

Abstract
Introduction
Antibiotic overuse drives antibiotic resistance. The optimal duration of antibiotic therapy for Gram-negative bacteraemia (GNB), a common community and hospital-associated infection, remains unknown and unstudied via randomised controlled trials (RCTs).
Methods and analysis
This investigator-initiated, multicentre, non-inferiority, informatics-based point-of-care RCT will randomly assign adult hospitalised patients receiving microbiologically efficacious antibiotic(s) for GNB to (1) 14 days of antibiotic therapy, (2) 7 days of therapy or (3) an individualised duration determined by clinical response and 75% reduction in peak C reactive protein (CRP) values. The randomisation will occur in equal proportions (1:1:1) on day 5 (±1) of efficacious antibiotic therapy as determined by antibiogram; patients, their physicians and study investigators will be blind to treatment duration allocation until the day of antibiotic discontinuation. Immunosuppressed patients and those with GNB due to complicated infections (endocarditis, osteomyelitis, etc) and/or non-fermenting bacilli (Acinetobacter spp, Burkholderia spp, Pseudomonas spp) Brucella spp, Fusobacterium spp or polymicrobial growth with Gram-positive organisms will be ineligible. The primary outcome is incidence of clinical failure at day 30; secondary outcomes include clinical failure, all-cause mortality and incidence of Clostridiumdifficile infection in the 90-day study period. An interim safety analysis will be performed after the first 150 patients have been followed for ≤30 days. Given a chosen margin of 10%, the required sample size to determine non-inferiority is roughly 500 patients. Analyses will be performed on both intention-to-treat and per-protocol populations.
Ethics and dissemination
Ethics approval was obtained from the cantonal ethics committees of all three participating sites. Results of the main trial and each of the secondary endpoints will be submitted for publication in a peer-reviewed journal.
Trial registration number
This trial is registered at www.clinicaltrials.gov (NCT03101072; pre-results).
Keywords: Bacteraemia, Gram-negative, Duration, Antibiotic Therapy, Point-of-care Randomization
Strengths and limitations of this study.
This large, multicentre randomised controlled trial will test the non-inferiority of shorter antibiotic courses for Gram-negative bacteraemia and thus provide clinicians with much-needed data from randomised patients on how to safely reduce antibiotic consumption.
This trial provides proof-of-concept and safety data for individualising antibiotic durations in the treatment of Gram-negative bacteraemia.
The trial will pilot electronic health record-guided patient-identification, randomisation and data-transfer procedures at its principal site.
Two substudies will explore (1) patient recall after oral versus written consent and (2) the external validity of the trial’s findings by observationally following the outcomes of excluded patients, respectively
The lack of full blinding (eg, with dummy for short-course arms) is a limitation to the study design.
Introduction and rationale
Antibiotic resistance is growing at an alarming rate and is now considered by many governments to be one of the most serious global threats of the 21st century.1–3 The key driver of antibiotic resistance is antibiotic overuse,4 and patients who receive extended courses of broad-spectrum antibiotics are at significantly higher risk for later infections with difficult-to-treat, multidrug-resistant bacteria.5 6
No randomised controlled trial (RCT) evaluating the optimal duration of therapy for Gram-negative bacteraemia (GNB), a frequent community-associated and hospital-associated infection, has been published. Traditionally, guidelines have somewhat arbitrarily recommended long antibiotic courses of 2 weeks, even though patients with no structural complications may recover after only 5 days of therapy.7 Direct evidence is mounting that longer antibiotic courses leave patients at risk of acquiring difficult-to-treat multiresistant organisms. In one RCT comparing 8 to 15 days of antibiotic therapy for ventilator-associated pneumonia, multiresistant pathogens emerged significantly less frequently in those who had received 8 days of antibiotics.6 Indeed, given rising concerns over resistance, many physicians have reduced antibiotic durations for uncomplicated GNB to 7 days with no apparent untoward consequences.8
Although these shorter durations have not been directly studied, there is nonetheless mounting evidence that they do not increase patients’ risk of relapse or other complications: several randomised studies evaluating the optimal duration of antibiotic therapy for pyelonephritis,9 pneumonia,10–12 peritonitis13 14 and surgical site infections15 have included patients with concurrent bloodstream infections (both Gram negative and positive). These have compared durations as short as 5 days with longer (7 or 14 days) durations, and none demonstrated differences in the subset of patients with bacteraemia in clinical or microbiological outcomes.7 Nonetheless, none of these trials aimed specifically to assess the equivalence or the non-inferiority of shorter versus longer durations of antibiotic therapy.
Antibiotic durations could also be individualised, guided by clinical response as measured by objective markers, including inexpensive biomarkers such as C reactive protein (CRP).16 This acute phase protein is a reliable and highly sensitive marker of inflammation across different patient populations and infections.17–19 While procalcitonin has been studied in more than 20 RCTs as a biomarker to guide the duration of antibiotic therapy in severe infections20 21 and has indeed proved the concept of biomarker-guided therapy, observational and randomised studies16 22 have demonstrated no substantial differences in the ability of these two markers to reflect improvement (or worsening) in the clinical course of severe infections. Indeed, an RCT comparing the two markers head-to-head for guiding antibiotic therapy duration in sepsis found that a procalcitonin-based protocol was not superior to a CRP-based protocol, while no difference in morbidity or mortality was observed.16 CRP is substantially less expensive and more accessible than procalcitonin across various clinical settings: many community hospitals do not offer or routinely perform the procalcitonin assay.
The increasing equipoise with regard to varying treatment durations, the high incidence of Gram-negative bacteraemia (GNB), the relative ease of its diagnosis and the high stakes of antibiotic overconsumption in an ageing population combine to make antibiotic use for GNB an appropriate subject of study for a point-of-care (POC) randomised trial. We hypothesise that shorter antibiotic courses for GNB reduce antibiotic treatment days without increasing relapse rate or mortality.
Objectives
The trial’s primary objective is to determine whether shorter antibiotic courses (5–7 days) are non-inferior to a 2-week antibiotic course in the treatment of GNB. Secondary objectives are to determine whether antibiotic durations can be safely determined via a simple algorithm employing clinical and laboratory (CRP) markers, whether shorter antibiotic courses for GNB will result in a decrease in antibiotic days, incidence of Clostridium difficile infection, emergence of bacterial resistance and length of hospital stay.
