

Abstract
A noninvasive method for molecular imaging of the activity of different signal transduction pathways and the expression of different genes in vivo would be of considerable value. It would aid in understanding the role specific genes and signal transduction pathways have in various diseases, and could elucidate temporal dynamics and regulation at different stages of disease and during various therapeutic interventions. We developed and assessed a method for monitoring the transcriptional activation of endogenous genes by positron-emission tomography (PET) imaging. The HSV1-tk/GFP (TKGFP) dual reporter gene was used to monitor transcriptional activation of p53-dependent genes. A retrovirus bearing the Cis-p53/TKGFP reporter system was constructed in which the TKGFP reporter gene was placed under control of an artificial cis-acting p53-specific enhancer. U87 glioma and SaOS-2 osteosarcoma cells were transduced with this retrovirus and used to establish xenografts in rats. We demonstrated that DNA damage-induced up-regulation of p53 transcriptional activity correlated with the expression of p53-dependent downstream genes, such as p21, in U87 (wild-type p53), but not in SaOS-2 osteosarcoma (p53 −/−) cells. We showed that PET, with [124I]FIAU (2′-fluoro-2′-deoxy-1-β-d-arabinofuranosyl-5-[124I]iodouracil) and the Cis-p53TKGFP reporter system, is sufficiently sensitive to image the transcriptional regulation of genes in the p53 signal transduction pathway. These imaging results were confirmed by independent measurements of p53 activity and the expression levels of downstream genes (e.g., p21) by using conventional molecular-biological assays. PET imaging of p53 transcriptional activity in tumor xenografts by using the Cis-p53TKGFP reporter system may be useful in assessing novel therapeutic approaches.
In the original paper by Tjuvajev et al. (1), we described a paradigm for noninvasive imaging of transgene expression that requires the appropriate combination of “reporter gene” and “reporter substrate.” The reporter gene product (an enzyme) selectively converts a reporter substrate to a metabolite that is trapped within the transduced cell. This imaging paradigm is an in vivo radiotracer enzyme assay, and it can be used to image the expression and regulation of different endogenous genes and signal transduction pathways. We established and validated model systems in tissue culture and in experimental animals by using HSV1-tk (herpes simplex virus type 1 thymidine kinase) as a reporter gene and radiolabeled 2′-fluoro-2′-deoxy-1-β-d-arabinofuranosyl-5-iodouracil (FIAU) as a reporter substrate (1). Highly specific images of HSV1-tk expression in experimental animal tumors were obtained noninvasively by using radioiodinated [131I]FIAU and a clinical γ camera system (2–4), or [124I]FIAU and positron-emission tomography (PET) (5). Similar results have been demonstrated using 18F-labeled acycloguanosine analogues, such as 8-fluoro-ganciclovir (6), 8-fluoropenciclovir, (7) and 9-[(3-fluoro-1-hydroxy-2-propoxy)methyl]guanine and 9-(4-fluoro-3-hydroxymethylbutyl)guanine (8) as reporter substrates and wild-type or mutant HSV1-tk as the reporter gene (8). Also, we demonstrated that HSV1-tk imaging could be used to monitor and quantitatively assess the expression of a second gene that is cis-linked to the reporter gene by an internal ribosome entry site (IRES) sequence (4). This observation was subsequently confirmed by other investigators (9).
In this paper we assess the feasibility of PET to image the transcriptional activation of p53-dependent genes in vivo. We selected the p53 tumor suppressor gene as a model gene for our imaging studies because it is the most commonly mutated gene in human cancer (10, 11). Studies were performed using two different human tumor xenograft models in rats. These included the U87 human glioma that expresses wild-type p53 (12) and the SaOS-2 osteosarcoma that has a deletion of the p53 gene in both alleles (13). Both tumor cell lines were transduced with retrovirus bearing the Cis-p53/TKGFP reporter system in which the TKGFP reporter gene is placed under control of an artificially constructed cis-acting p53-specific enhancer. We demonstrated that DNA damage-induced up-regulation of p53 transcriptional activity can be imaged and correlates with the expression of p53-dependent downstream genes, such as the p21, in Cis-p53/TKGFP-transduced U87 glioma cells. We show that up-regulation of p53 can be monitored by fluorescent microscopy in vitro and in situ, and by PET imaging of xenografts in vivo. This was accomplished by using a dual-modality reporter gene, TKGFP, which is a fusion between the HSV1-tk and eGFP (enhanced green fluorescent protein) genes (14).
Materials and Methods
Generation of Cis-p53/TKGFP and TKGFP Reporter Vectors.
