

Abstract
5′-Methylthioadenosine/S-adenosylhomocysteine nucleosidase (MTAN) is a bacterial enzyme that catalyzes the hydrolysis of the N-ribosidic bond in 5′-methylthioadenosine (MTA) and S-adenosylhomocysteine (SAH). MTAN activity has been linked to quorum sensing pathways, polyamine biosynthesis, and adenine salvage. Previously, the coding sequence of Rv0091 was annotated as a putative MTAN in Mycobacterium tuberculosis. Rv0091 was expressed in Escherichia coli, purified to homogeneity, and shown to be a homodimer, consistent with MTANs from other microorganisms. Substrate specificity for Rv0091 gave a preference for 5′-deoxyadenosine relative to MTA or SAH. Intrinsic kinetic isotope effects (KIEs) for the hydrolysis of [1′-3H], [1′-14C], [5′-3H2], [9-15N], and [7-15N]MTA were determined to be 1.207, 1.038, 0.998, 1.021, and 0.998, respectively. A model for the transition state structure of Rv0091 was determined by matching KIE values predicted via quantum chemical calculations to the intrinsic KIEs. The transition state shows a substantial loss of C1′–N9 bond order, well-developed oxocarbenium character of the ribosyl ring, and weak participation of the water nucleophile. Electrostatic potential surface maps for the Rv0091 transition state structure show similarity to DADMe-immucillin transition state analogues. DADMe-immucillin transition state analogues showed strong inhibition of Rv0091, with the most potent inhibitor (5′-hexylthio-DADMe-immucillinA) displaying a Ki value of 87 pM.
Graphical abstract
Mycobacterium tuberculosis is the causative agent of tuberculosis (TB), a leading cause of death among infectious diseases.1 In 2014, 9.6 million cases of TB were reported, and 1.5 million people died of TB-related infections. Accordingly, the continued rise of multidrug-resistant, extensively drug-resistant, and totally drug-resistant strains of M. tuberculosis has become a major public health concern.2 These challenges highlight the pressing need for new antitubercular therapies with novel targets and mechanisms of action.
5′-Methylthioadenosine/S-adenosylhomocysteine nucleosidases (MTANs) are involved in S-adenosylmethionine (SAM)-related pathways, including methylation, polyamine biosynthesis, adenine salvage, the biosynthesis of quorum sensing autoinducers and radical reactions involving SAM.3,4 MTANs catalyze the hydrolysis of the N-ribosidic bond in 5′-methylthioadenosine (MTA), S-adenosylhomocysteine (SAH), and 5′-deoxyadenosine (5′-dAdo; Figure 1). The absence of MTANs in human metabolism makes this protein class an appealing target for antibacterials that function through blocking the quorum-sensing pathway or SAM recycling.
Figure 1.

The MTAN reaction. MTANs catalyze the hydrolysis of the N-ribosidic bond of 5′-methylthioadenosine (MTA), S-adenosylhomocysteine (SAH), and 5′-deoxyadenosine (5′-dAdo) to produce methylthioribose (MTR), S-ribosylhomocysteine (SRH), and 5′-deoxyribose (5′-dR), respectively. MTAN: 5′-methylthioadenosine nucleosidase.
