

Abstract
Azacitidine (AzaC) mitigates Graft vs. Host Disease (GvHD) in both murine preclinical transplant models and in human clinical trials while maintaining a robust Graft vs. Leukemia (GvL) effect. Previous studies have failed to investigate the role of natural Tregs on the mitigation of GvHD by AzaC; instead focusing on the generation of suppressive regulatory T cells (Tregs, CD4+CD25+FOXP3+) through the in vivo conversion of alloreactive donor T effectors (Teff CD4+ CD25-, FOXP3-) and the direct anti-proliferative effects of AzaC on allogeneic T cells. Using B6.Foxp3DTR/GFP mice in which Tregs can be specifically ablated through administration of Diphtheria toxin, we demonstrate that nTregs are required in the donor graft for AzaC to optimally protect against GvHD and that nTregs, unlike T effs (Teffs, CD3+FOXP3-), are resistant to the anti-proliferative effects of AzaC. Gene expression analysis identified the potent cell cycle inhibitor, p21, was significantly upregulated in Teffs but not nTregs after treatment with AzaC. Furthermore, we demonstrate that Teffs deficient in p21 are less sensitive to the antiproliferative effects of AzaC. These results, demonstrate that nTregs are essential for AzaC to fully protect against GvHD and have important clinical implications for future clinical trials testing AzaC as a novel method of GvHD prophylaxis in man.
Keywords: Allogeneic Bone Marrow Transplantation, Graft versus Host Disease, GvHD, Azacitidine, Treg
Introduction
Allogeneic hematopoietic stem cell transplantation (allo-HSCT) is the primary treatment for high risk and relapsed hematological malignancies. However, the desired graft versus leukemia (GvL) effect is frequently accompanied by graft versus host disease (GvHD) in which donor lymphocytes target host tissues, often resulting in significant morbidity and mortality. Azacitidine (AzaC) has great potential in the post allo-HSCT setting and has been shown to safely mitigate GvHD in both murine preclinical transplant models and human clinical trials while also maintaining a robust GvL effect (1-3).
Regulatory T cells (Tregs, CD4+CD25+FOXP3+), a subset of T cells essential for the maintenance of self-tolerance, have been studied extensively in the field of HSCT(4). Tregs, defined by their exclusive expression of FOXP3, have been shown to reduce GvHD by suppressing allo-reactive T cells, without diminishing the beneficial GvL effect (5). The FOXP3 locus in both humans and mice, is highly methylated at CpG dinucleotides and silenced in Teffs while the same locus is unmethylated and transcriptionally active in Tregs (6, 7). Thus, hypomethylation of the FOXP3 locus in Teffs by small molecule DNA methyltransferase 1 inhibitors such as the cytidine analogue AzaC, is an attractive strategy for increasing Tregs. However, while AzaC is a potent hypomethylating agent, AzaC is also directly incorporated into RNA disrupting both nuclear and cytoplasmic mRNA metabolism, resulting in dysregulation of protein synthesis and the inhibition of cell growth/proliferation (8).
We have previously published data suggesting AzaC reduces GvHD through the in vivo conversion of alloreactive donor CD4+CD25-FOXP3- cells into suppressive Tregs (CD4+CD25+FOXP3+) via the pharmacologic hypomethylation of the Foxp3 promoter (1). However, the effect of AzaC on natural Tregs (nTregs) present in the donor graft has yet to be investigated. In this study we explore the effect of AzaC on nTregs and define an additional mechanism for the GvHD mitigating effects of AzaC following allo-transplantation.
Methods
Mice
The B6.Foxp3DTR/GFP (H-2Kb, CD45.2+)(9) and B6.Foxp3GFP (H-2Kb, CD45.2+) mice were obtained from Alexander Rudensky and Tim Ley, respectively. Balb/c (H-2Kd, CD45.2+), C57BL/6 (B6; H-2Kb, CD45.1+), DsRED expressing mice (B6.Cg-Tg(CAG-DsRed*MST)1Nagy/J) and p21-/- mice (B6.129S6(Cg)-Cdkn1atm1Led/J) were purchased from the Jackson Laboratory (Bar Harbor, ME). Animal care and euthanasia procedures were approved by the Washington University School of Medicine Animal Studies Committee.
T cell purification and nTreg depletion
T cells were purified from murine splenocytes using magnetic isolation. Splenocytes were labelled using the pan T isolation kit II, (Miltenyi Biotec, Germany) and separated using the AutoMACS Pro (Miltenyi) in accordance with the manufactures instructions. To deplete nTregs, the Miltenyi pan T isolation kit II was supplemented with 1.5 μg of biotinylated anti-CD25 antibody (Clone 7D4 BD Pharminogen™) per 1×107 splenocytes. An additional wash step was included prior to incubation with magnetics beads to remove any unbound antibody. Magnetic separation was performed using the AutoMACS Pro (program Deplete_S). We found the GFP in B6.Foxp3DTR/GFP mice to be relatively dim, thus, the purity of Treg depleted T cells was confirmed by intracellular FOXP3 staining (FOXP3-PECy5.5 clone FJK-16s; eBioscience). A representative example of the purity obtained following magnetic depletion of Tregs during T cells isolation can be found in supplemental Figure 2.
Allo-HSCT
Allo-HSCT was performed as previously described with the following modifications (1). To induce GvHD, 1×107 pan T cells or 1×107 pan T cells depleted of nTregs, from either B6.Foxp3DTR/GFP or B6.Foxp3GFP (hemizygous males, H-2Kb, CD45.2+) mice, were injected via the lateral tail vein on day 11 post T cell depleted bone marrow (TCD-BM) infusion (B6 CD45.1+) into lethally irradiated (TBI 925cGy) allogeneic recipients (Balb/c). Transplanted mice were injected intraperitoneally (i.p.) with either PBS (carrier) or AzaC (2 mg/kg, Sigma Aldrich, St Louis, MO) on days +15,+17,+19, and +21, followed by DT (Sigma Aldrich) or PBS on days +16,+18,+20 (DT 10 μg/kg) and +22 (DT 50 μg/kg, Figure 1b). Depletion of Tregs by DT was confirmed by flow cytometry (Figure 1c).
Figure 1. Depletion of Foxp3DTR/GFP Tregs by DT reduces the protective effect of AzaC in an allotransplant model.

