

Abstract
To monitor the structural integrity of therapeutic proteins, hydrogen–deuterium exchange mass spectrometry (HDX-MS) is increasingly utilized in the pharmaceutical industry. The successful outcome of HDX-MS analyses depends on the sample preparation conditions, which involve the rapid digestion of proteins at 0 °C and pH 2.5. Very few proteases are able to withstand such harsh conditions, with pepsin being the best-known exception, even though its activity is also strongly reduced at 0 °C. Here, we evaluate the usage of a prolyl endopeptidase from Aspergillus niger (An-PEP) for HDX-MS. What makes this protease very attractive is that it cleaves preferentially the hardest to digest amino acid, proline. To our surprise, and in contrast to previous reports, An-PEP activity was found optimal around pH 2.5 and could be further enhanced by urea up to 40%. Under typical HDX-MS conditions and using small amounts of enzyme, An-PEP generated an equivalent number of peptides as pepsin, as exemplified by using the two model systems tetrameric human hemoglobin (Hb) and human IgG4. Interestingly, because An-PEP peptides are shorter than pepsin-generated peptides, higher sequence resolution could be achieved, especially for Pro-containing protein regions in the alpha subunit of Hb, revealing new protected Hb regions that were not observed with pepsin. Due to its Pro-preference and resistance to low pH, we conclude that An-PEP is an archetype enzyme for HDX-MS, highly complementary to pepsin, and especially promising for structural studies on Pro-rich proteins or proteins containing Pro-rich binding domains involved in cellular signaling.
Biological processes are governed by the function of proteins and their noncovalent interactions with ligands, which can range from small molecules (metabolites and cofactors) to nucleic acids (DNA and RNA) and other proteins. With the advent of electrospray ionization,1 several mass spectrometry (MS)-based methods have been developed to investigate protein assemblies and their interactions.2 These entail chemical surface labeling techniques such as hydrogen–deuterium exchange MS (HDX-MS), cross-linking MS, limited proteolysis MS, and also direct mass analysis of intact proteins and protein complexes, the latter (re)defined as native mass spectrometry.3 HDX-MS is based on the principle that protein amide groups constantly exchange hydrogens from water in solution. For HDX-MS, hydrogens in proteins can be typically divided into three groups: (i) hydrogens in thiol, amino, and carboxyl groups in amino acid side chains that exchange too fast to be monitored, (ii) aliphatic hydrogens that do no exchange at all, while (iii) amide hydrogens in the peptide backbone exchange with specific and measurable rates.4 When a hydrogen is replaced by a deuterium, which has a higher mass, the measured mass differences can reveal solvent-exposed protein regions or regions that remain unaffected because they are buried within the protein structure. As protein–ligand interactions modify the exchange rates of amide protons, these measurements can provide information about protein structure and dynamics; therefore, HDX-MS has been used for the study of membrane protein interactions,5−7 antibodies,8 and even protein assemblies9 and viruses.10,11
As HDX-MS follows primarily a peptic-centric (i.e., bottom-up) approach, it employs proteases for the generation of peptides prior to MS analysis. To minimize the exchange of the deuterium-labeled protein surface back to hydrogen, digestion needs to take place at low pH and temperature, typically 2.5 and at 0 °C, respectively.12 For this reason and because of its broad specificity, pepsin is commonly used in HDX-MS even though alternative proteases have been used. These include the protease type XIII from Aspergillus saitoi, protease type XVIII from Rhizhopus species,13−15 and the plant Nepenthes gracilis proteases nepenthesin I16 and II.17 Like pepsin, all of these proteases are not specific to one amino acid but can cleave at multiple residues. Under typical HDX-MS conditions, pepsin cleaves mostly after Met, Leu, and Phe, while protease type XIII and XVIII additionally cleave at the positively charged His, Lys, and Arg. The nepenthesins have an even broader specificity. To improve the digestion efficiency in HDX workflows and allow for complete sequence coverage with many overlapping peptides and no autolysis products, proteases have been immobilized on chromatography resins and often used in tandem. Such efforts have led to better results with more peptides and higher sequence resolution.14,18,19
The efficiency of a protease to generate peptides spanning the whole protein sequence depends strongly on the presence of suitable cleavage sites equally distributed alongside the protein sequence. This ensures the digestion of medium-sized peptides with good ionization properties required for ESI. Pepsin does not process disordered protein regions adequately because these have limited hydrophobic residues (e.g., Met, Leu, and Phe) and often contain many Pro or charged amino acids.20 To overcome this, Ray et al.16 employed nepenthesin I and performed the HDX analysis of the protein XRCC4. Even though cleavage at Pro was not particularly pronounced, nepenthesin I achieved a superior coverage of the disordered region compared to pepsin for this protein. Here, we characterize the performance of a genuine Pro-specific peptidase from Aspergillus niger the Prolyl endopeptidase (An-PEP)21,22 in HDX-MS applications. Currently, due to its Pro preference, An-PEP is used in tests for the alleviation of celiac disease symptoms.23−25 An-PEP is an acidic protease that displays high thermostability and in addition to its preference for Pro, Ala and other amino acids are also cleaved albeit to a lesser extend.21,26 Here, activity-based profiling using an intact fluorescent protein revealed that An-PEP has a pH optimum near 2 and that low concentration of urea boosts proteolytic activity by 40% at 0 °C. We demonstrate that, during HDX analysis of the hemoglobin tetramer and IgG4 (using a mutant lacking the hinge region), An-PEP generated a number of peptides comparable to that of pepsin albeit with very distinct and shorter sequences. The complementarity of the two enzymes was apparent in the generated sequence maps showing that different regions of the proteins were sequenced with variable resolution in each digest. Our results on the solvent exposure of the studied proteins agree with published crystallographic data but also extend our knowledge for regions that could not have been previously sequenced in detail using pepsin alone. Due to its Pro-preference and resistance to low pH, we conclude that An-PEP is a suitable enzyme for HDX-MS, highly complementary to pepsin, and especially promising for structural studies on Pro-rich proteins (PRPs) or proteins containing Pro-rich binding domains involved in cellular signaling.
