

Abstract
Aim
To pilot enhanced safety surveillance of seasonal influenza vaccine meeting the European Medicines Agency (EMA) requirement to rapidly detect a significant increase in the frequency or severity of adverse events of interest (AEIs), which may indicate risk from the new season’s vaccine.
Study design
A prospective passive enhanced safety surveillance combining data collection from adverse drug reaction (ADR) cards with automated collection of pseudonymised routinely collected electronic health record (EHR) data. This study builds on a feasibility study carried out at the start of the 2015/2016 influenza season. We will report influenza vaccine exposure and any AEIs reported via ADR card or recorded directly into the EHR, from the commencement of influenza vaccination and ends as specified by EMA (30 November 2016).
Setting
Ten volunteer English general practices, primarily using the GSK influenza vaccines. They had selected this vaccine in advance of the study.
Participants
People who receive a seasonal influenza vaccine, in each age group defined in EMA interim guidance: 6 months to 5 years, 6–12 years, 13–17 years, 18–65 years and >65 years.
Outcome measures
The primary outcome measure is the rate of AEIs occurring within 7 days postvaccination, using passive surveillance of general practitioner (GP) EHR systems enhanced by a card-based ADR reporting system. Extracted data will be presented overall by brand (Fluarix Tetra vs others), by age strata and risk groups. The secondary outcome measure is the vaccine uptake among the subjects registered in the enrolled general practices.
Ethics and dissemination
Ethical approval was granted by the Proportionate Review Sub-committee of the North East—Newcastle & North Tyneside 2 on 5 August 2016. The study received approval from the Health Research Authority on 1 September 2016. We will produce an interim analysis within 8 weeks, and an end-of-study report, which will be submitted to peer-reviewed journals.
Keywords: EPIDEMIOLOGY, Public health, BIOTECHNOLOGY & BIOINFORMATICS, PREVENTIVE MEDICINE, PRIMARY CARE, Adverse events
Strengths and limitations of this study.
This study sets out the first methods for near-real-time enhanced passive surveillance of seasonal influenza vaccine using routinely collected data.
Customised adverse drug reaction cards may enhance reporting over standard passive surveillance, which may result in under-reporting of less severe symptoms not requiring medical attendance.
The methods outlined in this study have the potential to be expanded to other brands.
The practice recruitment is intended to have wide and representative coverage of England.
The data of the participating patients will be thoroughly protected by means of a pseudonymising algorithm that allowed removal of strong identifiers.
This feasibility study has not been powered or designed to detect rare events or detect significant statistical differences of adverse event rates across brands.
We are also exploring the feasibility of using rates of adverse events in non-vaccinated patients as a basis for comparison.
Introduction
EMA guidance
In response to a recent expansion of national vaccination programmes in European Union (EU) member states, the European Medicines Agency (EMA) has released interim guidance on enhanced safety surveillance for seasonal influenza vaccines in the EU.1 This set out new standards for surveillance that all marketing authorisation holders (MAHs) providing vaccines in the EU must address. The key objective of the EMA enhanced safety surveillance is to rapidly detect a significant increase in the frequency and/or severity of expected reactions (local, systemic or allergic reactions) that may indicate a potential or more serious risk as exposure to the vaccine increases.
Since 2015, European regulatory requirements to evaluate the safety and immunogenicity of seasonal influenza vaccines in small-scale clinical trials were withdrawn.2 Such trials had insufficient power to adequately evaluate safety concerns arising from annual formulation changes (eg, adverse events occurring at a rate of 1%–2%). These clinical trials are to be replaced by enhanced, preferably active, safety monitoring and vaccine effectiveness assessments.
The EMA Interim Guidance on enhanced safety surveillance for seasonal influenza vaccines in the EU suggested that there would be three options envisioned for enhanced surveillance:
Enhanced active surveillance (post-authorisation safety studies): active follow-up of a cohort of children and adults for 7 days after immunisation for reactogenicity end points/adverse events.
Enhanced passive surveillance: rapidly estimate vaccine usage and facilitate adverse drug reaction (ADR) reporting in order to determine reporting rate as a surrogate of incidence of the adverse events of interest (AEI).
Data mining or other use of electronic health record (EHR)/ computerised medical record (CMR).
We opted for enhanced passive surveillance because while highly computerised medical record systems maximise the likelihood of reliably capturing the AEIs, we felt this needed enhancement through the use of customised ADR cards. These cards were preprinted with the categories of possible adverse events to facilitate the reporting and the subsequent coding of events. They also contained a specific tick box when no AEIs were experienced leading to a reasonably acceptable return rate.
We expect that by enhancing surveillance with a customised card and encouraging patients to directly report their symptoms, we will more reliably detect a greater number of events. The proposed approach was designed to meet the EMA enhanced passive surveillance definition.
The present collaborative pilot study between marketing authorisation holder GlaxoSmithKline Biologicals (GSK) and the Clinical Informatics and Health Outcomes Research Group at the University of Surrey builds on the lessons learnt from the pilot study (EPI-FLU-045 VS UK) implemented during the 2014/2015 influenza season and aims to address the EMA commitment for enhanced safety surveillance of seasonal vaccines in Europe. We will begin data collection on 1 September 2016, and the analysis will be completed on 31 March 2017.
The EPI-FLU-045 VS UK pilot study showed that the proposed surveillance setting in the UK was suitable to rapidly detect and evaluate potential new safety concerns each influenza season. The primary purpose of the 2016/2017 pilot study is to improve the combination of an ADR card-based system and the use of routine data to collect adverse events following vaccination with seasonal influenza vaccines.