We have described the TKGFP fusion gene and generation of the TKGFP reporter vector (14). For Cis-p53/TKGFP reporter vector, TKGFP cDNA was obtained from the plasmid pTKGFP (14) and placed into the plasmid p53-Luc (Stratagene) (15), replacing the luciferase gene under control of 15 tandem repeats of –TGCCTGGACTTTGCCTGG- sequence that contains two p53-specific binding sites (underlined) (15). Briefly, the luciferase gene located between the EcoRI and XbaI sites was removed from the p53-Luc plasmid, the EcoRI end was blunted, and the NheI–XbaI fragment from the plasmid pTKGFP containing the TKGFP fusion gene with a blunted NheI end was ligated with the p53-Luc backbone. The BamHI-NotI fragment was taken from the resulting plasmid containing the p53 enhancer element, TATA-box, and the TKGFP fusion gene, and placed into the multiple-cloning site of the retroviral vector pLXSN (CLONTECH) 5′ to the neomycin-resistance gene that is under control of the simian virus 40 promoter. The NheI–NheI fragment containing the pBR322 backbone (which is a part of the pLXSN vector) and most of the viral 3′ long terminal repeat (LTR) was replaced with the same fragment from the Moloney murine leukemia virus retroviral vector dSFG-Ntp (16) containing the pUC19 backbone and a crippled 3′LTR. The resulting plasmid termed DXS53TGN is shown in Fig. 1A. DXS53TGN was transfected into the H29 transient retroviral producer cell line (17). After transfection of H29 cells, the tetracycline block was removed to allow for retroviral vector production. The Cis-p53/TKGFP retrovirus-containing medium was collected for 4 consecutive days and stored at −80°C.
Figure 1.

Validation of Cis-p53/TKGFP reporter system in cell cultures. (A) Structure of the DXS53TGN retroviral vector bearing the Cis-p53/TKGFP reporter system. This vector has a mutation in the 3′LTR that renders the silencing of its promoter activity after duplication as 5′LTR during integration. The expression of the TKGFP gene is regulated by an artificial promoter containing multiple tandem repeats of a p53-specific DNA-binding motif. Constitutive expression of the neomycin-resistance gene (Neo) is driven by the simian virus 40 early immediate promoter, allowing for the selection of stably transduced cells. Fluorescence microscopy and fluorescence-activated cell sorting (FACS) analysis of a transduced U87p53/TKGFP cell population in the noninduced (control) state (B and C), and 24 h after a 2-h treatment with N,N′-bis(2-chloroethyl)-N-nitrosourea (BCNU) at 40 μg/ml (D and E). (F) Immunoblot analysis for total p53, activated p53 (Ser15 phosphorylated), p21, and TKGFP protein levels in U87p53/TKGFP cells in the noninduced state (0) and after treatment with different doses of etoposide (5–40 μg/ml), 40 μg/ml BCNU, or 400 mJ of UV radiation. (G) Reverse transcription (RT)-PCR analysis for p21 and TKGFP mRNA levels performed in the same samples as shown in F. The levels of phospho-p53, total p53, p21, and TKGFP proteins increase after etoposide treatment in a dose-dependent manner; similar increases were observed for BCNU and UV treatments (data not shown). (H) TKGFP expression in different cell populations as measured by the radiotracer assay. The FIAU/thymidine (TdR) ratio is low in wild-type U87 cells (negative control) and in noninduced U87p53/TKGFP cells. In contrast, BCNU-treated U87p53/TKGFP cells had a significantly higher FIAU/TdR accumulation ratio (higher TKGFP expression), which was within the range observed in RG2TKGFP cells that constitutively express TKGFP.
Tumor Cells and Transduction.
The U87 and SaOS-2 cell lines were obtained from The American Type Culture Collection. The U87 human glioblastoma cells (wild-type p53; see ref. 12) were grown in DMEM with nonessential amino acids and 10% FCS (HyClone). Human osteosarcoma SaOS-2 cells (p53−/−) (13) were also grown in McCoy's modified medium (Life Technologies, Gaithersburg, MD) with 10% FCS. The RG2 rat glioma cells were provided by D. Bigner (Duke University Medical Center, Durham, NC) and grown in DMEM with 10% FCS. The retroviral transient producer cell line H29 (17) was maintained in DMEM supplemented with 10% FCS, puromycin (2 μg/ml), and tetracycline (1 μg/ml). The U87 and SaOS cells were transduced with the Cis-p53/TKGFP vector by incubating 50% confluent tumor cell cultures with the vector-containing medium for 12 h in the presence of hexadimethrine bromide (8 μg/ml). Mixed populations of transduced U87p53/TKGFP and SaOSp53/TKGFP cells were obtained by selection in corresponding media supplemented with 500 μg/ml of G418 (Life Technologies). The RG2TKGFP cells that constitutively express TKGFP reporter gene were described (14).
Selection of Transduced Tumor Cell Clones.