Experimental Section
Proteins
Human hemoglobin and porcine pepsin were purchased from Sigma-Aldrich and used without further purification. AN-PEP was provided by after purification from A. niger using ion-exchange chromatography with a 10 mm MonoQ column (GE Healthcare) according to the protocol provided by the vendor DSM. IgG4Δhinge mutant was a gift from Genmab (Utrecht, The Netherlands), which has been described in detail previously.27
Activity Assays
Activity of An-PEP was measured using the fluorescence protease assay kit (Pierce-Thermo Scientific) and because An-PEP is an acidic protease, we used the Twining method.28 A detailed description of this procedure is included as Supporting Information.
HDX-MS Analysis
A detailed description of this analysis is included as Supporting Information. In short, An-PEP solution was prepared in 50 mM citric acid pH 2.5 and pepsin in 0.1% formic acid pH 2.5. IgG4Δhinge27 and human Hb were prepared in phosphate-buffered saline buffer pH 7.4. Sixty picomoles of Hb or IgG4Δhinge were diluted 30-fold to either D2O (a 99.9% pure solution of D2O, pD 7.4) for the deuterated samples or water pH 7 for the nondeuterated controls. Deuteration proceeded at room temperature (RT) for different time points, and the reaction was quenched by 1:1 dilution into an ice-cold solution of 4 M urea and 200 mM TCEP at a final pH of 2.5. An-PEP was kept at RT for 10 min to equilibrate and immediately after quenching of the H/D exchange, digestion was performed with either An-PEP or pepsin in solution at a 1:1 molar ratio (1 uM final concentration) in the quench conditions for 3 min at 0 °C. Peptide mixtures were then immediately injected into a nano-Acquity UPLC system with HDX technology coupled to a Waters Xevo QTof G2 instrument, and each sample was analyzed in triplicate. Peptides were sequenced using MSE data acquisition and identified with ProteinLynx Global Server 2.5 software at 1% false discovery rate (FDR). Uptake of deuterium for each peptide was calculated compared to the nondeuterated control samples using Waters DynamX 3.0.0 software.
Results and Discussion
Proline residues in proteins are particularly difficult to digest by most proteases because of their cyclic nature. However, different Pro-targeting enzymes do exist. Pro aminopeptidases and carboxypeptidases can remove Pro residues from the protein termini.29 Depending on the size of the containing amino acid chain, Pro can also be processed in shorter substrates by oligopeptidases and finally in large proteinaceous substrates by a specific class of endoproteases (PEP).30 These proteases are divided into different classes, including serine and metalloproteases. PEPs are characterized by a wide range of properties with respect to temperature and ionic strength, demonstrating a pH profile with a characteristic double sigmoidal curve that peaks at pH 6 and 8.31 In contrast, the PEP from A. niger, used here, displays an acidic pH optimum (pH 4–5) and it is therefore particularly interesting for HDX-MS applications.
An-PEP Activity
Protease activities are commonly detected using short fluorescent synthetic peptides. The same approach is also used for measuring in vitro the activity of recombinant proteases. However, the successful outcome of such tests depends on the degree of protease specificity toward the amino acid that is coupled to the fluorophore. Besides Pro, An-PEP is known to also cleave Ala and also other amino acids, albeit with lower efficiency.21,26 Therefore, we reasoned that the use of a full protein, in this case FTC-casein, would be a much more suitable probe for An-PEP activity than Pro-ending peptides. To our surprise and in contrast to previous reports using Pro-ending peptides,21,26 we observed that An-PEP activity shows two optima at pH ∼2 and ∼6. This pattern of double pH optima has been previously reported also for neutral PEPs31 but not for an acidic PEP. The highest substrate hydrolysis occurred at pH 2, and a 60% lower activity was observed at pH 6 (Figure 1A). The same profile was also observed at 0 °C, even though the enzymatic activity was reduced compared to that at pH 2 at RT. To prime the PEP activity and achieve a better proteolytic digestion at low temperature, we kept An-PEP at RT for 10 min to equilibrate prior to addition to the casein substrate. Compared to 0 °C, An-PEP pre-equilibration enhanced digestion by more than 20% and reached a final activity level of 40% at pH 2 compared to that at RT (Figure 1A).
Figure 1.
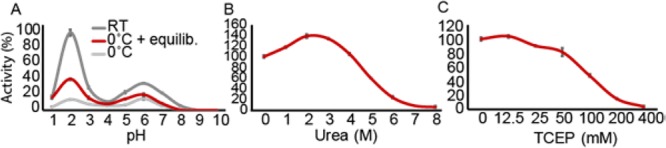
An-PEP activity profiling. (A) Effect of pH on An-PEP activity at RT or 0 °C with or without pre-equilibration at RT. The effect of (B) urea and (C) TCEP on An-PEP activity at pH 2 and at 0 °C after pre-equilibration. Values in panels B and C are normalized to the maximal activity of An-PEP at pH 2 and 0 °C after pre-equilibration (panel A). Error bars represent the standard deviation of the mean of triplicate measurements. Activity is expressed as relative fluorescence units at 30 °C and absorbance at 480 nm after correction for background fluorescence.
