn1p may shorten its half-life.
Figure 8.
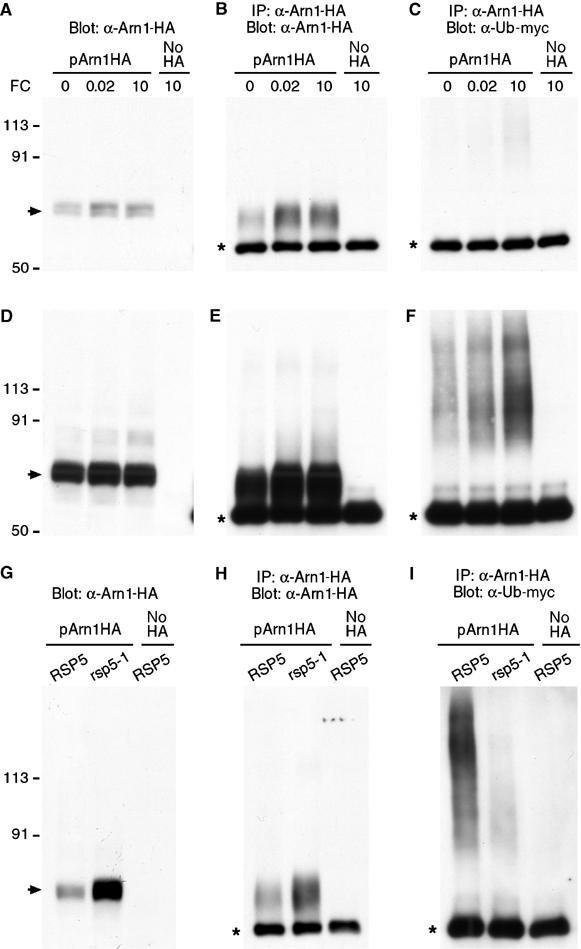
FC-induced, Rsp5-dependent ubiquitination of Arn1p. Strains YPH499 (A–F), LHY23 (G–I, rsp5-1), and LHY291 (G–I, RSP5) were transformed with either pMET Arn1-HA (pArn1HA) or the empty vector pRS416 (no HA) and Yep105, which expresses myc-tagged ubiquitin from the CUP1 promoter. Transformed strains were grown in synthetic complete medium without methionine and with 100 μM copper to induce expression of Arn1-HA and myc-ubiquitin, respectively. FC (μM) was added at the indicated concentrations for 2 h (A–F) or cultures were shifted to 37°C with the addition of 5 μM FC for 2 h (G–I). Then, an equivalent number of cells was harvested from each culture. Cells were lysed and an aliquot was subjected to Western blotting with anti-HA antibody (A, D, G). Membranes were purified from the remaining lysate, detergent solubilized, and subjected to immunoprecipitation with anti-HA antibody. Immunoprecipitates were then subjected to Western blotting using anti-HA antibodies to detect Arn1-HA (B, E, H) or anti-myc antibodies to detect myc-ubiquitin (C, F, I). Panels D–F are an overexposure of blots depicted in panels A–C to more clearly demonstrate ubiquitinated species. Panel I is approximately five-fold overexposed with respect to panel H. The arrow indicates unmodified Arn1-HA. The asterisk indicates immunoglobulin heavy chain.
Discussion
The receptor domain of Arn1p
Alanine substitution of conserved residues within the last extracytosolic loop of Arn1p resulted in the loss of FC transport activity, the loss of low- and high-affinity FC binding activities, and a failure to relocalize to the plasma membrane in the presence of FC. The last extracytosolic loop of Arn1p contains five aromatic amino-acid residues that are conserved among the Arn transporters with specificity for FC. These residues were selected because the crystal structures of the Escherichia coli FC receptor, FhuA, and the FC binding protein, FhuD, indicate that aromatic amino acids lining the FC binding pockets contribute to the important electrostatic interactions that ‘extract' FC from the surrounding solvent (Ferguson et al, 1998; Clarke et al, 2000). Arginine and glutamine residues also contribute to FC binding, and these residues were mutated as well. Nine of the 10 alanine substitution mutants examined in these studies exhibited defects in activity, a somewhat surprising result given that only six of the amino-acid side chains of LacY are irreplaceable for transport function (Abramson et al, 2003a). Here, mutations were made in pairs and groups, with the intention of disrupting the potential FC binding site rather than identifying individual residues that interact with FC. Our findings support a model in which the last extracytosolic domain of Arn1p contains or affects the high- and low-affinity FC binding sites. The binding of FC to this domain alters the localization of the transporter, directing it to the plasma membrane. Alanine substitutions in other regions of Arn1p were associated with loss of FC transport activity and variable defects in FC binding and trafficking. The magnitude of the binding defects in the Arn1p mutants did not consistently reflect the magnitude of the transport defects in the corresponding mutant. This likely reflects differences in the capacity of the mutants to undergo the major conformational changes that are required during a transport cycle. Mutations that affect FC binding at either the high- or low-affinity sites may also affect this conformational change to varying extents. Thus, Arn1p represents a bifunctional protein in which transporter and receptor activities are combined.