(a) Balb/c mice were infused with 1×107 B6.Foxp3DTR/GRP pan T cells 11 days after an initial T cell depleted (TCD) bone marrow (BM, CD45.1) transplant. They were subsequently treated with AzaC alone (AzaC+PBS), DT alone (PBS+DT), neither drug (PBS+PBS), or both drugs (AzaC+DT). Some mice received irradiation, but no cells (irr only), only TCD BM (BM only), or BM along with DT (BM+ DT). (b) Treatment with 2 mg/kg of AzaC up-regulates FOXP3+ Tregs (PBS vs AzaC, Day 19 spleen) p=0.0017). DT (10 μg/kg) depletes Tregs in both DT and DT+AzaC treated mice (PBS vs. DT p=0.003, PBS vs. AzaC+DT p<0.001, Day 19 spleen). (c) Mice treated with AzaC had a significantly higher survival rate than mice treated with both AzaC and DT (p<0.0001), and had higher BM derived B220+ B cells (d, p=0.04) CD3+ T cells (e, p<0.0001). Data represents a pool of two or more independent experiments.
Gastrointestinal histopathology
Intestines were flushed and fixed in 10% formalin. Tissue sections of intestine were prepared and stained with hematoxylin and eosin by the Division of Comparative Medicine, Washington University School of Medicine, St. Louis, MO and graded by a pathologist in blinded fashion for acute GvHD according to the Lerner grading system (10, 11). A scale from 0 to 4 was used where histopathologic changes were identified as follows: 1, mild; 2, moderate; 3, severe; and 4, maximal. Additional information can be found in Supplemental Figure 1.
In vivo proliferation assay
B6.Foxp3DTR/GFP pan T cells (CD45.2+) were labeled with carboxyfluorescein diacetate, succinimidyl ester (CFSE) at a final concentration of 300 nM as previously described (1). Allo-HSCT was performed as previously described but with the following modifications. Transplanted mice were injected (i.p.) with either PBS (carrier) or AzaC (2 mg/kg) on days +15 and +17 followed by injection (i.p.) of DT (10 μg/kg) or PBS on days +16 and +18. Splenocytes were harvested on day 19 and analyzed using flow cytometry. Experiments investigating the differential effect of AzaC on Teffs and Tregs were performed as above but with the following modifications. Balb/c mice were infused with 9.5×106 B6 CD45.1 pan T cells, depleted of nTregs (using anti-CD25 antibodies) and labelled with CFSE, and 5×105 FOXP3+ Tregs (B6 CD45.2) labelled with 1 μM violet proliferation Dye 450 (BD biosciences VPD450), 11 days after an initial BM transplant. Transplanted mice were injected (i.p.) with either PBS or AzaC (2 mg/kg) on days +15 and +17. Mice were sacrificed on Day 19 and the spleens harvested for analysis by flow cytometry.
In vitro proliferation assay
T cells depleted of nTregs (depleted using anti-CD25 antibodies during purification) were purified from the spleens of WT and p21-/- mice and labelled with (CFSE) at a final concentration of 300 nM as previously described (1). T cells were activated for 36 hrs in the presence of anti-CD3/CD28 beads (bead:cell 1:1; Invitrogen) and Xcyte medium supplemented with L-glutamine (4 mM), penicillin (100 U/mL), streptomycin (100 μg/mL), and human recombinant IL-2 (hIL-2; 10 U/mL). The activated T cells were incubated in the presence of AzaC (1 μM) (Sigma-Aldrich) or PBS for an additional 2 days.
In vitro T cell culture for RNA analysis
To distinguish between nTregs and AzaC generated Tregs, nTregs were purified from the spleens of B6.Foxp3GFP mice and Treg depleted T cells were obtained from B6. Foxp3GFP × B6.CAGDSRED. T cells were co-cultured at a 1:10 ratio of nTreg to Teff and were activated for 2 days in the presence of anti-CD3/CD28 beads (bead:cell 1:1; Invitrogen) and Xcyte medium supplemented with L-glutamine (4 mM), penicillin (100 U/mL), streptomycin (100 μg/mL), and human recombinant IL-2 (hIL-2; 500 U/mL). The activated T cells were incubated in the presence of AzaC (1 μM) (Sigma-Aldrich) or PBS for an additional 2 days. Cells were sorted using FACS Aria II (BD) to isolate nTregs (CD4+DSRED-FOXP3GFP+), CD4+ Teffs (CD4+DSRed+FOXP3GFP-), CD8+ Teffs (CD8+DSRed+FOXP3GFP-).
Microarray analysis
RNA was extracted from T cells using the RNA nucleospin XS Kit (Macheray-Nagel, Düren Germany). RNA were assessed with Qubit Fluorometer quantitation (Life Technologies) and Agilent Bioanalyzer RNA 6000 Pico Kit prior to input into Affymetrix Genechip WT Pico Kit (San Carlos, CA) and the Mouse Gene 2.0 ST array (Affymetrix, Santa Clara, CA) following the manufacturer's recommendations.
Data was analyzed with the Affymetrix GeneChip Command Console. Microarray expression data was processed using Command Console (Affymetrix, Inc) and the raw (.CEL) files generated were analyzed using Expression Console software with Affymetrix default RMA Gene analysis settings (Affymetrix, Inc). Probe summarization (Robust Multichip Analysis, RMA), quality control analysis, and probe annotation were performed according to recommended guidelines (Expression Console Software, Affymetrix, Inc.). Normalized data was imported into Partek® Discovery Suite software (version 6.6, Partek Inc., St. Louis, MO). Differential expression was determined by a 1-way ANOVA model producing a p-value, fold change and mean ratio. Genes upregulated >1.5 fold after treatment with AzaC were included in gene ontology (GO) analysis. GO analysis was performed using PANTHER (12). Pathways with p<0.05 were considered significant. The data discussed in this publication have been deposited in NCBI's Gene Expression Omnibus and are accessible through GEO Series accession number GSE92841. www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE92841
Quantitative real time polymerase chain reaction (qPCR)
QPCR was performed on the Applied Biosystems StepOnePlus Real-Time System (Thermo fisher) using pre-designed TaqMan® Gene Expression Assays (Life Technologies) (18S RNA Mm03928990 and p21 Mm04205640) according to manufacturer's instructions. The ΔΔCT method was used to calculate changes in fold expression, and results were analyzed with Graphpad Prism 6.
Flow cytometry analysis
The following antibodies were used: FOXP3-PECy5 (clone FJK-16s; eBioscience), H-2Kb-APC, CD4-e450, CD3-PECy7, B220-APCCy7 and CD45.2 PE (BD Pharmingen). Cell viability was determined using 7-Aminoactinomycin D (life technologies) or Fixable Viability Stain 510 (BD Horizon). Cells were analyzed on either a FACScan (BD Biosciences) or Gallios cytometer (Beckman coulter).
