

Abstract
Chemotherapeutic efficacy can be greatly improved by developing nanoparticulate drug delivery systems (nano-DDS) with high drug loading capacity and smart stimulus-triggered drug release in tumor cells. Herein, we report a novel redox dual-responsive prodrug-nanosystem self-assembled by hydrophobic small-molecule conjugates of paclitaxel (PTX) and oleic acid (OA). Thioether linked conjugates (PTX-S-OA) and dithioether inserted conjugates (PTX-2S-OA) are designed to respond to the redox-heterogeneity in tumor. Dithioether has been reported to show redox dual-responsiveness, but we find that PTX-S-OA exhibits superior redox sensitivity over PTX-2S-OA, achieving more rapid and selective release of free PTX from the prodrug nanoassemblies triggered by redox stimuli. PEGylated PTX-S-OA nanoassemblies, with impressively high drug loading (57.4%), exhibit potent antitumor activity in a human epidermoid carcinoma xenograft. This novel prodrug-nanosystem addresses concerns related to the low drug loading and inefficient drug release from hydrophobic prodrugs of PTX, and provides possibilities for the development of redox dual-sensitive conjugates or polymers for efficient anticancer drug delivery.
Keywords: Paclitaxel, small-molecule prodrug, single thioether, redox dual-responsive, prodrug nanoparticles
Graphical abstract
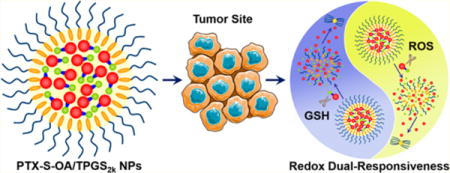
Nanoparticulate drug delivery systems (nano-DDS) for anticancer drug delivery have experienced tremendous development in the past few decades.1–3 However, only a few of these formulations could be actually applied in the clinics. Paclitaxel (PTX), a unique antimitotic agent, is no exception. Although efforts have been made to improve the therapeutic efficacy and reduce the adverse effect of PTX injection (Taxol) by utilizing nano-DDS, very few of them can be applied in a clinical setting.4,5 Additionally, the clinical outcomes of those commercially available PTX nanoformulations are not as satisfactory as expected. Drawbacks of the conventional PTX nano-DDS include low drug loading capacity (usually less than 10%), the risk of premature drug leakage in blood circulation, high tendency to crystallize during storage, and potential biomaterials-induced toxicity.2 In addition to nanotechnology, prodrug formulations have also been extensively studied for PTX.5,6 One of the most successful examples is the conjugate of docosahexaenoic acid (DHA) and PTX (PTX-DHA, Figure S1A), which has entered into clinical trials.7–10 However, the outcomes of the recent phase III clinical trials of PTX-DHA conjugate fell short of expectations, probably due to the extremely low release rate of PTX in the tumor site.10 In another study, it was also reported that almost no free PTX was released from a very similar conjugate (PTX-linoleic acid) after 24 h incubation in PBS and plasma.11 Therefore, it seems clear that a rapid and selective release of free PTX in tumor site from prodrugs could be of crucial importance for rational design of hydrophobic PTX-fatty acid conjugates.
The redox potential gradient between normal cells and tumor cells has been extensively explored for designing stimulus-sensitive nano-DDS.12 Compared to normal cells, tumor cells contain a strongly reducing environment due to the overproduction of intracellular glutathione (GSH).13 Some tumor cells also simultaneously overproduce reactive oxygen species (ROS), leading to increased oxidative stress.14,15 In addition, tumor cells are characteristically heterogeneous in terms of redox potential difference.16,17 Overproduced GSH and ROS may exist in different tumors or even coexist in different regions in the same tumor. Different GSH/ROS levels have also been found at different growth stages of one tumor.16,17 However, most stimulus-sensitive nano-DDS were simply designed to respond to only one of the redox stimulus (GSH or ROS),18 resulting in partial and limited therapeutic efficacy. A dithioether linked amphiphilic conjugate of oligo (ethylene glycol) (OEG) and 7-ethyl-10-hydroxyl-camptothecin (SN38) had been previously reported to respond to the redox heterogeneity in tumor, abbreviated as OEG-2S-SN38 (see the chemical structure in Figure S1B).19 The nanoassemblies of OEG-2S-SN38 conjugates could release SN38 quickly when triggered by either GSH or ROS.19 Drug release from OEG-2S-SN38 occurred via thiolysis by GSH or oxidation of the thioether to hydrophilic sulfoxide or sulfone by ROS.19,20 Inspired by these findings, we proposed that a dithioether linker may also facilitate the rapid and selective drug release from the hydrophobic conjugates of PTX and fatty acids. Furthermore, according to the redox-responsive mechanism of dithioether,19,20 we hypothesized that single thioether may have better efficacy than dithioether as a redox dual-sensitive linkage. We proposed that the single thioether close to the ester bond conjugated to SN38 played the key role in controlling the redox-sensitivity, rather than the sulfur atom that is further away from the ester bond (Figure S1B). In addition to the long distance, dithioether would consume twice as much GSH or ROS as single thioether does.