Although the structure of Arn1p and the mechanism of FC transport are unknown, the structure and mechanism of action of two related MFS transporters, GlpT and LacY, are known in great detail (Abramson et al, 2003b; Huang et al, 2003). The amino-acid homology between GlpT and LacY is limited, yet their crystal structures and predicted mechanism of action are very similar, and other MFS transporters, such as Arn1p, are likely to exhibit similar structures and mechanisms. Both GlpT and LacY exhibit dyadic symmetry, as they can be divided into similar amino- and carboxyl-terminal halves, each containing a bundle of six membrane-spanning α-helices. The two bundles come together to form a central pore that extends from the cytosolic face of the protein to a point approximately midway across the membrane, and a single substrate binding site is buried deep within this pore. Although structures have only been determined for transporters in this inward-open conformation, an analogous outward-open conformation exists. Substrate may be translocated across the membrane by alternation between the outward-open and inward-open conformations, and it is the binding of substrate that triggers the conformational change. The dyadic symmetry of GlpT and LacY is very likely retained in the first 12 TMDs of Arn1p, but the last two TMDs, the last extracytosolic loop, and the carboxyl terminus form a unique structural domain. The results presented here support a model in which this domain coordinates the intracellular trafficking of Arn1p with the presence of its transport substrate, a feature of the ARN transporters that is not shared by the prokaryotic members of the MFS.
Proposed mechanism for FC transport
Arn1p is present in the endosome when neither the high-affinity site (the FC receptor site, in blue) nor the low-affinity site (the FC transport site, in green) contains bound FC (in red) (see model, Figure 9A). The transporter may be in the inward-open conformation (shown) or the outward-open conformation. When FC binds at the receptor site (Figure 9B), a conformational change in the carboxyl terminus occurs (Figure 9C) that allows Arn1p to relocalize to the plasma membrane (Figure 9D). This FC-induced conformational change may also stabilize the outward-open conformation of Arn1p and thereby allow for proton binding. The movement of a proton down its concentration gradient provides the energy to transport substrates in many MFS transporters (Goffeau et al, 1997). LacY is thought to bind a proton in the absence of oligosaccharide substrates (Abramson et al, 2003a), and proton binding in the endosome or on the plasma membrane may precede FC binding at the transport site of Arn1p.
Figure 9.
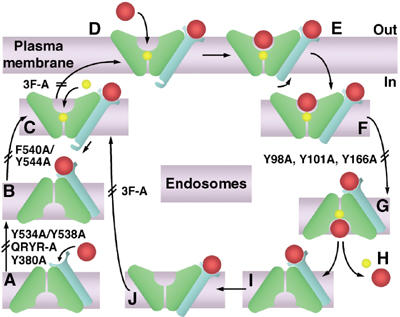
Proposed mechanism for trafficking of Arn1p and uptake of FC. Apo-Arn1p is sorted to recycling endosomes (A). FC enters the endosome through fluid-phase endocytosis and, upon binding to the receptor site (B), triggers a conformational change in the cytosolic tail and stabilization of the outward-open conformation (C), then proton binding and relocalization to the plasma membrane (D). Upon binding of a second molecule of FC in the transport site (E), Arn1p undergoes endocytosis (F), which triggers a shift to the inward-open conformation (G), wherein FC and the proton are released (H). FC bound to the receptor site (I) can then again trigger stabilization of the outward-open conformation (J) and conformational change in the cytosolic tail (C). The transport domains (green) and receptor domain (blue) of Arn1p, iron-bound FC (red), and proton (yellow) are shown. Double bars mark the steps that were inhibited by the indicated mutations.