The RCGP RSC network
The Clinical Informatics Research Group, in the Department of Clinical and Experimental Medicine at the University of Surrey, is the home of the data and analysis hub for the Royal College of General Practitioners Research and Surveillance Centre (RCGP RSC). The RCGP RSC provides a national primary care surveillance system and is supported by Public Health England (PHE). The RCGP RSC network of practices has a membership designed to give representative coverage of 1.5%–2% of the English population.3 The RCGP RSC has been described as the gold standard sentinel network.
The most important work of the RCGP RSC network is its influenza surveillance; many practices have been involved in this work for decades.4 Data are uploaded from the network on a weekly basis to a secure sever, with the possibility to switch the frequency of the release to a twice-weekly upload during epidemics. The methods developed by the University of Surrey will be used in this passive enhanced safety surveillance study, with a focus on reporting on adverse events.
Seasonal influenza vaccination in England
Seasonal influenza vaccines present several specific challenges for pharmacovigilance. These include immunisation in large population cohorts in a relatively short and fixed time period each year, and multiplicity of vaccine products on the market with the need to conduct product-specific safety surveillance. In the UK, the 2015/2016 influenza plan recommended the following groups to be vaccinated5 :
People aged 65 years or over (based on age on 31 March 2016);
People aged from 6 months to less than 65 years with a medical condition: chronic (long-term) respiratory disease, such as severe asthma; chronic obstructive pulmonary disease or bronchitis; chronic heart disease, such as heart failure; chronic kidney disease, stages 3–5; chronic liver disease; chronic neurological disease, such as Parkinson’s disease or motor neuron disease, or a learning disability; diabetes; splenic dysfunction; immunocompromised due to disease (such as HIV/AIDS) or treatment (such as cancer treatment);
All pregnant women (including those women who become pregnant during the influenza season);
All those aged 2, 3 and 4 years (but not 5 years or older) on 31 August 2015 (ie, date all children of school years 1 and 2 age through locally commissioned arrangements);
Primary school-aged children in areas that participated in primary school pilots in 2014/2015;
People living in long-stay residential care homes or other long-stay care facilities;
People who are in receipt of a carer’s allowance, or those who are the main carer of an older or disabled person;
Household contacts of immunocompromised individuals.
The list above is not exhaustive, and the healthcare practitioner should apply clinical judgement to take into account the risk of influenza exacerbating any underlying disease.
Expansion of national vaccination has created a greater need for timely information and reassurance on the balance of risks and benefits for those receiving the vaccines. The collaborative pilot study is conceived in response to the EU requirements triggered by the EMA’s call for enhanced safety surveillance in Europe. The continuation of the pilot study in the 2016/2017 season will help to build a framework for passive enhanced safety surveillance in England, but will also contribute to an EU-wide programme of enhanced safety surveillance for seasonal influenza vaccines.
Research methods
This published protocol is a summary of the full protocol, submitted for ethical approval, and the long version is available as an online supplementary material.
bmjopen-2016-015469supp001.pdf (3MB, pdf)
Objectives and end points
Primary objective
To estimate on a weekly basis the crude and cumulative incidence rate of AEIs within 7 days following vaccination with a seasonal influenza vaccine, using passive surveillance of general practitioner (GP) EHR systems enhanced by a card-based ADR reporting system. Extracted data will be presented overall, by brand (Fluarix Tetra vs others), by EMA defined age strata and UK Department of Health Chief Medical Officer (CMO)- specified risk groups.
Secondary objective
To estimate on a weekly basis the vaccine uptake among the subjects registered in the enrolled GP practices, by age strata (6 months to 5 years; 6–12 years; 13–18 years; ≥18–65 years; >65 years) and CMO-specified risk groups.
Primary end points
Occurrence and onset dates of AEIs within 7 days postvaccination reported using a card-based ADR reporting system in vaccinated patients overall, by brand (also indicating those for whom brand data are unavailable), by age strata (6 months to 5 years; 6–12 years; 13–17 years; 18–65 years; >65 years) and CMO-specified risk groups. AEIs will be presented by categories depending of the nature of the event.
Fever or other febrile illness
Local reactions
General reaction (eg, fatigue and myalgia)
Secondary end points
Seasonal influenza vaccination status among the subjects registered in the enrolled GP practices, vaccine brand, by age strata (6 months to 5 years; 6–12 years; 13–18 years; ≥18–65 years; >65 years) and CMO-specified risk groups and date of vaccine administration collected in the CMR system.
Study design
Study setting and population
The proposed pilot study (EPI-FLU-046 VS UK) is to follow a cohort of patients who would be exposed to seasonal influenza vaccination in the months between 1 September 2016 and 30 November 2016. The final data collection will occur on 10 January 2017 to allow for any delays in records of up to 6 weeks.
Invitation letters will be sent to GP practices ordering mainly GSK’s Fluarix Tetra vaccine for the 2016/2017 season, and evenly representative of geographical locations and systems. Practices will be reimbursed for their involvement in this study, according to the National Institute of Health Research guidelines for industry-sponsored studies.6
For this pilot project, routinely collected primary care data from up to 10 GP practices will be extracted, to provide passive surveillance. However, this passive surveillance is enhanced by all practices additionally using a card-based ADR reporting system. In last year’s study, the card-based ADR reporting system used the Yellow Card developed by the UK Medicines and Healthcare products Regulation Agency. We have developed a more specific ADR card for use in this study, which will be distributed to practices. The new ADR card is preprinted with the categories of likely AEIs the EMA require surveillance for, to facilitate the recording of AEIs and to make their coding into the GP EHR system easier. There was also a tick box for no AEI.