To test our hypothesis, two novel redox dual-sensitive PTX-fatty acid conjugates (PTX-S-OA and PTX-2S-OA) were designed and synthesized by conjugating OA to PTX via single thioether bond and dithioether bond as linkages, respectively (Figures 1A and S2). An ester bond linked conjugate (PTX-OA) was utilized as nonsensitive control. The chemical structures of PTX-S-OA and PTX-2S-OA were confirmed by MS, 1H NMR, and IR (Figures S3 and S4).
Figure 1.
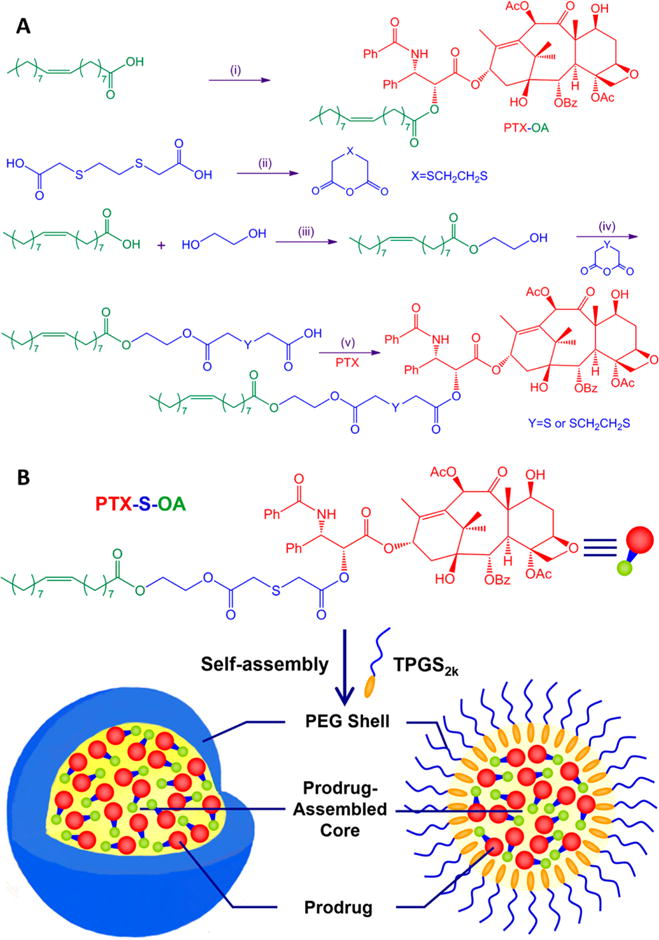
(A) Synthesis of conjugates. (i) PTX, DCC, DMAP, r.t.; (ii) acetic anhydride; (iii) pTsOH, reflux; (iv) EDCI, HOBt, DMAP, r.t.; (v) EDCI, HOBt, r.t. (B) Schematic representation of the preparation of PEGylated prodrug NPs of PTX-S-OA.
One-step nanoprecipitation method was applied to prepare prodrug nanoparticles (NPs).21 When these hydrophobic conjugates dissolved in ethanol were injected into deionized water, assembly spontaneously occurred. Although the self-assembly of hydrophobic PTX conjugates into NPs have been reported elsewhere,21–23 the detailed self-assembly mechanism is still unclear now. However, PTX precipitated immediately when it was used to perform the same nanoprecipitation process. Therefore, one thing for sure is that the hydrophobic moieties conjugated to PTX play key roles in the self-assembly, e.g., the alkyl chains of OA, isoprene unit, squalene chain, and vitamin E (Figure S5). Interestingly, what they all have in common is that all these moieties have double bond or phenyl group in their chemical structures (Figure S5). As a result, the unsaturated alkyl chains or phenyl group may facilitate the intermolecular π–π stacking with the planar structures of PTX (Figure S6). In addition, the unsaturated alkyl chain of OA would provide flexible steric structures of the hydrophobic conjugates (Figure S6). Therefore, the sufficient structural flexibility and possible intermolecular π–π stacking would facilitate thermodynamic feasible status with the lowest possible energy state during the self-assembly of the PTX conjugates.
To achieve long systemic circulation in blood, tocopheryl polyethylene glycol 2000 succinate (TPGS2k, 15% w/w) was added to the prodrug in ethanol prior to precipitation. Dynamic light scattering (DLS) was utilized to determine the particle size and zeta potential. The average diameter of prodrug NPs was around 100 nm (Figure S7), and the zeta potential was determined to be −30 to −50 mV (Table S1). Transmission electron microscopy (TEM) images also revealed the successful fabrication of prodrug NPs with regularly spherical-shaped structures (Figure S8). The PEGylated prodrug NPs (PTX-OA/TPGS2k NPs, PTX-2S-OA/TPGS2k NPs, and PTX-S-OA/TPGS2k NPs) showed significantly higher drug loading efficiency (54.9–66.5% of free PTX, w/w) compared to conventional nanoformulations of PTX (usually less than 10%, w/w).4,5 High drug loading efficiency will greatly facilitate chemotherapeutic efficiency and reduce excipient-associated toxicities.