Single cell-derived clones of transduced U87p53/TKGFP and SaOSp53/TKGFP tumor cells with optimal properties for the reporter system (low background and high inducibility), were obtained by using fluorescence-activated cell sorting (FACS). Clones with very low background fluorescence were obtained and propagated in duplicate 96-well plates. The clones in one plate were treated with N,N′-bis(2-chloroethyl)-N-nitrosourea (BCNU; 40 μg/ml) for 2 h to activate the p53 pathway as described elsewhere (18). After the BCNU treatment, the clones were assessed for TKGFP fluorescence by microscopy every hour, and the brightest fluorescing clones were selected. The fluorescence intensity in the selected clones was quantified by FACS. Clones with the lowest background and the highest BCNU-inducible fluorescence were selected and propagated further. The background and BCNU-inducible TKGFP fluorescence was reassessed after expansion in each clone, and a clone with optimal characteristics was selected and used in further experiments.
Fluorescent Microscopy and FACS Analysis.
The level of TKGFP reporter gene expression was assessed by fluorescent microscopy (Nikon Eclipse TS-100) and quantified by FACS (FACStarplus; Becton Dickinson) using 488-nm excitation and 525-nm emission filters (19). The average fluorescence in viable cells was used as a quantitative measure of the TKGFP reporter gene expression.
Radiotracer Assay for TKGFP Expression in Vitro.
The level of TKGFP expression in different cell populations was assessed by using the [14C]FIAU and tritiated thymidine ([3H]dThd) radiotracer assay as described (1).
Reverse Transcription (RT)-PCR for TKGFP and p21.
For TKGFP, the specific HSV1-tk primers were 5′-ATGGCTTCGTACCCCTG-3′ and 5′-AAGGTCGGCGGGATGAG-3′. For p21, a commercial primer kit was used (Stratagene, catalog no. 720092). For reference, the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) levels were assessed by using a commercial kit (Stratagene, catalog no. 302047). Total RNA from the tumor samples was obtained by using the RNeasy kit (Qiagen, Chatsworth, CA). The RT-PCR procedures were performed by using the single-tube system (Stratagene) according to the manufacturer's protocol. The amplification products were separated by electrophoresis in 2% agarose/ethidium bromide gel, visualized under UV light (302 nm), and analyzed by using a digital densitometry system (MCID; Imaging Research; St. Catharines, ON, Canada).
Western Blot Analysis.
Cells or tumor tissue samples were washed with ice-cold PBS and mechanically lysed in a buffer containing 10 mM Tris⋅HCl (pH 7.5), 1 mM DTT, 20% (vol/vol) glycerol, 1 mM EDTA, and protease inhibitor mixture (Sigma; 100 μl/100 ml buffer). The samples were centrifuged at 4°C for 5 min and the supernatant was mixed with SDS sample buffer 1:2 and boiled for 5 min. A 10-μl sample subjected to SDS/PAGE (10%) electrophoresis and transferred to Immobilon-P poly(vinylidene difluoride) (PVDF) membrane (Millipore). Antibodies against p53 (Ab-6; Calbiochem; dilution, 1:1000), p53 phosphorylated at Ser-15 (Ser-15; New England Biolabs; dilution, 1:500), and p21/WAF1/Cip1/Sdi1/Pic1 (Ab-11; Santa Cruz Biotechnology; dilution, 1:200) were used. Anti-HSV1-TK mouse monoclonal antibody 4C8 (dilution, 1:500) was obtained from W. Summers (Yale University, New Haven, CT). Immunochemical detection of these proteins was performed by using the chemiluminescence ECL kit (Amersham Pharmacia). Protein concentration in samples was assayed by using the Bio-Rad Protein Assay kit (Bio-Rad).
Generation of s.c. Tumor Xenografts in Rats.
The experimental protocol was approved by the Institutional Animal Care and Use Committee of the Memorial Sloan–Kettering Cancer Center. s.c. xenografts were produced in rnu/rnu rats (Frederick Cancer Research and Development Center) by s.c. injection of 106 tumor cells in 100 μl of PBS while the animals were under anesthesia (ketamine 87 mg/kg and xylazine 13 mg/kg, i.p.) as follows: U87p53TKGFP, right shoulder (test); wild-type U87, left shoulder (negative control); RG2TKGFP, left thigh (positive control). Studies were conducted when the tumors were ≈15 mm in diameter. Generation of SaOS and SaOSp53TKGFP xenografts in rnu/rnu rats was unsuccessful, and is consistent with previous reports (13).
[124I]FIAU Synthesis.
124I was produced on a CS-15 cyclotron employing the (p, n) nuclear reaction in an enriched tellurium-124 oxide target. After irradiation, the target was subjected to dry distillation to release the trapped radioiodide, and 124I was isolated in a small volume of phosphate buffer as described (20). No-carrier-added synthesis of [124I]FIAU was performed with a stannylated precursor, 2′-fluoro-2′-deoxy-1-β-d-arabinofuranosyl-5-(tri-n-butyltin)-uracil (FTBSnAU), which was allowed to react with NaI[124I] in the presence of a mixture of acetic acid and 30% hydrogen peroxide. After quenching with sodium metabisulfite, the [124I]FIAU was isolated on a C-18 Sep-Pak cartridge system, and eluted with methanol. After evaporation of methanol, the product was reconstituted in a sterile, pyrogen-free, physiological saline (with 5% ethanol added) and passed through a sterile 0.22-μm filter (Millipore). The radiochemical purity was determined by radio-TLC (Sigma-Aldrich silica gel plates, eluent: ethyl acetate/acetone/water 14:8:1; [124I]FIAU exhibiting an Rf of 0.61). Radio-TLC plates were analyzed with a System 200 Scanner equipped with an Auto Changer 1000 (BioScan, Inc., Washington, DC). The radiochemical purity was 97 ± 2%; [124I]iodide was the only contaminant. The specific activity of the initial 124I was found to be >30 Ci/mmol (1 Ci = 37 GBq) (20), so that the specific activity of [124I]FIAU was estimated to be close to this value.
