

Abstract
Population genetic structure using nine polymorphic nuclear microsatellite loci was assessed for the tiger shark (Galeocerdo cuvier) at seven locations across the Indo-Pacific, and one location in the southern Atlantic. Genetic analyses revealed considerable genetic structuring (FST > 0.14, p < 0.001) between all Indo-Pacific locations and Brazil. By contrast, no significant genetic differences were observed between locations from within the Pacific or Indian Oceans, identifying an apparent large, single Indo-Pacific population. A lack of differentiation between tiger sharks sampled in Hawaii and other Indo-Pacific locations identified herein is in contrast to an earlier global tiger shark nDNA study. The results of our power analysis provide evidence to suggest that the larger sample sizes used here negated any weak population subdivision observed previously. These results further highlight the need for cross-jurisdictional efforts to manage the sustainable exploitation of large migratory sharks like G. cuvier.
Keywords: tiger shark, Galeocerdo cuvier, population structure, microsatellite loci, Indo-Pacific Ocean
1. Introduction
Migratory species are described as entire populations (or any geographically separate part of a population), whose members cyclically and predictably cross one or more national jurisdictional boundaries [1]. In the marine environment, defining sharks as migratory species requires considerable knowledge of individual movement patterns, as well as an understanding of contemporary population structure. As with all wild populations, the population structure of sharks is determined in genetic terms by genetic drift, mutation and selection [2,3]. Countering the influence of these microevolutionary forces is gene flow, which mediates genetic isolation through the successful reproduction of immigrants in situ [4]. In highly migratory marine species, the expectation is that signatures of genetic differentiation are less pronounced, as their large dispersal capabilities permit ongoing population connectivity, even at a global scale [5]. However this is not always the case, with evidence of genetic structuring for some migratory shark species at regional geographical scales, due to the restriction of gene flow by large oceanic expanses, phylogeographic boundaries, or thermal barriers [3,6–9]. Identifying these subpopulations within the broader distribution of migratory sharks is a critical step in delineating the stocks, which need to be identified for potential conservation and management of the species [3].
The tiger shark (Galeocerdo cuvier) (Péron and Lesueur, 1822) is the largest of the Carcharhiniformes, with a circumglobal distribution in tropical and warm temperate coastal and pelagic waters. Across the Indo-Pacific Ocean basin tiger sharks are targeted by a range of commercial, recreational, artisanal and shark control fishing operations [10–14]. Despite these pressures, little is known about the population size throughout this region, with long-term catch rate trends providing the only potential estimate of abundance to date [15]. Satellite tracking studies across the Indo-Pacific have documented round trip migrations of over 5000 km, with individuals tagged off the Australian east coast recording movements across the Coral Sea to New Caledonia [16,17] and Papua New Guinea [18]. Research based in Western Australia has also shown broad-scale movements into the Northern Territory and Indonesian waters [19], and potentially across the broader Indian Ocean [20]. In contrast to these broad movement patterns, tiger sharks tagged off Hawaii appear to exhibit more restricted movements within the Hawaiian island group [21–23]. A feature of all of these studies is the return of tiger sharks to specific areas on a regular basis, which has thus far been attributed to seasonal feeding excursions rather than fidelity to mating or parturition grounds, the latter of which has not yet been identified in this species. Indeed, the ability to identify philopatry and sex-biased dispersal through satellite tracking studies of tiger sharks has been hampered by the need to capture and tag adequate numbers of animals from both sexes across their full size range. Further, as female tiger sharks are likely to have a triennial reproductive cycle in the Pacific [24], identifying a return to natal grounds would require several years of satellite tracking data, which remains problematic due to restrictions on tag battery life, retention and biofouling.
Different management and monitoring regimes of neighbouring jurisdictions, coupled with a lack of fishing regulation in international waters, generate significant threats to migratory shark species [25]. For tiger sharks this is of particular concern, with a recent global study by Bernard et al. [9] reporting nDNA genetic differentiation between western Atlantic and Indo-Pacific Ocean basins, indicating wide-ranging populations that inhabit and move through a variety of fishing jurisdictions, Exclusive Economic Zones (EEZ) and international waters. Bernard et al. [9] also suggested there may be limitations to the connectivity of tiger sharks across the central Pacific Ocean, with the tiger sharks sampled in the Hawaiian archipelago assigned to a separate population.
Using nine nDNA microsatellite loci previously developed for the species [26], the current study focuses on tiger shark population connectivity and dynamics in the Indo-Pacific region. Specifically, our aims were to: (i) detect population genetic structuring across the eastern Indian and central and western Pacific Oceans using large sample sizes to provide good statistical power, and (ii) test the expectation that tiger sharks from Hawaii assign to a separate population from the broader Indo-Pacific region.
2. Methods
2.1. Sample collection, extraction methods and genotyping
Fin clips or muscle tissue were collected from G. cuvier caught in the western Pacific Ocean off the east coast of Australia (New South Wales, NSW; Queensland, QLD; the Coral Sea, COR; New Caledonia, NCL). Samples from the central Pacific Ocean were collected off Hawaii (HAW), while eastern Indian Ocean samples were obtained off Western Australia (WA), with additional northern Australian samples from the Northern Territory (NT). Out-group samples were obtained from Brazil (BRA) in the Atlantic Ocean (figure 1). All samples were collected between 2002 and 2015. As a guide to sample sizes required for the study, the theoretical statistical power required to detect genetic differentiation among populations was estimated using POWSIM [27]. Tissue was stored in 95% ethanol or 20% dimethyl sulfoxide (DMSO) solution. Genotypes were obtained using nine microsatellite loci previously developed for the species (see [26]). Sixty-nine juvenile (birth–200 cm TL), 69 sub-adult (200–300 cm TL), 96 adult (300 cm TL+), and 121 (size not recorded) G. cuvier (159 female, 107 male and 89 sex unknown) totalling 355 individuals were genotyped. Juvenile, sub-adult and adult sharks were sampled from QLD, NSW, NT and NCL, while predominantly adult sharks were sampled in WA and HAW (figure 2). Apart from during transit to Australia, all samples were stored at 4°C or below until laboratory processing.