Previously, we reported models for the transition state (TS) structures of MTANs from Escherichia coli,5 Streptococcus pneumonia,6 and Neisseria meningitidis7 using a combination of experimental kinetic isotope effect (KIE) measurements and computational methods. Intrinsic KIEs report on bond vibrational changes between the ground state (GS) and TS of chemical reactions and can be used as boundary constraints on quantum chemical calculations to model the structure of reactants at the TS. Such analyses provide information that can be leveraged in the design of chemically stable analogues that mimic features of the TS structure.4,8–10 TS analogues designed for the MTAN reaction function as extremely potent inhibitors with Kd values in the picomolar (pM) and femtomolar (fM) range.11–13 Importantly, these inhibitors have been shown to disrupt quorum sensing pathways in E. coli and Vibrio cholerae14 and inhibit growth in Helicobacter pylori by blocking menaquinone biosynthesis.15
The Rv0091 protein from M. tuberculosis was annotated previously as a putative bifunctional MTA/SAH nucleosidase.16 Herein, we report the expression, purification, and kinetic characterization of Rv0091. We demonstrate that the preferred substrate for Rv0091 is 5′-dAdo, and the protein exhibits less efficient nucleosidase activity with MTA and SAH substrates. A model of the Rv0091 TS structure for MTA hydrolysis was produced by matching KIE values predicted via quantum chemical calculations to a family of intrinsic KIEs. This model reveals that the Rv0091 TS structure and DADMe-immucillin TS analogues share similar electrostatic distributions. The potential of DADMe-immucillin TS analogues to modulate Rv0091 activity was investigated via in vitro inhibition assays.
RESULTS AND DISCUSSION
Oligomeric State and Substrate Specificity for Rv0091
A synthetic gene was designed for Rv0091 (NCBI GenBank: CCP42816.1) and purchased from DNA2.0 Inc. in a pJexpress414 expression vector. Rv0091 was heterologously expressed in E. coli with an N-terminal His6 tag and purified to homogeneity via Ni2+-affinity chromatography. The purified protein was analyzed via SDS-PAGE to reveal a protein band of a molecular weight consistent with the predicted monomer size for Rv0091 (29.4 kDa; Figure 2). To determine the oligomeric state of Rv0091, the purified protein was treated with glutaraldehyde to enable intersubunit cross-linking.17 SDS-PAGE analysis of the cross-linked protein revealed a band consistent with a dimeric oligomerization state (Figure 2). These data are consistent with previous reports for MTANs from other organisms, which have been characterized as homodimers in X-ray crystallography.14,18–20
Figure 2.

SDS-PAGE analysis of purified monomeric and cross-linked Rv0091. Rv0091 was purified to homogeneity, and SDS-PAGE analysis revealed a band consistent with the predicted mass (29.4 kDa) of the monomeric protein. The pure protein was treated with 0.1% to 2% glutaraldehyde to allow for intersubunit cross-linking. A band consistent with a dimeric oligomerization state was observed for the cross-linked protein. Lane 1, protein molecular weight standards; lane 2, Rv0091 + 0.1% glutaraldehyde; lane 3, Rv0091 + 0.5% glutaraldehyde; lane 4, Rv0091 + 2% glutaraldehyde; lane 5, Rv0091 before cross-linking.
Recently, a 5′-methylthioadenosine phosphorylase (MTAP) was identified in M. tuberculosis21 and M. smegmatis.22 MTAPs catalyze the phosphorolysis of MTA to adenine and 5′-methylthioribose-1′-phosphate. Prior to these reports, MTAPs were believed to be absent from bacteria, and the biological reason why M. tuberculosis and M. smegmatis expresses both MTAP and MTAN is currently unknown. We explored the substrate specificity of Rv0091 by determining the kinetic constants with MTA, SAH, and 5′-dAdo.23 These experiments indicate that the preferred substrate for Rv0091 is 5′-dAdo, which displayed a specificity constant (kcat/KM) 7-fold greater than MTA and 700-fold greater than SAH (Table 1). MTA was reported as the preferred substrate for M. tuberculosis MTAP,21 and the addition of phosphate to the Rv0091 reaction mixture did not enhance the rate of MTA hydrolysis. These data indicate Rv0091 does not exhibit MTAP activity and suggests that Rv0091 primarily functions as a 5′-dAdo nucleosidase.
Table 1.