PET Imaging and Quantitation.
One day before [124I]FIAU administration, the animals received a 2-ml i.p. injection of a 0.9% NaI solution to block the thyroid uptake of radioactive iodide (small amounts of contaminant). [124I]FIAU was administered i.v., 300 μCi per animal. PET imaging was performed on an Advance PET tomograph (General Electric) 24 h after tracer administration to allow for sufficient clearance of body background radioactivity. The 24-h biological clearance improved the signal-to-noise ratio and specificity of the images.
Animals were anesthetized by i.p. injection of ketamine (87 mg/kg) and xylazine (13 mg/kg). Transmission and emission scans were obtained in two-dimensional mode; the emission scans were corrected for random counts, dead time, and attenuation. The duration of the transmission and emission scans was 15 and 45 min, respectively. Images were reconstructed by using an iterative reconstruction algorithm (5), yielding slice thickness of 4.2 mm and a transaxial resolution element size of 4 mm2. Radioactivity concentration in tumors (% dose per g) was measured from the PET images as described (5).
To estimate tumor dosimetry of [124I]FIAU and to confirm the PET radioactivity measurements, the individual tumors, muscle, and venous blood were sampled, weighed, and assayed for 124I radioactivity by using a Packard 5500 γ spectrometer. A portion of each tumor tissue sample was used for the assessments of p21 and TKGFP gene expression with RT-PCR. The cryosections of the remaining portion of each tumor sample were used to assess p53 activity status and p21 expression with immunohistochemistry, and TKGFP expression with fluorescence microscopy.
Immunohistochemistry for p21 and Activated p53.
Anti-p21/WAF mouse monoclonal antibody F-5 (Santa Cruz Biotechnology) and anti-phospho-p53(15Ser) rabbit polyclonal antibody (BioLabs, NJ) were used as primary antibodies in combination with the anti-mouse and anti-rabbit Vectastain Elite ABC immunohistochemistry kits (Vector Laboratories). Counterstaining was performed with eosin.
Results
Characterization of the Selected U87p53/TKGFP and SaOSp53/TKGFP Clones in Vitro.
U87p53/TKGFP and SaOSp53/TKGFP cells were seeded into 150 × 25 mm plates and grown until 60% confluent. The cells were then exposed to media with different concentrations of etoposide (5–40 μg/ml), BCNU (0.1–100 μg/ml), or to UV (50–800 mJ), and assessed every hour for p53 pathway induction by monitoring TKGFP expression with fluorescence microscopy and FACS. There was some background p53 activity (TKGFP fluorescence) in a few of the nontreated U87/Cis-p53/TKGFP cells that was probably cell cycle-associated (Fig. 1 B and C). Two peaks of TKGFP expression were observed: one at 4–6 h and another at about 24 h after induction with etoposide, BCNU, or UV (data not shown).
U87p53/TKGFP cells were also assessed for activation of the p53 pathway and for TKGFP expression 24 h after exposure to different concentrations of etoposide (5–40 μg/ml) or BCNU (40 μg/ml) for 2 h, or exposed to UV (400 mJ). U87p53/TKGFP cells treated with 40 μg/ml of etoposide or 40 μg/ml BCNU for 2 h, or those that received 400 mJ of UV radiation, had optimal activation of the p53 signaling pathway and levels of TKGFP expression (Fig. 1, D and E). Significant increases in p53 phosphorylation and total p53 protein expression were observed (Fig. 1F). Increased p53 activity caused up-regulation of p21 and TKGFP gene expression, as manifested by increases in corresponding mRNA (Fig. 1G) and protein levels (Fig. 1F). Increased TKGFP mRNA and protein levels in BCNU-treated U87p53/TKGFP cells corresponded with significantly higher levels of [14C]FIAU accumulation (FIAU/dThd ratio 0.20 ± 0.03), compared with that in nontreated U87p53/TKGFP cells (0.04 ± 0.02; Fig. 1H). A FIAU/dThd ratio of 0.1 or greater has previously been shown by us to be adequate for noninvasive imaging (1–3). In contrast, SaOSp53/TKGFP cells did not have any background p53 activity and did not show any induction of the p53 signaling pathway by etoposide, BCNU, or UV treatment, as demonstrated by the absence of TKGFP fluorescence on fluorescence microscopy and FACS, by nondetectable levels of TKGFP mRNA, and by a very low [14C]FIAU accumulation, which was similar to that in nontransduced SaOS cells (data not shown).