Patients will be provided with the appropriate ADR reporting card and invited to return the card to the GP practices within 7 days, but not later than 14 days, postvaccination.7 To protect confidentiality, this ADR card will be returned to the practice, and data from it will be recorded in that patient’s EHR. The data will be used to estimate proportions of AEIs among influenza-vaccinated individuals.
Inclusion criteria
All patients receiving a seasonal influenza vaccine between 1 September 2016 and 30 November 2016 in 1 of the 10 volunteer practices are eligible for inclusion in the analysis. The main inclusion criteria for practices is that they state their principal vaccine supplier will be GSK.
Exclusion criteria
Patients who have explicitly opted out of data sharing will be excluded from the analysis. We will identify these patients using the opt-out codes within GP information systems where the patients have made an explicit choice to opt out; patients will be informed of their option to opt out via posters in the practices and information sheets accompanying the ADR cards.
Sample size calculation
The average practice size in England and Wales is 7034,8 and we estimate that data will be collected on a population of approximately 70 340 patients (across 10 practices). In the period from September to December 2015, the seasonal influenza vaccine uptake for those over 65 years old was 71.0%; for those in a clinical risk group aged 6 months to 65 years, the uptake was 45.1%; and for pregnant women, it was 42.3%. We have estimated influenza vaccine uptake using the coverage estimates published by PHE.9
The minimum needed target population to be medically followed by the GPs is estimated at 50 000 subjects (approximately 5000 per practice). We expect to enrol at least 5000 vaccinated subjects with 7 days of follow-up after vaccination (as per EMA interim guidance request). This sample size estimation sets out to estimate the probability to observe at least one AEI in the study population and evaluate the level of ‘certainty’ around this finding; this is over the 14-week period of enhanced surveillance (1 September to 30 November 2016).
We have not taken into account any effect of clustering in our surveillance study design or power calculation. Similarities, or homogeneity, between subjects in clusters reduce the variability of their responses, compared with that expected from a random sample.
The cluster effect has not been taken into consideration in the calculation of the sample size. It may increase the relative SE (RSE) and thus decrease the precision around the proportions presented below. Nevertheless, this will be accounted for during the analysis.
Table 1 shows the 95% CI, the probability of observing at least one AEI during the study period in the study cohort and the RSE for a range of scenarios in terms of cohort size, vaccine coverage and expected probability of AEI.10 With an overall sample size of a minimum of about 50 000 subjects medically followed by the enrolled GP practices, a follow-up period of 14 weeks, a vaccine coverage of 5%, 10% or 20% and an expected probability of AEI varying from 0.01% to 20%, the corresponding probability to observe at least one event in our study population varies from 2% to 100%, and the associated RSE varies from 2.0% to 200% depending on the scenario.
Table 1.
CIs, RSE and probability to observe at least one AEI according to expected probabilities of occurrence of AEI
| Expected population medically followed by the enrolled practices |
Vaccine coverage (%) | Vaccinated subjects | Subjects with events | Expected proportion of subjects with ≥1 AEI reported (%) | Lower 95% CL (%) |
Upper 95% CL (%) |