Kinetic Parameters for Rv0091 with 5′-dAdo, MTA, and SAHa
| substrateb | kcat (s−1) | KM (µM) | kcat/KM (M−1 s−1) |
|---|---|---|---|
| 5′-dAdo | 0.47 ± 0.02 | 10.9 ± 1.5 | 43.4 × 103 |
| MTA | 0.023 ± 0.001 | 3.7 ± 0.4 | 6.3 × 103 |
| SAH | 0.004 ± 0.001 | 63.1 ± 7.0 | 0.06 × 103 |
Parameters obtained using a luciferase-based assay23 at 25 °C.
Adenosine and 2′-deoxyadenosine showed less substrate activity than MTA and were not further characterized.
5′-dAdo, 5′-deoxyadenosine; MTA, 5′-methylthioadenosine; SAH, S-adenosylhomocysteine.
The catalytic efficiencies (kcat/KM) for Rv0091 with 5′-dAdo and MTA (Table 1) are lower than those of E. coli MTAN (kcat-MTA/KM-MTA = 2.3 × 106 M−1 s−1; kcat-5′dAdo/KM-5′dAdo = 0.8 × 106 M−1 s−1).23 The low catalytic efficiency of enzymes from M. tuberculosis, such as Rv0091, has been attributed to the slow doubling time of M. tuberculosis (24 h) as compared to E. coli (20 min).24
Determination of Intrinsic KIEs for MTA Hydrolysis
KIEs for the hydrolysis of MTA by Rv0091 were measured via the competitive radiolabel approach9,10 using MTA substrates with isotopic labels incorporated at sensitive or remote positions (Table 2). Isotope effects on enzymatic reactions measured via internal competition provide V/K KIEs, which report on all steps from substrate binding, up to and including the first irreversible chemical step.25 The most valuable information for interrogating TS structure is derived from intrinsic KIEs, which reflect the chemical step alone.26 For a given isotope x, the intrinsic KIE on an enzymatic reaction (xk) can be extracted from the xV/K KIE using Northrop’s equation (eq 1),25 when forward commitment (Cf), reverse commitment (Cr), and the equilibrium isotope effect (xKeq) are known.
| (1) |
Table 2.
Kinetic Isotope Effects for the Hydrolysis of MTA by Rv0091
| heavy MTA | light MTA | type of KIE | V/K KIEs | intrinsic KIEsc | calculated KIEsd |
|---|---|---|---|---|---|
| [1′-14C] | [5′-3H2] | primary | 1.035 ± 0.004b | 1.038 ± 0.005 | 1.039 |
| [1′-3H] | [5′-14C]a | α-secondary | 1.194 ± 0.009 | 1.207 ± 0.010 | 1.209 |
| [9-15N, 5′-14C] | [5′-3H2] | primary | 1.019 ± 0.006b | 1.021 ± 0.007 | 1.023 |
| [7-15N, 5′-14C] | [5′-3H2] | β-secondary | 0.998 ± 0.004b | 0.998 ± 0.005 | 0.985 |
| [5′-3H2] | [5′-14C] | remote | 0.998 ± 0.008 | 0.998 ± 0.009 | 0.990 |
The KIE on the remote [5′-14C] label is assumed to be unity.
Experimental values were corrected for the remote [5′-3H2] KIE using the expression KIE = KIEobs × [5′-3H2] KIE.
Intrinsic KIEs were determined by correcting V/K values using eq 2 assuming Cf = 0.079.
Calculated values (Gaussian 09, RB3LYP/6-31g(d) theory) corresponds to those for the final TS model shown in Figure 4.