Methods and analyses
Study design and setting
This investigator-initiated, analyst-blinded, POC RCT will enrol 500 hospitalised adult patients diagnosed with community-acquired or hospital-acquired GNB. The trial will take place at the Geneva University Hospitals (HUG; principal site), the Centre Hospitalier Universitaire Vaudois (CHUV) and the Cantonal Hospital St Gallen (KSSG; figure 1). The HUG, CHUV and KSSG perform roughly 59 000, 43 000 and 35 000 admissions per year, respectively; each has both a microbiology laboratory and a team of consulting infectious disease physicians available at all times.
Figure 1.
Trial sites: the HUG is the principal site; CHUV and KSSG are participating peripheral sites. CHUV, Centre Hospitalier Universitaire Vaudois; HUG, Geneva University Hospitals; KSSG, Catonal Hospital St Gallen.
Study population and entry criteria
Potential study patients will be identified via both the laboratory and the electronic health record (EHR): in all of these hospital systems, the microbiology laboratory is required to report daily all positive blood cultures to the infectious disease consult team.
Inclusion criteria
Age ≥18 years
The presence of GNB in at least one blood culture bottle
Treatment with a microbiologically efficacious antibiotic
Exclusion criteria
Immunosuppression (including HIV infection with CD4 cell count ≤500/µL, haematopoietic stem-cell transplantation in the first month after transplantation and at any time before engraftment, neutropaenia in the 48 hours prior to randomisation, receipt of high-dose steroids (>40 mg prednisone or its equivalent) daily for >2 weeks) in the 2 weeks prior to randomisation
- GNB due to the following complicated infections:
- Endocarditis or other endovascular infection without a removable focus;
- Necrotising fasciitis;
- Osteomyelitis or septic arthritis;
- Confirmed prostatitis;
- Undrainable abscess or other unresolved sources requiring surgical intervention (eg, cholecystitis) at the time of enrolment;
- Central nervous system infections;
- Empyema;
GNB due to non-fermenting bacilli (Acinetobacter spp, Burkholderia spp, Pseudomonas spp) Brucella spp, Fusobacterium spp or polymicrobial growth with Gram-positive organisms;
Fever (≥38°C) or haemodynamic instability in the 24 hours prior to recruitment.
Intervention
With day 1 defined as the first day of appropriate (microbiologically efficacious per antibiogram results) antibacterial therapy, patients will be randomised 1:1:1 on day 5 (±1) to one of the following three arms (figure 2):
‘Fixed long’ antibiotic course of 14 days (control arm);
‘Fixed short’ antibiotic course of 7 days (first intervention arm);
‘Individualised’ antibiotic course: starting on day 5, therapy will be discontinued after the patient has been afebrile for 48 hours and the CRP level has decreased from its peak by at least 75% (second intervention arm).
Figure 2.
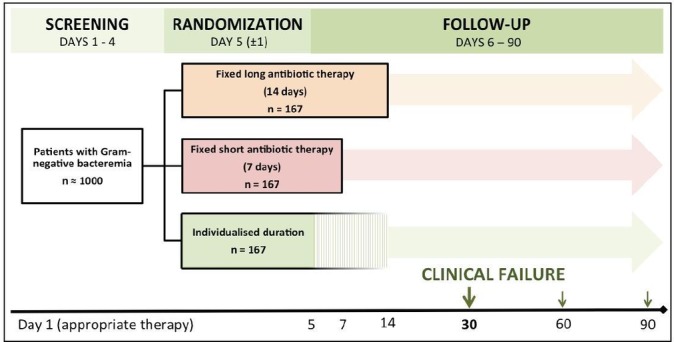
Trial flow: day 1 is defined as the first day of microbiologically appropriate antibiotic therapy. Patients will be randomised on day 5 (±1) and followed until day 90.
The rationale for our ‘fixed-short’ treatment arm of 7 days derives from several observational studies suggesting the safety of shorter antibiotic durations (5–10 days) for patients with bacteraemia.7 23 24
CRP algorithm for the individualised-duration arm
The rationale for the individualised algorithm’s specific use of a 75% reduction in peak CRP values is based on an RCT of patients in intensive care units (ICUs) with severe sepsis or septic shock with or without bacteraemia.16 That study demonstrated that an even more restrictive algorithm (antibiotic stop once the CRP has decreased by ≥50% if peak CRP was ≥100 mg/L or once CRP is less than 25 mg/L if peak CRP was <100 mg/L) was safe and effective to reduce antibiotic use. This study additionally used a 7-day maximum duration of antibiotic therapy for non-bacteraemic patients while bacteraemic patients received at least 7 days of antibiotics.16 Our individualised algorithm is slightly adapted in analogy to procalcitonin-based algorithms, which have been successfully tested in several RCTs and used ≥80% decreases of procalcitonin to discontinue antibiotic therapy.20 25–28 Our slight modifications take into account the slower decrease of CRP values compared with procalcitonin after resolution of an infection27 29 and incorporate an additional safety margin compared with the study by Oliveira et al, which treated bacteraemic patients differentially.16 Of note, if the CRP value has not decreased by 75% by day 14, the marker will no longer be used to guide the duration of therapy. In these cases, the duration will be determined by clinical judgement per usual practice.
Choice of antibiotic(s) and mode of administration
In all arms, the choice and mode of administration of antibiotic(s) will be left to the patient’s attending physician and consulting infectious disease specialist and thus will follow usual standards of care, determined primarily by the three sites’ local institutional antibiotic therapy guidelines and antimicrobial resistance patterns. (Resistance prevalences in these hospitals and communities are similar, and the sites’ treatment guidelines contain no significant discrepancies with respect to acceptable antibiotic treatment standards of uncomplicated GNB.)
De-escalation (from a broad-spectrum to a more narrow-spectrum antibiotic), switches from intravenous to oral antibiotic therapy or from intermittent to continuous infusions or vice-versa, will be allowed per current standard practice. Study investigators will not interfere with these clinical decisions but will collect detailed data on all therapeutic management for later subanalyses. Although in Geneva randomisation and subsequent discontinuation of antibiotic therapy according to treatment arm will occur through the EHR, at all sites study investigators will also not interfere with attending physicians’ decisions to prolong therapy should a patient’s clinical condition worsen. (See sections on statistical analysis and sample size calculation.)
Randomisation
We determine day 5 as the appropriate randomisation point because this is the usual timing of the study’s and clinicians’ essential question:
Now that my patient has been stabilized and appears to be improving, when can I safely discontinue antibiotic therapy?