Monitoring Transcriptional Activation of p53-Regulated Genes by PET Imaging in Vivo.
We assessed the efficacy of the Cis-p53/TKGFP reporter system in vivo with [124I]FIAU and PET. rnu/rnu rats bearing s.c. xenografts in multiple sites were studied: U87p53/TKGFP (test); wild-type U87 (negative control), and RG2TKGFP+ (positive control). Half of the animals (n = 6) were injected with BCNU (40 mg/kg, i.p.) to induce the p53 pathway in the xenografts. The animals were injected i.v. with [124I]FIAU 24 h after BCNU administration, and imaged with PET 24 h later.
PET images of [124I]FIAU accumulation obtained in nontreated (control) rats revealed minimal accumulation of [124I]FIAU in both the U87p53/TKGFP and wild-type U87 xenografts (Figs. 2A and 3), whereas substantial accumulation was observed in RG2TKGFP (positive control) xenografts (Figs. 2B and 3). In animals treated with BCNU (40 mg/kg i.p.), a significantly higher accumulation of [124I]FIAU was observed in U87p53/TKGFP xenografts as compared with wild-type U87 xenografts (Fig. 2C and 3; P < 0.05, paired t test), and as compared with nontreated U87p53/TKGFP xenografts (Figs. 2 A–C and 3; P < 0.05, unpaired t test). These results indicate that BCNU-induced activation of the p53 signal transduction pathway in U87p53/TKGFP xenografts can be imaged with PET.
Figure 2.

PET imaging of endogenous p53 activation. Transaxial PET images through the shoulder (A and C) and pelvis (B and D) of two rats are shown; the images are color-coded to the same radioactivity scale (% dose/g). A nontreated animal is shown on the left, and a BCNU-treated animal is shown on the right. Both animals had three s.c. tumor xenografts: U87p53TKGFP (test) in the right shoulder, U87 wild-type (negative control) in the left shoulder, and RG2TKGFP (positive control) in the left thigh. The nontreated animal on the left shows localization of radioactivity only in the positive control tumor (RG2TKGFP); the test (U87p53TKGFP) and negative control (U87wt) tumors are at background levels. The BCNU-treated animal on the right shows significant radioactivity localization in the test tumor (right shoulder) and in the positive control (left thigh), but no radioactivity above background in the negative control (left shoulder).
Figure 3.

Tissue radioactivity and tissue-to-muscle ratios. Two groups of animals are compared; nontreated (solid bars, n = 6) and BCNU-treated (40 mg/kg, i.p., shaded bars, n = 6). Tissue radioactivity (% dose/g; A) and the tissue-to-muscle radioactivity ratio (B) are shown; error bars are ± SD. Significant differences between BCNU-treated and nontreated animals (unpaired t test, P < 0.05) are indicated by *. Significant differences between the test (U87p53TKGFP) and negative control (U87wt) xenografts (paired t test, P < 0.05), in either BCNU-treated or nontreated animals, is indicated by †.
[124I]FIAU clearance from blood was somewhat slower in BCNU-treated animals, as reflected by the higher and more variable 24-h plasma levels (Fig. 3A). This difference in plasma clearance is most likely related to BCNU toxicity and is reflected in muscle and wild-type U87 tissue levels as well. Therefore, the xenograft-to-muscle radioactivity ratio was calculated to normalize the data to body background activity. BCNU treatment also had a significant negative effect on the constitutive expression of TKGFP in the RG2TKGFP xenografts (positive control).
Fluorescent microscopic analysis of tumor tissue sections from the animals that were imaged with PET demonstrated a low incidence of fluorescing (TKGFP expressing) cells in nontreated U87p53TKGFP xenografts (Fig. 4A). In contrast, the U87p53TKGFP xenografts from the animals that were treated with BCNU had a high density of fluorescing cells (Fig. 4B) that was comparable to that in the RG2TKGFP xenografts (positive control).
Figure 4.