As the Rv0091 reaction was irreversible under the conditions used to measure KIEs, Cr is assumed to be zero. As such, eq 1 is reduced to eq 2 whereby xk can be extracted from the xV/K KIE using Cf alone:
| (2) |
The V/K and intrinsic KIEs for the hydrolysis of MTA by Rv0091 are reported in Table 2. Experimental KIEs were corrected for remote effects to provide the V/K KIEs, and intrinsic values were determined using eq 2 where Cf = 0.079. The value of Cf for Rv0091 was measured experimentally using the substrate trapping method developed by Rose,27 as previously described (Figure S1).6
The primary C1′ KIE for N-ribosyltransferases is sensitive to atomic motion along the reaction coordinate and provides information for differentiating SN1 and SN2 reactions. For a fully dissociative SN1 reaction, with complete loss of bond order to the leaving group and no participation of the incoming nucleophile at the TS, a primary 1′-14C KIE near unity would be predicted.9 For SN1 mechanisms in which partial bond order to either the leaving group or the nucleophile is retained at the TS, 1′-14C KIEs would range from 1.01 to 1.03. By contrast, primary 1′-14C KIEs for associative SN2 mechanisms range from 1.08 to 1.13. The 1′-14C KIE measured for MTA hydrolysis by Rv0091 was 1.038 ± 0.005 (Table 2), which suggests either the N9 of the adenine leaving group and/or the oxygen of the nucleophilic water (Ow) retain partial bond order with the C1′ position at the TS. The primary 9-15N KIE reports on the loss of C1′–N9 bond order and rehybridization of the N9 position. A value of 1.021 ± 0.007 was obtained for the primary 9-15N KIE on the Rv0091 reaction (Table 2), which indicates that the C1′–N9 bond undergoes significant, but not full, loss of bond order at the TS.
The α-secondary 1′-H KIE for N-ribosyltransferases reports directly on changes in hybridization at the C1′ position and reflects the degree of oxocarbenium character retained by the ribose ring in the TS structure.28 A highly dissociative TS, with considerable oxocarbenium character, will typically display 1′-3H KIEs in the range of 1.06 to 1.22.9 By contrast, an associative TS with less oxocarbenium character will display 1′-3H KIE values ranging from 1.00 to 1.03. The 1′-3H KIE measured for hydrolysis of MTA by Rv0091 was 1.207 ± 0.010 (Table 2), which suggests a TS structure with significant oxocarbenium character. Finally, the β-secondary 7-15N KIE, which reports on the protonation state at the N7 position of adenine, was measured as 0.998 ± 0.005. Previous studies suggest that intrinsic 7-15N KIEs near unity for the MTAN reaction indicate protonation of the N7 position at the TS.11
Computational Models of the Rv0091 TS Structure
To investigate the mechanism of MTA hydrolysis by Rv0091, we developed a model of the Rv0091 TS structure using intrinsic KIEs as experimental boundaries for density functional theory (DFT) calculations. As there is no structural information available for the conformation of MTA bound to Rv0091, the input geometry for the TS structure search was taken from the coordinates for 5′-methylthiotubercidin bound to E. coli MTAN (PDB: 1NC1).20 A family of energy-minimized TS structures was generated by constraining the C1′–N9 bond distance along the reaction coordinate as detailed below. Predicted KIEs were calculated (Gaussian 09, RB3LYP/6-31g(d) theory)29 from the scaled vibrational frequencies of the optimized structures for MTA in the GS and at the TS. The optimized structure of MTA in the GS was generated without constraints using water as an implicit solvent (polarizable continuum model) and was identical for all KIE calculations.
Previous studies on the TS structures of MTAN variants from E. coli,5 S. pneumonia,6 and N. meningitides7 found MTA hydrolysis proceeds through a DN*AN process wherein C1′–N9 bond cleavage and C1′–Ow bond formation occur in a stepwise manner (Figure 3). Accordingly, initial efforts to model the Rv0091 TS structure considered two possible DN*AN manifolds: (1) a DN‡*AN mechanism in which cleavage of the C1′–N9 ribosidic bond to form an oxocarbenium intermediate is rate-limiting and (2) a DN*AN‡ mechanism in which the oxocarbenium intermediate is in equilibrium with MTA and nucleophilic attack of water is rate-limiting (Figure 3).
Figure 3.