General principles and peripheral site randomisation procedures
Randomisation will be based on investigator-blinded blocks of randomly varying size to protect against potential predictability of treatment assignments. Blocks will contain 3, 6, 9 or 12 allocations. Randomisation will be stratified by study site, given that the three sites do not share a common EHR system and that site launches will be staggered (HUG will be the first site to launch; CHUV and KSSG will follow). For randomisation at HUG, see the next section. For randomisation at CHUV and KSSG, a statistician not involved in the study analysis will produce the randomisation list prior to the initiation of the study; the blocks’ order will be generated by use of a computer-based randomised number system, and the allocation implemented by means of sealed, opaque envelopes. The statistician will keep copies of the randomisation list.
POC randomisation through the electronic health record at HUG
In POC trials, the extent of the EHR’s involvement in randomisation is variable: on one end of the spectrum, the EHR simply provides an automatic alert to a prescribing physician making her aware of the existence of the POC trial and the patient’s likely eligibility for it while simultaneously alerting a study investigator of the patient. At the other end of the spectrum, the EHR provides such alerts and, according to a physician-triggered EHR workflow, ultimately performs the actual randomisation with treatment assignment.
In the present study, through the work of information technology (IT) specialists at HUG, this principal study site will implement a fully EHR-integrated process (figure 3) using HUG’s ‘Dossier patient informatique’ (DPI), with an initial, early alert to the study team at the moment the positive blood culture is registered in the EHR and automatic alerts for the treating physician at POC and study personnel once the patient enters the eligibility window. Study personnel will be notified to (1) provide information to the patient, (2) verify study inclusion criteria and then (3) approve the EHR-based randomisation.
Figure 3.
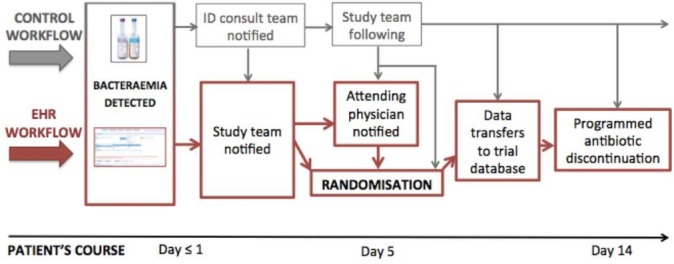
Electronic-healthcare record workflow for patient identification, randomisation and follow-up in Geneva. The EHR workflow is outlined in red, the control (‘back-up’) workflow in grey. Grey arrows indicate safety valves; these cover all points at which the EHR workflow could malfunction. In this hypothetical case, the patient has been randomised to the control arm (antibiotic therapy duration of 14 days). EHR, electronic health record.
At the moment of randomisation, the EHR will (1) automatically place a note of participation in the medical record and (2) establish the duration of the patient’s antibiotic prescription (7 or 14 days or the individualised duration, which will also be determined within the EHR, as it houses temperature and CRP data). On the predetermined day of antibiotic discontinuation, both physicians and nurses will receive alerts regarding that discontinuation. These alerts will be repeated until the physician electronically signs his acknowledgement of the discontinuation.
The control workflow
Importantly, the current study focus of GNB provides an ideal occasion for piloting and testing this process since, in reality, the study team will already be well aware of all patients hospital-wide with any GNB, given the established daily communication between microbiology laboratories and infectious disease teams described above. There will thus be a ‘control’ workflow shadowing the automated EHR workflow at all times, with safety valves present at all workflow nodes (figure 3).
The EHR-determined randomisation procedure
The EHR will be programmed to randomise using the same principles described above: randomisation will be based on randomly varying blocks with 3, 6, 9 or 12 treatment duration assignments. The bioinformatics specialist leading the informatics component of this trial will keep a copy of the EHR’s randomisation list, as will a designated, unblinded member of the study team (control workflow).
Blinding
‘Ad terminum’ blinding of patients, attending physicians and investigators
The blinding of patients, hospital staff and designated trial investigators to assigned treatment duration will be key to avoiding the introduction of bias in the follow-up/management of patients between study arms. It is conceivable that the knowledge that one’s patient will receive a shorter course of antibiotics could result in defensive medicine, with that patient’s receiving ‘special treatment’ such as additional surveillance blood cultures and/or imaging tests just before antibiotic discontinuation. For this reason, we will carry blinding of patients, treating hospital staff and designated investigators through the furthest study point possible. These parties will be blinded to treatment assignment from randomisation until antibiotic discontinuation (‘ad terminum’). Thus, in the fixed short arm, all parties will be blinded until day 7, and in the individualised arm until the clinical requirements for discontinuation have been met. In the effort to avoid unblinding by the process of elimination after day 7, attending physicians and nurses will not be made aware of the specific algorithm defining criteria for antibiotic discontinuation in the individualised group. They will thus be allowed to view all CRP results, but they will not be able to predict when an individualised duration will end.
Blinded outcomes assessment and data analysis
Clinical data on all included patients will be collected regularly by study personnel and recorded in the electronic case report form (eCRF) and database (Secutrial V.4.8). HUG data managers, experienced in generating blinded reports, will then provide data exports to a blinded outcomes assessor and a blinded data analyst; these exports will contain recoded (‘scrambled’) study identification numbers and no information on treatment assignments, allowing for both fully blinded outcomes assessment and data analysis.
Lifting the blind
Because (1) no experimental therapy will be given and (2) treating physicians will have the right to override the patient’s treatment duration assignment in the case of clinical worsening, it is not anticipated that any early lifting of the blind will be necessary. Nonetheless, instructions and the means to access the randomisation list overnight and on weekends will be available to the infectious disease physician on call should early lifting of the blind be deemed necessary.
Primary and secondary outcome measures
The primary outcome will be the clinical failure rate in all arms at day 30. Clinical failure is defined by the presence of at least one of the following:
Relapse: a recurrent bacteraemia due to the same bacterium occurring from the day of treatment cessation and through day 30;
Local suppurative complication that was not present at infection onset (eg, renal abscess in pyelonephritis, empyema in pneumonia);
Distant complications of the initial infection, defined by growth of the same bacterium causing the initial bacteraemia (as determined by antibiotic susceptibility profiling);
The restarting of Gram-negative-directed antibiotic therapy after its initial discontinuation due to clinical worsening suspected to be due to the initial infecting organism and for which there is no alternate diagnosis/pathogen suspected;
Death due to any cause through day 30.