Although the conjugates of PTX and OA could self-assemble into NPs in deionized water without any surfactant (Figure S8), non-PEGylated formulations showed poor stability in PBS due to the highly hydrophobic surface. As shown in Figure S9, the hydrated sheath of non-PEGylated prodrug NPs was destroyed by the salts in PBS and precipitated. To address this issue, a small amount of TPGS2k was added to prepare PEGylated prodrug NPs for improved stability. As shown in Figure S10A, the PEGylated prodrug NPs showed good colloidal stability in PBS (pH 7.4) supplemented with 10% FBS at 37 °C for 48 h. In addition, these PEGylated prodrug NPs remained stable after being stored for three months at 4 °C (Figure S10B).
We then investigated the in vitro redox dual-responsivity of PTX-S-OA and PTX-2S-OA, with the expectation that the redox responsiveness of single thioether linkage would increase when compared with dithioether linkage. As shown in Figure 2, there was almost no PTX released from PTX-OA/TPGS2k NPs after incubation in PBS (pH 7.4) with H2O2 (a prevailing ROS simulatant) or dithiothreitol (DTT, a prevailing GSH simulatant). In comparison, both PTX-S-OA/TPGS2k NPs and PTX-2S-OA/TPGS2k NPs exhibited redox-responsive drug release in the presence of two opposite stimuli, as expected (Figure 2B–E). More importantly, PTX-2S-OA/TPGS2k NPs exhibited quite a slower release rate, with ~46% of the total amount of prodrug hydrolyzed in the presence of 10 mM H2O2 over 12 h (Figure 2C,D). In contrast, more than 90% of PTX-S-OA was hydrolyzed within 6 h under the same conditions (Figure 2B,D). In addition, PTX-S-OA/TPGS2k NPs released drug at a faster rate in the presence of 10 mM DTT than PTX-2S-OA/TPGS2k NPs. More than 50% of the total amount of PTX was released from PTX-S-OA/TPGS2k NPs in 48 h (Figure 2E). In contrast, only 26% of PTX-2S-OA was hydrolyzed under the same conditions (Figure 2E). The in vitro release results confirmed our hypothesis that the thioether bond would provide a distinct advantage in redox dual-responsivity over the dithioether bond in terms of redox dual-responsivity.
Figure 2.
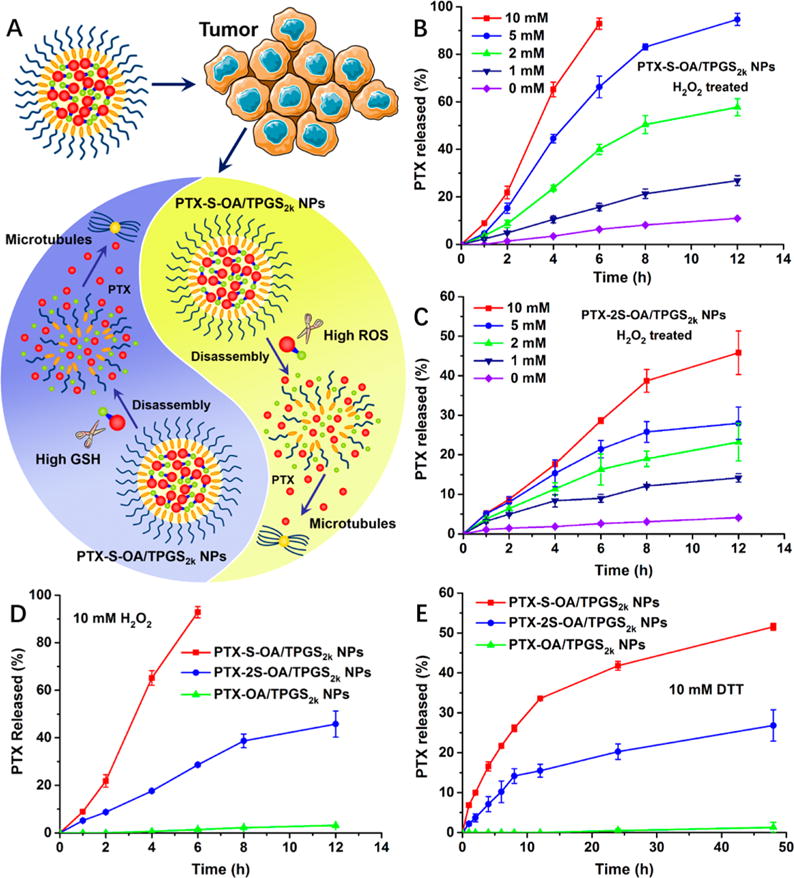
(A) Schematic representation of redox dual-responsive drug release of prodrug NPs in the presence of two opposite stimuli within tumor cells. PTX release from (B) PTX-S-OA/TPGS2k NPs and (C) PTX-2S-OA/TPGS2k NPs in the presence of various concentrations of H2O2. PTX release from PTX-S-OA/TPGS2k NPs, PTX-2S-OA/TPGS2k NPs, and PTX-OA/TPGS2k NPs in the presence of (D) 10 mM H2O2 and (E) 10 mM DTT (n = 3).