Probability to observe ≥1 AEIs in the study population (%) |
Associated RSE (%) |
| 50 000 | 20 | 10 000 | 2 000 | 20.00 | 19.2 | 20.8 | 100.00 | 2.0 |
| 50 000 | 20 | 10 000 | 1500 | 15.00 | 14.3 | 15.7 | 100.00 | 2.4 |
| 50 000 | 20 | 10 000 | 1 000 | 10.00 | 9.4 | 10.6 | 100.00 | 3.0 |
| 50 000 | 20 | 10 000 | 500 | 5.00 | 4.6 | 5.4 | 100.00 | 4.4 |
| 50 000 | 20 | 10 000 | 400 | 4.00 | 3.6 | 4.4 | 100.00 | 4.9 |
| 50 000 | 20 | 10 000 | 200 | 2.00 | 1.7 | 2.3 | 100.00 | 7.0 |
| 50 000 | 20 | 10 000 | 100 | 1.00 | 0.8 | 1.2 | 100.00 | 9.9 |
| 50 000 | 20 | 10 000 | 10 | 0.10 | 0.0 | 0.2 | 99.95 | 31.6 |
| 50 000 | 20 | 10 000 | 9 | 0.09 | 0.0 | 0.2 | 99.88 | 33.3 |
| 50 000 | 20 | 10 000 | 8 | 0.08 | 0.0 | 0.2 | 99.70 | 35.3 |
| 50 000 | 20 | 10 000 | 7 | 0.07 | 0.0 | 0.1 | 99.27 | 37.8 |
| 50 000 | 20 | 10 000 | 6 | 0.06 | 0.0 | 0.1 | 98.27 | 40.8 |
| 50 000 | 20 | 10 000 | 5 | 0.05 | 0.0 | 0.1 | 95.96 | 44.7 |
| 50 000 | 20 | 10 000 | 4 | 0.04 | 0.0 | 0.1 | 90.85 | 50.0 |
| 50 000 | 20 | 10 000 | 3 | 0.03 | 0.0 | 0.1 | 80.09 | 57.7 |
| 50 000 | 2 | 10 000 | 2 | 0.02 | 0.0 | 0.1 | 59.40 | 70.7 |
| 50 000 | 20 | 10 000 | 1 | 0.01 | 0.0 | 0.1 | 26.42 | 100.0 |
| 50 000 | 10 | 5 000 | 250 | 5.00 | 4.4 | 5.6 | 100.00 | 6.2 |
| 50 000 | 10 | 5 000 | 200 | 4.00 | 3.5 | 4.6 | 100.00 | 6.9 |
| 50 000 | 10 | 5 000 | 100 | 2.00 | 1.6 | 2.4 | 100.00 | 9.9 |
| 50 000 | 10 | 5 000 | 50 | 1.00 | 0.7 | 1.3 | 100.00 | 14.1 |
| 50 000 | 10 | 5 000 | 5 | 0.10 | 0.0 | 0.2 | 95.96 | 44.7 |
| 50 000 | 10 | 5 000 | 4.5 | 0.09 | 0.0 | 0.2 | 93.90 | 47.1 |
| 50 000 | 10 | 5 000 | 4 | 0.08 | 0.0 | 0.2 | 90.85 | 50.0 |
| 50 000 | 10 | 5 000 | 3.5 | 0.07 | 0.0 | 0.2 | 86.42 | 53.4 |
| 50 000 | 10 | 5 000 | 3 | 0.06 | 0.0 | 0.2 | 80.09 | 57.7 |
| 50 000 | 10 | 5 000 | 2.5 | 0.05 | 0.0 | 0.2 | 71.28 | 63.2 |
| 50 000 | 10 | 5 000 | 2 | 0.04 | 0.0 | 0.1 | 59.40 | 70.7 |
| 50 000 | 10 | 5 000 | 1.5 | 0.03 | 0.0 | 0.1 | 44.22 | 81.6 |
| 50 000 | 10 | 5 000 | 1 | 0.02 | 0.0 | 0.1 | 26.42 | 100.0 |
| 50 000 | 10 | 5 000 | 0.5 | 0.01 | 0.0 | 0.1 | 9.02 | 141.4 |
| 50 000 | 5 | 2 500 | 125 | 5.00 | 4.2 | 5.9 | 100.00 | 8.7 |
| 50 000 | 5 | 2 500 | 100 | 4.00 | 3.3 | 4.8 | 100.00 | 9.8 |
| 50 000 | 5 | 2 500 | 50 | 2.00 | 1.5 | 2.6 | 100.00 | 14.0 |
| 50 000 | 5 | 2 500 | 25 | 1.00 | 0.6 | 1.5 | 100.00 | 19.9 |
| 50 000 | 5 | 2 500 | 12.5 | 0.50 | 0.3 | 0.9 | 100.00 | 28.2 |
| 50 000 | 5 | 2 500 | 2.5 | 0.10 | 0.0 | 0.3 | 71.29 | 63.2 |
| 50 000 | 5 | 2 500 | 2.25 | 0.09 | 0.0 | 0.3 | 65.76 | 66.6 |
| 50 000 | 5 | 2 500 | 2 | 0.08 | 0.0 | 0.3 | 59.41 | 70.7 |
| 50 000 | 5 | 2 500 | 1.75 | 0.07 | 0.0 | 0.3 | 52.22 | 75.6 |
| 50 000 | 5 | 2 500 | 1.5 | 0.06 | 0.0 | 0.3 | 44.22 | 81.6 |
| 50 000 | 5 | 2 500 | 1.25 | 0.05 | 0.0 | 0.3 | 35.54 | 89.4 |
| 50 000 | 5 | 2 500 | 1 | 0.04 | 0.0 | 0.2 | 26.42 | 100.0 |
| 50 000 | 5 | 2 500 | 0.75 | 0.03 | 0.0 | 0.2 | 17.33 | 115.5 |
| 50 000 | 5 | 2 500 | 0.5 | 0.02 | 0.0 | 0.2 | 9.02 | 141.4 |
| 50 000 | 5 | 2 500 | 0.25 | 0.01 | 0.0 | 0.2 | 2.65 | 200.0 |
AEI, adverse event of interest; RSE, relative SE.
Table 2 shows the evolution by week of the 95% CI, the cumulative probability of observing at least one AEI in the study cohort and the RSE in the course of the study for a range of scenarios in term of cohort size, vaccine coverage and probability of AEI of 1%. With an overall sample size of a minimum of about 50 000 subjects medically followed by the enrolled GP practices, a follow-up period of 14 weeks and a vaccine coverage of 5%, 10% or 20%, the corresponding probability to observe at least one event in our study population varies from 53% to 99% after week 1, and the associated relative SE varies from 53% to 37% depending on the scenario.
Table 2.