Secondary outcomes include the incidence of clinical failure at days 60 and 90; all-cause mortality at days 30, 60 and 90; the total number of antibiotic days; the incidence of antibiotic-related adverse events through day 90 (including C. difficile infection, a common by-product of antibiotic overconsumption); the incidence of the emergence of bacterial resistance in those with recurrence; the number of patients in each arm whose assigned antibiotic duration was ‘overridden’ by physicians in the absence of clinical failure (and the reasons for these deviations) and length of hospital stay. Cost-effectiveness and other health-economic analyses will also be performed.
Additional subgroup analyses will be performed for main causative organisms, resistance patterns, involved organ systems, antibiotic regimens including single versus combination therapy and de-escalation. Moreover, risk factors for clinical failure will be determined, such as age, antibiotic choice, anatomic focus of primary infection, comorbidity status, infection acquisition type (community vs nosocomial), severity of illness at the time of diagnosis and so on.
Other definitions
Bacteraemias will be categorised as nosocomial if the first positive sample is taken ≥48 hours after hospital admission; otherwise they will be categorised as community acquired. Additionally, if the patient has been admitted to hospital in the preceding 30 days, transferred from another healthcare facility (eg, long-term care unit), is receiving chronic dialysis or has metastatic cancer, their bacteraemia will be considered healthcare associated.30
Severity of illness at the time of bacteraemia onset will be defined by the Quick SOFA Score (qSOFA), which can be determined for all patients (including non-ICU patients). The score consists of three variables and has a maximum of three points (one point each for systolic blood pressure ≤100 mm Hg, respiratory rate ≥22 breaths/min and any altered mental state (Glasgow Coma Scale <15)); the presence of ≥2 points is associated with higher risks for mortality and extended ICU stay.31
Study schedule
After randomisation, patients will be followed for a total of 90±21 days. An important principle of POC trials is that patients be allowed to remain in their normal clinical setting; they are followed non-invasively for outcomes data. The only additional laboratory test that may be requested is later-phase CRP measurements if these have not already been ordered by treating physicians (on days 8±2 and 12±2 in patients whose antibiotic therapy was not already discontinued).
On days 30, 60 and 90, clinical data necessary for determining the primary and secondary outcomes listed above will be collected. If on these days the patient is no longer hospitalised, he will be contacted by study staff by telephone and interviewed according to a structured questionnaire including the clinical information in the following section. In the event that a patient reports clinical worsening at the time of follow-up, he will be asked to come for an in-person visit at the HUG’s Policlinique des maladies infectieuses, or respective outpatient clinics at CHUV and KSSG, respectively. The study’s schedule of assessments is shown in table 1.
Table 1.
Schedule of assessments. Day 1 is the first day of microbiologically efficacious antibiotic therapy
| Study visit/observation point | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
| Screening | Randomisation | Follow-up | |||||
| Timeline (study day) | 0–5 | 5 | 8 | 12 | 30 | 60 | 90 |
| Window period (days) | ±1 | ±1 | ±2 | ±2 | ±7 | ±14 | ±21 |
| Informed consent | X | (X) | (X) | (X) | |||
| Entry criteria | X | ||||||
| CRP measurement* (2 mL blood) | X | X | |||||
| AEs reviewed | X | X | |||||
| SAEs reviewed | X | X | X | X | X | ||
| Other outcomes data collected | X | X | X | ||||
*The CRP will not be requested in patients whose antibiotic therapy has already been discontinued.
AE, adverse events; CRP, C reactive protein; SAE, serious adverse events.
Interim analyses
An interim analysis for safety will be performed after roughly 150 patients have reached 30 days of follow-up (described in more detail below). At this time, and if determined necessary, a non-blinded investigator will also assess the performance of the CRP as a marker for guiding durations. If the marker is proving either impractical (eg, logistically difficult to obtain) or its algorithm difficult to follow (eg, CRP initially decreases but not quite by 75%, then rises again due to another inflammatory process), an alteration in the algorithm or in the arm itself will be explored. Any recommended changes will undergo review by all study investigators and methodologists, and any proposed amendment to the protocol will be submitted to the central ethics committee per usual routine.
Safety ‘checkpoint’ analyses
An initial safety ‘checkpoint’ analysis will be performed after 150 patients (roughly one-third of the target) have reached the 30-day follow-up mark to assess whether arms with shorter therapy durations (fixed short duration of 7 days and individualised arm) could potentially result in worse clinical outcomes, specifically with increased clinical failure. The outcomes assessment and data analysis for this assessment will be done in blinded fashion. If, however, results show a significant difference in outcomes in any arm, the blind will be lifted for the data analyst. (Blinding and unblinding of data will occur with the aid of the data manager, as described above.) Given the relatively small number of patients to be included in the first safety analysis (necessarily reducing power), statistical support in the interpretation of the data will be provided by study statisticians.
Unblinded results will be forwarded to an independent Safety Monitoring Board (SMB) for review. Should patients in either of the intervention arms demonstrate significantly worse clinical outcomes, recommendations made by the SMB will be followed. Other safety checkpoints may be scheduled depending on the outcome of this first fixed analysis.
Adverse events
An adverse event (AE) is any untoward medical occurrence in a subject that may occur during or after administration of a pharmaceutical product and does not necessarily have a causal relationship with the intervention. An AE can therefore be any unfavourable and unintended sign (including an abnormal laboratory finding), symptom or disease temporally associated with the study intervention, whether or not considered related to the study intervention.
For the purposes of this study, whose focus is not on the choice of antibiotic but on its duration and within which only postmarket non-experimental antibiotics with well-established safety profiles will be used, only AE considered possibly, probably or certainly related to the antibiotic(s) being administered for the GNB will be recorded into the eCRF. The AE will thus be recorded during and in the 2 days following discontinuation of antibiotic therapy targeting the GNB. After that point and for the remainder of the study period, only symptomatic C. difficile infection and serious adverse events will be recorded in the eCRF.
Sample size calculation
Previous observational studies and RCTs including patients with bacteraemia10 16 30 32 have demonstrated clinical failure rates between 10% and 30% in settings with access to broad-spectrum antibiotics and resistance prevalences similar to those in Switzerland. The primary reason for failure in these and other studies is the lack of appropriate antibiotic therapy (either due to antimicrobial resistance or a delay in therapy initiation). Because this trial’s inclusion criteria require 5 days of microbiologically adequate antibiotic therapy at the time of randomisation, we assume the upper limit of ‘success’ or, inversely, the clinical failure will be 10% in both the control and intervention arms. To establish non-inferiority, we will allow a difference up to 10% in the primary outcome. The chosen margin is wide because the expected gain from reducing the use of antibiotics is significant (decreased odds for antibiotic resistance, reduction in treatment adverse events, reduced medical costs by shorter length of hospital stay).9 Furthermore, as we will have excluded immunosuppressed patients and those with complicated infections, there will be decreased risk for life-threatening events and serious deterioration.