General mechanisms for the hydrolysis of MTA by Rv0091. The Rv0091 reaction is represented as two possible mechanisms in which the transformations are considered as their elementary steps. For MTA hydrolysis, the AN step refers to the association of the water nucleophile and the DN step refers to dissociation of the adenine leaving group. If the reaction proceeds through a concerted bimolecular TS, the mechanism is termed ANDN. If dissociation of the leaving group precedes association of the nucleophile and the reaction proceeds through a stepwise process via discrete transition states, the reaction is termed DN*AN. For DN*AN processes, a superscript “‡” is used to denote the rate-limiting step, e.g., DN‡*AN or DN*AN‡. MTA, 5′-methylthioadenosine; MTR, 5′-methylthioribose.
To examine the DN‡*AN mechanism, TS structures were generated by fixing the C1′–N9 bond of MTA at increasing lengths from 1.6 to 3.0 Å in 0.2 Å increments. Predicted KIEs were calculated and TS structures yielding close matches to the intrinsic values were refined by varying the C1′–N9 bond along the reaction coordinate in 0.01 Å steps. The TS structure providing the closest match to the intrinsic KIEs was identified at a C1′–N9 distance of 2.78 Å, although this model matched only the primary 1′-14C KIE within experimental error (Figure S2). The influence of charge accumulation on the leaving group was investigated by a parallel set of calculations to those detailed above in which the N7 position of adenine was protonated at the TS. Protonation at the N7 position activates the adenine for scission of the C1′–N9 bond by neutralizing negative charge build-up on the leaving group and is a common TS feature among N-ribosyltransferases,10 including E. coli5 and N. meningitides7 MTANs. In this TS structure search, the best match to intrinsic KIEs was found at a C1′–N9 distance of 2.42 Å (Figure S3). Relative to the TS structures in which adenine was modeled as an anionic leaving group, the TS structure including N7 protonation provided a better match to intrinsic KIEs. The primary 1′-14C and 9-15N KIEs were well within experimental error of the intrinsic values, and the α-secondary 1′-3H and β-secondary 7-15N were just outside these boundaries (Figure S3).
The feasibility of a DN*AN‡ mechanism was also considered, in which the oxocarbenium intermediate is in equilibrium with MTA prior to the rate-limiting addition of water (Figure 3). For DN*AN‡ mechanisms, observed KIEs are the product of the equilibrium isotope effect (EIE) for the first step and the KIE for the second step (Figure 3).28 The EIEs for oxocarbenium formation were calculated for both neutral (N7 protonated) and anionic (N7 deprotonated) forms of adenine (Figure S4). Predicted KIEs were then calculated for TS structures involving nucleophilic attack of water on the oxocarbenium intermediate with C1′–Ow bond distances from 1.7 to 2.7 Å. Interestingly, the predicted 9-15N EIEs for both neutral and anionic forms of adenine were within experimental error of the intrinsic KIEs (Figure S4). However, the product of the EIEs and KIEs (for the nucleophilic attack of water) did not agree with the intrinsic values for the 1′-14C or 1′-3H KIEs (Figure S4).
The preliminary DN‡*AN model for MTA hydrolysis by Rv0091 (Figure S3) was refined through the addition of low bond order to the nucleophilic water. The TS structure providing the best match to intrinsic values (Table 2) had a C1′–N9 distance of 2.45 Å and a C1′–Ow distance of 2.70 Å (Figure 4). Interestingly, the C1′–Ow distance of 2.70 Å in this TS model is similar to the distance between the 1′ position of the aminoribitol ring and the bound active site water (2.75 Å) in the binary complex of 5′-methylthio-DADMe-immucillinA (MT-DADMe-ImmA) and E. coli MTAN (PDB: 1Y6Q; Figure S5).30 The slight discrepancy between the predicted 7-15N KIE and corresponding intrinsic value (Table 2) likely results from hydrogen bonding interactions with Asp220, which is predicted by homology modeling to interact with the N7 position of adenine,31 but was not included in this TS model. This final model is most consistent with a dissociative ANDN TS displaying significant loss of C1′–N9 bond order, low bond order to the incoming nucleophile, and substantial oxocarbenium character of the ribosyl ring. The C1′–N9 distance of 2.45 Å corresponds to a 0.74 loss of bond order at the TS, and the nucleophilic water shares a bond order of 0.074 with C1′ (Table S1). Oxocarbenium character of the ribosyl ring at the TS is supported by a short O4′–C1′ bond (1.42 Å in MTA versus 1.27 Å at the TS), which corresponds to an increase of 0.468 in bond order relative to the GS (Table S1). In addition, natural bond orbital (NBO) analysis indicates the relative positive charge increases by 0.182 for C1′ and 0.178 for O4′ at the TS (Table S2).