Figure 1.

Sampling locations and number of G. cuvier tissue samples obtained from the Indo-East Indian (IEI, blue), the Central-West Pacific (CWP, black) and the Atlantic Ocean (BRA, red). Pink open circle denotes the Torres Strait land bridge.
Figure 2.

Length-frequency distribution of G. cuvier from which tissues were sampled in the Indo-Pacific region. No length measurements were recorded for Coral Sea samples.
DNA extraction was performed using either a QIAGEN DNeasy blood and tissue extraction kit following the manufacturer's protocols (QIAGEN Inc., Valencia, CA, USA), or a salting out method [28]. Loci were optimized into polymerase chain reaction (PCR) multiplexes using a fluorescently labelled M13 primer. For each locus, one primer stock consisting of forward and reverse primer pairs, and the corresponding M13 primer (Fam, Vic, Ned or Pet) was used. Primer stocks consisted of 3 µl of 100 µM forward primer, 30 µl of 100 µM reverse primer and 30 µl of 100 µM of M13-fluro labelled primer. To achieve optimal amplification across all loci, primer stock proportions were experimentally determined (see electronic supplementary material, table S1). Multiplex 1 consisted of loci tgr_1157, tgr_212, tgr_47, tgr_233 and tgr_348; and multiplex 2 consisted of tgr_1033, tgr_1185, tgr_891 and tgr_943. The samples were amplified using a 14 µl PCR mixture containing 10 ng of genomic DNA, 8 µl of master mix (1x Kapa Buffer A, 1.5% DMSO, 0.18 mM dNTP, 0.25 M Betaine, 0.8 units/reaction Taq), and 4 µl of primer mix (3 µl forward, 30 µl reverse, 30 µl M13-fluoro, 87 µl H2O). Loci were amplified in 2 multiplexes using the following protocols: Multiplex 1: 94°C for 2 min, followed by 12 cycles of 94°C for 15 s, 56°C for 30 s and 72°C for 45 s, with the annealing temperature reduced by 0.5°C each cycle (touchdown cycle). The reaction was then exposed to 23 cycles of 94°C for 15 s, 50°C for 30 s and 72°C for 45 s. Multiplex 2: 94°C for 2 min, followed by 10 cycles of 94°C for 15 s, 60°C for 30 s and 72°C for 45 s, reduced by 0.5°C each cycle. The reaction was then exposed to 23 cycles of 94°C for 15 s, 55°C for 30 s and 72°C for 45 s. The reactions for multiplex 1 and 2 completed at 72°C for 7 min before being held at 4°C until required for further analysis. Amplicons were diluted 60-fold, before being further diluted (1 in 5) in formamide containing LIZ-500 size standard (Applied Biosystems, Foster City, CA, USA) and then gel separated by capillary electrophoresis (Applied Biosystems 3130xl) following the manufacturer's recommendation.
2.2. Statistics
Alleles were sized against an internal size standard (GeneScan-500 LIZ) before being scored using GeneMapper v. 5. Genotypes were checked for scoring errors using Micro-Checker v. 2.2.3 [29]. We tested for Hardy–Weinberg equilibrium (HWE) using the Markov chain Monte Carlo (MCMC) method in GENEPOP v. 4.1.3 [30], with 100 000 steps, 100 batches and 10 000 subsequent iterations. We also tested for linkage disequilibrium (LD) among pairs of loci using an exact test based on a Markov chain method as implemented in GENEPOP, in both cases using Bonferroni to correct for multiple tests (p < 0.05) [31]. If significant departures from HWE or LD were found at one or more locus by population comparisons, then they were excluded from further analysis. To ensure that duplicate samples were not inadvertently included a Queller and Goodnight [32] relatedness test was performed in GenAlEx v. 6.5. Individuals that were identical across the nine microsatellite loci were treated as duplicates and excluded from further analysis.
Population pairwise standard diversity indices were estimated using GenAlEx v. 6.5, which included number of alleles (Na), number of effective alleles (Ne), expected (He) and observed heterozygosities (Ho), and inbreeding coefficient (Fis) [33]. To investigate the degree of subdivision between locations, pairwise FST values was estimated using Arlequin v. 3.11 [34] with the level of significance calculated by pseudo-replication (10,100) of individuals between locations; and Jost's Dest (hereafter Dest) was determined using GenAlEx v. 6.5 (999 iterations; [33,35]). Genetic populations were defined by locations (or groups of locations) that did not have significantly different FST values from one another, while showing significant variation to other sampled populations. An analysis of molecular variance (AMOVA) was also undertaken using a hierarchical approach in Arlequin v. 3.11, where population clusters were validated to ensure that the maximum amount of variance among groups of samples (regions) was maintained. Comparisons were made between the genotypes reported from Hawaii in Bernard et al. [9] and those herein to evaluate the presence of a separate Hawaiian population. To ensure the allele frequency bins were uniform between the studies at each locus, a comparison of dominant alleles was undertaken, and homology was determined for alleles having frequencies above 10% using allele sizes and per study frequencies. Two loci (tgr_348 and tgr_891) were unable to be reliably compared between the studies and were therefore excluded from the pairwise comparison analyses.
Bayesian cluster analysis was performed in STRUCTURE [36] on the microsatellite allele frequencies. STRUCTURE was run 10 independent times on the microsatellite dataset for the potential number of groups (K) ranging from K = 1 to 8. The run assumed an admixture ancestor model with a burn-in length of 100 000 MCMC steps, followed by simulation set at 1 000 000 repetitions. To estimate the number of groups (K) an ad hoc approach was taken by obtaining the mean posterior probability of the data ΔK using STRUCTURE Harvester [37]. To visualize clustering across runs of K, structure outputs were submitted to the CLUMPAK pipeline [38].