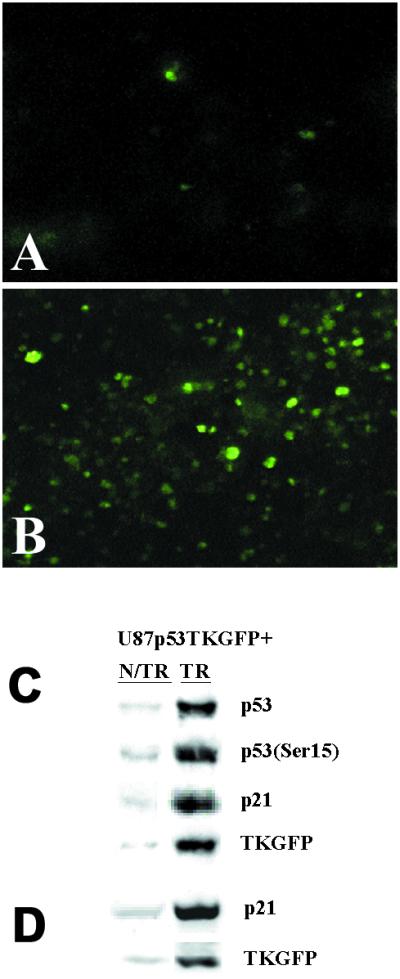
Assessment of Cis-p53/TKGFP reporter system in U87p53/TKGFP s.c. tumor tissue samples. Fluorescence microscopy images of U87p53/TKGFP s.c. tumor samples obtained from nontreated rats (A) and rats treated with 40 mg/kg BCNU i.p. (B). The same U87p53/TKGFP s.c. tumor samples obtained from nontreated (N/TR) and BCNU-treated (TR) animals were assessed for the levels of activated p53 (Ser-15 phosphorylated), total p53 protein, p21, and TKGFP proteins by immunoblot analysis (C); and for the levels of p21 and TKGFP mRNAs by RT-PCR (D).
As compared with nontreated U87p53/TKGFP xenografts, BCNU-treated U87p53/TKGFP xenografts had increased phospho-p53 and total p53 protein levels, increased p21 and TKGFP mRNA, and corresponding protein levels (Fig. 4 C and D). Immunohistochemical analysis of tumor tissue sections demonstrated increased density of both the phospho-p53-positive cells and p21-positive cells in BCNU-treated U87p53/TKGFP xenografts as compared with nontreated U87p53/TKGFP xenografts. Both, the phospho-p53- and p21-positive immunohistochemical staining colocalized with TKGFP-fluorescing cells (data not shown).
Discussion
The development of methods that allow imaging at the molecular-genetic level or allow visualization of different steps in various signal transduction pathways is a new research endeavor. However, there are practical limitations to the development of large numbers of unique probes for direct imaging of each gene or each step of different signal transduction pathways. These and other limiting factors stimulated the development of several reporter gene-based assay systems. These reporter systems can monitor the regulation of gene expression at the level of transcription (21, 22) or posttranscription (23), can monitor the activity of different transcription factors (24), and can assess protein–protein interactions in different signal transduction pathways (24). In our opinion, a more reasonable approach would be to develop imaging methods that use one (or a few) well-characterized probes for reporter gene-based systems that allow for the detection and quantitation of different molecular-biological processes.
We selected the p53 gene as a model gene for in vivo molecular-genetic imaging studies. The p53 tumor suppressor is the most commonly mutated gene in human cancer (10, 11). In normal cells under physiological conditions, the tumor-suppressive p53 protein is expressed at low concentrations and has a short half-life because of rapid turnover due to ubiquitination and proteolysis (26). p53 becomes stabilized and activated in response to a number of stressful stimuli, including exposure to DNA-damaging agents, hypoxia, nucleotide depletion, or oncogene activation (22). The activation of p53 allows it to carry out its function as a tumor suppressor through a number of growth-controlling endpoints (27). These endpoints include cell cycle arrest, apoptosis, senescence, differentiation, and anti-angiogenesis (27). In its role as a tumor suppressor, p53 serves as a “guardian” of the genome and a “gatekeeper” for growth and division, by regulating critical checkpoints in response to the distinct stress stimuli (11, 27).
In this paper we describe the results of a merger between one of the molecular-biologic reporter-gene based systems, the cis-reporter system (21, 22), and an established method for noninvasive imaging of HSV1-tk reporter gene expression with [124I]FIAU and PET (1–5). A recently developed dual-modality reporter gene, TKGFP (14), was used in these studies to provide additional optical (fluorescence) imaging and FACS analysis data. The TKGFP gene was introduced into the retroviral reporter vector and placed under the control of a p53-specific enhancer element, so that TKGFP reporter gene expression would be transcriptionally up-regulated along with several other endogenous genes upon activation (phosphorylation) of p53 protein by different signal transduction pathways. A significant increase in TKGFP expression over the baseline was observed in U87p53/TKGFP cells treated with known activators of p53 pathway (BCNU, etoposide, and UV radiation). A biphasic pattern of p53 activation after BCNU induction observed at 4–6 h and at about 24 h is in agreement with reported data (23, 28).
There are several groups of genes that are transcriptionally regulated by p53, including p21 (cell cycle arrest), MDM2, GADD45 and GoS8 (G0/G1switch) (22), 14-3-3 protein σ1 (G2 arrest, repair) (22); bax, Fas/APO1, KILLER/DR5, IGF-BP3, PIG3, and PAG608 (apoptosis) (22, 28); and GD-AiF and TSP1 (anti-angiogenesis) (22, 28). p21 is one of the most important proteins in the p53 pathway, acting immediately downstream and mediating many of the actions of p53 (22, 30, 31). We demonstrated that the level of TKGFP reporter gene mRNA paralleled that of the p21 mRNA in both the noninduced and induced states. Similarly, a correlation was observed between TKGFP and p21 protein levels in a dose-dependent manner. These results in cell cultures demonstrate that the Cis-p53/TKGFP reporter system adequately reflects the activity of the p53 signal transduction pathway and the status of endogenous p53-dependent transcriptionally regulated genes, such as p21.