Protein digestion prior to HDX-MS is done under mild denaturing and reducing conditions, very shortly at 0 °C and typically at pH ∼2.5. Under these conditions, the back-exchange of deuterium to hydrogen is minimized,12 but also, most proteolytic enzymes become inactive. This is the main reason why only a few acidic proteases have been used to date in HDX-MS experiments. Notably pepsin, even though it works best at 37 °C, remains active enough at 0 °C to digest proteins within a few minutes and is thus commonly used in HDX-MS. An-PEP is an interesting protease for HDX-MS because it has a cleavage preference very different than that of pepsin and, in this respect, the two enzymes could be very complementary. Seeing that An-PEP remains sufficiently active at low temperature and acidic conditions, we implemented more HDX-MS experimental settings in our tests and analyzed An-PEP activity levels at increasing concentrations of urea or TCEP at 0 °C and acidic pH (2.5). As before, An-PEP was pre-equilibrated at RT. Interestingly, we found that at these conditions, urea (until 3 M) enhances the previously seen optimal An-PEP activity at pH 2 by approximately 20–40% (Figure 1B), probably due to the unfolding of the substrate. In fact, at custom HDX-MS conditions such as 0 °C, pH ∼2, and 2 M urea, the enhancing effect of urea was about 40%. TCEP had an overall inhibitory effect on An-PEP (Figure 1C) and at HDX-MS conditions (100 mM TCEP), An-PEP activity reached 50%. Collectively, our observations showed that, under these conditions, An-PEP remains at least 50% active and thus very efficient for protein digestion in the typical HDX-MS workflow.
An-PEP Performance during HDX-MS
Biopharmaceuticals (i.e., protein or nucleic acid-based pharmaceutical substances, which are produced by means other than direct extraction from a native biological source) require high structural consistency during development. To monitor the structural integrity of therapeutic proteins, HDX-MS has gained popularity within the pharmaceutical industry. The advantages of this method include high sensitivity for conformational changes, relatively low sample amount, no protein size restrictions, and completion of the analysis within days. Currently, pepsin is commonly used in the HDX-MS pipeline, but digestion does not always lead to complete protein coverage; thus, alternative proteases may be beneficial.
To investigate the utility of An-PEP for HDX-MS, we used two model systems that range in size and Mw from 290 to 1304 residues or from 64 (tetrameric hemoglobin) to 146 kDa (dimeric assembly of IgG4) and benchmarked the performance of An-PEP against pepsin. The engineered 146 kDa antibody was a variant of IgG4, earlier used in native MS studies27 in which the hinge region had been removed (IgG4Δhinge), allowing for the occurrence of mono- and dimeric IgG4 molecules.27 After LC–MS/MS, the resulting mixture of deuterated peptides (Supplementary Table 1) was used to analyze enzyme specificity and the solution-phase properties of the protein samples and to compare the performance of the two enzymes.
Even though the number of identified deuterated peptides differed slightly for both proteins, the sequence coverage that was achieved with An-PEP compared well with pepsin and was higher than 90% for both hemoglobin (Hb) and IgG4Δhinge (Table 1). In fact, An-PEP generated more Hb peptides, while pepsin generated more IgG4Δhinge peptides. Even though these results did not affect sequence coverage, variable amino acid redundancy values (i.e., times that a given amino acid is mapped by different peptides) were observed between digests, which indicates that certain protein regions were digested more efficiently by one protease than the other. As such, An-PEP sequenced deeper the α subunit of Hb, while pepsin performed better for the light chain of the IgG4Δhinge molecule, which demonstrates that the results obtained with these proteases are highly complementary. Consequently, the overlap in peptide identifications within the different protein digests was less than 12% (Supplementary Figure 1A), and the An-PEP-generated peptides were on average shorter than pepsin-generated peptides (Supplementary Figure 1B), which is a feature that in principle may increase sequence resolution during HDX-MS. In summary, An-Pep performed similar or even better than pepsin under the used experimental conditions using low amounts of enzyme and represents a valuable alternative to pepsin for HDX-MS. Furthermore, our results endorse that also in HDX-MS analysis the use of multiple proteases can lead to superior results, as is well-known for bottom-up (phospho)proteomics.32−34
Table 1. Descriptive Results of the HDX-MS Analysis of Hemoglobin and IgG4Δhinge with An-PEP or Pepsin.
| Hemoglobin | ||||||||
|---|---|---|---|---|---|---|---|---|
| unique
peptides |
sequence coverage |
redundancy |
||||||
| α subunit | β subunit | total | α subunit | β subunit | total | α subunit | β subunit | |
| An-PEP | 30 | 8 | 38 | 99 | 92 | 96 | 4 | 1.7 |
| pepsin | 13 | 14 | 27 | 89 | 99 | 95 | 2.8 | 2.6 |
| IgG4Δhinge | ||||||||
|---|---|---|---|---|---|---|---|---|
| unique
peptides |
sequence coverage |
redundancy |
||||||
| light chain | heavy chain | total | light chain | heavy chain | total | light chain | heavy chain | |
| An-PEP | 19 | 69 | 88 | 88 | 92 | 91 | 2.1 | 3.5 |
| pepsin | 24 | 75 | 99 | 100 | 90 | 93 | 3.2 | 4.3 |
An-PEP Specificity
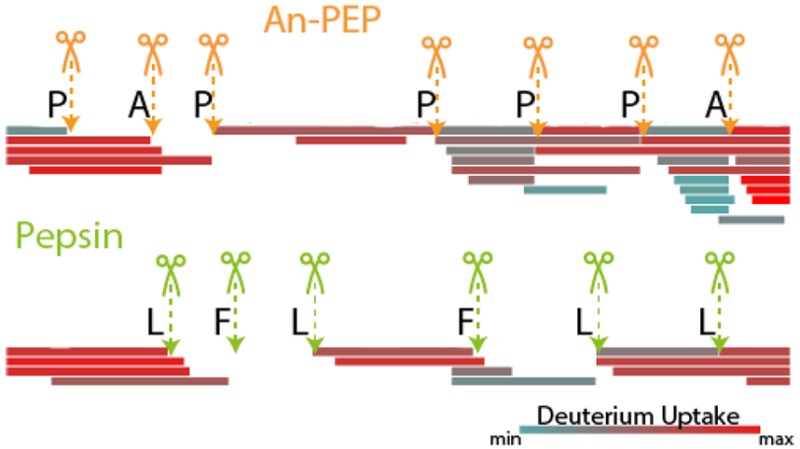
The specificity of An-PEP has been previously evaluated at pH 4–5 and, unlike pepsin, An-PEP preferentially cleaves C-terminal to Pro residues. In addition, cleavage at Ala and other amino acids is also observed.21,26 Here, we evaluated the An-PEP specificity at pH 2.5, i.e., under optimal HDX-MS conditions, and we confirm that also at this pH, An-PEP cleaves preferentially C-terminal to Pro (Figures 2A and B). In fact, more than 50% of prolines in the tested proteins were cleaved by An-PEP.