To identify the theoretical statistical power required to detect an FST differentiation among locations, pilot genotyping data from 96 randomly selected sharks was used in POWSIM [27]. Allele frequencies from the pilot data were used to simulate populations (differing by a range of user-defined overall FST values) to match populations sampled here from the three ocean basins (Central-West Pacific (CWP), Indo-East Indian (IEI), and the Atlantic). Three user-defined levels of divergence were simulated among populations; FST = 0.01, 0.005 and 0.0025. The following parameters were used to simulate the set of FST values: effective population sizes (Ne) = 1000; number of simulations = 1000; and generations of drift (t) = 20 (FST = 0.01), 10 (FST = 0.005) and 5 (FST = 0.0025). To test for the effect of sample size on the ability to detect significant population differentiation (α = 0.05) at a given FST value, tests were undertaken with sample sizes of 25, 50 and 100 per population. The degree of significant differentiation between populations for each replicate run at a set FST value was tested using Chi-squared and Fisher's probabilities.
3. Results
3.1. Power simulation
When the power simulation was run with sample sizes of 100 across all nine microsatellite loci, there was sufficient power to delineate population subdivision in greater than 98% of simulations across the three levels of genetic differentiation tested (FST = 0.01, 0.005, 0.0025). When halving the sample size from 100 to 50, the ability to detect differences at the lowest FST (0.0025) decreased by 35%. Minor reductions in the ability to detect population structure (100% to more than 97%) were also found at FST = 0.005 and 0.01 when simulated sample sizes were reduced to 50. When sample sizes were further decreased to 25, an FST of 0.01 could be detected in over 95% of simulations; however, detections of smaller FST (from 0.005 and 0.0025; 58% and 22%, respectively) were considered unreliable. To summarize, the ability to detect differences among populations with low levels of genetic structure was found to decrease considerably with reductions in sample size.
3.2. Microsatellite diversity
Screening of the samples using a Queller and Goodnight [32] relatedness test detected 17 duplicates in the NCL samples, which were subsequently removed from further analysis. Across all samples, the nine loci had an average of 9.8 alleles (range 2–26) and unbiased heterozygosity of 0.70 (range 0.18–0.94). The most polymorphic locus was Tgr_891 (mean = 11.3 alleles), followed by Tgr_348 (9.7) and Tgr_943 (8.0), while Tgr_212 was the least polymorphic (1.4 alleles) (table 1). Screening of genotypes detected no linkage disequilibrium for any population, and genotype proportions at all loci did not deviate from Hardy–Weinberg expectations following Bonferroni correction. Pairwise FST and Dest comparisons between locations indicated considerable genetic structuring between all Indo-Pacific locations and Brazil (FST values > 0.14, p < 0.001; Dest values > 0.29, p < 0.001). In contrast, no significant genetic differences were observed between locations from within the Pacific or Indian Oceans (FST range 0.000–0.004; Dest range 0.000–0.007) (table 2). As a result, collection locations within these regions were pooled for subsequent analysis by ocean basin. The ocean basin pools were the Central-West Pacific (CWP; consisting of NSW, QLD, COR, NCL, HAW), the Indo-East Indian (IEI; consisting of NT and WA) and the Atlantic Ocean (BRA). A comparison of FST and Dest values among these three ocean basins was unable to identify any significant difference between CWP and IEI, which meant the null hypothesis of genetic homogeneity was unable to be rejected (table 3). Lack of genetic differentiation among samples from CWP and IEI was further supported by AMOVA analysis. Differences among groupings were maximized and variation within groupings was minimized when the Indo-Pacific samples (i.e. CWP and IEI) were considered as a single population (among groupings = 14.72%, within = 82.5%), compared to two separate populations (among groupings = 1.65%, within = 95.17%). Differentiation between the samples from Brazil (Atlantic) and all other locations were confirmed, with significant differences associated with grouped samples from the CWP (FST = 0.151, p < 0.001; Dest = 0.321, p = 0.001) and IEI (FST = 0.145, p < 0.001; Dest = 0.317, p = 0.001).
Table 1.
Microsatellite marker diversity described by number of alleles (Na), number of effective alleles (Ne), observed (Ho) and expected heterozygosity (He), and inbreeding coefficient (Fis). n = number of individuals. Note that n in combined columns differs to total as not all loci could be genotyped per individual.