The up-regulation of p53 signaling pathway and TKGFP expression in U87p53/TKGFP xenografts observed after BCNU treatment by using PET imaging was confirmed by the analysis of tissue samples (i.e., increased levels of active p53 protein, up-regulation of p21 and TKGFP mRNAs and protein levels, increased density of TKGFP fluorescing tumor cells). These data demonstrate that the images of the Cis-p53/TKGFP reporter system activity obtained with [124I]FIAU and PET reflect the activity of the p53 signaling pathway and transcriptional status of p53-dependent downstream genes.
PET imaging of transcriptional activity of p53 in tumors by using the Cis-p53TK/GFP reporter system may be used to noninvasively assess treatment efficacy and to optimize the dosing of new drugs or to evaluate novel therapeutic approaches that depend on p53 activation. It should also be possible to generate transgenic mice carrying the Cis-p53/TKGFP reporter system. Cis-p53/TKGFP-transgenic mice could be crossed with other transgenic mouse models of cancer. Tumors developing in these hybrid animals could be used to noninvasively image the activity of the p53 pathway and the expression of p53-regulated downstream genes in different experimental settings.
In summary, we have demonstrated that noninvasive nuclear imaging of endogenous gene expression is feasible using a cis-reporter system. In particular, we have demonstrated that PET imaging with [124I]FIAU and the Cis-p53/TKGFP reporter system is sufficiently sensitive to detect the transcriptional up-regulation of genes in the p53 signal transduction pathway. Moreover, our PET imaging results were confirmed by independent measurements of p53 activity and the expression levels of downstream genes (e.g., p21). PET imaging of p53 transcriptional activity in tumors by using the Cis-p53/TKGFP reporter system may be used to assess new drugs or novel therapeutic approaches.
Acknowledgments
This work was supported by National Institutes of Health Grants P50 CA86438, RO1 CA69769, R24CA98023, and RO1 CA76117, and Department of Energy Grant FG03-86ER60407.
Abbreviations
- HSV1-tk
herpes simplex virus type 1 thymidine kinase
- GFP
green fluorescent protein
- PET
positron emission tomography
- FIAU
2′-fluoro-2′-deoxy-1-β-d-arabinofuranosyl-5-iodouracil
- dThd
thymidine
- FACS
fluorescence activated cell sorting
- BCNU
N,N′-bis(2-chloroethyl)-N-nitrosourea
- RT
reverse transcription
- LTR
long terminal repeat
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Tjuvajev J G, Stockhammer G, Desai R, Uehara H, Watanabe K, Gansbacher B, Blasberg R. Cancer Res. 1995;55:6126–6132. [PubMed] [Google Scholar]
- 2.Tjuvajev J G, Finn R, Watanabe K, Joshi R, Oku T, Kennedy J, Beattie B, Koutcher J, Larson S, Blasberg R. Cancer Res. 1996;56:4087–4095. [PubMed] [Google Scholar]
- 3.Tjuvajev J G, Chen S H, Joshi A, Joshi R, Guo Z S, Balatoni J, Ballon D, Koutcher J, Finn R, Woo S L, Blasberg R G. Cancer Res. 1999;59:5186–5193. [PubMed] [Google Scholar]
- 4.Tjuvajev J G, Joshi A, Callegari J, Lindsley L, Joshi R, Balatoni J, Finn R, Larson S, Sadelain M, Blasberg R. Neoplasia. 1999;1:315–320. doi: 10.1038/sj.neo.7900053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tjuvajev J G, Avril N, Oku T, Sasajima T, Miyagava T, Joshi R, Safer M, Beattie B, DiResta G, Daghighian F, et al. Cancer Res. 1998;58:4333–4341. [PubMed] [Google Scholar]
- 6.Gambhir S S, Herschman H R, Cherry S R, Barrio J R, Satyamurthy N, Toyokuni T, Phelps M E, Larson S M, Balatoni J, Finn R, et al. Neoplasia. 2000;2:118–138. doi: 10.1038/sj.neo.7900083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iyer M, Barrio J R, Namavari M, Bauer E, Satyamurthy M, Nguyen K, Toyokuni T, Phelps M E, Herschman H R, Gambhir S S. J Nucl Med. 2001;42:106–109. [PubMed] [Google Scholar]