Further assuming a one-sided type 1 error (α) of 0.025, a power (ß) of 0.80, an attrition (loss to follow-up) of ~5% and potential treatment switching of ~12%, 167 patients will be needed in each of the three arms to prove non-inferiority, making the total sample size 500.
Statistical analysis
We will perform the primary analysis on both the intention-to-treat (ITT) population (all patients randomised) and the per-protocol (PP) population (all patients adhering to the study protocol with no major deviations). While ITT analyses are critical in all studies, in non-inferiority trials PP analyses take on a particularly important role in the effort to avoid commission of a type 2 error.33 (One can imagine, for example, that non-adherence to the full 14-day antibiotic regimen could lead to this arm’s appearing generally less effective, thus lowering the bar for the two intervention arms.) In this particular study, however, the PP analysis will have another key role. As stated above, attending physicians will have the freedom to override their patients’ treatment duration assignments in the event of perceived clinical worsening. There may thus be some switching from intervention towards control arms (non-adherence to treatment assignment) with the risk of diluting the difference in clinical failure rates between the intervention groups and the control group and thus increasing the risk of incorrectly concluding non-inferiority of shorter antibiotic duration. The sample size has thus been increased to accommodate treatment switching of ~12% study wide or some 20 patients per group (see above).
Descriptive analyses with standard methods for randomised trials will be used to measure primary and secondary outcomes. Continuous variables will be compared between the three study arms with the use of Student's t-test or the Mann-Whitney U test, as appropriate; categorical variables will be compared with the χ2 test or Fisher’s exact test, as appropriate. To test the hypothesis of non-inferiority for the prespecified margin of 10 percentage points (see "Sample size calculation"), we will perform a generalised linear regression model with a log link and binomial distribution reporting risk differences of clinical failure between intervention arms compared with the control arm. The treatment assignment will be the main predictor with the control arm (‘fixed long’) as the reference and the model will be adjusted for the study centre.34 If some differences in the use of antibiotics are described between the three intervention arms, we will also adjust for it in the regression model. We will conclude non-inferiority of the ‘fixed short’ arm then of the ‘individualised’ arm compared with the ‘fixed long’ if the 95% upper bound is less than the 10 percentage points’ non-inferiority margin. With three centres, a mixed regression model is less appropriate. We will also present risk ratios or ORs with 95% CIs.
Missing data
Missing data will be taken into consideration using several methods, among them responder analysis, complete case analysis (modified ITT) and potentially multiple imputation. These sensitivity analyses will be used to validate study findings.
Data collection and management
Data management will be contracted to the Unité d’Investigation Clinique (UIC). Baseline and outcomes clinical data from primary source records will be entered into the eCRF for integration into the electronic database (SecuTrial platform). At the HUG site, in an effort to modernise data collection in this POC trial, the UIC data management team will work together with the DPI informatics team to pilot algorithms for the regular transfer of coded, postrandomisation clinical data directly from DPI into the SecuTrial database, reducing both staffing/resource use and the risk of manual transcription errors. At the peripheral sites (neither of which uses DPI), study investigators will enter data into SecuTrial.
The principal investigator will be responsible for overseeing the receipt, entering, cleaning, querying, analysis and storage of all data that accrue from the study by designated persons. For each set of data, quality control and triggers to computerised logic and/or consistency checks will be systematically applied to detect errors or omissions. After integration of all corrections in the complete set of data, the database will be locked and saved before being released for statistical analysis. Each step of this process will be monitored through the implementation of individual passwords and/or regular backups to maintain appropriate database access and to guarantee database integrity.
All data will be coded: patients’ data will be identified by a unique study identification number containing no personally identifiable information (PII) in the eCRF. A separate confidential file containing PII will be stored in a secured (locked) location in accordance with data protection requirements. Only the sponsor representative, study investigators, the study monitor and the Ethics Commission will be granted access to the records.
Observational substudies
Nested prospective observational cohort study on recall and understanding after oral versus written informed consent
Background and rationale: oral consent with witness testimony
In line with the recent advice of the Geneva Ethics Commission and with OClin Art.8 al.1b, patients consenting to participate in this study may provide oral consent when an independent witness can sign testimony to that consent. The background and justification for this informed consent model are detailed below. The decision to obtain oral or written consent will be left to the discretion of the including investigator and will depend on the clinical and cognitive state of the patient.
Very little is known about patient recall and understanding after oral versus written consent, whether that consent was granted for a research study or a clinical procedure; we therefore propose a nested study to compare these outcomes among the patients providing oral consent with those of patients providing written consent for participation in this study, as well as to outcomes of historical controls (patients providing written consent for participation in other trials, such as those followed by Chenaud et al.35 We know that recall and understanding in the weeks after written consent are not optimal; our hypothesis is that they will not differ much (will not be significantly worse) after oral consent.
Nested oral consent study design, setting and population
All patients approached for inclusion in the "Point-of-care, informatics-based randomised controlled trial for decreasing overuse of antibiotic therapy in Gram-negative bacteraemia" (PIRATE) study throughout its three trial sites will be eligible for participation in this nested, prospective observational cohort study. This substudy will begin and end in step with the larger PIRATE trial, thus patients are expected to be included from the spring of 2017 through the spring of 2019.
Nested oral consent study outcomes
The primary outcome is the percentage of patients in both groups who recall granting informed consent to participate in the PIRATE trial on day 30 (±7 days). Secondary outcomes will include the same endpoint on days 60 (±14 days) and 90 (±21 days), as well as the ability to recall the purpose and risks of the trial as stated in the information brochure at all named time-points. For these, the simple questionnaire in box 1 will be used. Another outcome will simply be the number of patients included in each arm, and the investigator’s cited reason for pursuing an oral versus written consent. Finally, we will perform correlation analyses for recall and understanding, looking at baseline demographic factors, other factors such as whether the patient asked a question during the initial information encounter,35 whether family members were present and whether an attending physician or family member signed the witness testimony.
Box 1. Informed consent - recall and understanding post-consent.
Do you recall agreeing to participate in this study?