Results
Diphtheria toxin-induced depletion of Tregs reduces the efficacy of azacitidine to mitigate GvHD
To assess the role of Tregs (both nTregs and AzaC generated Tregs) in the mitigation of GvHD by AzaC in vivo, we transplanted 1×107 B6.Foxp3DTR/GFP pan T cells (both Teffs and nTregs) into lethally irradiated Balb/c recipients and treated the mice with PBS, DT, AzaC or a combination of both AzaC and DT (Figure 1a). Treatment of recipient mice with DT resulted in the depletion of donor FOXP3+ Tregs. Furthermore, treatment with AzaC resulted in a 2.5 fold increase in Tregs after only two treatments of AzaC (Figure 1b). Treated of mice with both AzaC and DT resulted in the depletion of both nTregs and AzaC generated Tregs.
Mice treated with either PBS or DT alone developed severe GvHD and survived an average of 26 and 24 days respectively. Due to the severity of GvHD induced in this major mismatched mouse model of bone marrow transplantation, no significant difference was observed in survival between mice treated with PBS and depleted of nTregs using DT. In sharp contrast to the PBS and DT treated groups, >90% of the AzaC treated mice survived the full length of the study (100 days, Figure 1c), displayed no visible GvHD related symptoms, and gained weight at a similar rate as the bone marrow only controls. The treatment of mice with AzaC+DT showed prolonged survival compared to PBS treated mice (p<0.001) but in contrast to the AzaC group, had significantly greater mortality (median survival = 47.5 days, p<0.0001). Furthermore, the AzaC alone group had higher percentages of both donor bone marrow derived (CD45.1+) B220+ B cells, (Figure 1d, p=0.04) and CD3+ T cells (Figure 1e, p<0.0001) than the AzaC+DT group, which is consistent with less clinical GvHD (1). AzaC treated mice also had significantly less gastrointestinal GvHD compared to PBS treated mice (p=0.016) when graded in a blinded fashion by a pathologist (Supplemental Figure 1).
To confirm that our observations were specific for Treg depletion and not due to non-specific effects of DT, we repeated the experiment, but substituted B6.Foxp3DTR/GFP T cells with DT insensitive B6.Foxp3GFP pan T cells. No significant difference in survival, percentages of CD3+ T cells, B220+ B cells, and FOXP3+ CD4 T cells were observed between the AzaC and AzaC+DT groups in this control experiment (Figure 2), confirming that the observed effect of DT in the B6.Foxp3DTR/GFP model was specific to Treg ablation, and not as a result of DT toxicity.
Figure 2. The effect of DT upon GvHD is specific to B6.Foxp3DTR/GFP mice.

Mice were transplanted with 1 ×107 Foxp3GFP pan T substituted in place of 1 ×107 B6.Foxp3DTR/GFP and treated as outlined in the material and methods. There was no significant difference in survival (a), percentage of donor bone marrow derived CD3+ T cells (b) or B220+ B cells (c) when comparing AzaC+PBS to AzaC+DT treated mice. FACS performed on peripheral blood, Day 19. Data shown is combined from two independent experiments.
Tregs depletion attenuates the effect of AzaC in vivo
To determine the extent to which Tregs are involved in the suppression of allogeneic T cell proliferation by AzaC, we performed an in vivo proliferation assay using 1 × 107 CFSE labeled B6.Foxp3DTR/GFP donor pan T cells (Figure 3a). When we gated on donor CD4+ T cells (CD4+/CD45.2+/H-2b+) we found that AzaC-treated mice had significantly higher percentages of CFSE+ CD4+ T cells than AzaC+DT treated mice (p=0.04) indicating that Tregs contribute to suppression of the Teff proliferation in vivo after AzaC treatment (Figure 3b). However, AzaC could still partially suppress the proliferation of CFSE-labeled Teff cells in the absence of Tregs (p=0.04). One representative experiment is shown in Figure 3c. This figure demonstrates the complete elimination of FOXP3+ CD4 T cells in both mice treated with DT and in those mice treated with both DT plus AzaC. In addition, the effects of AzaC in vivo can be clearly seen to increase Tregs. Treatment with AzaC after transplant results in significantly decreased proliferation of donor CD4 T cells through the direct antiproliferative effect of AzaC and through the suppressive effect of Tregs.
Figure 3. The anti-proliferative properties of AzaC upon allogeneic T cells are, in part, mediated by FOXP3+ Tregs.

(a) B6.Foxp3DTR/GFP pan T cells (CD45.2) were labeled with CFSE at a final concentration of 300 nM as previously described (1). Allo-HSCT was performed as described previously. Transplanted mice were injected (i.p.) with either PBS or AzaC (2 mg/kg) on days +15 and +17 followed by injection (i.p.) of DT (10 ug/kg) or PBS on days +16 and +18. Splenocytes were harvested on day 19 and analyzed using flow cytometry. (b and c) CFSE labeled allogeneic donor T cells proliferate significantly less in mice treated with AzaC compared to PBS treated mice (p<0.001) (b) Splenocytes on day 19 post allo-HSCT, gated on H2Kb+ CD45.2+ CD4+. Suppression of proliferation calculated as a percentage of CFSE+ cells as a percentage of total FOXP3- CD4+ donor T cells. In addition, AzaC treated mice had significantly less donor T cell proliferation when compared to mice treated with both AzaC+DT (p=0.04). Data shown is combined from three independent experiments. (c) One representative plot from each group.
nTregs are required for AzaC to mitigate GvHD
To determine the importance of nTregs on the mitigation of GvHD by AzaC, experiments were conducted using pan T cells from B6.Foxp3DTR/GFP mice depleted of nTregs using anti-CD25 antibodies prior to infusion (confirmed by intracellular staining for FOXP3; Supplementary figure 2) into an allo-HSCT model of GvHD (B6 to Balb/c Figure 4a). AzaC+DT treated mice (in vivo elimination of AzaC-generated Tregs) survived significantly longer compared to PBS treated mice (Figure 4b) suggesting direct inhibition of Teffs by AzaC enhanced survival and reduced GvHD (p < 0.0001). Furthermore, AzaC+DT treated mice had significantly shorter survival and increased GvHD compared to mice treated with AzaC alone (p=0.02). These data suggest that in vivo conversion of alloreactive T cells into suppressive Tregs is required for the amelioration of GvHD. Most importantly, however, those mice infused with nTreg-depleted T cell grafts and subsequently treated with AzaC + PBS had dramatically increased GvHD and decreased survival compared to those mice treated with AzaC + PBS who received nTreg replete T cell grafts (P= 0.0004, Figure 4c) demonstrating the requirement of nTreg in the donor T cell grafts for the full GvHD mitigating effect of AzaC.