Figure 2.
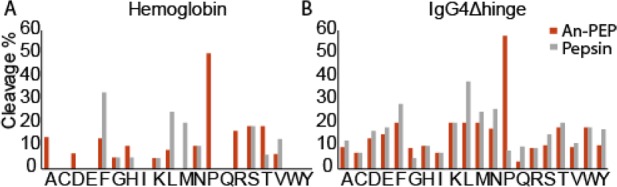
Amino acid frequency at cleavage position P1(Schechter and Berger nomenclature35) in An-PEP and pepsin digestions of (A) hemoglobin and (B) IgG4Δhinge during HDX-MS analysis. Cleavage percentage was calculated relative to the frequency of each amino acid in the respective protein.
On the contrary, such events were very rare (Figure 2B) or even absent (Figure 2A) in the digests of pepsin. Other amino acids, including Ala, were also cleaved by An-PEP, but in all such cases, cleavage was lower than 20%. In our data set, pepsin cleaved primarily at Phe, Leu, and Met by more than 30% in addition to other amino acids. Compared to the strong An-PEP preference for Pro (>50%), pepsin is found to be a less specific protease than An-PEP at pH 2.5.
Although HDX-MS is a peptic-centric mass spectrometry-based method, it is not ideal for the analysis of hundreds of proteins and thousands of resulting peptides within a single LC–MS run and is therefore not suited for the detailed analysis of protease specificity. For this, we performed the digestion of the Escherichia coli proteome at pH 2.5 and analyzed the peptides with bottom-up proteomics (Supplementary Tables 2 and 3). As with the two HDX-MS test cases, E. coli proteins were also cleaved predominantly at Pro (Supplementary Figures 2A and B). Ala was more frequently cleaved in the bottom-up than in the HDX-MS setup probably because during the first, the digestion was performed longer to overcome the higher sample complexity. Overall, we did not observe any major determinants of An-PEP specificity besides Pro or Ala at position P1. For Pro-cleaved peptides, no other sequence determinants were observed and, interestingly, Pro was never found at P1′ (Supplementary Figure 2C). Also in Ala-cleaved peptides, Pro at P1′ was not favorable for cleavage (Supplementary Figure 2D). For both Pro and Ala-cleaved peptides, a relative preference for acidic residues was seen at P1′, while the basic Lys is somewhat frequent at the nonprime side of Ala-cleaved peptides (Supplementary Figure 2D). These results are in agreement with recent reports.36
Next, after inspection of peptides containing a missed cleavage site (Supplementary Figure 3), we observed that Pro-missed sites were 8 times fewer than Ala-missed sites, which shows that Pro is more efficiently cleaved than Ala. Considering the sequence context in Pro and Ala-missed cleavages, we observed that all basic residues (Arg, Lys, and His) at P2′ have a negative effect on An-PEP cleavage efficiency. Also, even though we would assume that Pro would be present at position P1′ in the missed cleavage iceLogos, we were not able to see this information because the used software takes into account only the natural abundance of amino acids in individual protein positions and does not consider the frequency of Pro-Pro or Ala-Pro motifs in the proteome.37
Structural Analysis of Human Hemoglobin by HDX-MS
Human hemoglobin (Hb) is a globular tetramer consisting of two heterodimers of α and β subunits (αβ). The α and β subunits consist of seven to eight α-helices (A–H), respectively (Figure 3 and Supplementary Figure 4). Each of the four Hb subunits contains a heme group where oxygen can bind and therefore Hb is the oxygen carrier protein in red blood cells. Besides its biological role, Hb is routinely used as a model system for intact protein analysis with MS. Here, we analyzed D2O uptake in Hb after digestion with either An-PEP or pepsin (Supplementary Table 1) to assess solvent accessibility and compared the results obtained by using the two proteases. Overall, the solution-phase properties of Hb were found to be similar using either An-PEP or pepsin and in agreement with previously published HDX-MS38 and crystallographic data39 for bovine Hb (Figure 3). In fact, segments in helices αG and βG showed significant protection as these elements are deeply buried to form anchoring points at the hydrophobic intersubunit interface.38,39 On the contrary, the C-terminus of the α subunit and the N-terminus of the β subunit are the most solvent-accessible regions of the protein (Figure 3 and Supplementary Figure 4). Despite the similarities in the overall D2O uptake between the two digests, a few differences were observed. Those included the N-terminus of the α subunit (region I), helix αH (region II), helix βC (region III), and helix βF−βG (region IV) (Figure 3 and Supplementary Figure 4). Because An-PEP and pepsin generated very distinct peptides in terms of sequence and length (Supplementary Figure 1), protein segments were mapped with overlapping peptides of variable length in each of these digests. For the Hb regions I and II, An-PEP generated peptides smaller than those of pepsin, enabling better sequence resolution of the corresponding segment. This revealed short protected sequence segments that in pepsin maps were masked by the overall high deuterium uptake of long peptides. Consequently, the superior sequence resolution that was here achieved with An-PEP for the N-terminal segment of the αA helix (area I) and its overlaying segment of aH helix (area II) has not been previously reported with pepsin-based HDX data38 and revealed another solvent-protected region of Hb. On the other hand, at the β subunit in regions III and IV, An-PEP did not generate sufficient peptides, probably because there are not many prolines present in this region to create peptides of suitable length. Thus, pepsin-based data allowed for a better, yet again not optimal, view of this subunit with a few more peptides than An-PEP (Figure 3 and Supplementary Figure 4). Overall, the combined analysis of Hb using An-PEP and pepsin confirmed the known features of the Hb quaternary structure. Furthermore, due to its Pro preference, An-PEP also allowed a more in depth investigation of the solution-phase properties of Hb regions where prolines are found (region II). For this model system we conclude that the performance of An-PEP for HDX-MS is at least on par with pepsin, offering additionally deeper sequencing of Pro-containing regions.