Figure 4.

Calculated model for the Rv0091 TS structure. TS structure model of MTA hydrolysis catalyzed by Rv0091 was generated by matching KIEs predicted for calculated transition states (RB3LYP/6-31g(d) theory) to intrinsic KIEs. Features of the Rv0091 TS structure include a C1′–N9 bond distance of 2.45 Å, a C1′–Ow bond distance of 2.70 Å, protonation at the N7 position of adenine, and significant oxocarbenium character. TS, transition state; MTA, 5′-methylthioadenosine; KIEs, kinetic isotope effects; Ow, water.
Previous reports indicate the MTAN reaction for other species proceeds via highly dissociative (E. coli5 and S. pneumonia6) and early (N. meningitides7) DN*AN transition states (Figure 3). As such, inclusion of the nucleophilic water in the Rv0091 TS model (Figure 4) distinguishes it from that of other MTAN variants, though dissociative ANDN mechanisms are common among N-ribosyltransferases.28,32–35 A feature of note in the Rv0091 TS model is the Ow–C1′–N9 angle is 150.4° rather than the 180° (Figure 4), which would be predicted for optimal orbital overlap in a pure SN2 reaction. Accordingly, the partial bond orders present in this model dictate a nonlinear TS for the most favorable orbital overlap. The term “nucleophile-assisted SN1” has been invoked to describe SN1 mechanisms in which low bond order must be included to the nucleophile in order to match predicted KIEs with experimental values.36 As the C1′–Ow bond order is low (0.074; Table S1) in the Rv0091 TS model, it is plausible that the active site water could be preassociated in the TS structure via stabilizing interactions with the oxocarbenium species, but not directly participating in a concerted displacement reaction. Preassociation of a nucleophilic water within electron-reorganization distance of the C1′ reactive center is consistent with mass spectrometry experiments carried out with MT-DADMe-ImmA and E. coli MTAN, where the enzyme, inhibitor, and nucleophilic water remain associated in the gas phase.37
TS Analogues Are Picomolar Inhibitors of Rv0091
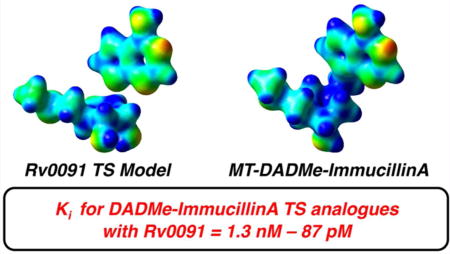
The first- and second-generation immucillins were developed as TS analogue inhibitors for N-ribosyltransferase reactions.38–40 We sought to compare the electrostatics of the Rv0091 TS structure with those of 5′-methylthio-immucillinA (MT-ImmA; a first generation analogue) and MT-DADMe-ImmA (a second generation analogue; Figure 5). EPS maps extrapolated from single-point energy calculations (RB3LYP/6-31g(d) theory)29 are presented in Figure 5 for the Rv0091 TS model, and optimized structures of MT-ImmA and MT-DADMe-ImmA. Comparison of the EPS maps reveals that the geometry and charge localization observed in the Rv0091 TS (Figure 5a) is more similar to that of MT-DADMe-ImmA (Figure 5b) than to MT-ImmA (Figure 5c). Consistent with these models, the Ki for MT-DADMe-ImmA with Rv0091 (1.5 nM) was 56-fold more potent than that of MT-ImmA (Ki = 85 nM).