The redox dual-responsive mechanism of PTX-S-OA was illustrated in Figure 3B. For GSH triggered drug release, it has been reported that the thiolysis process, initiated by the thiol group of GSH, facilitated the drug release from prodrugs.19 Drug release in response to ROS may take place in three steps (Figure 3): (i) oxidation of the thioether to hydrophilic sulfone;20 (ii) hydrolysis of oleic acid 2-hydroxyethyl ester; (iii) release of active PTX molecule. After the thioether was oxidized to a hydrophilic sulfone, the proximal ester bond would be more easily hydrolyzed. The oleic acid 2-hydroxyethyl ester was attacked first, due to its decreased steric hindrance. Once the long lipophilic OA chain was removed, another ester bond conjugated with PTX could be readily hydrolyzed, and the free PTX molecules were released.
Figure 3.
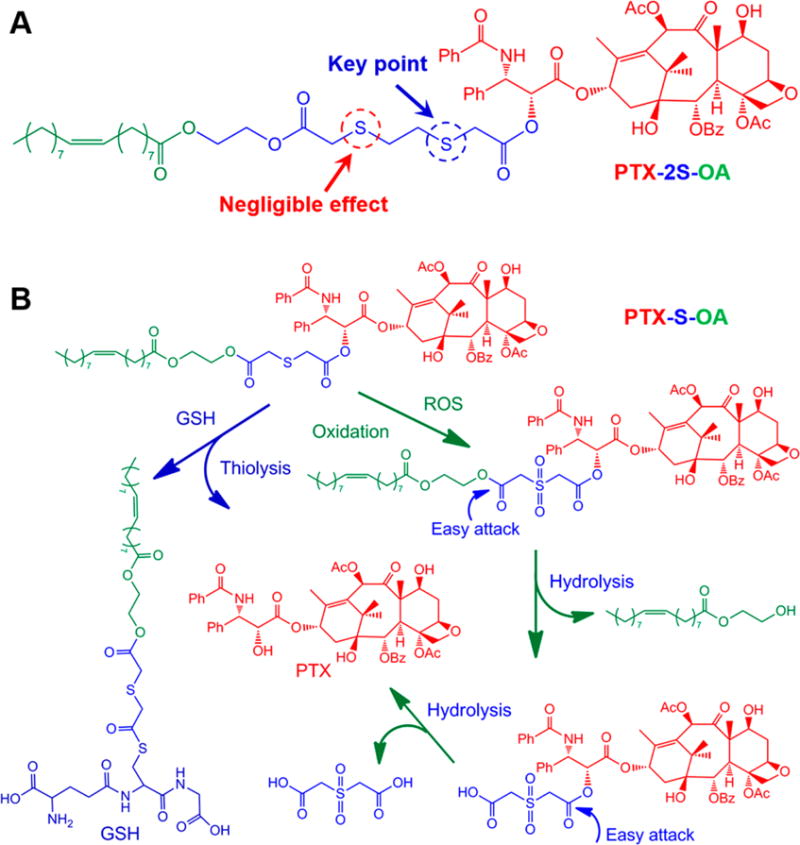
(A) Chemical structure of PTX-2S-OA. (B) Redox-sensitive drug release mechanism of PTX-S-OA triggered by GSH/ROS.19,20
The question of why OEG-2S-SN38 underwent rapid hydrolysis under redox conditions but PTX-2S-OA did not also begs consideration. The most obvious distinction between them is that OEG-2S-SN38 is an amphiphilic prodrug and that PTX-2S-OA is a hydrophobic conjugate. In addition, PTX is a much larger hydrophobic molecule than SN38. That means that the ester bond of OEG-2S-SN38 is much more susceptible to attack by water molecules than that of PTX-2S-OA, leading to faster release rate of SN38 from OEG-2S-SN38. The hydrophilic properties of OEG-2S-SN38 magnified the redox-responsive capability of dithioether. By comparison, the higher redox dual-responsivity of PTX-S-OA compared with PTX-2S-OA may be a result of the following: (i) the single thioether close to the ester bond conjugated with PTX is the key point to initiate the GSH/ROS-triggered drug release (Figure 3A); (ii) dithioether consumes twice as much GSH/ROS as single thioether does; (iii) the dithioether linker further extend the hydrophobic chain of prodrug (Figure 3), leading to less efficient attack of water molecules; and (iv) the ethylidene between the two thioether bonds in PTX-2S-OA increase the hydrophobicity of the surrounding environment around the thioether (Figure 3A), which was detrimental to the interaction with GSH and ROS. As a result, single thioether showed much higher redox dual-responsivity than dithioether.