Figure 3.

Deuterium uptake in human Hb following 1 min of incubation mapped onto the Hb heterodimer. Segments that show high deuterium uptake are colored in red, and low uptake segments are in cyan. Protein regions that could not be mapped with deuterated peptides are in white. Helices are denoted following the accepted annotations as A–H for each subunit, and regions I–IV that showed varying uptake levels between digests are marked with segmented lines. Insets illustrate the deuterated peptides that were identified for selected regions. Source of the PDB structure: 2HHB42
Structural analysis of IgG4Δhinge by HDX-MS
Human immunoglobulin G (IgG) is a protein of 146 kDa that consists of two heavy (H) and two light (L) chains interlinked via a flexible hinge region. The complete molecule consists of the Fab and the Fc regions (Figure 4A). Antigens are bound to the Fab region of IgG, which is formed by a piece of the H chain linked to the full L chain. In each of the H and L chains in the Fab region, there is a variable (VL or VH) and a constant domain (CL or CH1). The Fc region consists of the CH2 and CH3 domains of the H chain and mediates the interaction of IgG with proteins and receptors on the cell surface.
Figure 4.

Deuterium uptake data following incubation of IgG4Δhinge for 10 s in D2O mapped onto the IgG4Δhinge half body structure. Segments that show high D2O uptake are colored in red, and low uptake segments are in cyan. Protein regions that were not mapped are in white. (A) Schematic IgG4Δhinge domain representation adapted to the orientation of the molecule as presented in the figure. Light chain in pink with variable (VL) and constant (CL) domains. Heavy chain in purple with variable (VH) and constant domains (CH1–CH3). D2O uptake deduced from (B) An-PEP and (C) pepsin-derived peptides. Regions that displayed varying uptake levels are marked with segmented lines in the An-PEP map and highlighted in yellow. Insets illustrate the deuterated peptides that were identified for each region in the two digests. For simplicity of the figure, the protein regions that are discussed in detail in the text are named only in the pepsin map. The here presented IgG4Δhinge structure was modeled based on the available Pembrolizumab structure43 (PDB: 5DK3).
In this study, we focused on IgG4, a subclass of human antibodies that can be functionally monovalent in vivo40 and therefore can dynamically exchange half molecules (heavy–light chain pair) in a mechanism called Fab-arm exchange (FAE).41 To date, there is only one complete crystal structure available for a IgG443 and, due to the limitation of crystallographic data in predicting adequately the properties of a protein in solution, we set out to investigate IgG4 properties with HDX-MS. The analyzed IgG4 antibody was depleted of the hinge region (IgG4Δhinge), and this prevented the interheavy chain disulfide bond formation and dimerization, allowing for a dynamic equilibrium of mono- and dimeric molecules.27
Deuterium uptake data (Supplementary Table 1) were mapped onto a structural model of IgG4Δhinge that we modeled based upon the structure of the IgG4 Pembrolizumab.43 In contrast to IgG1 monoclonal antibodies, crystallographic data have shown that the IgG4 molecule is very compact and has an affinity for Fc receptors and complement C1q overall lower than those of IgG1 and IgG3.43−46 Interactions with Fc receptors at the FG loop (for IgG4Δhinge: Lys318-Ile324) and the CH2 loop (for IgG4Δhinge: Pro236-Pro249) take place at the solvent-exposed regions of CH2 domain closely located to the hinge region (Figures 4B and C). Another exposed site where the glycan attaches to the CH3 domain is at Asn383. Accordingly, in both An-PEP and pepsin HDX-MS uptake maps for IgG4Δhinge, our data indicated the same solvent-exposed regions as seen in crystallographic data of Pembrolizumab (Figures 4B and C and Supplementary Figure 5). Again, as seen in the HDX-MS analysis of Hb (Figure 3), the uptake data derived from the two digests are in close agreement, and the observed differences are due to differences in peptide length. An-PEP allowed for a complete coverage of the CH2 loop and led to higher sequence resolution in the CH2–CH3 interdomain loop, while pepsin generated smaller overlapping peptides in the CH3 strand and helix (Leu344–Asn354) following the CH2–CH3 interdomain loop. Pepsin-generated peptides also revealed the exposure of the loop bearing Asn383, where a glycan is attached in Pembrolizumab and confirmed the crystallographic data. Furthermore, our An-PEP data revealed another exposed region in the VL domain of the light chain. From our HDX-MS data on Hb and IgG4Δhinge, we conclude that the structural properties of both analyzed proteins were in close agreement in each digestion and with the previously reported crystallographic data. An-PEP and pepsin generated peptides of different sequence and length and mapped protein regions with distinct resolution (Supplementary Figure 4 and 5). Also, protein segments that were not sequenced with one protease were identified with the other (Supplementary Figure 6). Combination of the sequence information derived with each protease led to higher sequence coverage for both analyzed proteins.