Figure 4. Depletion of AzaC generated B6.Foxp3DTR/GFP Tregs by DT reduces the protective effect of AzaC in an allotransplant model depleted of natural Tregs.

(a) Balb/c mice were lethally irradiated and infused with 1×107 B6.Foxp3DTR/GFP pan T cells, depleted of nTregs (using anti-CD25 antibodies), 11 days after TCD-BM transplant (CD45.1). Mice were subsequently treated with AzaC alone (AzaC+PBS), DT alone (PBS+DT) neither drug (PBS+PBS), or both drugs (AzaC+DT). Some mice received irradiation and only TCD-BM (BM only) (b) AzaC prolongs the survival of mice transplanted with 1×107 pan T cells depleted of nTregs when compared to mice treated with both AzaC and DT. ǂ The effect of AzaC generated Tregs. (p=0.02) ǂǂ The direct antiproliferative effects of AzaC upon Teffs (p<0.0001). (c) nTregs are required in the donor graft for optimal mitigation of GvHD by AzaC. Lethally irradiated Balb/c were infused with either 1×107 nTreg depleted pan T (TRD) or 1×107 nTreg replete pan T prior to treatment with PBS or AzaC on days +15, +17, +19 and +21. Mice receiving nTreg depleted pan T cells had significantly reduced survival when compared to mice receiving nTreg replete pan T following treatment with AzaC (pan T AzaC vs. TRD AzaC, p=0.0004). Data combined from multiple independent experiments.
nTregs are insensitive to the anti-proliferative effects of AzaC
Our data suggest that nTreg, as well as AzaC generated Tregs, contribute to the expanded Treg population after treatment with AzaC in vivo after a fully MHC mismatched allo-HSCT. We then asked if Teffs and nTregs were equally sensitive to the anti-proliferative effects of AzaC. Purified Teffs (CD3+ CD25- CD45.1) and nTregs (CD4+ CD25+ CD45.2) from B6 donors were labeled with the cellular proliferation dyes, CFSE and VPD450 respectively (allowing us to track both simultaneously in the same mouse) and transplanted into lethally irradiated Balb/c recipients on day +11 post allo-HSCT (Figure 5a) followed by treatment with AzaC (2 mg/kg) or PBS on days +15 and +17. Mice receiving AzaC (2 mg/kg) had significantly reduced Teff proliferation compared to PBS treated mice (CFSE+ PBS 5.63% vs. AzaC 14.33% p=0.035, Figure 5c). In contrast however, nTreg proliferation was completely insensitive to AzaC treatment in vivo (CFSE+ PBS 21.43% vs. AzaC 19.64%, p=0.15, Figure 5b). These data suggest that AzaC treatment increases the percentage of Tregs after transplant by preferentially inhibiting Teff proliferation, while allowing nTregs to expand unimpeded. These data are consistent with the results shown in Figure 4 and suggest that both conversion of Teff to Treg and a differential inhibition of Teff proliferation over nTreg proliferation by AzaC contribute to the GvHD sparing effects of AzaC in vivo.
Figure 5. Natural Tregs are insensitive to the anti-proliferative effects of AzaC compared to T effectors in vivo.

(a) Balb/c mice were infused with 9.5×106 B6 CD45.1 pan T cells, depleted of nTregs (using anti-CD25 antibodies) and labelled with CFSE, and 5×105 FOXP3+ Tregs (B6 CD45.2) labelled with VPD450, 11 days after an initial BM transplant. Transplanted mice were injected (i.p.) with either PBS or AzaC (2 mg/kg) on days +15 and +17 before spleens were harvested for analysis on Day 19. (b) FOXP3+ Tregs showed no significant difference in the % of VPD450 negative cells between PBS and AzaC treated mice, suggesting no significant inhibition on Treg proliferation with AzaC. (c) Within the same mice, AzaC significantly inhibited Teff proliferation compared to PBS treated mice (p=0.035).
AzaC significantly increases expression of p21 in Teff but not nTreg
To elucidate the mechanism of Treg insensitivity to the antiproliferative effects of AzaC, we performed array-based gene expression analyses using mRNA obtained from nTregs and CD4+ Teffs after in vitro culture with anti-CD3/CD28 in the presence or absence of 1 μM AzaC (the approximate in vivo peak concentration of AzaC in our mouse experiments). These revealed 507 genes significantly dysregulated in nTregs following treatment with AzaC compared to 487 genes significantly dysregulated in CD4+ Teff. Gene ontology analysis (PANTHER 9.0) was used to identify genes in pathways responsible for the antiproliferative effects of AzaC on Teffs. Genes upregulated by AzaC (>1.5 fold) in CD4 Teffs following treatment with AzaC were significantly enriched in the pathway 'negative regulation of cell proliferation (p=0.022, Table 1 and Supplementary Table 1). Conversely, genes upregulated (>1.5 fold) by AzaC in nTregs were significantly associated with the positive regulation of T cell activation (p=0.0025) (Table 2 and Supplementary Table 2).
Table 1.
Expression profile of genes related to the negative regulation of cell proliferation. CD4 AzaC Vs. PBS >1.5 Fold
| Gene symbol | Gene ID | Fold Change | Gene description | |
|---|---|---|---|---|
|
| ||||
| CD4 AzaC vs CD4 PBS | nTreg AzaC vs nTreg PBS | |||
| Serpine2 | NM_009255 | 4.85 | 2.75 | serpin peptidase inhibitor |
| p21 | NM_007669 | 3.87 | 1.26 | cyclin-dependent kinase inhibitor 1A |
| Foxp3 | NM_001199347 | 2.96 | 1.28 | forkhead box P3 |
| Ptpn14 | NM_008976 | 2.85 | 1.86 | protein tyrosine phosphatase, non-receptor type 14 |
| Cdk6 | NM_009873 | 2.60 | 1.93 | cyclin-dependent kinase 6 |
| Ifitm3 | NM_025378 | 2.21 | 1.36 | interferon induced transmembrane protein 3 |
| tnf | NM_001278601 | 2.17 | 2.07 | tumor necrosis factor |
Table 2.