| Tgr_1033 | Tgr_1157 | Tgr_1185 | Tgr_212 | Tgr_233 | Tgr_348 | Tgr_47 | Tgr_891 | Tgr_943 | ||
|---|---|---|---|---|---|---|---|---|---|---|
| QLD | Na | 4 | 10 | 6 | 3 | 24 | 18 | 5 | 19 | 11 |
| (n=69) | Ne | 1.964 | 6.283 | 3.582 | 1.646 | 7.081 | 9.338 | 1.808 | 12.253 | 8.056 |
| He | 0.441 | 0.750 | 0.706 | 0.348 | 0.912 | 0.924 | 0.485 | 0.939 | 0.853 | |
| Ho | 0.491 | 0.841 | 0.721 | 0.392 | 0.859 | 0.893 | 0.447 | 0.918 | 0.876 | |
| Fis | 0.101 | 0.108 | 0.021 | 0.114 | −0.062 | −0.035 | −0.086 | −0.023 | 0.026 | |
| NSW | Na | 5 | 10 | 5 | 3 | 26 | 21 | 3 | 17 | 13 |
| (n=81) | Ne | 2.275 | 5.717 | 3.125 | 1.391 | 7.772 | 12.189 | 1.547 | 11.685 | 8.258 |
| He | 0.560 | 0.825 | 0.680 | 0.281 | 0.871 | 0.918 | 0.354 | 0.914 | 0.879 | |
| Ho | 0.605 | 0.838 | 0.738 | 0.309 | 0.888 | 0.911 | 0.300 | 0.901 | 0.877 | |
| Fis | −0.080 | −0.015 | −0.085 | −0.098 | −0.019 | 0.007 | 0.151 | 0.014 | 0.003 | |
| COR | Na | 4 | 9 | 5 | 3 | 18 | 16 | 3 | 15 | 13 |
| (n=36) | Ne | 2.185 | 6.234 | 3.871 | 1.536 | 6.863 | 9.138 | 1.885 | 11.187 | 9.554 |
| He | 0.556 | 0.800 | 0.781 | 0.361 | 0.886 | 0.912 | 0.417 | 0.943 | 0.853 | |
| Ho | 0.542 | 0.840 | 0.742 | 0.349 | 0.854 | 0.891 | 0.470 | 0.911 | 0.895 | |
| Fis | −0.024 | 0.047 | −0.053 | −0.034 | −0.037 | −0.024 | 0.113 | −0.035 | 0.047 | |
| NT | Na | 4 | 10 | 6 | 3 | 24 | 21 | 4 | 17 | 12 |
| (n=62) | Ne | 1.858 | 3.883 | 3.834 | 1.418 | 7.586 | 10.030 | 1.659 | 12.160 | 7.742 |
| He | 0.452 | 0.758 | 0.661 | 0.306 | 0.836 | 0.869 | 0.344 | 0.967 | 0.952 | |
| Ho | 0.462 | 0.742 | 0.739 | 0.295 | 0.868 | 0.900 | 0.397 | 0.918 | 0.871 | |
| Fis | 0.022 | −0.021 | 0.105 | −0.039 | 0.037 | 0.035 | 0.133 | −0.054 | −0.093 | |
| BRA | Na | 3 | 5 | 4 | 2 | 9 | 9 | 2 | 12 | 8 |
| (n=8) | Ne | 2.510 | 3.122 | 3.048 | 1.508 | 7.538 | 7.538 | 1.753 | 9.846 | 7.000 |
| He | 0.625 | 0.750 | 0.875 | 0.429 | 0.857 | 1.000 | 0.625 | 0.875 | 1.000 | |
| Ho | 0.602 | 0.680 | 0.672 | 0.337 | 0.867 | 0.867 | 0.430 | 0.898 | 0.857 | |
| Fis | −0.039 | −0.103 | −0.302 | −0.273 | 0.012 | −0.153 | −0.455 | 0.026 | −0.167 | |
| NCL | Na | 4 | 8 | 6 | 3 | 15 | 15 | 3 | 13 | 10 |
| (n=22) | Ne | 1.699 | 4.500 | 3.868 | 1.340 | 7.056 | 8.647 | 2.066 | 10.127 | 7.670 |
| He | 0.400 | 0.810 | 0.810 | 0.286 | 0.952 | 0.857 | 0.476 | 0.850 | 0.952 | |
| Ho | 0.411 | 0.778 | 0.741 | 0.254 | 0.858 | 0.884 | 0.516 | 0.901 | 0.870 | |
| Fis | 0.027 | −0.041 | −0.092 | −0.125 | −0.110 | 0.031 | 0.077 | 0.057 | −0.095 | |
| WA | Na | 5 | 9 | 6 | 4 | 22 | 19 | 3 | 17 | 12 |
| (n=56) | Ne | 2.047 | 4.938 | 3.269 | 1.524 | 8.267 | 11.521 | 1.863 | 12.398 | 6.938 |
| He | 0.526 | 0.807 | 0.649 | 0.298 | 0.930 | 0.895 | 0.439 | 0.982 | 0.839 | |
| Ho | 0.512 | 0.797 | 0.694 | 0.344 | 0.879 | 0.913 | 0.463 | 0.919 | 0.856 | |
| Fis | −0.029 | −0.012 | 0.065 | 0.133 | −0.058 | 0.020 | 0.053 | −0.068 | 0.019 | |
| HAW | Na | 7 | 10 | 4 | 3 | 14 | 13 | 4 | 15 | 12 |
| (n=21) | Ne | 1.869 | 5.765 | 3.139 | 1.156 | 5.345 | 9.660 | 2.353 | 11.025 | 8.791 |
| He | 0.571 | 0.667 | 0.579 | 0.095 | 0.619 | 0.938 | 0.550 | 0.905 | 0.950 | |
| Ho | 0.465 | 0.827 | 0.681 | 0.135 | 0.813 | 0.896 | 0.575 | 0.909 | 0.886 | |
| Fis | −0.229 | 0.193 | 0.150 | 0.294 | 0.238 | −0.046 | 0.043 | 0.005 | −0.072 | |
| Combined | n | 353 | 352 | 347 | 354 | 350 | 341 | 351 | 347 | 349 |
| (n=355) | Mean Na | 4.500 | 8.875 | 5.250 | 3.000 | 19.000 | 16.500 | 3.375 | 15.625 | 11.375 |
| Mean Fis | −0.032 | 0.024 | −0.023 | −0.019 | −0.001 | −0.020 | 0.004 | −0.010 | −0.041 |
Table 2.