CIs RSE cumulative probability to observe at least one AEI by week associated with a probability of occurrence of event of 1%
| Week | Expected population medically followed by the enrolled practices |
Cumulative vaccine coverage after 14 weeks (%) | Cumulative no of vaccinated subjects | Cumulative no of subjects reported ≥1 AEIs |
Average proportion of AEI reported (%) | Lower 95% CL (%) |
Upper 95% CL (%) |
Cumulative probability to observe at least one event (%) |
Associated RSE (%) |
| 1 | 50 000 | 20 | 714 | 7 | 1.00 | 0.4 | 2.0 | 99.37 | 37.2 |
| 2 | 50 000 | 20 | 1 428 | 14 | 1.00 | 0.5 | 1.6 | 100.00 | 26.3 |
| 3 | 50 000 | 20 | 2 142 | 21 | 1.00 | 0.6 | 1.5 | 100.00 | 21.5 |
| 4 | 50 000 | 20 | 2 857 | 28 | 1.00 | 0.7 | 1.4 | 100.00 | 18.6 |
| 5 | 50 000 | 20 | 3 571 | 35 | 1.00 | 0.7 | 1.4 | 100.00 | 16.7 |
| 6 | 50 000 | 20 | 4 285 | 42 | 1.00 | 0.7 | 1.3 | 100.00 | 15.2 |
| 7 | 50 000 | 20 | 5 000 | 50 | 1.00 | 0.7 | 1.3 | 100.00 | 14.1 |
| 8 | 50 000 | 20 | 5 714 | 57 | 1.00 | 0.8 | 1.3 | 100.00 | 13.2 |
| 9 | 50 000 | 20 | 6 428 | 64 | 1.00 | 0.8 | 1.3 | 100.00 | 12.4 |
| 10 | 50 000 | 20 | 7 142 | 71 | 1.00 | 0.8 | 1.3 | 100.00 | 11.8 |
| 11 | 50 000 | 20 | 7 857 | 78 | 1.00 | 0.8 | 1.2 | 100.00 | 11.2 |
| 12 | 50 000 | 20 | 8 571 | 85 | 1.00 | 0.8 | 1.2 | 100.00 | 10.7 |
| 13 | 50 000 | 20 | 9 285 | 92 | 1.00 | 0.8 | 1.2 | 100.00 | 10.3 |
| 14 | 50 000 | 20 | 10 000 | 100 | 1.00 | 0.8 | 1.2 | 100.00 | 9.9 |
| 1 | 50 000 | 10 | 357 | 3 | 1.00 | 0.2 | 2.4 | 87.26 | 52.7 |
| 2 | 50 000 | 10 | 714 | 7 | 1.00 | 0.4 | 2.0 | 99.37 | 37.2 |
| 3 | 50 000 | 10 | 1 071 | 10 | 1.00 | 0.4 | 1.7 | 99.98 | 30.4 |
| 4 | 50 000 | 10 | 1 428 | 14 | 1.00 | 0.5 | 1.6 | 10000 | 26.3 |
| 5 | 50 000 | 10 | 1 785 | 17 | 1.00 | 0.6 | 1.5 | 100.00 | 23.6 |
| 6 | 50 000 | 10 | 2 142 | 21 | 1.00 | 0.6 | 1.5 | 100.00 | 21.5 |
| 7 | 50 000 | 10 | 2 500 | 25 | 1.00 | 0.6 | 1.5 | 100.00 | 19.9 |
| 8 | 50 000 | 10 | 2 857 | 28 | 1.00 | 0.7 | 1.4 | 100.00 | 18.6 |
| 9 | 50 000 | 10 | 3 214 | 32 | 1.00 | 0.7 | 1.4 | 100.00 | 17.6 |
| 10 | 50 000 | 10 | 3 571 | 35 | 1.00 | 0.7 | 1.4 | 100.00 | 16.7 |
| 11 | 50 000 | 10 | 3 928 | 39 | 1.00 | 0.7 | 1.4 | 100.00 | 15.9 |
| 12 | 50 000 | 10 | 4 285 | 42 | 1.00 | 0.7 | 1.3 | 100.00 | 15.2 |
| 13 | 50 000 | 10 | 4 642 | 46 | 1.00 | 0.7 | 1.3 | 100.00 | 14.6 |
| 14 | 50 000 | 10 | 5 000 | 50 | 1.00 | 0.7 | 1.3 | 100.00 | 14.1 |
| 1 | 50 000 | 5 | 178 | 1 | 1.00 | 0.0 | 3.1 | 53.24 | 74.6 |
| 2 | 50 000 | 5 | 357 | 3 | 1.00 | 0.2 | 2.4 | 87.26 | 52.7 |
| 3 | 50 000 | 5 | 535 | 5 | 1.00 | 0.3 | 2.2 | 97.04 | 43.0 |
| 4 | 50 000 | 5 | 714 | 7 | 1.00 | 0.4 | 2.0 | 99.37 | 37.2 |
| 5 | 50 000 | 5 | 892 | 8 | 1.00 | 0.4 | 1.8 | 99.87 | 33.3 |
| 6 | 50 000 | 5 | 1 071 | 10 | 1.00 | 0.4 | 1.7 | 99.98 | 30.4 |
| 7 | 50 000 | 5 | 1 250 | 12 | 1.00 | 0.5 | 1.7 | 100.00 | 28.1 |
| 8 | 50 000 | 5 | 1 428 | 14 | 1.00 | 0.5 | 1.6 | 100.00 | 26.3 |
| 9 | 50 000 | 5 | 1 607 | 16 | 1.00 | 0.6 | 1.6 | 100.00 | 24.8 |
| 10 | 50 000 | 5 | 1 785 | 17 | 1.00 | 0.6 | 1.5 | 100.00 | 23.6 |
| 11 | 50 000 | 5 | 1 964 | 19 | 1.00 | 0.6 | 1.5 | 100.00 | 22.5 |
| 12 | 50 000 | 5 | 2 142 | 21 | 1.00 | 0.6 | 1.5 | 100.00 | 21.5 |
| 13 | 50 000 | 5 | 2 321 | 23 | 1.00 | 0.6 | 1.5 | 100.00 | 20.7 |
| 14 | 50 000 | 5 | 2 500 | 25 | 1.00 | 0.6 | 1.5 | 100.00 | 19.9 |
AEI, adverse event of interest; RSE, relative SE.
Data sources
In this passive enhanced safety surveillance, there are two data sources: general practice EHR data, providing passive surveillance, and ADR cards completed by patients providing the enhanced component. The ADR cards are being returned to the patient’s own practice to ensure confidentiality. The data from these cards would also be coded into the EHR and uploaded weekly (figure 1).
Figure 1.