Mutant Y534A/Y538A exhibited a loss of the FC receptor site, while partially maintaining the FC transport site, suggesting that either this mutation stabilized the outward-open conformation in the absence of FC binding, or that FC binding was not necessary for Arn1p to adopt the outward-open conformation. This mutation did, however, prevent both FC receptor binding and plasma membrane relocalization. Mutant QRYR-A may have had an effect similar to that of Y534A/Y538A, with defects in both the FC receptor site and relocalization. Mutant F540A/Y544A exhibited no defect in the receptor site, but the transport site was not detectable and this mutant did not relocalize to the plasma membrane with FC. The F540A/Y544A mutation may have resulted in the stabilization of the inward-open conformation, wherein FC binding did not elicit the conformational change in the carboxyl terminus and the transport site remained inaccessible to external FC.
Binding of FC to the receptor site was necessary, but not sufficient for the FC-induced relocalization of Arn1p to the plasma membrane. Mutation of phenylalanine residues in the carboxyl terminus had no effect on the binding of FC to the receptor or transport sites, yet mutant 3F-A failed to relocalize to the plasma membrane upon binding FC. Sorting of membrane-bound cargo proteins within the secretory pathway is accomplished through a variety of mechanisms, one of which is through the interaction of clathrin adaptor proteins (such as the heterotetrameric AP complexes and the monomeric GGAs) with short peptide sequences in the cytosolic termini of cargo proteins (Aridor and Traub, 2002; Bonifacino, 2004). FC binding to the receptor site of Arn1p may result in a conformational change in the carboxyl terminus that exposes one of these targeting sequences and allows Arn1p to be sorted into exocytic vesicles destined for the plasma membrane.
When Arn1p is located on the plasma membrane in the outward-open conformation, Arn1p can bind a second molecule of FC at the transport site (Figure 9E). Binding of FC at the transport site triggers the internalization of Arn1p (Figure 9F), perhaps through another conformational change at the carboxyl terminus and/or through a post-translational modification such as ubiquitination or phosphorylation. We found that Arn1p was ubiquitinated in an Rsp5p-dependent manner. The increase in ubiquitination at high concentrations of FC suggests that ubiquitin may be involved in the FC-induced internalization. In yeast, uracil binding can trigger both the internalization of the Fur4p uracil permease from the plasma membrane and the direct sorting of Fur4p from the Golgi to the vacuole (Blondel et al, 2004). Ubiquitination via the Rsp5p ubiquitin ligase is required for both processes, and studies are underway to determine the precise role of ubiquitin in Arn1p trafficking. Once Arn1p reaches the endosome with FC bound at both sites, the transporter shifts to the inward-open configuration (Figure 9G), with the release of both FC and proton (Figure 9H). The loss of FC uptake activity in the Y98A, Y101A, and Y166A mutants may be due to defects in this shift to the inward-open conformation.
FC uptake through Arn1p is inhibited when the endocytosis of plasma membrane proteins is blocked (Kim et al, 2002); this inhibition may be due to a requirement for the lipid environment of the endosome rather than that of the plasma membrane to facilitate the change to the inward-open conformation. The plasma membrane of yeast is rich in sterols and sphingolipids, while intracellular membranes are rich in phospholipids. Numerous integral membrane proteins exhibit requirements for specific lipids or sterols in order to maintain full activity (Opekarova and Tanner, 2003). Interestingly, LacY requires the phospholipid phosphatidyl ethanolamine for activity. Alternatively, the requirement for endocytosis prior to FC translocation may reflect a requirement for the proton-rich environment of the acidified endosome, which might suggest that proton binding occurs after Arn1p is internalized.