Do you recall the purpose of this study? (per information brochure: “The purpose of this research study is to compare 14 days of antibiotic therapy to either 7 days or an “individualized” number of days…for their efficacy and safety.”
Do you recall the risks of the study? (per information brochure: “Rarely an additional blood draw may be needed to continue to measure the response to therapy. Blood draws can lead to bruising and pain at the point of puncture. The total loss of blood (2 ml, or half a teaspoon) is not higher than for a blood donation and thus not enough to cause medical problems in people with no underlying illness. An unknown risk cannot be excluded.”)
Substudy statistical considerations
Given the hypothesis of non-inferiority in recall after oral versus written consent, a presumed recall after consent in the control (written) arm of roughly 80%,35 and assuming a significance (alpha) level and power of 5% and 80%, respectively, roughly 198 patients would be needed in each arm to demonstrate non-inferiority with a margin of 10%, a sample size achievable given the context of the larger PIRATE trial. Nonetheless, we appreciate that we will ultimately be relying on a convenience sample. This is because we cannot confirm that PIRATE trial inclusions by oral and written consent will occur at a 1:1 rate; patients will not be randomised to either mode of consent.
Descriptive statistics will be used to describe patient characteristics and measure recall outcomes in each arm. Continuous variables will be compared with the use of Student's t-test or the Mann–Whitney U test, as appropriate; categorical variables will be compared with the χ2 test or Fisher’s exact test, as appropriate. Logistic or log binomial regression models, where appropriate, will be used for the correlation analyses described above.
Observational study on excluded patients’ clinical outcomes
Background and rationale
Traditional RTCs have historically excluded patients who are ‘too old,’ ‘too sick’ and ‘too comorbid’; these exclusions reduce their external validity and thus the relevance of their results for clinicians dealing with real patients. Paul et al recently provided a striking example of this problem: they observed the clinical outcomes of the 220 patients who were not included in a RCT comparing vancomycin to trimethoprim–sulfamethoxazole for severe methicillin-resistant Staphylococcus aureus infections in comparison with those of the 252 patients who were included. Most patients were excluded because of an inability or unwillingness to provide written informed consent. The clinical failure rate in this group was 80%, while only 33% of included patients experienced clinical failure. Within the trial, mortality in the vancomycin group was non-significantly lower (mortality OR 0.76, 95% CI 0.36 to 1.62), but among excluded patients, mortality was significantly higher with vancomycin treatment (OR 2.63, 95% CI 1.04 to 6.64).36
As described in the first pages of this protocol, POC randomised trials seek a greater inclusiveness and should thus theoretically provide stronger external validity. One particular aspect of the PIRATE trial is its option to allow witnessed, oral consent from patients who are too sick and/or tired to be able or willing to hand-sign a consent form. We hope that this will allow for a more inclusive trial and thus more methodologically robust and applicable outcome data. We therefore propose to follow excluded patients’ clinical outcomes (‘EPCO’), as Paul et al did, in an observational cohort study, but with the hypothesis that in this case, outcomes will be less divergent among included versus non-included patients.
EPCO study design, setting and population
All patients approached for but not included in the PIRATE trial will be eligible for participation in this prospective, multicentre observational cohort study. This study will begin and end in step with the larger PIRATE trial, thus patients are expected to be included from the spring of 2017 through the spring of 2019.
EPCO study outcomes
The primary outcome will be the same as that of the PIRATE trial: the rate of clinical failure rate, as defined above, at day 30±7 days among excluded patients receiving 7 versus 14 days (vs other durations) of antibiotic therapy. Secondary outcomes will include the median duration of antibiotic therapy, length of hospital stay and the reasons for exclusion (consent, patient characteristics and infection characteristics). We will further assess associations, if any, between baseline patient and infection characteristics and willingness or ability to provide informed consent, whether oral or written.37–41 Given the difficulties of obtaining follow-up information in this type of population, patients’ clinical outcomes will be followed only through day 30±7 (with day 1 being the first day of microbiologically appropriate antibiotic therapy).
EPCO study statistical considerations
As described above, we estimate that included patients will experience a clinical success rate between approximately 80% and 90%. Assuming a significance (alpha) level of 5%, power of 80% and attrition (inability to glean follow-up information) of roughly 20%, approximately 90 patients will be needed to enable the detection of at least a 20% difference in the primary outcome rate between the observation cohort and the PIRATE RCT.
Descriptive statistics will be used to describe patient and infection characteristics. Continuous variables will be compared with the use of Student's t-test or the Mann-Whitney U test, as appropriate; categorical variables will be compared with the χ2 test or Fisher’s exact test, as appropriate. Unadjusted ORs for the comparison between the different antibiotic durations in each cohort (observational and PIRATE trial) will be computed with 95% CIs and compared using the Breslow-Day test.
Data handling for the EPCO observational study
No data from excluded patients will be entered into the PIRATE eCRF and database. EPCO patients will be identified by means of an EPCO study number, and no personally identifying information will be transcribed into the EPCO CRF, which will be stored under lock and key, independently and apart from PIRATE data. EPCO data will be entered into a separate, password-protected electronic database that will serve the EPCO study only.
Monitoring
External monitoring will be performed according to International Conference on Harmonisation (ICH) Good Clinical Practice (GCP) by the Unité d’investigation Clinique of the HUG. Following a monitoring plan and written standard operating procedures (SOP), the monitors will verify that the clinical trial is conducted and data are generated, documented and reported in compliance with the protocol, GCP and the applicable regulatory requirements. The investigating team will provide direct access to all trial-related source data, documents and reports for the purpose of monitoring and auditing by the sponsor and inspection by local and regulatory authorities.
Ethics and dissemination
Study investigators will ensure that this study is conducted according to the principles of the latest revision of the Declaration of Helsinki (Fortaleza, Brazil, October 2013) and in full conformity with the ICH GCP, the requirements of the Swiss Human Research Act (HRA;810.30, 2011) and the Swiss ordinance on clinical trials (ClinO; 810.305, 2013) and local regulatory requirements.
Informed consent
All patients will be informed of the study by means of a written information brochure, per usual ICH standards, with full details of the study including its risks and benefits (see information brochure, V.1.1, dated 3 August 2017) before enrolment. In accordance with the recent advice of the Geneva Ethics Commission, and in line with ClinO 810.305, we will allow consenting patients to grant oral consent when written consent cannot be given due to bodily or cognitive reasons and when an independent witness can provide signed testimony to that oral consent. The substudy detailed above will compare patients’ postconsent recall and understanding of the study after oral with written consent.