Pairwise FST values (above diagonal) and Dest values (below diagonal) for G. cuvier across eight sampling locations. Significant results were accepted at p < 0.01 and denoted by *, with p-value listed in parentheses.
| QLD | NSW | COR | NT | NCL | WA | HAW | BRA | |
|---|---|---|---|---|---|---|---|---|
| QLD | — | −0.001 | −0.008 | −0.002 | 0.003 | 0.003 | 0.001 | 0.146* |
| (0.820) | (0.766) | (0.891) | (0.216) | (0.054) | (0.405) | (0.000) | ||
| NSW | −0.003 | — | −0.003 | 0.000 | 0.003 | 0.003 | −0.004 | 0.152* |
| (0.808) | (0.891) | (0.487) | (0.261) | (0.036) | (0.847) | (0.000) | ||
| COR | −0.003 | −0.007 | — | 0.002 | 0.002 | 0.000 | 0.001 | 0.167* |
| (0.631) | (0.926) | (0.225) | (0.252) | (0.451) | (0.559) | (0.000) | ||
| NT | −0.006 | −0.001 | 0.003 | — | 0.004 | 0.002 | 0.000 | 0.143* |
| (0.944) | (0.644) | (0.306) | (0.162) | (0.190) | (0.541) | (0.000) | ||
| NCL | 0.005 | 0.005 | 0.001 | 0.006 | — | 0.000 | 0.001 | 0.167* |
| (0.276) | (0.237) | (0.457) | (0.218) | (0.423) | (0.441) | (0.000) | ||
| WA | 0.007 | 0.007 | −0.001 | 0.003 | 0.001 | — | −0.001 | 0.151* |
| (0.076) | (0.053) | (0.529) | (0.243) | (0.437) | (0.514) | (0.000) | ||
| HAW | 0.002 | −0.008 | 0.000 | −0.002 | 0.000 | −0.001 | — | 0.157* |
| (0.396) | (0.873) | (0.436) | (0.561) | (0.434) | (0.507) | (0.000) | ||
| BRA | 0.321* | 0.32* | 0.336* | 0.319* | 0.331* | 0.315* | 0.298* | — |
| (0.001) | (0.001) | (0.001) | (0.001) | (0.001) | (0.001) | (0.001) |
Table 3.
Pairwise FST values (above diagonal) and Dest values (below diagonal) for G. cuvier across three grouped sampling locations. Significant results were accepted at p < 0.01 and denoted by *, with p-value listed in parentheses.
| Central-West Pacific | Indo-East Indian | Atlantic | |
|---|---|---|---|
| Central-West Pacific | — | 0.000 (0.135) | 0.151* (0.000) |
| Indo-East Indian | 0.002 (0.192) | — | 0.145* (0.000) |
| Atlantic | 0.321* (0.001) | 0.317* (0.001) | — |
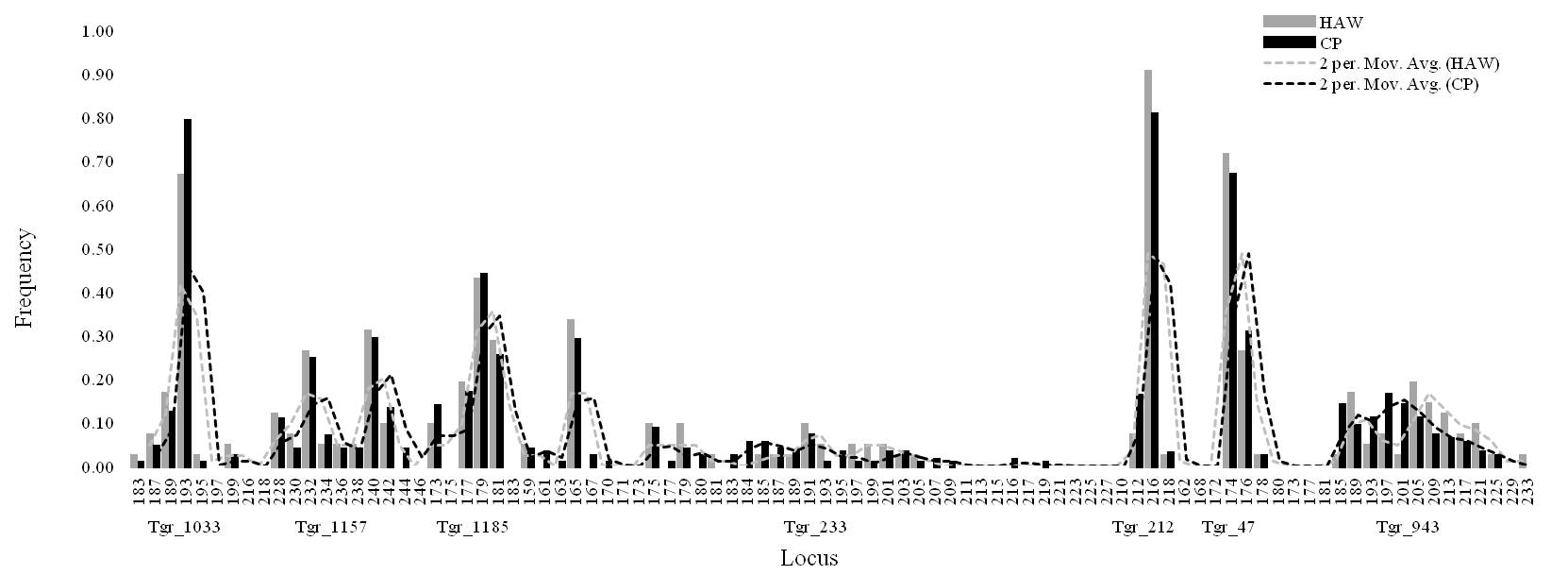
Pairwise comparisons were also made between the Hawaiian genotypes from this study, and the Central Pacific (CP) genotypes reported in Bernard et al. [9]. No significant differences were observed between Hawaiian samples from both datasets. The similarity between datasets was reflected in the allele frequency distributions between datasets (see electronic supplementary material).
Bayesian STRUCTURE analysis of microsatellite genotypes supported the groupings identified by FST. The ΔK method in STRUCTURE Harvester identified the K = 2 as the most likely number of populations. Visual clustering of individuals using CLUMPAK revealed that all samples from the Indian and Pacific Ocean were assigned to the same group, while the second group was made up of samples from Brazil. To investigate the Bayesian structuring of individuals within the Indo-Pacific, samples from Brazil were removed before the remaining samples were re-analysed. When subsequent STRUCTURE analysis including only the Indo-Pacific samples was run, the ΔK suggested only one discernible grouping was present within the data.