Expression profile of genes related to positive regulation of T cell activation. nTreg AzaC Vs. PBS >1.5 Fold
| Gene symbol | Gene ID | Fold Change | Gene description | |
|---|---|---|---|---|
|
| ||||
| CD4 AzaC vs CD4 PBS | nTreg AzaC vs nTreg PBS | |||
| Cd74 | NM_001042605 | 1.95 | 3.06 | invariant polypeptide of major histocompatibility complex |
| H2-Aa | NM_010378 | 1.19 | 2.56 | histocompatibility 2, class II antigen A, alpha |
| Xbp1 | NM_013842 | 1.38 | 2.37 | X-box binding protein 1 |
| Adam8 | NM_001291066 | 1.24 | 2.28 | ADAM metallopeptidase domain 8 |
| Myb | NM_001198914 | 1.45 | 2.21 | myeloblastosis oncogene |
| Icosl | NM_015790 | 1.29 | 2.05 | inducible T-cell co-stimulator ligand |
| Cd86 | XM_006521741 | 0.93 | 1.99 | B7.2 |
| Hsph1 | NM_013559 | 1.54 | 1.78 | heat shock 105kDa/110kDa protein 1 |
| Cd274 | NM_021893 | 1.45 | 1.64 | PD-L1 |
| Il7r | NM_008372 | 0.66 | 1.50 | interleukin 7 receptor |
While the genes Ptpn14, CDK6, and Ifitm3, all associated with the negative proliferation in CD4+ Teffs, were also somewhat upregulated in nTregs treated with AzaC, there is a greater increase in expression of these genes in CD4+ Teffs treated with AzaC than nTregs. Of particular note, our expression analysis suggests that the cell dependent kinase inhibitor, p21, a potent inhibitor of cell cycle progression (13), is significantly upregulated by AzaC in CD4+ Teffs (3.87 fold AzaC vs. PBS) but not in nTregs (1.26 fold AzaC vs. PBS). To validate our findings, RNA expression analysis was performed using qRT-PCR with RNA extracted from CD4+Teffs, CD8+ Teffs, and nTregs after treatment with AzaC. AzaC treatment resulted in a 3.4 fold increase of p21 expression in CD4+ Teffs (FACS sorted to remove AzaC generated Tregs) (AzaC vs. PBS p<0.0012), a 6.65 fold increase in CD8 Teffs (AzaC vs. PBS p<0.0027) and a 2 fold (non-significant) reduction in expression (AzaC vs. PBS p=0.35) in nTregs when compared to PBS treated controls (Figure 6a).
Figure 6. p21-/- attenuates the antiproliferative effects of AzaC.
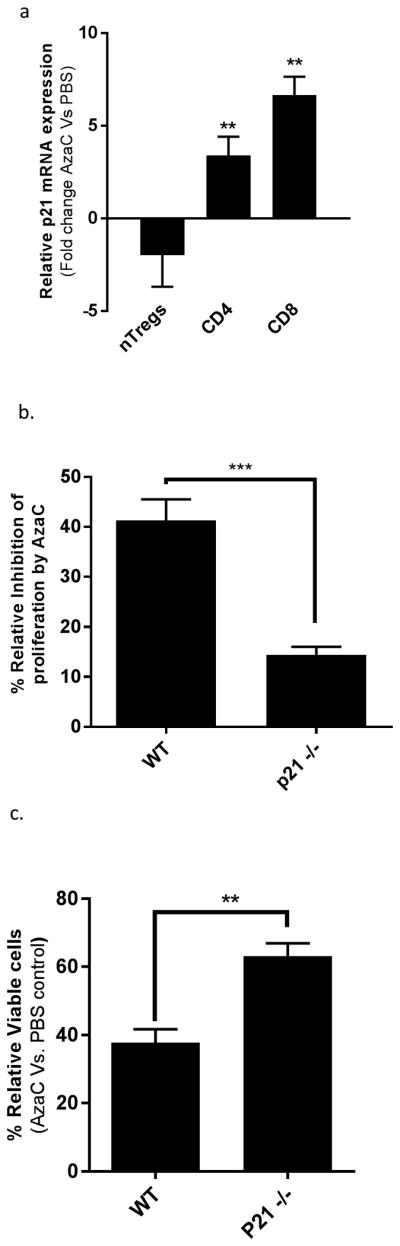
(a) p21 mRNA is significantly upregulated in CD4+ and CD8+ Teff following treatment with AzaC. Teffs were isolated from the spleens of B6. Foxp3GFP × B6.CAGDSRED and nTregs were isolated from B6. Foxp3GFP. Cells were co-cultured at a 1:10 ratio of nTregs to Teffs for 2 days in the presence of anti-CD3/CD28 beads (bead:cell 1:1; Invitrogen) and Xcyte medium supplemented with L-glutamine (4 mM), penicillin (100 U/mL), streptomycin (100 μg/mL), and human recombinant IL-2 (hIL-2; 500 U/mL). The activated T cells were cultured with AzaC (1 μM) or PBS for an additional 2 days. Cells were sorted using FACS Aria II (BD) to isolate nTregs (CD4+DSRED-FOXP3GFP+), CD4+ Teffs (CD4+DSRed+FOXP3GFP-), and CD8+ Teffs (CD8+DSRed+FOXP3GFP-) prior to RNA extraction. QPCR was performed on the Applied Biosystems StepOnePlus Real-Time System using pre-designed TaqMan® Gene Expression Assays (18S RNA Mm03928990 and p21 Mm04205640). Relative fold changes in expression were determined using the ΔΔCT method. AzaC treatment resulted in a 3.4 fold increase of p21 expression in CD4+ Teffs (FACS sorted to remove AzaC converted Tregs) (AzaC vs. PBS p<0.0012), a 6.65 fold increase in CD8+ Teffs (AzaC vs. PBS p<0.0027) and a 2 fold (non-significant) reduction in expression (AzaC vs. PBS p=0.35) in nTregs when compared to PBS treated controls. (b) Teffs from WT and p21-/- mice were labelled with CFSE as previously described. T cells were activated in Xctye media, supplemented with hIL-2 (10 U/mL), for 36 hrs in the presence of anti- CD3/CD28 antibody coated beads (bead:cell 1:1). Activated T cells were cultured in the presence of AzaC (1 μM) for an additional 48 prior to analysis by flow cytometry. Relative inhibition is defined as the percentage decrease of CFSE- cells in AzaC treated cells relative to PBS control. P21-/- Teffs were significantly less sensitive to the antiproliferative effects of AzaC when compared to WT Teffs (p21-/- 14.44% vs. WT 41.82% p= 0.0005). (c) Teff counts relative to PBS control in Teffs purified from WT and p21-/- mice. Activated T cells were cultured in the presence of AzaC (1 μM) for 24 hrs. Cells were stained with Cellometer ViaStain™ AOPI Staining solution and cell counts quantified using the Nexelcom automated cell counter. AzaC reduced the total number of viable WT Teffs to 37.77% (±2.254) relative to PBS control compared to 63.13% (±2.168) in AzaC treated p21-/- Teffs (p = 0.0013)
p21 null Teff are less sensitive to the antiproliferative effects of AzaC
In order to determine the effect of p21 in Teffs treated with AzaC, we performed in vitro proliferation assays in which CFSE labelled Teffs (CD3+CD25-) obtained from either WT or p21-/- mice were activated with anti-CD3/CD28 beads and cultured in the presence of PBS or 1 μM AzaC. AzaC reduced the capacity of WT Teffs to proliferate by 41.28% as determined by CFSE dilution assays compared to WT Teffs treated with PBS. However, p21-/- Teffs were significantly less sensitive than WT Teffs to the anti-proliferative effects of AzaC (p=0.0005), with only a 14% reduction in proliferation of p21-/- Teffs treated with AzaC compared to p21-/- Teffs treated with PBS (Figure 6b). Additionally, AzaC reduced the total number of viable WT Teffs to 37.77% (± 2.254) of PBS control cells compared to 63.13% (± 2.168) in AzaC treated p21-/- Teffs (WT vs. p21-/- p = 0.0013, Figure 6c). These data suggest that AzaC may differentially regulate cell proliferation in nTregs vs. Teffs through the differential regulation of p21 expression.