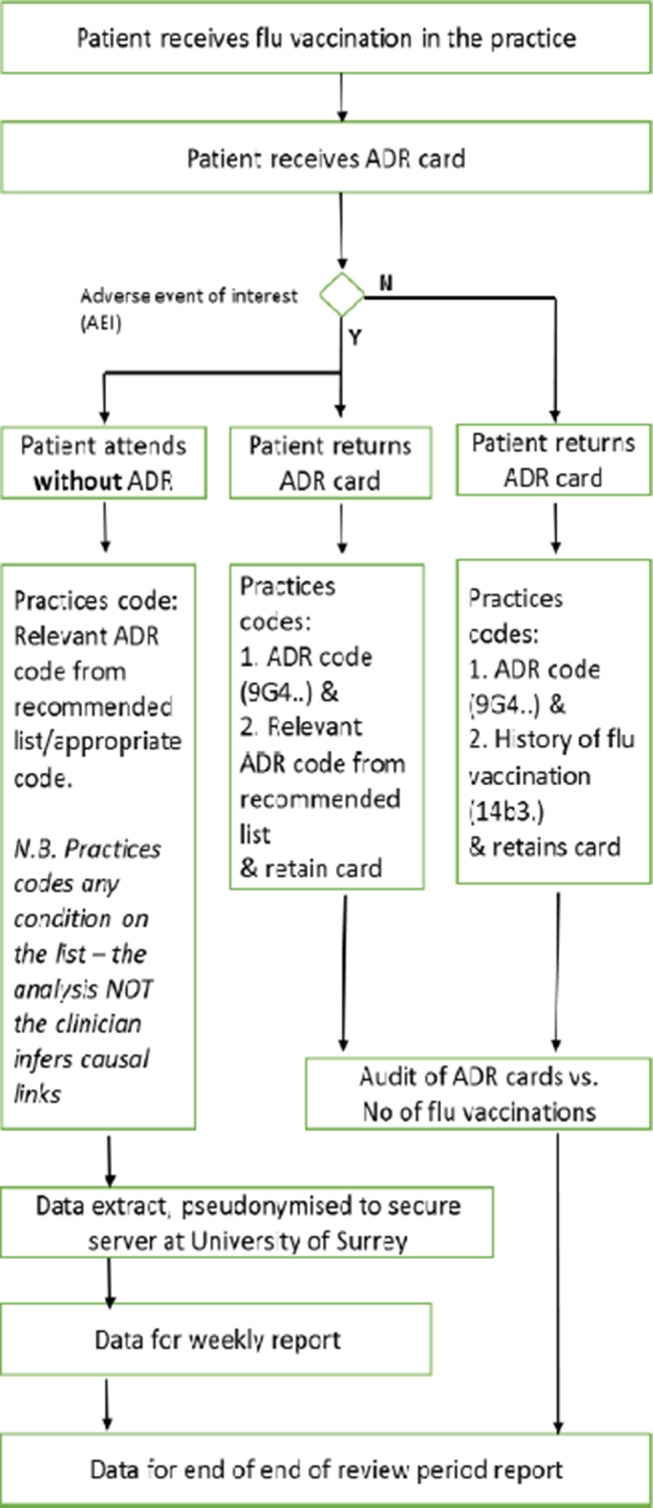
Data capture flowchart : patient receiving influenza vaccination in the surgery.
General Practice EHR data recorded by the practice team. Weekly data about vaccine exposure and any subsequent AEIs will be uploaded (anonymised) to the University of Surrey. The EHR data contains both AEIs recorded by the practice team as well as data reported to the practice on an ADR by a vaccinated patient.
ADR cards completed by patients. Among the 10 participating GP practices, patients who are vaccinated against influenza will be provided ADR cards. These ADR cards, customised following practice feedback to match EMA requirements, collect AEIs reported after the receipt of influenza vaccination.
These data originating from the two sources (patient completed ADR card or practice recorded) will be then imported (anonymised) into the secure servers of the University of Surrey. The final dataset will therefore combine data routinely collected for all patients registered with the 10 participating sites and data collected form the ADR cards and encoded during the 2016/2017 influenza season. In addition to the adverse event data, we will extract demographics, vaccination status and relevant comorbidities.
We will only extract coded data, that is, where the GP or other health professional codes a disease or symptom into the EHR system.11 The overwhelming majority of the large volume of research that has come out of UK primary care is based on coded data.12 The richness of primary care data are such that we anticipate being able to detect important AEIs.13 We will request practices to use the relevant Read code for ADR notifications, when recording data from a returned card (Read Code: 9G4 Adverse drug reaction notification).
Data analyses
We will interpret coded data by the creation of ontologies that we will map to case definitions, where available. However, we do not have the in-depth descriptions required for case definition found, for example, in clinical trials. We will be inferring meaning from brief clinical coded information, though we have considerable experience of this and will have the opportunity to confirm with practices and practitioners how to interpret their clinical records.
Statistical analysis will consist primarily of descriptive statistics: rates and proportions for categorical data and summary statistics for continuous variables. Confidence intervals will be calculated; however, due to the effects of clustering and practice differences in this relatively small pilot, these are likely to be wide. All statistical analyses will be conducted using R Studio.
Analyses of the primary objective
All analyses will be carried out by overall, by brand (Fluarix Tetra vs others), by age strata (6 months to 5 years; 6–12 years; 13–17 years; 18–65 years; >65 years) and CMO-specified risk groups.
To estimate on a weekly basis the crude incidence rate of AEIs within 7 days,
the denominator will consist of the number vaccinated subjects receiving a vaccination card and reaching 7 days of follow-up postvaccination during the week of interest and cumulatively since the beginning of the study.