4. Discussion
Analysis of genetic homogeneity with microsatellites across all locations in this study revealed two genetically independent populations; an Indo-Pacific group composed of all samples collected in the IEI and the CWP, and the Brazilian sample from the western Atlantic. The absence of genetic differentiation between the east and west Australian coasts is consistent with previously reported movement data, with evidence of migrating sharks from both coasts moving into adjoining Northern Territorial waters (see [18,19]). Evidence of tiger shark movement through the Torres Strait land bridge [18] suggests that the relatively shallow waters (less than 15 m deep between Cape York, QLD and Papua New Guinea) do not act as a barrier to movements, and possibly gene flow, as it does for other fish species [39]. A high level of genetic connectivity throughout the Indo-Pacific region was also reported by Bernard et al. [9] in their global study, with little differentiation observed between sharks sampled from the east and west Australian coasts, the Andaman Sea (southeast Asia) and as far west as the eastern seaboard of South Africa.
Our investigation suggests genetic connectivity among tiger sharks from the Indo-Pacific extends into the Pacific Ocean as far as Hawaii (central Pacific). This finding was in contrast to that identified in the global study by Bernard et al. [9], which reported significant differences between Hawaii and all other global sites surveyed, including those from the Indo-Pacific. The STRUCTURE analysis by Bernard et al. [9] may explain the differences observed between the two studies. Despite ΔK suggesting K = 2 throughout all global samples, when K = 3 was assumed by Bernard et al. [9], a few individuals from the southwest Pacific (eastern Australia) assigned to a third cluster, which was subsequently reported as evidence of a genetically distinct Hawaiian population. Although Bernard et al. [9] demonstrated a weak signal for population subdivision comparing samples from Eastern Australia (n = 21) and Hawaii (n = 65) (FST = 0.01, Dest = 0.02), the low statistical power due to the small sample size from Eastern Australia reduces the likelihood that the statistically significant finding actually reflects a true effect [40]. Moreover, the power analysis completed herein demonstrates that the ability to detect differences among tiger shark populations with low levels of genetic structure was found to decrease considerably with reductions in sample size less than 25. Ecological studies undertaking satellite tracking across the Hawaiian archipelago have shown that tiger sharks in this region do exhibit regionally localized movements (e.g. [21,23,41]). Notwithstanding, there is a confirmed record of a conventionally tagged tiger shark from Hawaii being captured in Mexico, approximately 5000 km east of its tagging location (C. Meyer 2016, personal communication). This recapture indicates that tiger sharks have the ability to cross the large oceanic expanses, with the possibility of promoting gene flow across the broader Pacific Ocean. Acquiring and analysing additional tissue samples from the eastern Pacific region, along with further research using alternative genetic markers such as mtDNA and single nucleotide polymorphisms (SNP) in nuclear DNA is needed to confirm the single population. The level of heterozygosity of microsatellite loci reported herein suggests that variation in SNP loci should also be high. The development of SNPs for G. cuvier is likely to increase the power to detect genetic variation, even if sample sizes are small [42].
Continued advances in tagging technology that allow for multi-year data collection from mature sharks, and further developments in molecular analyses, is likely to improve our understanding of how partial migration, sex-biased dispersal and reproductive mixing influences population structure and connectivity of tiger sharks within and among global ocean basins. Given the evidence of exploitation-driven declines in G. cuvier across the world [14,15,43,44], continued assessments are vital to inform population-level management and conservation efforts across the entire distribution of this oceanodromous species.
Supplementary Material
{kind=link}
Supplementary Material
{kind=link}
Supplementary Material
{kind=link}
Acknowledgements
We wish to acknowledge the contribution of the genetic work by Sean Corley from the Animal Genetics Laboratory, University of Queensland, as well as those who donated G. cuvier tissues, including Will Macbeth, Dr Lindsay Marshall, Grant Johnson, Danillo Pinhal, Dr Will White, Dr Chris Dudgeon, Dr Jenny Giles, Dr Thomas Vignaud, Dr Serge Planes, Luciana Ferreira, Dr Adam Barnett, Dr Rory McAuley, Dr James True, Shin Arunrugstichai, Dr Kim Holland, Dr Carl Meyer and Dr Sabine Wintner. We also thank the Queensland Shark Control Program contractors Paddy Dimond, Mark Cawthray, Greg Pearce, Craig Newton, Geoff Bachmann, Bruce Mortley and Mal Paskin who provided specimens for this research.
Ethics
All procedures were approved by the University of Queensland Animal Ethics Committee (CMS/300/08/DPI/SEAWORLD and CMS/326/11/DPI), the Department of Primary Industries and Fisheries (permit nos. 100541, 165491 and 56095) and the Department of Environment and Resource Management (permit nos. QS2009/GS001, QS2010/MAN26 and QS2010/GS059).
Data accessibility
Tiger shark genotypes are available through the UQ eSpace (http://espace.library.uq.edu.au/view/UQ:613874).
Authors' contributions
B.J.H. and J.R.O. conceived the project. B.J.H., E.E.N., J.R.O. and S.L.M. extracted DNA. B.J.H. and S.M.W. arranged genotyping, completed analyses and wrote the paper. B.J.H., S.M.W. and J.R.O. discussed the results, and all authors contributed to the revision of the final manuscript.
Competing interests
We declare we have no competing interests.
Funding
B.J.H. received funding to undertake the project from the New South Wales Department of Primary Industries and the Mohammed Bin Zayed Species Conservation Fund.