Conclusions
Here, we investigated the properties of the Prolyl endopeptidase from A. niger, An-PEP, with respect to HDX-MS and showed that An-PEP can digest proteins very efficiently, within a few minutes, at the optimal pH (2.5) and temperature (0 °C) required. Using two human proteins (Hb and IgG4Δhinge) as model systems, we showed that HDX-MS is a powerful technique to study protein structures with an emphasis on solvent accessibility. The An-PEP genuine preference for cleavage after Pro residues makes this protease very distinct from pepsin and allows for a superior view of Pro-containing protein regions. Such regions are for instance present in intrinsically unstructured proteins20,47 but also in so-called Pro-rich motifs that are recognized by other protein domains involved in signaling pathways, including the SH3, phosphotyrosine-binding, and WW domains.48 The here acquired deuterium uptake data were in good agreement with previous structural data on Hb and IgG4 structures obtained by crystallography. In particular, the An-PEP derived exchange data allowed a more in-depth investigation of the solution-phase properties of Hb regions where Pro residues are included. For this reason, the combined use of An-PEP and pepsin in HDX-MS may offer an enhanced and more detailed view of protein structures.
Additionally, the calculation of deuterium uptake in HDX workflows could become simpler with the use of prolyl-specific proteases. As deuterium incorporation is not possible at Pro due to its lack of a backbone amide hydrogen, peptides that carry an N-terminal Pro do not suffer from N-terminal back exchange to hydrogen, which is very fast and therefore needs to be deducted from the overall peptide uptake during the calculation. In this respect, a protease that cleaves N-terminally to Pro and generates Pro-starting peptides would simplify uptake calculation in HDX-MS analyses, but to our knowledge, no such protease has been identified yet. Another acidic Pro-specific protease, Neprosin,49 from the Nepenthes plant genus, has been recently used for proteomics and histone mapping.36
However, this protease has an optimum temperature of 37–50 °C, which probably restricts it from usage in HDX-MS applications. Here, we conclude that, due to its optimal activity at pH ∼2.5, which can be even further enhanced by adding 1–2 M urea, An-PEP represents a novel protease with ideal opportunities for HDX-MS investigations, providing structural insights that are complementary to pepsin digestion. In our initial experiments, we already showed that An-PEP performs on par with pepsin regarding equivalent number of peptides when used on relatively large protein structures such as the tetrameric human hemoglobin and the dimeric IgG4. Further optimization such as on column enzyme-immobilization and optimized fragmentation schemes for An-PEP peptides using electron-transfer dissociation, electron-transfer and higher-energy collision dissociation, or ultraviolet photodissociation fragmentation may make An-PEP the protease of choice for future HDX-MS experiments. Especially, protein regions rich in Pro residues such as those frequently present in intrinsically disordered proteins and binding domains involved in signaling may represent ideal systems to investigate by An-PEP and HDX-MS.
Acknowledgments
We thank all members of the Biomolecular Mass Spectrometry and Proteomics Laboratory at Utrecht University for fruitful discussions, and especially Anja Boumeester, Philip Lössl, and Michiel van de Waterbeemd for their help with HDX-MS and modeling the structure of IgG4Δhinge and Pymol. L.T. acknowledges EMBO for a Long-Term Fellowship (LTF 776-2013). We thank Aran Labrijn and Janine Schuurman (Genmab, The Netherlands) for providing the IgG4Δhinge construct and The Netherlands Organization for Scientific Research (NWO) for the funding of the large-scale proteomics facility Proteins@Work (Project 184.032.201), a part of The Netherlands Proteomics Centre.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.7b01161.
Experimental details; Figure S1, global differences between An-PEP and pepsin-generated peptides; Figure S2, An-PEP specificity derived from E. coli lysate digestion; Figure S3, missed cleavage sequence information; Figures S4 and S5, deuterium uptake heat maps for Hb and IgG4Δhinge proteins, respectively; Figure S6, sequence coverage maps; Table S2, descriptive numbers of E. coli proteome analysis with An-PEP (PDF)
Supplementary Table 1: Deuterium uptake data for the identified peptides with HDX-MS analysis (XLSX)
Supplementary Table 3: E. coli protein and peptide identifications (XLSX)
Author Contributions
A.J.R.H. and L.T. conceived the idea. L.T. designed and performed the experiments, analyzed the data, and wrote the manuscript. M.A. and M.O. supplied the An-PEP and provided input to the study. All authors have read and approved the final version of the manuscript.
The authors declare the following competing financial interest(s): M.A. and M.O. are DSM employees. DSM sells An-PEP for food applications.