Discussion
AzaC has been shown to be a safe and effective therapy for both the treatment of patients with MDS and for the reduction of GvHD in the clinic (3, 14-16). In addition to reducing GvHD, AzaC has been shown to be well tolerated post HSCT and effective at maintaining a robust anti-leukemic effect (3, 16-18). However, in order to optimize treatment it is essential to understand the mechanisms resulting in the mitigation of GvHD.
We demonstrate that nTregs present in the donor graft are required for AzaC to optimally protect against GvHD and, unlike Teffs, are not inhibited by treatment with AzaC in vivo after allogeneic stem cell transplantation. Furthermore, we demonstrate that AzaC generated Tregs and the direct antiproliferative effect of AzaC on Teffs are only partially effective at mitigating GvHD in the absence of nTregs. This expands on previous work which attributed the GvHD mitigating effect of AzaC, either to the in vivo conversion of CD4+ Teffs into Tregs (1) or to the direct antiproliferative effect of AzaC on all allo-reactive donor T cells, including Tregs (2).
Using a similar model to that used in this study (B6.Foxp3DTR/GFP → Balb/c) Ganguly et al. elucidated the mechanism of post-transplant high dose cyclophosphamide (PTCy) mediated GvHD protection (19). Ganguly et al. concluded that the clinical benefit on acute GvHD seen in patients receiving post-transplant high dose PTCy after haploidentical transplant is largely dependent on the presence of nTregs in the graft and that nTregs in these grafts are relatively insensitive to the antiproliferative effects of PTCy. These results are similar to data presented in this study, using AzaC. AzaC, like PTCy, preferentially inhibits Teff proliferation compared to Treg proliferation, but additionally converts Teffs to Tregs via hypomethylation of the Foxp3 promoter. AzaC directly induces the enhanced expression of FOXP3 as well as other genes required for Tregs to exert their suppressive properties (1). Further work will be required to determine the effects of combining AzaC with drugs such as PTCy, the mTOR inhibitor rapamycin (sirolimus) and mycophenylate to enhance Treg expansion while at the same time, inhibiting Teff proliferation (19, 20).
In patients undergoing allo-HSCT for treatment of AML, Goodyear et al observed a 3 fold increase in Tregs in patients treated with AzaC (36 mg/m2 on day +42 post HSCT, and administered daily for 5 consecutive days every 28 days for 10 cycles) compared to controls during the first three cycles (3). During later time points (cycles 6 and 9), however, there was no observable difference in the percentage of Tregs between AzaC treated patients and controls. Increasing Tregs by in vivo conversion of Teffs into Tregs and the inhibition of Teffs relative to Tregs may only occur during the first months following transplantation when T cells are rapidly proliferating. AzaC is incorporated into both RNA and DNA and, like decitabine, results in DNMT1 depletion/inhibition and DNA hypomethylation which occurs during DNA replication. Furthermore, transcriptional activity of expanding cells is also subject to AzaC mediated mRNA dysregulation and significant reduction in protein synthesis. Thus cells undergoing rapid expansion are more sensitive to the toxic effects of AzaC resulting in fewer cells in S and G2M (2). Interestingly, the percentages of Tregs in the control patients of the Goodyear study increased comparative to the AzaC treated patients at the later time points (cycle 6 and 9) suggesting that this rapid increase in Tregs relative to Teffs in AzaC treated patients in the early stage following transplantation may be vital for AzaC to exert its protective effect. Thus, administration of AzaC at the earliest tolerated time point may further mitigate GvHD in patients receiving allo-HSCT. We are currently undertaking clinical studies to test this hypothesis in both recipients of matched unrelated donor (MUD) transplants and those undergoing haploidentical stem cell transplants hoping to identify both the earliest tolerated time point for AzaC administration following BMT and the impact of early AzaC treatment on acute and chronic GvHD (Clinical trials IDs # NCT01747499 and NCT02750254 respectively).
Consistent with our in vivo data, Costantini et al. reported that human Tregs are less sensitive to AzaC than Teffs in vitro (21). Our study is the first report to demonstrate that nTregs can proliferate normally in the presence of AzaC in vivo, while Teff proliferation is inhibited and this differential sensitivity is essential for the GvHD mitigating effect of AzaC. Our data also suggest that, in addition to AzaC's differential effect on the proliferation of Teffs vs. Tregs, it also enhances the conversion of Teffs into Tregs presumably via hypomethylation of the Foxp3 locus and subsequent expression of this forkhead transcription factor. Although beyond the scope of the current study, it is conceivable that nTregs in the donor graft may also enhance the in vivo conversion of CD4+CD25- Teffs into suppressive AzaC generated Tregs. In contrast to Teffs, Tregs produce high quantities of TGF-β, which, in combination with AzaC might increase both the number of inducible Tregs (iTregs) and the stability of the hypomethylated Foxp3 promoter (22-24). This suggests that the influence of nTregs in the donor graft may aid in the expansion and or differentiation of Teff to Treg beyond what would be predicted from the direct suppression of alloreactive Teffs.
Gene expression analysis of AzaC treated CD4 Teffs (sorted to exclude AzaC Tregs) and nTregs revealed genes associated with the negative regulation of cell proliferation were upregulated in CD4 Teffs after treatment with AzaC, while genes positively associated with T cell activation were upregulated in nTregs. This finding not only validates our observation that nTregs are less sensitive to the antiproliferative effect of AzaC but also provides insight into the potential mechanism of the differential sensitivity of nTregs and Teffs to AzaC.