The numerator will encompass all vaccinated subjects reporting at least one AEI within 7 days following vaccination with a seasonal influenza vaccine.
Ethics and dissemination
Ethical review
In ‘Defining Research’ (http://www.hra.nhs.uk/documents/2013/09/defining-research.pdf), the National Research Ethics Service (NRES) guidance suggests that surveillance does not require formal review by a Research Ethics Committee. The research team will however seek an opinion from the NRES’s Proportional Review system to check if formal approval from a NHS Research Ethics Committee (REC) is needed prior to the commencement of the study, as well as Section 251 approval. Ethical approval was granted by the Proportionate Review Sub-committee of the North East—Newcastle & North Tyneside 2 REC on 5 August 2016 (REC reference: 16/NE/0271). Section 251 application was not deemed necessary by the Health Research Authority and the study received approval on 1 September 2016 (IRAS ID: 2 11 560).
Data extraction and data management
The method and governance procedure has been developed by the University of Surrey, using an approved provider, Apollo Medical Software Solutions. Alternatively, we will use another approved data extraction supplier or securely extract the relevant study data ourselves using standard data extraction tools such as Morbidity Information Query Export Syntax (MIQUEST), a Department of Health sponsored data extract tool. Data extractions will be conducted in accordance with the Research Group’s standard operating procedures in data extraction, pseudonymisation and transfer.
All data are strongly encrypted by a combination of symmetric and asymmetric encryption algorithms, Triple DES1 and RSA 10242 before transmission, and utilises public and private key pairs unique to each project. Data are pseudonymised as near to source as possible. Pseudonymisation is applied at this stage to allow for backwards identification should there be a need to do so as part of an ethically approved study.
Pseudonymisation is a process that involves the removal of all personal identifiers from data—such as name, date of birth and so on. However, there is a risk that if data are linked to other data, a person might be identified.14 Therefore, although all identifiers are removed, we keep data encrypted during transfer and on a secure network that meets NHS Information Governance standards to minimise the risk of reidentification. Pseudonymisation is the standard approach for this type of surveillance. A legally binding definition of pseudonymisation has been introduced into European law15 on the recommendation of the European Data Protection Supervisor.16
All data processing and analysis in the present proposed study will be conducted within the secure IT environment of the Clinical Informatics Research Group, at the University of Surrey. The information security policies and procedures of the Research Group have been approved by the NHS Health and Social Care Information Centre as meeting Information Governance Toolkit standards.17 The University of Surrey is registered with the Information Commissioner’s Office Data Protection Register and is compliant with the Data Protection Act and other legislations.
In line with the principle of the Data Protection Act 1998, data subjects will be informed of the uses of their data in this study. Participating GP practices will be asked to display project information in their website, and project information posters in reception areas, from when the practice has consented to take part in the study and until the study is completed. We will respect the codes in the data indicating that a patient does not wish to have their record available for research; we will, however, seek to report the number of patients within a practice who have chosen to opt out.
No personally identifiable information, such as NHS numbers, postcodes, dates of birth and so on, will be available to GSK, third parties or disclosed in publications. Additionally, no patient-level data will be sent to GSK to remove any possibility that any individual patient might be reidentified. GSK will also be blinded to practice identities and the locality at which any AEI occurs, other than where the patient gives consent or on their own chooses to report any condition in line with best practice.
Safety reporting, including routine pharmacovigilance
This study’s primary end points are safety related. However, it will be clearly communicated to participating practices that the study does not replace AEI reporting that would occur as part of routine practice. If a GP felt an AEI merited reporting, they should do so in whatever way they would generally do so. If the team at the University of Surrey becomes aware of a serious adverse event (SAE) experienced by a study participant, the SAE should be reported to GSK within 24 hours of awareness, in writing. An SAE is defined as any untoward medical occurrence that
Results in death,
Life-threatening (where the participant is at risk of death),
Requires hospitalisation or prolongation of existing hospitalisation,
Results in disability/incapacity (where there is a substantial disruption of a person’s ability to conduct normal life functions),
Important medical events: events that may not be immediately life threatening or result in death or hospitalisation but may jeopardise the study participant or may require medical or surgical intervention to prevent one of the other outcomes listed in the above definition.
Dissemination and public register disclosure
The outputs from the research will be disseminated primarily through peer-reviewed papers in high-impact journals within the domains of primary care, surveillance, vaccines and infectious diseases.18 19 We will present findings at relevant seminars and conferences. The University of Surrey, in accordance with GSK policy, will post a summary of the study protocol and results within 12 months of study completion and following review and comment by GSK on GSK’s Clinical Study Register (accessible at http://www.gsk-clinicalstudyregister.com and at www.clinicaltrials.gov).
Supplementary Material
Footnotes
Contributors: SdeL
developed the study design and is the main writer of the protocol.
GDS
helped with the study design development and contributed to the writing.
AC
contributed to the methods and analysis section of this protocol.
FH
contributed to the sample size and analysis section of this protocol.
IY
helped develop the practice recruitment methods and contributed to writing the protocol.
FL
reviewed the protocol and contributed to the writing.
RB
helped develop the data flow structure and contributed to writing the protocol.
FF
extensively reviewed the protocol and contributed to the writing.
KS
reviewed the protocol and contributed to the writing.
TC
contributed to the ethical implications and background section of this protocol.
Funding: GlaxoSmithKline Biologicals was the funding source and was involved in study design and interpretation, study number 202056.