Regulation of transporter activity through intracellular trafficking
The controlled intracellular trafficking of transporters constitutes an important means of regulating transporter activity. Numerous yeast transporters, such as the zinc transporter Zrt1 and the uracil transporter Fur4, undergo ubiquitin-mediated endocytosis and degradation in the vacuole in response to elevated levels of their respective substrates (Gitan and Eide, 2000; Blondel et al, 2004). In yeast, the substrate-induced recruitment of a transporter to the plasma membrane may be unique to the ARN transporters, but the regulated exocytosis of mammalian transporters is well recognized. The copper transporter defective in Menkes disease is recruited from the trans-Golgi network to the plasma membrane in the presence of elevated copper levels, and the related Wilson disease copper transporter also undergoes copper-induced alterations in trafficking (Petris et al, 1996; Hung et al, 1997). A detailed understanding of these processes may permit the pharmacological modulation of transport processes in the future.
Materials and methods
Yeast strains and media
All strains were derived from YPH499 (MATa ura3-52 lys2-801 ade2-101 trp1-Δ63 his3-Δ200 leu2-Δ1). CWY101, the yeast strain in which all four ARN transporters have been deleted (MATa ura3-52 lys2-801 ade2-101 trp1-Δ63 his3-Δ200 leu2-Δ1 arn1Δ∷HISG arn2Δ∷HISG arn3Δ∷HISG arn4Δ∷HISG) (Yun et al, 2001), was used for FC binding assays. CWY121, the congenic strain from which all four ARN transporters and FET3 have been deleted (MATa ura3-52 lys2-801 ade2-101 trp1-Δ63 his3-Δ200 leu2-Δ1 arn1Δ∷HISG arn2Δ∷HISG arn3Δ∷HISG arn4Δ∷HISG fet3∷HIS3) (Yun et al, 2000b), was used for FC uptake assays and plate assays. The strain YPH499 FET3-HA FTR1-myc, which constitutively overexpresses integrated copies of HA-tagged FET3 and myc-tagged FTR1 under the control of the phosphoglycerate kinase promoter (Yun et al, 2000a), was used in the fractionation of organelles and Western blotting. The congenic parent strain YPH499 was used for immunofluorescence. Yeast strains were grown at 30°C under agitation in YPD as the rich medium and in complete synthetic defined medium. Iron-poor medium contained 10 μM ferrous ammonium sulfate and 1 mM ferrozine. For ubiquitination studies, the strains CWY101 and LHY23 (MATa ura3 leu2 trp1 bar1 GAL rsp5-1) and the congenic RSP5 parent LHY291 (Dunn and Hicke, 2001), and ATCC strain 15511 (MATalpha his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 ydr069CΔ∷KanMX) and the congenic parent BY4742 were transformed with pMet Arn1-HA (Kim et al, 2002) and Yep105, a high-copy-number plasmid that expresses myc-tagged ubiquitin from the CUP1 promoter (Ellison and Hochstrasser, 1991). Transformed strains were grown in synthetic complete medium without methionine and with 100 μM copper to induce expression of Arn1-HA and myc-ubiquitin, respectively.
Site-directed mutagenesis
Site-directed mutagenesis was performed using the QuikChange XL Site-Directed Mutagenesis Kit according to the manufacturer's instructions (Stratagene). The entire ARN1 gene carrying a carboxyl-terminal, triple-HA tag was subcloned into the single-copy plasmid pRS316 (Sikorski and Hieter, 1989). Sequences of the synthetic oligonucleotides are given in Table II. PCR-amplified DNA was treated with DpnI to remove the nonmutated parental DNA template and used to transform E. coli XL10-gold strain. The promoters and coding regions of mutant proteins were sequenced. Each of the mutant alleles of Arn1p was subcloned into the multicopy plasmid pRS426 (Sikorski and Hieter, 1989), which were used for FC binding affinity assays.
FC-iron uptake and plate assays
FC-iron uptake and plate assays were performed as described previously (Yun et al, 2000a) in cells transformed with the wild-type or mutant alleles of ARN1 in the single-copy plasmid. For the plate assay, modified synthetic complete media were used in which copper and iron were omitted and 1 μM copper sulfate and 100 μM bathophenanthroline sulfonate (BPS) were added. Cells were suspended in water at 2 × 106 cells/ml, plated in serial 10-fold dilutions on solid media containing 10 μM FC, and incubated at 30°C for 3 days prior to imaging. For the uptake assay, [55Fe]FC at 1.0 μM was added to cells for 20 min.