Ethics committee review
The protocol, IC, and all other study documents have received full approval from the Geneva Cantonal Ethics Commission (lead) as well as the Ethics Commissions of the peripheral sites in Lausanne and St Gallen. Modifications to the protocol will be submitted to these ethics commissions as formal amendments before being implemented.
Dissemination
A manuscript with the results of the primary study will be published in a peer-reviewed journal. Separate manuscripts will be written on each of the substudies and on some secondary outcomes, and these will also be submitted for publication in peer-reviewed journals.
On completion of the trial, and after publication of the primary manuscript, data requests can be submitted to the researchers at the HUG.
Supplementary Material
Acknowledgments
The authors thank Drs Bernard Hirschel, Benedikt Huttner and Thomas Perneger for their thoughtful advice on design and implementation.
Footnotes
Contributors: AH, WA, PYB, EvD, SH and LK designed the study; AH, WA and AGA wrote the main randomised controlled trial protocol; AR assisted in the design of the EPCO substudy and EvD in the design of the informed consent nested study; AH wrote the protocol manuscript. All authors approved the manuscript.
Funding: Swiss National Science Foundation, 74th National Research Program 'Smarter Health Care' (no. 407440_167359) protocol version 1.2, dated 27.03.2017.
Competing interests: None declared.
Ethics approval: Geneva Cantonal Ethics Commission.
Provenance and peer review: Not commissioned; peer reviewed for ethical and funding approval prior to submission.
References
- 1. Huttner A, Harbarth S, Carlet J, et al. Antimicrobial resistance: a global view from the 2013 World Healthcare-Associated Infections Forum. Antimicrob Resist Infect Control 2013;2:31. 10.1186/2047-2994-2-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. WHO Antimicrobial Fact Sheet. 2015. http://www.who.int/mediacentre/factsheets/fs194/en/.
- 3. Swiss Federal Office of Public Health: responses to the antimicrobial resistance threat. 2015. http://www.bag.admin.ch/themen/internationales/11287/.
- 4. Holmes AH, Moore LS, Sundsfjord A, et al. Understanding the mechanisms and drivers of antimicrobial resistance. Lancet 2016;387:176–87. 10.1016/S0140-6736(15)00473-0 [DOI] [PubMed] [Google Scholar]
- 5. Zarkotou O, Pournaras S, Tselioti P, et al. Predictors of mortality in patients with bloodstream infections caused by KPC-producing Klebsiella pneumoniae and impact of appropriate antimicrobial treatment. Clin Microbiol Infect 2011;17:1798–803. 10.1111/j.1469-0691.2011.03514.x [DOI] [PubMed] [Google Scholar]
- 6. Chastre J, Wolff M, Fagon JY, et al. PneumA Trial Group. Comparison of 8 vs 15 days of antibiotic therapy for ventilator-associated pneumonia in adults: a randomized trial. JAMA 2003;290:2588–98. 10.1001/jama.290.19.2588 [DOI] [PubMed] [Google Scholar]
- 7. Havey TC, Fowler RA, Daneman N. Duration of antibiotic therapy for bacteremia: a systematic review and meta-analysis. Crit Care 2011;15:R267. 10.1186/cc10545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mermel LA, Allon M, Bouza E, et al. Clinical practice guidelines for the diagnosis and management of intravascular catheter-related infection: 2009 Update by the Infectious Diseases Society of America. Clin Infect Dis 2009;49:1–45. 10.1086/599376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sandberg T, Skoog G, Hermansson AB, et al. Ciprofloxacin for 7 days versus 14 days in women with acute pyelonephritis: a randomised, open-label and double-blind, placebo-controlled, non-inferiority trial. Lancet 2012;380:484–90. 10.1016/S0140-6736(12)60608-4 [DOI] [PubMed] [Google Scholar]
- 10. Tellier G, Niederman MS, Nusrat R, et al. Clinical and bacteriological efficacy and safety of 5 and 7 day regimens of telithromycin once daily compared with a 10 day regimen of clarithromycin twice daily in patients with mild to moderate community-acquired pneumonia. J Antimicrob Chemother 2004;54:515–23. 10.1093/jac/dkh356 [DOI] [PubMed] [Google Scholar]
- 11. File TM, Mandell LA, Tillotson G, et al. Gemifloxacin once daily for 5 days versus 7 days for the treatment of community-acquired pneumonia: a randomized, multicentre, double-blind study. J Antimicrob Chemother 2007;60:112–20. 10.1093/jac/dkm119 [DOI] [PubMed] [Google Scholar]
- 12. Dunbar LM, Wunderink RG, Habib MP, et al. High-dose, short-course levofloxacin for community-acquired pneumonia: a new treatment paradigm. Clin Infect Dis 2003;37:752–60. 10.1086/377539 [DOI] [PubMed] [Google Scholar]
- 13. Runyon BA, McHutchison JG, Antillon MR, et al. Short-course versus long-course antibiotic treatment of spontaneous bacterial peritonitis. A randomized controlled study of 100 patients. Gastroenterology 1991;100:1737–42. [DOI] [PubMed] [Google Scholar]
- 14. Chaudry ZI, Nisar S, Ahmed U. Short course of antibiotic treatment in spontaneous bacterial peritonitis: a randomized controlled study. Journal of the College of Physicians and Surgeons Pakistan 2000;10:284–8. [Google Scholar]
- 15. Hepburn MJ, Dooley DP, Skidmore PJ, et al. Comparison of short-course (5 days) and standard (10 days) treatment for uncomplicated cellulitis. Arch Intern Med 2004;164:1669–74. 10.1001/archinte.164.15.1669 [DOI] [PubMed] [Google Scholar]
- 16. Oliveira CF, Botoni FA, Oliveira CR, et al. Procalcitonin versus C-reactive protein for guiding antibiotic therapy in sepsis: a randomized trial. Crit Care Med 2013;41:2336–43. 10.1097/CCM.0b013e31828e969f [DOI] [PubMed] [Google Scholar]
- 17. Adamina M, Steffen T, Tarantino I, et al. Meta-analysis of the predictive value of C-reactive protein for infectious complications in abdominal surgery. Br J Surg 2015;102:590–8. 10.1002/bjs.9756 [DOI] [PubMed] [Google Scholar]