References
- 1.Fowler S. 2014. The conservation status of migratory sharks, 30pp Bonn, Germany: UNEP/CMS Secretariat. [Google Scholar]
- 2.Geraghty PT, Williamson JE, Macbeth WG, Wintner SP, Harry AV, Ovenden JR, Gillings MR. 2013. Population expansion and genetic structure in Carcharhinus brevipinna in the Southern Indo-Pacific. PLoS ONE 9, e94738 (doi:10.1371/journal.pone.0075169) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ashe JL, Feldheim KA, Fields AT, Reyier EA, Brooks EJ, O'Connell MT, Skomal G, Gruber SH, Chapman DD. 2015. Local population structure and context-dependent isolation by distance in a large coastal shark. Mar. Ecol. Prog. Ser. 520, 203–216. (doi:10.3357/meps11069) [Google Scholar]
- 4.Palumbi SR. 1994. Genetic divergence, reproductive isolation and marine speciation. Annu. Rev. Ecol. Syst. 25, 547–572. (doi:10.1146/annurev.es.25.110194.002555) [Google Scholar]
- 5.DeWoody JA, Avise JC. 2000. Microsatellite variation in marine, freshwater and anadromous fishes compared with other animals. J. Fish Biol. 56, 461–473. (doi:10.1111/j.1095-8649.2000.tb00748.x) [Google Scholar]
- 6.Benavides MT, et al. 2011. Global phylogeography of the dusky shark Carcharhinus obscurus: implications for fisheries management and monitoring the shark fin trade. Endangered Species Res. 14, 13–22. (doi:10.3354/esr00337) [Google Scholar]
- 7.Blower DC, Pandolfi JM, Bruce BD, Gomez-Cabrera MdC, Ovenden JR. 2012. Population genetics of Australian white sharks reveals fine-scale spatial structure, transoceanic dispersal events and low effective population sizes. Mar. Ecol. Prog. Ser. 455, 229–244. (doi:10.3354/meps09659) [Google Scholar]
- 8.Dudgeon CL, et al. 2012. A review of the application of molecular genetics for fisheries management and conservation of sharks and rays. J. Fish Biol. 80, 1789–1843. (doi:10.1111/j.1095-8649.2012.03265.x) [DOI] [PubMed] [Google Scholar]
- 9.Bernard AM, Feldheim KA, Heithaus MR, Wintner SP, Wetherbee BM, Shivji MS. 2016. Global population genetic dynamics of a highly migratory, apex predator shark. Mol. Ecol. 25, 5312–5329. (doi:10.1111/mec.13845) [DOI] [PubMed] [Google Scholar]
- 10.Pepperell JG. 1992. Trends in the distribution, species composition and size of sharks caught by gamefish anglers off south-eastern Australia, 1961–90. Aust. J. Mar. Freshw. Res. 43, 213–225. (doi:10.1071/MF9920213) [Google Scholar]
- 11.Wetherbee BM, Lowe CG, Crow GL. 1994. A review of shark control in Hawaii with recommendations for future research. Pac. Sci. 48, 95–115. (doi:hdl.handle.net/10125/2202) [Google Scholar]
- 12.Macbeth WG, Geraghty PT, Peddemors VM, Gray CA. 2009. Observer-based study of targeted commercial fishing for large shark species in waters off northern New South Wales: final report to the Northern Rivers Catchment Management Authority, Project No. IS8-9-M-2, Fisheries Final Report Series 114. Cronulla, NSW, Australia: Industry and Investment NSW.
- 13.Liston J, Clark G, Alexander D. 2011. Pacific island heritage: archaeology, identity and community. Canberra, ACT, Australia: ANU E Press, The Australian National University. [Google Scholar]
- 14.Reid DD, Robbins WD, Peddemors VM. 2011. Decadal trends in shark catches and effort from the New South Wales Shark Meshing Program 1950 to 2010. Mar. Freshw. Res. 62, 676–693. (doi:10.1071/MF10162) [Google Scholar]
- 15.Holmes BJ, Sumpton WD, Mayer DG, Tibbetts IR, Neil DT, Bennett MB. 2012. Declining trends in annual catch rates of the tiger shark (Galeocerdo cuvier) in Queensland, Australia. Fish. Res. 129–130, 38–45. (doi:dx.doi.org/10.1016/j.fishres.2012.06.005) [Google Scholar]
- 16.Holmes BJ, Pepperell JP, Griffiths SP, Jaine FRA, Tibbetts IR, Bennett MB. 2014. Tiger shark (Galeocerdo cuvier) movement patterns and habitat use determined by satellite tagging in eastern Australian waters. Mar. Biol. 161, 2645–2658. [Google Scholar]
- 17.Werry JM, Planes S, Berumen ML, Lee KA, Braun CD, Clua E. 2014. Reef-fidelity and migration of tiger sharks, Galeocerdo cuvier, across the Coral Sea. PLoS ONE 9, e83249 (doi:10.1371/journal.pone.0083249) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fitzpatrick R, Thums M, Bell I, Meekan M, Stevens JD, Barnett A. 2012. A comparison of the seasonal movements of tiger sharks and green turtles provides insights into their predator-prey relationship. PLoS ONE 7, e51927 (doi:10.1371/journal.pone.0051927) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ferreira LC, Thums M, Meeuwig JJ, Vianna GMS, Stevens J, McAuley R, Meekan MG. 2015. Crossing latitudes—long-distance tracking of an apex predator. PLoS ONE 10, e0116916 (doi:10.1371/journal.pone.0116916) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heithaus MR, Wirsing AJ, Dill LM, Heithaus LI. 2007. Long-term movements of tiger sharks satellite-tagged in Shark Bay, Western Australia. Mar. Biol. 151, 1455–1461. (doi:10.1007/s00227-006-0583-y) [Google Scholar]
- 21.Holland KN, Wetherbee BM, Lowe CG, Meyer CG. 1999. Movements of tiger sharks (Galeocerdo cuvier) in coastal Hawaiian waters. Mar. Biol. 134, 665–673. (doi:10.1007/s002270050582) [Google Scholar]