We next compared the in vitro cytotoxicity of PTX-S-OA/TPGS2k NPs and PTX-2S-OA/TPGS2k NPs in three human cancer cell lines, namely, a human epidermoid carcinoma cell line (KB-3–1), a human lung carcinoma cell line (H460), and a human ovarian carcinoma cell line (OVCAR-8). An MTT assay was used to determine cell viability after 48 or 72 h. The IC50 values were calculated and summarized in Table S2. Compared to Taxol, these prodrug NPs were less effective in killing cancer cells in vitro due to the delayed release of the active PTX molecule (Figures 4 and S11). Among these prodrug NPs, PTX-S-OA/TPGS2k NPs showed much greater cytotoxicity than PTX-2S-OA/TPGS2k NPs and PTX-OA/TPGS2k NPs. Additionally, the released PTX by the end of the incubation of KB-3–1 cells with the prodrug NPs was determined. As shown in Figure S12, much more PTX molecules were released from PTX-S-OA/TPGS2k NPs than that from PTX-2S-OA/TPGS2k NPs and PTX-OA/TPGS2k NPs at both 48 and 72 h. The in vitro cytotoxicity results were consistent with the results of the in vitro release experiment, which further confirmed that single thioether had better redox dual-responsivity than dithioether. In addition, the in vitro cytotoxicity results indicated that a rapid and selective release of active PTX molecule within tumor cells will facilitate the drug-induced apoptosis of tumor cells.
Figure 4.
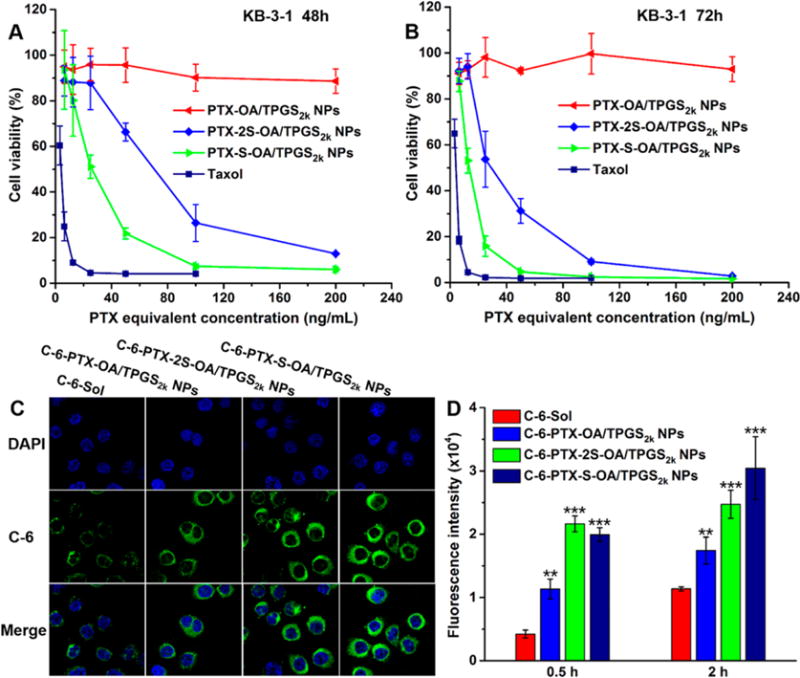
Cell viability treated with various concentrations of Taxol and prodrug NPs after (A) 48 h and (B) 72 h treatment. (C) Confocal laser scanning microscopy (CLSM) images of KB-3–1 cells incubated with free C-6 or C-6-labeled prodrug NPs for 2 h. (D) Cellular uptake in KB-3–1 cells after incubation with free C-6 or C-6-labeled prodrug NPs for 0.5 and 2 h by flow cytometry. Difference from C-6-Sol group, **P < 0.01, ***P < 0.001.
The cellular uptake of prodrug NPs was investigated in KB-3–1 cells. Coumarin-6 (C-6) was coassembled with prodrugs to form C-6-labeled prodrug NPs. As shown in Figures 4C,D and S13, the cellular uptake of free C-6 and C-6-labeled prodrug NPs was time-dependent, and C-6-labeled prodrug NPs displayed significantly higher intracellular fluorescence intensity than that of free C-6 at either 0.5 or 2 h.