The cyclin dependent kinase inhibitor, p21, is a critical regulator of cell proliferation inhibiting cell cycle transition from G0G1 to S phase through cyclin binding (25) and the inhibition of DNA replication through binding to PCNA (26, 27). Balomenos et al, reported that p21 is an important regulator of T cell proliferation, demonstrating that p21 null CD4+ and CD8+ T cells proliferated more rapidly than WT T cells under conditions of prolonged stimulation, and furthermore, deletion of p21 promoted cell cycle progression from G0G1 to S phase (28). Knockout of p21 in T cells exacerbated alloreactivity and cardiac graft rejection, while overexpression of p21 inhibited allograft rejection (29), highlighting the importance of p21 in regulating alloreactive T cells. Interestingly, this phenotype is only observed in vivo, but not in vitro (28, 29). We found that p21 is upregulated in Teffs populations, but not in Tregs after treatment with AzaC, and that p21-/- Teffs are less sensitive to the antiproliferative effects of AzaC than WT Teffs. This observation is consistent with the finding of Sanchez-Abarca et al, in which G0G1 cell cycle arrest was observed following the treatment of T cells with AzaC. (2) In addition, 5-aza-2-deoxycytidine (decitabine), a member of the same family of nucleoside analogues as AzaC, has previously been shown to upregulate p21 in myeloid leukemia (30). Together, these data suggest that AzaC may inhibit Teff proliferation, but not nTreg proliferation, in part through p21 mediated cell cycle arrest.
In conclusion, the data presented here suggests the effect of AzaC on GvHD in vivo is dependent on the presence of nTregs in the donor T cell graft and that these nTregs, relative to Teff, are relatively insensitive to the anti-proliferative effects of AzaC, resulting in rapid changes in the ratio of Teff/Treg in vivo. These results coupled with the direct effect of AzaC on promoting the in vivo conversion of Teff to Treg have important clinical implications in future trials testing AzaC as a novel method of GvHD prophylaxis, for enhancing solid organ transplantation and for the treatment of autoimmune diseases.
Supplementary Material
Acknowledgments
We thank Chyi Hsieh and Alexander Rudensky the for B6.Foxp3DTR/GFP mice, Tim Ley for providing the B6.Foxp3GFP. We thank Michael Rettig and Linda Eissenberg for critical reading of the manuscript.
Funding Sources: NCI SPORE P50 CA171063-01, and Bryan Campbell Foundation
Footnotes
Contribution: M.L.C, J.C., M.A.S, and J.F.D designed and analysed the experiments and wrote the paper; M.L.C., J.C. and J.R. performed the animal studies. DK performed FACS sorting and RNA purifications. K.V. performed pathological histology. All authors discussed the results and commented on the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
References
- 1.Choi J, Ritchey J, Prior JL, Holt M, Shannon WD, Deych E, Piwnica-Worms DR, DiPersio JF. In vivo administration of hypomethylating agents mitigate graft-versus-host disease without sacrificing graft-versus-leukemia. Blood. 2010;116:129–139. doi: 10.1182/blood-2009-12-257253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sanchez-Abarca LI, Gutierrez-Cosio S, Santamaria C, Caballero-Velazquez T, Blanco B, Herrero-Sanchez C, Garcia JL, Carrancio S, Hernandez-Campo P, Gonzalez FJ, Flores T, Ciudad L, Ballestar E, Del Canizo C, San Miguel JF, Perez-Simon JA. Immunomodulatory effect of 5-azacytidine (5-azaC): potential role in the transplantation setting. Blood. 2010;115:107–121. doi: 10.1182/blood-2009-03-210393. [DOI] [PubMed] [Google Scholar]
- 3.Goodyear OC, Dennis M, Jilani NY, Loke J, Siddique S, Ryan G, Nunnick J, Khanum R, Raghavan M, Cook M, Snowden JA, Griffiths M, Russell N, Yin J, Crawley C, Cook G, Vyas P, Moss P, Malladi R, Craddock CF. Azacitidine augments expansion of regulatory T cells after allogeneic stem cell transplantation in patients with acute myeloid leukemia (AML) Blood. 2012;119:3361–3369. doi: 10.1182/blood-2011-09-377044. [DOI] [PubMed] [Google Scholar]
- 4.Schneidawind D, Pierini A, Negrin RS. Regulatory T cells and natural killer T cells for modulation of GvHD following allogeneic hematopoietic cell transplantation. Blood. 2013;122:3116–3121. doi: 10.1182/blood-2013-08-453126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Edinger M, Hoffmann P, Ermann J, Drago K, Fathman CG, Strober S, Negrin RS. CD4+CD25+ regulatory T cells preserve graft-versus-tumor activity while inhibiting graft-versus-host disease after bone marrow transplantation. Nature medicine. 2003;9:1144–1150. doi: 10.1038/nm915. [DOI] [PubMed] [Google Scholar]
- 6.Baron U, Floess S, Wieczorek G, Baumann K, Grutzkau A, Dong J, Thiel A, Boeld TJ, Hoffmann P, Edinger M, Turbachova I, Hamann A, Olek S, Huehn J. DNA demethylation in the human FOXP3 locus discriminates regulatory T cells from activated FOXP3(+) conventional T cells. Eur J Immunol. 2007;37:2378–2389. doi: 10.1002/eji.200737594. [DOI] [PubMed] [Google Scholar]
- 7.Floess S, Freyer J, Siewert C, Baron U, Olek S, Polansky J, Schlawe K, Chang HD, Bopp T, Schmitt E, Klein-Hessling S, Serfling E, Hamann A, Huehn J. Epigenetic control of the foxp3 locus in regulatory T cells. PLoS biology. 2007;5:e38. doi: 10.1371/journal.pbio.0050038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li LH, Olin EJ, Fraser TJ, Bhuyan BK. Phase specificity of 5-azacytidine against mammalian cells in tissue culture. Cancer research. 1970;30:2770–2775. [PubMed] [Google Scholar]
- 9.Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nature immunology. 2007;8:191–197. doi: 10.1038/ni1428. [DOI] [PubMed] [Google Scholar]
- 10.Heymer B. Histopathological manifestations of acute GvHD. Berlin, Heidelberg, New York: Springer-Verlag; 2002. Clinical and diagnostic pathology of Graft-versus-host Disease; pp. 52–83. [Google Scholar]