Supplementary Material
References
- Fenn J. B.; Mann M.; Meng C. K.; Wong S. F.; Whitehouse C. M. Science 1989, 246 (4926), 64–71. 10.1126/science.2675315. [DOI] [PubMed] [Google Scholar]
- Lössl P.; van de Waterbeemd M.; Heck A. J. EMBO J. 2016, 35 (24), 2634–2657. 10.15252/embj.201694818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leney A. C.; Heck A. J. R. J. Am. Soc. Mass Spectrom. 2017, 28 (1), 5–13. 10.1007/s13361-016-1545-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englander S. W.; Downer N. W.; Teitelbaum H. Annu. Rev. Biochem. 1972, 41 (1), 903–924. 10.1146/annurev.bi.41.070172.004351. [DOI] [PubMed] [Google Scholar]
- Chung K. Y.; Rasmussen S. G. F.; Liu T.; Li S.; DeVree B. T.; Chae P. S.; Calinski D.; Kobilka B. K.; Woods V. L.; Sunahara R. K. Nature 2011, 477 (7366), 611–615. 10.1038/nature10488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla A.; Futrell J. J. Mass Spectrom. 2000, 35 (9), 1069–1090. . [DOI] [PubMed] [Google Scholar]
- Rostislavleva K.; Soler N.; Ohashi Y.; Zhang L.; Pardon E.; Burke J. E.; Masson G. R.; Johnson C.; Steyaert J.; Ktistakis N. T.; Williams R. L. Science 2015, 350 (6257), aac7365. 10.1126/science.aac7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose R. J.; van Berkel P. H. C.; van den Bremer E. T. J.; Labrijn A. F.; Vink T.; Schuurman J.; Heck A. J. R.; Parren P. W. H. I. MAbs 2013, 5 (2), 219–228. 10.4161/mabs.23532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanman J.; Lam T. T.; Emmett M. R.; Marshall A. G.; Sakalian M.; Prevelige P. E. Nat. Struct. Mol. Biol. 2004, 11 (7), 676–677. 10.1038/nsmb790. [DOI] [PubMed] [Google Scholar]
- Wang L.; Lane L. C.; Smith D. L. Protein Sci. 2001, 10 (6), 1234–1243. 10.1110/ps.100101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bereszczak J. Z.; Rose R. J.; van Duijn E.; Watts N. R.; Wingfield P. T.; Steven A. C.; Heck A. J. R. J. Am. Chem. Soc. 2013, 135 (17), 6504–6512. 10.1021/ja402023x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englander J. J.; Rogero J. R.; Englander S. W. Anal. Biochem. 1985, 147 (1), 234–244. 10.1016/0003-2697(85)90033-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cravello L.; Lascoux D.; Forest E. Rapid Commun. Mass Spectrom. 2003, 17 (21), 2387–2393. 10.1002/rcm.1207. [DOI] [PubMed] [Google Scholar]
- Rey M.; Man P.; Brandolin G.; Forest E.; Pelosi L. Rapid Commun. Mass Spectrom. 2009, 23 (21), 3431–3438. 10.1002/rcm.4260. [DOI] [PubMed] [Google Scholar]
- Zhang H.-M.; Kazazic S.; Schaub T. M.; Tipton J. D.; Emmett M. R.; Marshall A. G. Anal. Chem. 2008, 80 (23), 9034–9041. 10.1021/ac801417d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey M.; Yang M.; Burns K. M.; Yu Y.; Lees-Miller S. P.; Schriemer D. C. Mol. Cell. Proteomics 2013, 12 (2), 464–472. 10.1074/mcp.M112.025221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M.; Hoeppner M.; Rey M.; Kadek A.; Man P.; Schriemer D. C. Anal. Chem. 2015, 87 (13), 6681–6687. 10.1021/acs.analchem.5b00831. [DOI] [PubMed] [Google Scholar]
- Mayne L.; Kan Z.-Y.; Sevugan Chetty P.; Ricciuti A.; Walters B. T.; Englander S. W. J. Am. Soc. Mass Spectrom. 2011, 22 (11), 1898–1905. 10.1007/s13361-011-0235-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Pan H.; Smith D. L. Mol. Cell. Proteomics 2002, 1 (2), 132–138. 10.1074/mcp.M100009-MCP200. [DOI] [PubMed] [Google Scholar]
- Dunker A. K.; Lawson J. D.; Brown C. J.; Williams R. M.; Romero P.; Oh J. S.; Oldfield C. J.; Campen A. M.; Ratliff C. M.; Hipps K. W.; Ausio J.; Nissen M. S.; Reeves R.; Kang C.; Kissinger C. R.; Bailey R. W.; Griswold M. D.; Chiu W.; Garner E. C.; Obradovic Z. J. Mol. Graphics Modell. 2001, 19 (1), 26–59. 10.1016/S1093-3263(00)00138-8. [DOI] [PubMed] [Google Scholar]
- Edens L.; Dekker P.; van der Hoeven R.; Deen F.; de Roos A.; Floris R. J. Agric. Food Chem. 2005, 53 (20), 7950–7957. 10.1021/jf050652c. [DOI] [PubMed] [Google Scholar]
- Janssen G.; Christis C.; Kooy-Winkelaar Y.; Edens L.; Smith D.; van Veelen P.; Koning F. PLoS One 2015, 10 (6), e0128065. 10.1371/journal.pone.0128065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepniak D.; Spaenij-Dekking L.; Mitea C.; Moester M.; de Ru A.; Baak-Pablo R.; van Veelen P.; Edens L.; Koning F. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 291 (4), G621–G629. 10.1152/ajpgi.00034.2006. [DOI] [PubMed] [Google Scholar]