The pharmacokinetic profiles of Taxol, PEGylated prodrug NPs, and non-PEGylated prodrug NPs were studied in Sprague–Dawley (SD) rats. For direct comparison of different nanoformulations, the sum of PTX and prodrug molar concentration–time curves were summarized in Figure S14, and the respective prodrug and PTX molar concentration–time curves were exhibited in Figures S15–S17. The pharmacokinetics parameters were calculated and summarized in Table S3. As shown in Figure S14, free PTX was rapidly cleared from blood due to its short half-life. In addition, the non-PEGylated formulations exhibited an even shorter blood circulation time than that of Taxol due to the poor stability and the highly hydrophobic surface of NPs, which could be easily phagocytosed by the reticuloendothelial system (RES). By contrast, PEGylated prodrug NPs exhibited significantly prolonged retention in blood. In addition to the improved colloidal stability, the PEG chains on the outer surface of prodrug NPs played an important role in avoiding identification and phagocytosis by RES.24 As displayed in Table S3, the half-life (t1/2) and the mean residence time (MRT) of both prodrugs and PTX were significantly extended in the groups of PEGylated prodrug NPs when compared with Taxol and non-PEGylated formulations, indicating the prolonged blood-circulation by PEGylation.
The ex vivo biodistribution of prodrug NPs was studied in nude mice bearing KB-3–1 tumor. As shown in Figure S18, the ex vivo fluorescence images showed that DiR-labeled prodrug NPs exhibited significantly increased fluorescence intensity in tumor compared to free DiR. Negligible fluorescence signal of free DiR was found in tumors. By contrast, strong fluorescence signal was found in liver and tumors treated with DiR-labeled prodrug nanoformulations, and the fluorescent signal in tumors increased over time from 4 to 24 h. These results were well consistent with the pharmacokinetic studies described above. In addition to the long systemic circulation, the EPR effects of prodrug NPs also greatly facilitated the intratumoral accumulation of dye.
The favorable stability, drug release, cytotoxicity, and pharmacokinetics of PTX-S-OA/TPGS2k NPs make it a promising candidate for further clinical evaluation. The in vivo antitumor efficacy of prodrug NPs was evaluated in nude mice bearing KB-3–1 xenograft tumors. PBS, Taxol, PTX-OA/TPGS2k NPs, PTX-2S-OA/TPGS2k NPs, and PTX-S-OA/TPGS2k NPs were administrated intravenously every other day for a total of 5 injections at a dose equivalent to 8 mg kg−1 of PTX. As shown in Figure 5, PBS and PTX-OA/TPGS2k NPs groups showed a rapid increase in tumor volume (~1100–1300 mm3 at day 10). In contrast, PTX-2S-OA/TPGS2k NPs and Taxol were able to somewhat suppress tumor growth, but the tumor volume reached about 450–700 mm3 at day 10. As expected, mice treated with PTX-S-OA/TPGS2k NPs exhibited significantly delayed tumor progression, with almost no growth in tumor volume. Notably, DiR-labeled PTX-OA/TPGS2k NPs demonstrated comparably high intratumoral accumulation of DiR with DiR-labeled PTX-S-OA/TPGS2k NPs, but exhibited poor antitumor activity. These results further indicated that the low release rate of PTX from hydrophobic ester prodrugs greatly limited their chemotherapeutic efficacy, and a rapid and selective release of free PTX in tumor is of crucial importance.
Figure 5.
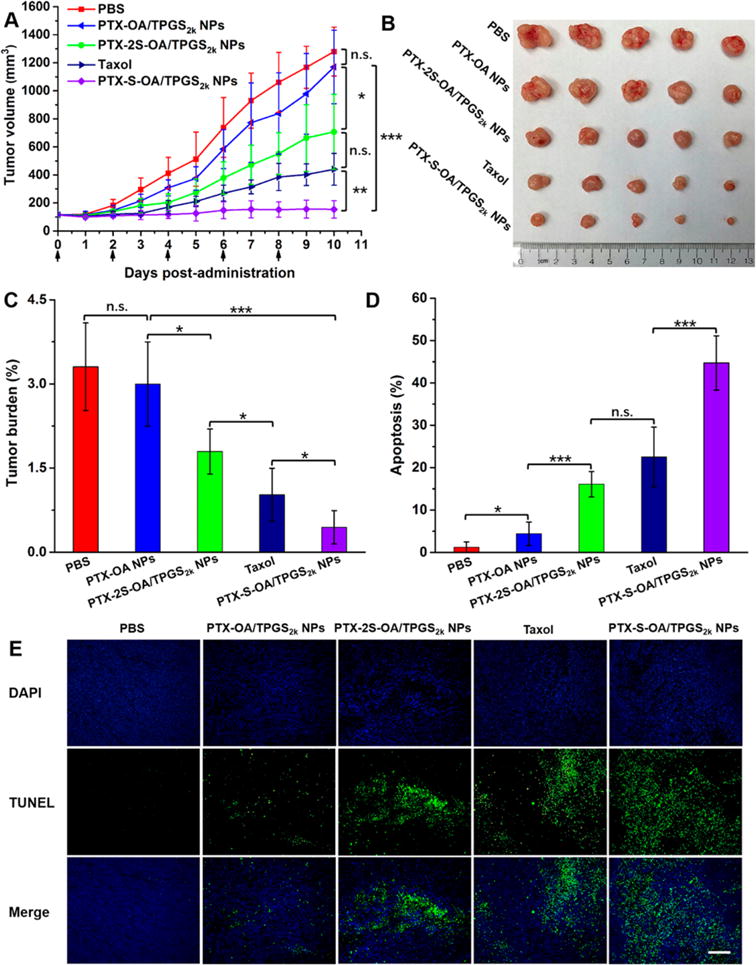
In vivo antitumor activity of prodrug NPs against KB-3–1 xenograft tumors (n = 5). (A) Tumor growth profiles treated with different formulations. (B) Images of tumors after the last treatment. (C) Tumor burden after the last treatment. (D) TUNEL assay quantitative results of tumor sections after the last treatment by ImageJ software. (E) TUNEL assay images of tumor sections after the last treatment, scale bar represents 500 μm. The data are presented as means ± SD, *P < 0.05, **P < 0.01, and *** P < 0.001. Scale bar represents 500 μm.