- 11.Choi J, Ziga ED, Ritchey J, Collins L, Prior JL, Cooper ML, Piwnica-Worms D, Dipersio JF. IFNgammaR signaling mediates alloreactive T-cell trafficking and GvHD. Blood. 2012;120:4093–4103. doi: 10.1182/blood-2012-01-403196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mi H, Muruganujan A, Casagrande JT, Thomas PD. Large-scale gene function analysis with the PANTHER classification system. Nature protocols. 2013;8:1551–1566. doi: 10.1038/nprot.2013.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brugarolas J, Moberg K, Boyd SD, Taya Y, Jacks T, Lees JA. Inhibition of cyclin-dependent kinase 2 by p21 is necessary for retinoblastoma protein-mediated G1 arrest after γ-irradiation. Proceedings of the National Academy of Sciences. 1999;96:1002–1007. doi: 10.1073/pnas.96.3.1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pleyer L, Burgstaller S, Girschikofsky M, Linkesch W, Stauder R, Pfeilstocker M, Schreder M, Tinchon C, Sliwa T, Lang A, Sperr WR, Krippl P, Geissler D, Voskova D, Schlick K, Thaler J, Machherndl-Spandl S, Theiler G, Eckmullner O, Greil R. Azacitidine in 302 patients with WHO-defined acute myeloid leukemia: results from the Austrian Azacitidine Registry of the AGMT-Study Group. Annals of hematology. 2014;93:1825–1838. doi: 10.1007/s00277-014-2126-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Breccia M, Molica M, Zacheo I, Alimena G. Azacitidine for myelodysplastic patients aged > 65 years: a review of clinical efficacy. Expert opinion on pharmacotherapy. 2014;15:1621–1630. doi: 10.1517/14656566.2014.936849. [DOI] [PubMed] [Google Scholar]
- 16.de Lima M, Giralt S, Thall PF, de Padua Silva L, Jones RB, Komanduri K, Braun TM, Nguyen HQ, Champlin R, Garcia-Manero G. Maintenance therapy with low-dose azacitidine after allogeneic hematopoietic stem cell transplantation for recurrent acute myelogenous leukemia or myelodysplastic syndrome: a dose and schedule finding study. Cancer. 2010;116:5420–5431. doi: 10.1002/cncr.25500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Czibere A, Bruns I, Kroger N, Platzbecker U, Lind J, Zohren F, Fenk R, Germing U, Schroder T, Graf T, Haas R, Kobbe G. 5-Azacytidine for the treatment of patients with acute myeloid leukemia or myelodysplastic syndrome who relapse after allo-SCT: a retrospective analysis. Bone marrow transplantation. 2010;45:872–876. doi: 10.1038/bmt.2009.266. [DOI] [PubMed] [Google Scholar]
- 18.Jabbour E, Giralt S, Kantarjian H, Garcia-Manero G, Jagasia M, Kebriaei P, de Padua L, Shpall EJ, Champlin R, de Lima M. Low-dose azacitidine after allogeneic stem cell transplantation for acute leukemia. Cancer. 2009;115:1899–1905. doi: 10.1002/cncr.24198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ganguly S, Ross DB, Panoskaltsis-Mortari A, Kanakry CG, Blazar BR, Levy RB, Luznik L. Donor CD4+ Foxp3+ regulatory T cells are necessary for posttransplantation cyclophosphamide-mediated protection against GvHD in mice. Blood. 2014;124:2131–2141. doi: 10.1182/blood-2013-10-525873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zeiser R, Leveson-Gower DB, Zambricki EA, Kambham N, Beilhack A, Loh J, Hou JZ, Negrin RS. Differential impact of mammalian target of rapamycin inhibition on CD4+CD25+Foxp3+ regulatory T cells compared with conventional CD4+ T cells. Blood. 2008;111:453–462. doi: 10.1182/blood-2007-06-094482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Costantini B, Kordasti SY, Kulasekararaj AG, Jiang J, Seidl T, Abellan PP, Mohamedali A, Thomas NSB, Farzaneh F, Mufti GJ. The effects of 5-azacytidine on the function and number of regulatory T cells and T-effectors in myelodysplastic syndrome. Haematologica. 2013;98:1196–1205. doi: 10.3324/haematol.2012.074823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Polansky JK, Kretschmer K, Freyer J, Floess S, Garbe A, Baron U, Olek S, Hamann A, von Boehmer H, Huehn J. DNA methylation controls Foxp3 gene expression. Eur J Immunol. 2008;38:1654–1663. doi: 10.1002/eji.200838105. [DOI] [PubMed] [Google Scholar]
- 23.Nakamura K, Kitani A, Strober W. Cell contact-dependent immunosuppression by CD4(+)CD25(+) regulatory T cells is mediated by cell surface-bound transforming growth factor beta. The Journal of experimental medicine. 2001;194:629–644. doi: 10.1084/jem.194.5.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Levings MK, Bacchetta R, Schulz U, Roncarolo MG. The role of IL-10 and TGF-beta in the differentiation and effector function of T regulatory cells. International archives of allergy and immunology. 2002;129:263–276. doi: 10.1159/000067596. [DOI] [PubMed] [Google Scholar]
- 25.Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75:805–816. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- 26.Li R, Waga S, Hannon GJ, Beach D, Stillman B. Differential effects by the p21 CDK inhibitor on PCNA-dependent DNA replication and repair. Nature. 1994;371:534–537. doi: 10.1038/371534a0. [DOI] [PubMed] [Google Scholar]
- 27.Waga S, Hannon GJ, Beach D, Stillman B. The p21 inhibitor of cyclin-dependent kinases controls DNA replication by interaction with PCNA. Nature. 1994;369:574–578. doi: 10.1038/369574a0. [DOI] [PubMed] [Google Scholar]
- 28.Balomenos D, Martin-Caballero J, Garcia MI, Prieto I, Flores JM, Serrano M, Martinez AC. The cell cycle inhibitor p21 controls T-cell proliferation and sex-linked lupus development. Nat Med. 2000;6:171–176. doi: 10.1038/72272. [DOI] [PubMed] [Google Scholar]
- 29.Welling TH, Lu G, Csencsits K, Wood SC, Jarvinen L, Bishop DK. Regulation of alloimmune Th1 responses by the cyclin-dependent kinase inhibitor p21 following transplantation. Surgery. 2008;143:394–403. doi: 10.1016/j.surg.2007.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schmelz K, Wagner M, Dorken B, Tamm I. 5-Aza-2′-deoxycytidine induces p21WAF expression by demethylation of p73 leading to p53-independent apoptosis in myeloid leukemia. International journal of cancer Journal international du cancer. 2005;114:683–695. doi: 10.1002/ijc.20797. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.