FC binding assays
FC binding assays were performed as described (Moore et al, 2003) with the following modifications. The wild-type or mutant alleles of ARN1 in the multicopy plasmid were used to transform CWY101. Cells were resuspended in ice-cold assay buffer (5% glucose, 1 mM BPS, 20 mM sodium azide, 20 mM potassium fluoride, 50 mM sodium citrate, pH 6.5) at a concentration of 2 × 108 cells/ml. Cells were incubated with [55Fe]FC in serial 2.2-fold dilutions, then washed with 8 ml of assay buffer, and the [55Fe]FC retained on the cells was measured by scintillation counting. Assays were performed in triplicate, and the experiment was replicated two times. Data from a representative experiment are shown. Nonlinear regression analysis was performed using PRISM version 3.0 for the Macintosh (GraphPad Software, San Diego).
Fluorescence microscopy
The strain YPH499 was transformed with the wild-type and mutant alleles of ARN1 in the low-copy-number plasmid. Transformed strains were grown in iron-poor medium to mid-log phase and 100 nM FC was added for the final hour of culture. Cells were prepared for indirect immunofluorescence microscopy as described previously (Stearman et al, 1996; Yun et al, 2000a).
Subcellular organelle fractionation, Western blotting, and immunoprecipitation
Fractionation of membranes was performed essentially as described previously (Bagnat et al, 2001) with modifications. Briefly, 2 × 108 cells were subjected to glass bead lysis in buffer B (25 mM Tris pH 8.0, 5 mM EDTA), with protease inhibitors and 1 mM dithiothreitol. Lysates were centrifuged at 500 g for 5 min. For quantitative Western blotting, the supernatant was collected and membranes were pelleted by centrifugation at 14 000 g for 10 min. Membranes were resuspended in buffer B and the protein concentration was determined. For density gradient fractionation, the 500 g supernatant was collected and glycerol added to a final concentration of 20%. Samples (200 μl) were layered on top of sucrose step gradients (0.4 ml of 57% and 0.9 ml of 43% in buffer B). After centrifugation for 2 h at 100 000 g, the top 200 μl (cytoplasmic fraction) was discarded and seven 180 μl fractions were collected from top to bottom. SDS–PAGE and Western blotting were performed as described previously (Yun et al, 2000a). Immunoprecipitations were carried out as described (Philpott et al, 1998) with the following modifications. Membranes were collected from the 500 g supernatant by centrifugation at 100 000 g for 60 min at 4°C. Membranes were solubilized in lysis buffer containing 1% dodecylmaltoside (Anatrace), 50 mM Tris, pH 7.5, 0.3 M NaCl, 5 mM EDTA, 1 mM DTT, and protease inhibitors. Lysates were incubated with 1 μl of anti-HA antibody and immune complexes precipitated with protein G-Sepharose beads. Monoclonal antibody 9E10 was used at a dilution of 1:5000 for Western blotting to detect myc-tagged immunoprecipitates.
Acknowledgments
We thank Jerry Kaplan for critical reading of this manuscript and Susan Buchanan for helpful discussions. We also thank David Eide and Linda Hicke for their generous gifts of plasmids and strains.