- 22.Meyer CG, Clark TB, Papastamatiou YP, Whitney NM, Holland KN. 2009. Long-term movement patterns of tiger sharks Galeocerdo cuvier in Hawaii. Mar. Ecol. Prog. Ser. 381, 223–235. (doi:10.3354/meps07951) [Google Scholar]
- 23.Papastamatiou YP, Meyer CG, Carvalho F, Dale JJ, Hutchinson MR, Holland KN. 2013. Telemetry and random walk models reveal complex patterns of partial migration in a large marine predator. Ecol. Soc. Am. 94, 2595–2606. (doi:10.1890/12-2014.1) [DOI] [PubMed] [Google Scholar]
- 24.Whitney NM, Crow GL. 2007. Reproductive biology of the tiger shark (Galeocerdo cuvier) in Hawaii. Mar. Biol. 151, 63–70. (doi:10.1007/s00227-006-0476-0) [Google Scholar]
- 25.Dulvy NK, et al. 2008. You can swim but you can't hide: the global status and conservation of oceanic pelagic sharks and rays. Aquat. Conserv. 18, 459–482. (doi:10.1002/aqc.975) [Google Scholar]
- 26.Bernard AM, Feldheim KA, Shivji MS. 2015. Isolation and characterization of polymorphic microsatellite markers from a globally distributed marine apex predator, the tiger shark (Galeocerdo cuvier). Conserv. Genet. Resour. 7, 509–511. (doi:10.1007/s12686-014-0408-0) [Google Scholar]
- 27.Ryman N, Palm S. 2006. POWSIM: a computer program for assessing statistical power when testing for genetic differentiation. Mol. Ecol. Notes 6, 600–602. (doi:10.1111/j.1471-8286.2006.01378.x) [DOI] [PubMed] [Google Scholar]
- 28.Sunnucks P, Hales DF. 1996. Numerous transposed sequences of mitochondrial cytochrome oxidase I-II in aphids of the genus Sitobion (Hemiptera: Aphididae). Mol. Biol. Evol. 13, 510–524. (doi:10.1093/oxfordjournals.molbev.a025612) [DOI] [PubMed] [Google Scholar]
- 29.Van Oosterhout C, Hutchinson W, Wills D, Shipley P. 2004. Microchecker: software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 4, 535–538. (doi:10.1111/j.1471-8286.2004.00684.x) [Google Scholar]
- 30.Raymond M, Rousset F. 1995. An exact test for population differentiation. Evolution 49, 1280–1283. (doi:10.2307/2410454) [DOI] [PubMed] [Google Scholar]
- 31.Rice WR. 1989. Analysing tables of statistical tests. Evolution 43, 223–225. (doi:10.2307/2409177) [DOI] [PubMed] [Google Scholar]
- 32.Queller DC, Goodnight KF. 1989. Estimating relatedness using genetic markers. Evolution 43, 258–275. (doi:10.2307/2409206) [DOI] [PubMed] [Google Scholar]
- 33.Peakall R, Smouse PE. 2012. GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research—an update. Bioinformatics 28, 2537–2539. (doi:10.1093/bioinformatics/bts460) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Excoffier L, Laval G, Schneider S. 2005. Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol. Bioinformatics Online 1, 47–50. [PMC free article] [PubMed] [Google Scholar]
- 35.Jost L. 2008. GST and its relatives do not measure differentiation. Mol. Ecol. 17, 4015–4026. (doi:10.1111/j.1365-294X.2008.03887.x) [DOI] [PubMed] [Google Scholar]
- 36.Pritchard JK, Stephens M, Donnelly P. 2000. Inference of population structure using multilocus genotype data. Genetics 155, 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Earl DA, Vonholdt BM. 2012. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 4, 359–361. (doi:10.1007/s12686-011-9548-7) [Google Scholar]
- 38.Kopelman NM, Mayzel J, Jakobsson M, Rosenberg NA, Mayrose I. 2015. Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Resour. 15, 1179–1191. (doi:10.1111/1755-0998.12387) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mirams AGK, Tremi EA, Shields JL, Liggins L, Riginos C. 2011. Vicariance and dispersal across an intermittent barrier: population genetic structure of marine animals across the Torres Strait land bridge. Coral Reefs 30, 937–949. (doi:10.1007/s00338-011-0767-x) [Google Scholar]
- 40.Button KS, Loannidis JPA, Mokrysz C, Nosek BA, Flint J, Robinson ESJ, Munafó MR. 2013. Power failure: why small sample size undermines the reliability of neuroscience. Nat. Rev. Neurosci. 14, 365–376. (doi:10.1038/nrn3475) [DOI] [PubMed] [Google Scholar]
- 41.Meyer CG, Papastamatiou YP, Holland KN. 2010. A multiple instrument approach to quantifying the movement patterns and habitat use of tiger (Galeocerdo cuvier) and Galapagos sharks (Carcharhinus galapagensis) at French Frigate Shoals, Hawaii. Mar. Biol. 157, 1857–1868. (doi:10.1007/s00227-010-1457-x) [Google Scholar]
- 42.Willing E, Dreyer C, van Oosterhout C. 2012. Estimates of genetic differentiation measured by FST do not necessarily require large sample sizes when using many SNP markers. PLoS ONE 7, e42649 (doi:10.1371/journal.pone.0042649) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baum JK, Myers RA, Kehler DG, Worm B, Harley SJ, Doherty PA. 2003. Collapse and conservation of shark populations in the northwest Atlantic. Science 299, 389–392. (doi:10.1126/science.1079777) [DOI] [PubMed] [Google Scholar]
- 44.Myers RA, Baum JK, Shepherd TD, Powers SP, Peterson CH. 2007. Cascading effects of the loss of apex predatory sharks from a coastal ocean. Science 315, 1846–1850. (doi:10.1126/science.1138657) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Tiger shark genotypes are available through the UQ eSpace (http://espace.library.uq.edu.au/view/UQ:613874).