- Salden B. N.; Monserrat V.; Troost F. J.; Bruins M. J.; Edens L.; Bartholomé R.; Haenen G. R.; Winkens B.; Koning F.; Masclee A. A. Aliment. Pharmacol. Ther. 2015, 42 (3), 273–285. 10.1111/apt.13266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tack G. J.; van de Water J. M. W.; Bruins M. J.; Kooy-Winkelaar E. M. C.; van Bergen J.; Bonnet P.; Vreugdenhil A. C. E.; Korponay-Szabo I.; Edens L.; von Blomberg B. M. E.; Schreurs M. W. J.; Mulder C. J.; Koning F. World J. Gastroenterol. 2013, 19 (35), 5837. 10.3748/wjg.v19.i35.5837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebela M.; Rehulka P.; Kábrt J.; Rehulková H.; Ozdian T.; Raus M.; Franc V.; Chmelík J. J. Mass Spectrom. 2009, 44 (11), 1587–1595. 10.1002/jms.1667. [DOI] [PubMed] [Google Scholar]
- Rose R. J.; Labrijn A. F.; van den Bremer E. T. J.; Loverix S.; Lasters I.; van Berkel P. H. C.; van de Winkel J. G. J.; Schuurman J.; Parren P. W. H. I.; Heck A. J. R. Structure 2011, 19 (9), 1274–1282. 10.1016/j.str.2011.06.016. [DOI] [PubMed] [Google Scholar]
- Twining S. S. Anal. Biochem. 1984, 143 (1), 30–34. 10.1016/0003-2697(84)90553-0. [DOI] [PubMed] [Google Scholar]
- Rosenblum J. S.; Kozarich J. W. Curr. Opin. Chem. Biol. 2003, 7 (4), 496–504. 10.1016/S1367-5931(03)00084-X. [DOI] [PubMed] [Google Scholar]
- Gass J.; Khosla C. Cell. Mol. Life Sci. 2007, 64 (3), 345–355. 10.1007/s00018-006-6317-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- POLGAR L. Eur. J. Biochem. 1991, 197 (2), 441–447. 10.1111/j.1432-1033.1991.tb15930.x. [DOI] [PubMed] [Google Scholar]
- Peng M.; Taouatas N.; Cappadona S.; van Breukelen B.; Mohammed S.; Scholten A.; Heck A. J. Nat. Methods 2012, 9 (6), 524–525. 10.1038/nmeth.2031. [DOI] [PubMed] [Google Scholar]
- Guo X.; Trudgian D. C.; Lemoff A.; Yadavalli S.; Mirzaei H. Mol. Cell. Proteomics 2014, 13 (6), 1573–1584. 10.1074/mcp.M113.035170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giansanti P.; Aye T. T.; van den Toorn H.; Peng M.; van Breukelen B.; Heck A. J. R. Cell Rep. 2015, 11 (11), 1834–1843. 10.1016/j.celrep.2015.05.029. [DOI] [PubMed] [Google Scholar]
- Schechter I.; Berger A. Biochem. Biophys. Res. Commun. 2012, 425 (3), 497–502. 10.1016/j.bbrc.2012.08.015. [DOI] [PubMed] [Google Scholar]
- Schräder C. U.; Lee L.; Rey M.; Sarpe V.; Man P.; Zabrouskov V.; Larsen B.; Schriemer D. C. Mol. Cell. Proteomics 2017, 16 (6), 1–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colaert N.; Helsens K.; Martens L.; Vandekerckhove J.; Gevaert K. Nat. Methods 2009, 6 (11), 786–787. 10.1038/nmeth1109-786. [DOI] [PubMed] [Google Scholar]
- Sowole M. A.; Konermann L. J. Am. Soc. Mass Spectrom. 2013, 24 (7), 997–1005. 10.1007/s13361-013-0647-4. [DOI] [PubMed] [Google Scholar]
- Aranda R.; Cai H.; Worley C. E.; Levin E. J.; Li R.; Olson J. S.; Phillips G. N.; Richards M. P. Jr; Richards M. P. Proteins: Struct., Funct., Genet. 2009, 75 (1), 217–230. 10.1002/prot.22236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aalberse R. C.; Schuurman J. Immunology 2002, 105 (1), 9–19. 10.1046/j.0019-2805.2001.01341.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Neut Kolfschoten M.; Schuurman J.; Losen M.; Bleeker W. K.; Martínez-Martínez P.; Vermeulen E.; den Bleker T. H.; Wiegman L.; Vink T.; Aarden L. A.; De Baets M. H.; van de Winkel J. G. J.; Aalberse R. C.; Parren P. W. H. I. Science (Washington, DC, U. S.) 2007, 317, 5844. 10.1126/science.1144603. [DOI] [PubMed] [Google Scholar]
- Fermi G.; Perutz M. F.; Shaanan B.; Fourme R. J. Mol. Biol. 1984, 175 (2), 159–174. 10.1016/0022-2836(84)90472-8. [DOI] [PubMed] [Google Scholar]
- Scapin G.; Yang X.; Prosise W. W.; McCoy M.; Reichert P.; Johnston J. M.; Kashi R. S.; Strickland C. Nat. Struct. Mol. Biol. 2015, 22 (12), 953–958. 10.1038/nsmb.3129. [DOI] [PubMed] [Google Scholar]
- Jefferis R.; Lund J. Immunol. Lett. 2002, 82 (1–2), 57–65. 10.1016/S0165-2478(02)00019-6. [DOI] [PubMed] [Google Scholar]
- Radaev S.; Sun P. Mol. Immunol. 2002, 38 (14), 1073–1083. 10.1016/S0161-5890(02)00036-6. [DOI] [PubMed] [Google Scholar]
- Bruhns P.; Iannascoli B.; England P.; Mancardi D. A.; Fernandez N.; Jorieux S.; Daëron M. Blood 2009, 113, 3716. 10.1182/blood-2008-09-179754. [DOI] [PubMed] [Google Scholar]
- Dyson H. J.; Wright P. E. Nat. Rev. Mol. Cell Biol. 2005, 6 (3), 197–208. 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- Kay B. K.; Williamson M. P.; Sudol M. FASEB J. 2000, 14 (2), 231–241. [PubMed] [Google Scholar]
- Rey M.; Yang M.; Lee L.; Zhang Y.; Sheff J. G.; Sensen C. W.; Mrazek H.; Halada P.; Man P.; McCarville J. L.; Verdu E. F.; Schriemer D. C. Sci. Rep. 2016, 6, 30980. 10.1038/srep30980. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.