- 18. Lin KH, Wang FL, Wu MS, et al. Serum procalcitonin and C-reactive protein levels as markers of bacterial infection in patients with liver cirrhosis: a systematic review and meta-analysis. Diagn Microbiol Infect Dis 2014;80:72–8. 10.1016/j.diagmicrobio.2014.03.029 [DOI] [PubMed] [Google Scholar]
- 19. Torres A, Ramirez P, Montull B, et al. Biomarkers and community-acquired pneumonia: tailoring management with biological data. Semin Respir Crit Care Med 2012;33:266–71. 10.1055/s-0032-1315638 [DOI] [PubMed] [Google Scholar]
- 20. Nobre V, Harbarth S, Graf JD, et al. Use of procalcitonin to shorten antibiotic treatment duration in septic patients: a randomized trial. Am J Respir Crit Care Med 2008;177:498–505. 10.1164/rccm.200708-1238OC [DOI] [PubMed] [Google Scholar]
- 21. Wacker C, Prkno A, Brunkhorst FM, et al. Procalcitonin as a diagnostic marker for sepsis: a systematic review and meta-analysis. Lancet Infect Dis 2013;13:426–35. 10.1016/S1473-3099(12)70323-7 [DOI] [PubMed] [Google Scholar]
- 22. Uçkay I, Garzoni C, Ferry T, et al. Postoperative serum pro-calcitonin and C-reactive protein levels in patients with orthopedic infections. Swiss Med Wkly 2010;140:w13124. 10.4414/smw.2010.13124 [DOI] [PubMed] [Google Scholar]
- 23. Havey TC, Fowler RA, Pinto R, et al. Duration of antibiotic therapy for critically ill patients with bloodstream infections: A retrospective cohort study. Can J Infect Dis Med Microbiol 2013;24:129–37. 10.1155/2013/141989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Corona A, Wilson AP, Grassi M, et al. Prospective audit of bacteraemia management in a university hospital ICU using a general strategy of short-course monotherapy. J Antimicrob Chemother 2004;54:809–17. 10.1093/jac/dkh416 [DOI] [PubMed] [Google Scholar]
- 25. Bouadma L, Luyt CE, Tubach F, et al. PRORATA trial group. Use of procalcitonin to reduce patients' exposure to antibiotics in intensive care units (PRORATA trial): a multicentre randomised controlled trial. Lancet 2010;375:463–74. 10.1016/S0140-6736(09)61879-1 [DOI] [PubMed] [Google Scholar]
- 26. de Jong E, van Oers JA, Beishuizen A, et al. Efficacy and safety of procalcitonin guidance in reducing the duration of antibiotic treatment in critically ill patients: a randomised, controlled, open-label trial. Lancet Infect Dis 2016;16:819–27. 10.1016/S1473-3099(16)00053-0 [DOI] [PubMed] [Google Scholar]
- 27. Albrich WC, Harbarth S. Pros and cons of using biomarkers versus clinical decisions in start and stop decisions for antibiotics in the critical care setting. Intensive Care Med 2015;41:1739–51. 10.1007/s00134-015-3978-8 [DOI] [PubMed] [Google Scholar]
- 28. Schuetz P, Briel M, Christ-Crain M, et al. Procalcitonin to guide initiation and duration of antibiotic treatment in acute respiratory infections: an individual patient data meta-analysis. Clin Infect Dis 2012;55:651–62. 10.1093/cid/cis464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Reinhart K, Bauer M, Riedemann NC, et al. New approaches to Sepsis: molecular diagnostics and biomarkers. Clin Microbiol Rev 2012;25:609–34. 10.1128/CMR.00016-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fitzpatrick JM, Biswas JS, Edgeworth JD, et al. Gram-negative bacteraemia; a multi-centre prospective evaluation of empiric antibiotic therapy and outcome in English acute hospitals. Clin Microbiol Infect 2016;22:244–51. 10.1016/j.cmi.2015.10.034 [DOI] [PubMed] [Google Scholar]
- 31. Angus DC, Seymour CW, Coopersmith CM, et al. A framework for the Development and Interpretation of Different Sepsis definitions and clinical criteria. Crit Care Med 2016;44:e113–e121. 10.1097/CCM.0000000000001730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pittet D, Li N, Wenzel RP. Association of secondary and polymicrobial nosocomial bloodstream infections with higher mortality. Eur J Clin Microbiol Infect Dis 1993;12:813–9. 10.1007/BF02000400 [DOI] [PubMed] [Google Scholar]
- 33. Detry MA, Lewis RJ. The intention-to-treat principle: how to assess the true effect of choosing a medical treatment. JAMA 2014;312:85–6. 10.1001/jama.2014.7523 [DOI] [PubMed] [Google Scholar]
- 34. Food & Drug Administration. Guidance for Industry. E9 Statistical Principles for Clinical Trials 1998. http://www.fda.gov/downloads/drugs/./guidances/ucm073137.pdf. [Google Scholar]
- 35. Chenaud C, Merlani P, Luyasu S, et al. Informed consent for research obtained during the intensive care unit stay. Crit Care 2006;10:R170. 10.1186/cc5120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Paul M, Bronstein E, Yahav D, et al. External validity of a randomised controlled trial on the treatment of severe infections caused by MRSA. BMJ Open 2015;5:e008838 10.1136/bmjopen-2015-008838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Balajonda N, Bisanar TL, Mathew JP, et al. Determinants of a subject's decision to participate in clinical anesthesia research. Anesth Analg 2013;116:448–54. 10.1213/ANE.0b013e318277dd7d [DOI] [PubMed] [Google Scholar]
- 38. Gayet-Ageron A, Rudaz S, Perneger T. Biobank attributes associated with higher patient participation: a randomized study. Eur J Hum Genet 2016;25:31–6. 10.1038/ejhg.2016.132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Knoester PD, Belitser SV, Deckers CL, et al. Recruitment of a cohort of lamotrigine users through community pharmacists: differences between patients who gave informed consent and those who did not. Pharmacoepidemiol Drug Saf 2005;14:107–12. 10.1002/pds.992 [DOI] [PubMed] [Google Scholar]
- 40. Beebe TJ, Ziegenfuss JY, Jenkins SM, et al. Who doesn't authorize the linking of survey and administrative health data? A general population-based investigation. Ann Epidemiol 2011;21:706–9. 10.1016/j.annepidem.2011.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Huang N, Shih SF, Chang HY, et al. Record linkage research and informed consent: who consents? BMC Health Serv Res 2007;7:18. 10.1186/1472-6963-7-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.