PTX-induced apoptosis by each prodrug conjugate was measured using a TUNEL assay. As shown in Figure 5D,E, significant tumor cell apoptosis was observed in mice treated with Taxol, PTX-2S-OA/TPGS2k NPs, and PTX-S-OA/TPGS2k NPs. PTX-S-OA/TPGS2k NPs showed distinct superiority over Taxol and PTX-2S-OA/TPGS2k NPs. In addition, no significant change in body weight was observed in any of the mouse populations (Figure S19A). The hematological parameters (aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels, blood urea nitrogen (BUN), and creatinine) lacked any indication of toxicity (Figure S19B). Moreover, no noticeable histological changes were observed in H&E-stained tissue sections of heart, liver, spleen, lung, and kidney (Figure S20). These results indicated that PTX-S-OA/TPGS2k NPs, despite potent antitumor efficacy, showed no significantly nonspecific toxicity to major organs and tissues.
In summary, we successfully developed a novel redox dual-responsive prodrug-nanosystem (PTX-S-OA/TPGS2k NPs) assembled by hydrophobic small-molecule prodrugs with impressively high drug loading efficiency (over 50% of free PTX, w/w). The ester bond linked prodrug (PTX-OA) showed measurably slower drug release, resulting in poor antitumor efficacy. By contrast, redox dual-responsive prodrug-nanosystem presented notable advantages. Additionally, the single thioether linked prodrug (PTX-S-OA) was distinctly superior to the dithioether-inserted conjugate (PTX-2S-OA) in terms of redox dual-sensitive drug release and in vivo antitumor efficacy. To the best of our knowledge, the present study is the first in-depth investigation of how tumor-stimuli-responsive linkage variations in hydrophobic small-molecule prodrugs correlate with their in vivo antitumor activity. This study confirmed our initial hypothesis that the limited clinical outcomes of hydrophobic prodrug (PTX-DHA) can be attributed to the extremely slow hydrolysis rate of active PTX molecules. This novel prodrug-nanosystem, with impressively high drug loading and efficiently selective drug release within tumor site, provides possibilities for the development of redox dual-sensitive conjugates or polymers for efficient anticancer drug delivery.
Supplementary Material
Acknowledgments
This work was financially supported by the National Basic Research Program of China (973 Program, No. 2015CB932100), the National Nature Science Foundation of China (No. 81273450, 81373336, 81473164), the Program for New Century Excellent Talents in University (No. NCET-12-1015), and NIH grant CA149387. The first author thanks the China Scholarship Council (CSC) for financially supporting his study in the United States (File No. 201408210110).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.nano-lett.6b01632.
Synthesis of prodrugs; preparation and characterization of prodrug nanoassemblies; in vitro release experiments; cytotoxicity assays; cellular uptake; pharmacokinetics; biodistribution; in vivo antitumor and imaging studies (PDF)
Notes
The authors declare no competing financial interest.