Competing interests: SdeL and AC participate in a European consortium called IMOVE+ funded by Horizon 2020 to monitor seasonal influenza vaccine effectiveness across Europe. GDS reports he was employed by Business & Decision Life Sciences on behalf of GSK Vaccines at the time of the study and is now employed by the GSK group of companies. FH are employees of the GSK group of companies. GDS holds shares in the GSK group of companies as part of their employee remuneration.
Ethics approval: Health Research Authority -Research Ethics Committee reference: 16/NE/0271.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data sharing statement: Once the study is completed, we will publish all relevant aggregated results. Unpublished aggregated results could be made available, on request, by the authors.
References
- 1. European Medicines Agency (EMA) Pharmacovigilance Risk Assessment Committee (PRAC). Interim guidance on enhanced safety surveillance for seasonal influenza vaccines in the EU. London: EMA; 2014. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2014/04/WC500165492.pdf (accessed May 2017). [Google Scholar]
- 2. European Medicines Agency (EMA). Explanatory note on the withdrawal of the note for guidance on harmonization of requirements for influenza vaccines and of the core SmPC/PL for inactivated seasonal influenza vaccines. London: EMA, 2014. http://www.ema.europa.eu/ema/index.jsp?curl=pages/includes/document/document_detail.jsp?webContentId=WC500161022&mid=WC0b01ac058009a3dc (accessed May 2017). [Google Scholar]
- 3. Correa A, Hinton W, McGovern A, et al. Royal College of General Practitioners Research and Surveillance Centre (RCGP RSC) sentinel network: a cohort profile. BMJ Open 2016;6:e011092 10.1136/bmjopen-2016-011092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Royal College of General Practitioners (RCGP). Research and Surveillance Centre (RSC). http://www.rcgp.org.uk/clinical-and-research/our-programmes/research-and-surveillance-centre.aspx (accessed May 2017).
- 5. Public Health England, Department of Health, NHS England. Flu plan Winter 2015/16. London: PHE publications, 2015. https://www.gov.uk/government/uploads/system/uploads/attachment_data/file/418038/Flu_Plan_Winter_2015_to_2016.pdf (accessed May 2017). [Google Scholar]
- 6. National Institute for Health Research Clinical Research Network. Guide to the NIHR CRN Industry Costing Template: Costing Industry Sponsored Studies. London: NHS National Institute for Health Research, 2014. https://www.crn.nihr.ac.uk/wp-Content/uploads/Industry/GUIDE_to_Industry_Costing_Template-APRIL2014.pdf (accessed May 2017). [Google Scholar]
- 7. Asch DA, Jedrziewski MK, Christakis NA. Response rates to mail surveys published in medical journals. J Clin Epidemiol 1997;50:1129– 36. [DOI] [PubMed] [Google Scholar]
- 8. General and personal medical services. England: Health & Social Care Information Centre, 2014. http://www.hscic.gov.uk/catalogue/PUB13849/nhs-staf-2003-2013-gene-prac-rep.pdf [Google Scholar]
- 9. GOV.UK. Vaccine uptake guidance and the latest coverage data. https://www.gov.uk/government/collections/vaccine-uptake#seasonal-flu-vaccine-uptake (accessed May 2017).
- 10. Clopper CJ, Pearson ES. The use of confidence or fiducial limits illustrated in the case of the binomial. Biometrika 1934;26:404– 13. 10.1093/biomet/26.4.404 [DOI] [Google Scholar]
- 11. de Lusignan S, Codes deLS. Codes, classifications, terminologies and nomenclatures: definition, development and application in practice. Inform Prim Care 2005;13:65–9. 10.14236/jhi.v13i1.580 [DOI] [PubMed] [Google Scholar]
- 12. Kousoulis AA, Rafi I, de Lusignan S. The CPRD and the RCGP: building on research success by enhancing benefits for patients and practices. Br J Gen Pract 2015;65:54–5. 10.3399/bjgp15X683353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. de Lusignan S, van Weel C. The use of routinely collected computer data for research in primary care: opportunities and challenges. Fam Pract 2006;23:253–63. 10.1093/fampra/cmi106 [DOI] [PubMed] [Google Scholar]
- 14. National Health Service (NHS). Pseudonymisation and anonymisation of data policy , 2015. http://www.google.fr/url?url=http://www.nhsbsa.nhs.uk/Documents/NHSBSACorporatePoliciesandProcedures/Pseudonymisation_and_Anonymisation_of_Data_Policy.doc&rct=j&frm=1&q=&esrc=s&sa=U&ved=0ahUKEwjZrqPvz4fNAhWEB8AKHU0IBz4QFggUMAA&usg=AFQjCNF-JPX5zueirRlBylzoF-WAEDNkmg (accessed 09 Jun 2016.)
- 15. European Union, JUSTICE. Reform of EU data protection rules. http://ec.europa.eu/justice/data-protection/reform/index_en.htm (accessed May 2017).
- 16. EUR-lex. EDPS recommendations on the EU’s options for data protection reform. http://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A52015XX0912(01)#ntc14-C_2015301EN.01000101-E0014 (accessed May 2017).
- 17. Health and Social Care Information Centre (HSCIC). Information Governance Toolkit (IGT). https://www.igt.hscic.gov.uk/ (accessed May 2017).
- 18. MRC policy and guidance on sharing of research data from population and patient studies. 2011. http://www.mrc.ac.uk/Ourresearch/Ethicsresearchguidance/datasharing/Policy/index.htm (accessed May 2017).
- 19. Promoting Access to Public Research Data for Scientific, Economic and Social Development. 2007. http://www.oecd.org/sti/sci-tech/38500813.pdf (accessed May 2017).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
bmjopen-2016-015469supp001.pdf (3MB, pdf)