- 8.Gambhir S S, Barrio J R, Phelps M E, Iyer M, Namavari M, Satyamurthy N, Wu L, Green L A, Bauer E, MacLaren D C, Nguyen K, et al. Proc Natl Acad Sci USA. 1999;96:2333–2338. doi: 10.1073/pnas.96.5.2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu Y, Annala A J, Barrio J R, Toyokuni T, Satyamurthy N, Namavari M, Cherry S R, Phelps M E, Herschman H R, Gambhir S S. Nat Med. 2000;6:933–937. doi: 10.1038/78704. [DOI] [PubMed] [Google Scholar]
- 10.Hollstein M, Sidransky D, Vogelstein B, Harris C C. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- 11.Levine A J, Perry M E, Chang A, Silver A, Dittmer D, Wu M, Welsh D. Br J Cancer. 1994;69:409–416. doi: 10.1038/bjc.1994.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Van Meir E G, Kikuchi T, Tada M, Li H, Diserens A C, Wojcik B E, Huang H J, Friedmann T, de Tribolet N, Cavenee W K. Cancer Res. 1994;54:649–652. [PubMed] [Google Scholar]
- 13.Vihinen P, Riikonen T, Laine A, Heino J. Cell Growth Differ. 1996;7:439–447. [PubMed] [Google Scholar]
- 14.Jacobs A, Dubrovin M, Hewett J, Sena-Esteves M, Tan C-W, Slack M, Sadelain M, Breakfield X, Tjuvajev J G. Neoplasia. 1999;1:154–161. doi: 10.1038/sj.neo.7900007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kern S E, Kinzler K W, Bruskin A, Jarosz D, Friedman P, Prives C, Vogelstein B. Science. 1991;252:1708–1711. doi: 10.1126/science.2047879. [DOI] [PubMed] [Google Scholar]
- 16.Ory D S, Neugeboren B A, Mulligan R C. Proc Natl Acad Sci USA. 1996;93:11400–11406. doi: 10.1073/pnas.93.21.11400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Riviere I, Sadelain M. In: Gene Therapy Protocols: Methods in Molecular Biology. Robbins P, editor. Totowa, NJ: Humana; 1997. pp. 59–78. [Google Scholar]
- 18.Chen Z P, Wang G, Huang Q, Sun Z F, Zhou L Y, Wang A D, Panasci L C. J Neurooncol. 1999;44:7–14. doi: 10.1023/a:1006245724456. [DOI] [PubMed] [Google Scholar]
- 19.Cormack B P, Valdivia R H, Falkow S. Gene. 1996;173:33–38. doi: 10.1016/0378-1119(95)00685-0. [DOI] [PubMed] [Google Scholar]
- 20.Sheh Y, Koziorowski J, Balatoni J, Lom C, Dahl J R, Finn R D. Radiochimica Acta. 2000;88:169–173. [Google Scholar]
- 21.Tang H Y, Zhao K, Pizzolato J F, Fonarev M, Langer J C, Manfredi J J. J Biol Chem. 1998;273:29156–29163. doi: 10.1074/jbc.273.44.29156. [DOI] [PubMed] [Google Scholar]
- 22.Zhao R, Gish K, Murphy M, Yin Y, Notterman D, Hoffman W H, Tom E, Mack D H, Levine A J. Genes Dev. 2000;14:981–993. [PMC free article] [PubMed] [Google Scholar]
- 23.Chen C Y, Del Gatto-Konczak F, Wu Z, Karin M. Science. 1998;280:1945–1949. doi: 10.1126/science.280.5371.1945. [DOI] [PubMed] [Google Scholar]
- 24.Wotton D, Lo R S, Lee S, Massague J. Cell. 1999;97:29–39. doi: 10.1016/s0092-8674(00)80712-6. [DOI] [PubMed] [Google Scholar]
- 25.Gerber H P, Condorelli F, Park J, Ferrara N. J Biol Chem. 1997;272:23659–23667. doi: 10.1074/jbc.272.38.23659. [DOI] [PubMed] [Google Scholar]
- 26.Haupt Y, Maya R, Kazaz A, Oren M. Nature (London) 1997;387:296–299. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- 27.Chan T A, Hwang P M, Hermeking H, Kinzler K W, Vogelstein B. Genes Dev. 2000;14:1584–1588. [PMC free article] [PubMed] [Google Scholar]
- 28.Wu L, Levine A J. Mol Med. 1997;3:441–451. [PMC free article] [PubMed] [Google Scholar]
- 29.Wu G S, Burns T F, McDonald E R, Jiang W, Meng R, Krantz I D, Kao G, Gan D D, Zhou J Y, Muschel R, et al. Nat Genet. 1997;17:141–143. doi: 10.1038/ng1097-141. [DOI] [PubMed] [Google Scholar]
- 30.Yu J, Zhang L, Hwang P M, Rago C, Kinzler K W, Vogelstein B. Proc Natl Acad Sci USA. 1999;96:14517–14522. doi: 10.1073/pnas.96.25.14517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fiscella M, Zambrano N, Ullrich S J, Unger T, Lin D, Cho B, Merecer W E, Anderson C W, Appella E. Oncogene. 1994;9:3249–3257. [PubMed] [Google Scholar]