References
- 1.Shi J, Votruba AR, Farokhzad OC, Langer R. Nano Lett. 2010;10:3223–30. doi: 10.1021/nl102184c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Luo C, Sun J, Sun B, He Z. Trends Pharmacol Sci. 2014;35:556–66. doi: 10.1016/j.tips.2014.09.008. [DOI] [PubMed] [Google Scholar]
- 3.Aslan B, Ozpolat B, Sood AK, Lopez-Berestein G. J Drug Target. 2013;21:904–13. doi: 10.3109/1061186X.2013.837469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Luo C, Wang Y, Chen Q, Han X, Liu X, Sun J, He Z. Mini-Rev Med Chem. 2012;12:434–44. doi: 10.2174/138955712800493924. [DOI] [PubMed] [Google Scholar]
- 5.Zhang Z, Mei L, Feng SS. Expert Opin Drug Delivery. 2013;10:325–40. doi: 10.1517/17425247.2013.752354. [DOI] [PubMed] [Google Scholar]
- 6.Bildstein L, Dubernet C, Couvreur P. Adv Drug Delivery Rev. 2011;63:3–23. doi: 10.1016/j.addr.2010.12.005. [DOI] [PubMed] [Google Scholar]
- 7.Bradley MO, Webb NL, Anthony FH, Devanesan P, Witman PA, Hemamalini S, Chander MC, Baker SD, He L, Horwitz SB, Swindell CS. Clin Cancer Res. 2001;7:3229–38. [PubMed] [Google Scholar]
- 8.Wolff AC, Donehower RC, Carducci MK, Carducci MA, Brahmer JR, Zabelina Y, Bradley MO, Anthony FH, Swindell CS, Witman PA, Webb NL, Baker SD. Clin Cancer Res. 2003;9:3589–97. [PubMed] [Google Scholar]
- 9.Harries M, O’Donnell A, Scurr M, Reade S, Cole C, Judson I, Greystoke A, Twelves C, Kaye S. Br J Cancer. 2004;91:1651–5. doi: 10.1038/sj.bjc.6602196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bedikian AY, DeConti RC, Conry R, Agarwala S, Papadopoulos N, Kim KB, Ernstoff M. Ann Oncol. 2011;22:787–93. doi: 10.1093/annonc/mdq438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ke XY, Zhao BJ, Zhao X, Wang Y, Huang Y, Chen XM, Zhao BX, Zhao SS, Zhang X, Zhang Q. Biomaterials. 2010;31:5855–64. doi: 10.1016/j.biomaterials.2010.03.079. [DOI] [PubMed] [Google Scholar]
- 12.Karimi M, Ghasemi A, Sahandi Zangabad P, Rahighi R, Moosavi Basri SM, Mirshekari H, Amiri M, Shafaei Pishabad Z, Aslani A, Bozorgomid M, Ghosh D, Beyzavi A, Vaseghi A, Aref AR, Haghani L, Bahrami S, Hamblin MR. Chem Soc Rev. 2016;45:1457–501. doi: 10.1039/c5cs00798d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee FY, Vessey A, Rofstad E, Siemann DW, Sutherland RM. Cancer Res. 1989;49:5244–8. [PubMed] [Google Scholar]
- 14.Konstantinov AA, Peskin AV, Popova E, Khomutov GB, Ruuge EK. Biochim Biophys Acta, Bioenerg. 1987;894:1–10. doi: 10.1016/0005-2728(87)90206-4. [DOI] [PubMed] [Google Scholar]
- 15.Fang J, Seki T, Maeda H. Adv Drug Delivery Rev. 2009;61:290–302. doi: 10.1016/j.addr.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 16.Grek CL, Tew KD. Curr Opin Pharmacol. 2010;10:362–8. doi: 10.1016/j.coph.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marusyk A, Polyak K. Biochim Biophys Acta, Rev Cancer. 2010;1805:105–17. doi: 10.1016/j.bbcan.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yin Q, Shen J, Zhang Z, Yu H, Li Y. Adv Drug Delivery Rev. 2013;65:1699–715. doi: 10.1016/j.addr.2013.04.011. [DOI] [PubMed] [Google Scholar]
- 19.Wang J, Sun X, Mao W, Sun W, Tang J, Sui M, Shen Y, Gu Z. Adv Mater. 2013;25:3670–6. doi: 10.1002/adma.201300929. [DOI] [PubMed] [Google Scholar]
- 20.Xiao C, Ding J, Ma L, Yang C, Zhuang X, Chen X. Polym Chem. 2015;6:738–47. [Google Scholar]
- 21.Mura S, Zouhiri F, Lerondel S, Maksimenko A, Mougin J, Gueutin C, Brambilla D, Caron J, Sliwinski E, Lepape A, Desmaele D, Couvreur P. Bioconjugate Chem. 2013;24:1840–9. doi: 10.1021/bc400210x. [DOI] [PubMed] [Google Scholar]
- 22.Wang Y, Liu D, Zheng Q, Zhao Q, Zhang H, Ma Y, Fallon JK, Fu Q, Haynes MT, Lin G, Zhang R, Wang D, Yang X, Zhao L, He Z, Liu F. Nano Lett. 2014;14:5577–83. doi: 10.1021/nl502044x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Caron J, Maksimenko A, Wack S, Lepeltier E, Bourgaux C, Morvan E, Leblanc K, Couvreur P, Desmaele D. Adv Healthcare Mater. 2013;2:172–85. doi: 10.1002/adhm.201200099. [DOI] [PubMed] [Google Scholar]
- 24.Du XJ, Wang JL, Liu WW, Yang JX, Sun CY, Sun R, Li HJ, Shen S, Luo YL, Ye XD, Zhu YH, Yang XZ, Wang J. Biomaterials. 2015;69:1–11. doi: 10.1016/j.biomaterials.2015.07.048. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.