References
- Abramson J, Smirnova I, Kasho V, Verner G, Iwata S, Kaback HR (2003a) The lactose permease of Escherichia coli: overall structure, the sugar-binding site and the alternating access model for transport. FEBS Lett 555: 96–101 [DOI] [PubMed] [Google Scholar]
- Abramson J, Smirnova I, Kasho V, Verner G, Kaback HR, Iwata S (2003b) Structure and mechanism of the lactose permease of Escherichia coli. Science 301: 610–615 [DOI] [PubMed] [Google Scholar]
- Aridor M, Traub LM (2002) Cargo selection in vesicular transport: the making and breaking of a coat. Traffic 3: 537–546 [DOI] [PubMed] [Google Scholar]
- Askwith C, Eide D, Van Ho A, Bernard PS, Li L, Davis-Kaplan S, Sipe DM, Kaplan J (1994) The FET3 gene of S. cerevisiae encodes a multicopper oxidase required for ferrous iron uptake. Cell 76: 403–410 [DOI] [PubMed] [Google Scholar]
- Bagnat M, Chang A, Simons K (2001) Plasma membrane proton ATPase pma1p requires raft association for surface delivery in yeast. Mol Biol Cell 12: 4129–4138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blondel MO, Morvan J, Dupre S, Urban-Grimal D, Haguenauer-Tsapis R, Volland C (2004) Direct sorting of the yeast uracil permease to the endosomal system is controlled by uracil binding and Rsp5p-dependent ubiquitylation. Mol Biol Cell 15: 883–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino JS (2004) The GGA proteins: adaptors on the move. Nat Rev Mol Cell Biol 5: 23–32 [DOI] [PubMed] [Google Scholar]
- Byers BR, Arceneaux JEL (1998) Microbial iron transport: iron acquisition by pathogenic microorganisms. In Iron Transport and Storage in Microorganisms, Plants, and Animals, Sigel A, Sigel H (eds) Vol. 35, pp 37–66. New York: Marcel Dekker [PubMed] [Google Scholar]
- Clarke TE, Ku SY, Dougan DR, Vogel HJ, Tari LW (2000) The structure of the ferric siderophore binding protein FhuD complexed with gallichrome. Nat Struct Biol 7: 287–291 [DOI] [PubMed] [Google Scholar]
- Dancis A, Klausner RD, Hinnebusch AG, Barriocanal JG (1990) Genetic evidence that ferric reductase is required for iron uptake in Saccharomyces cerevisiae. Mol Cell Biol 10: 2294–2301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn R, Hicke L (2001) Domains of the Rsp5 ubiquitin-protein ligase required for receptor-mediated and fluid-phase endocytosis. Mol Biol Cell 12: 421–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupre S, Volland C, Haguenauer-Tsapis R (2001) Membrane transport: ubiquitylation in endosomal sorting. Curr Biol 11: R932–R934 [DOI] [PubMed] [Google Scholar]
- Ellison MJ, Hochstrasser M (1991) Epitope-tagged ubiquitin. A new probe for analyzing ubiquitin function. J Biol Chem 266: 21150–21157 [PubMed] [Google Scholar]
- Ferguson AD, Hofmann E, Coulton JW, Diederichs K, Welte W (1998) Siderophore-mediated iron transport: crystal structure of FhuA with bound lipopolysaccharide. Science 282: 2215–2220 [DOI] [PubMed] [Google Scholar]
- Georgatsou E, Alexandraki D (1994) Two distinctly regulated genes are required for ferric reduction, the first step of iron uptake in Saccharomyces cerevisiae. Mol Cell Biol 14: 3065–3073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitan RS, Eide DJ (2000) Zinc-regulated ubiquitin conjugation signals endocytosis of the yeast ZRT1 zinc transporter. Biochem J 346 (Part 2): 329–336 [PMC free article] [PubMed] [Google Scholar]
- Goffeau A, Park J, Paulsen IT, Jonniaux JL, Dinh T, Mordant P, Saier MH Jr (1997) Multidrug-resistant transport proteins in yeast: complete inventory and phylogenetic characterization of yeast open reading frames with the major facilitator superfamily. Yeast 13: 43–54 [DOI] [PubMed] [Google Scholar]
- Heymann P, Ernst JF, Winkelmann G (1999) Identification of a fungal triacetylfusarinine C siderophore transport gene (TAF1) in Saccharomyces cerevisiae as a member of the major facilitator superfamily. Biometals 12: 301–306 [DOI] [PubMed] [Google Scholar]
- Heymann P, Ernst JF, Winkelmann G (2000a) A gene of the major facilitator superfamily encodes a transporter for enterobactin (Enb1p) in Saccharomyces cerevisiae. Biometals 13: 65–72 [DOI] [PubMed] [Google Scholar]
- Heymann P, Ernst JF, Winkelmann G (2000b) Identification and substrate specificity of a ferrichrome-type siderophore transporter (Arn1p) in Saccharomyces cerevisiae. FEMS Microbiol Lett 186: 221–227 [DOI] [PubMed] [Google Scholar]
- Hicke L (2001) Protein regulation by monoubiquitin. Nat Rev Mol Cell Biol 2: 195–201 [DOI] [PubMed] [Google Scholar]
- Hu CJ, Bai C, Zheng XD, Wang YM, Wang Y (2002) Characterization and functional analysis of the siderophore-iron transporter CaArn1p in Candida albicans. J Biol Chem 277: 30598–30605 [DOI] [PubMed] [Google Scholar]
- Huang Y, Lemieux MJ, Song J, Auer M, Wang DN (2003) Structure and mechanism of the glycerol-3-phosphate transporter from Escherichia coli. Science 301: 616–620 [DOI] [PubMed] [Google Scholar]
- Hung IH, Suzuki M, Yamaguchi Y, Yuan DS, Klausner RD, Gitlin JD (1997) Biochemical characterization of the Wilson disease protein and functional expression in the yeast Saccharomyces cerevisiae. J Biol Chem 272: 21461–21466 [DOI] [PubMed] [Google Scholar]
- Kim Y, Yun CW, Philpott CC (2002) Ferrichrome induces endosome to plasma membrane cycling of the ferrichrome transporter, Arn1p, in Saccharomyces cerevisiae. EMBO J 21: 3632–3642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostova Z, Wolf DH (2003) For whom the bell tolls: protein quality control of the endoplasmic reticulum and the ubiquitin–proteasome connection. EMBO J 22: 2309–2317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesuisse E, Raguzzi F, Crichton RR (1987) Iron uptake by the yeast Saccharomyces cerevisiae: involvement of a reduction step. J Gen Microbiol 133: 3229–3236 [DOI] [PubMed] [Google Scholar]
- Lesuisse E, Simon-Casteras M, Labbe P (1998) Siderophore-mediated iron uptake in Saccharomyces cerevisiae: the SIT1 gene encodes a ferrioxamine B permease that belongs to the major facilitator superfamily. Microbiology 144: 3455–3462 [DOI] [PubMed] [Google Scholar]
- Moore RE, Kim Y, Philpott CC (2003) The mechanism of ferrichrome transport through Arn1p and its metabolism in Saccharomyces cerevisiae. Proc Natl Acad Sci USA 100: 5664–5669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neilands JB (1995) Siderophores: structure and function of microbial iron transport compounds. J Biol Chem 270: 26723–26726 [DOI] [PubMed] [Google Scholar]
- Opekarova M, Tanner W (2003) Specific lipid requirements of membrane proteins—a putative bottleneck in heterologous expression. Biochim Biophys Acta 1610: 11–22 [DOI] [PubMed] [Google Scholar]
- Petris MJ, Mercer JF, Culvenor JG, Lockhart P, Gleeson PA, Camakaris J (1996) Ligand-regulated transport of the Menkes copper P-type ATPase efflux pump from the Golgi apparatus to the plasma membrane: a novel mechanism of regulated trafficking. EMBO J 15: 6084–6095 [PMC free article] [PubMed] [Google Scholar]
- Philpott CC, Rashford J, Yamaguchi-Iwai Y, Rouault TA, Dancis A, Klausner RD (1998) Cell-cycle arrest and inhibition of G1 cyclin translation by iron in AFT1-1(up) yeast. EMBO J 17: 5026–5036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P (1989) A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122: 19–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stearman R, Yuan DS, Yamaguchi-Iwai Y, Klausner RD, Dancis A (1996) A permease-oxidase complex involved in high-affinity iron uptake in yeast (see comments). Science 271: 1552–1557 [DOI] [PubMed] [Google Scholar]
- Yun CW, Bauler M, Moore RE, Klebba PE, Philpott CC (2001) The role of the FRE family of plasma membrane reductases in the uptake of siderophore-iron in Saccharomyces cerevisiae. J Biol Chem 276: 10218–10223 [DOI] [PubMed] [Google Scholar]
- Yun CW, Ferea T, Rashford J, Ardon O, Brown PO, Botstein D, Kaplan J, Philpott CC (2000a) Desferrioxamine-mediated iron uptake in Saccharomyces cerevisiae. Evidence for two pathways of iron uptake. J Biol Chem 275: 10709–10715 [DOI] [PubMed] [Google Scholar]
- Yun CW, Tiedeman JS, Moore RE, Philpott CC (2000b) Siderophore-iron uptake in Saccharomyces cerevisiae. Identification of ferrichrome and fusarinine transporters. J Biol Chem 275: 16354–16359 [DOI] [PubMed] [Google Scholar]