Figure 5.

EPS maps for Rv0091 TS structure and immucillin TS analogues. EPS maps (red = partial negative; blue = partial positive) were extrapolated from single-point energy calculations (RB3LYP/6-31g(d) theory)29 and visualized in GaussView 5.0 (isovalue = 0.04). (a) Energy-minimized structure and EPS map for the TS structure of MTA hydrolysis by Rv0091. (b) Energy-minimized structure and EPS map for MTDADMe-ImmA. (c) Energy-minimized structure and EPS map for MT-ImmA. EPS, electrostatic potential surface; TS, transition state; MTA, 5′-methylthioadenosine; MT-DADMe-ImmA, 5′-methylthio-DADMe-immucillinA; MT-ImmA, 5′-methylthioimmucillin-A.
DADMe-ImmA TS analogues inhibit MTAN activity in the picomolar to femtomolar range in other bacterial MTANs and display increased binding affinity when functionalized with hydrophobic groups at the C5′ position.12,15 Crystallographic studies of DADMe-ImmA TS analogues with MTANs from Salmonella enterica18 and H. pylori15 revealed an elongated hydrophobic binding pocket proximal to the C5′ substituent of the bound immucillin scaffolds. Consistent with these findings, the most potent inhibition of Rv0091 was observed with DADMe-ImmA inhibitors bearing long alkyl groups at the C5′ position (Figure 6). For example, hexyl thioether 3 inhibited Rv0091 activity with a Ki of 87 pM, whereas methyl thioether 1 and butyl thioether 2 both displayed Ki values in the low nanomolar range (Figure 6).
Figure 6.

Inhibition of Rv0091 with DADMe-ImmA TS analogues. Inhibition of Rv0091 activity was monitored at 305 nm via a coupled assay with xanthine oxidase43 in which the adenine product was converted to 2,8-dihydroxyadenine. The Ki values were calculated using the Morrison equation for tight-binding inhibitors.44
Overall, DADMe-ImmA TS analogues inhibit Rv0091 less potently than they do other MTANs. For example, the Ki values for MT-DADMe-ImmA with E. coli MTAN and Rv0091 are 2 pM31 and 1.5 nM (Figure 6), respectively. This difference in binding affinity for MT-DADMe-ImmA is anticipated from the lower catalytic activity of Rv0091 relative to E. coli MTAN. Catalytic rate enhancement has been correlated with the affinity of TS analogues for their enzyme targets by applying the Wolfenden approximation.4,41,42 As such, the attenuated binding of DADMe-ImmA inhibitors with Rv0091 is consistent with the slow turnover of Rv0091 indicated by kcat. In addition, the DADMe-ImmA scaffold was designed to mimic N-ribosyltransferase reactions with highly dissociative TS structures.38–40 The crystal structure of MT-DADMe-ImmA in binary complex with E. coli MTAN reveals a distance of 2.8 Å between the N1′ position of the 3′-hydroxypyrrolidine ring and C9 of the 9-deazaadenine moiety.30 This inhibitor geometry is a good match to the TS structure of E. coli MTAN wherein adenine is 3.0 Å from C1′.31 By comparison, the Rv0091 TS structure is somewhat earlier with a C1′–N9 distance of 2.45 Å (Figure 4). Moreover, the protonated aminoribitol scaffold of the DADMe-ImmA analogues (Figure 6) is designed to mimic the oxocarbenium species resulting from near full dissociation of adenine. The earlier TS of Rv0091 (relative to E. coli MTAN) would result in less electropositive charge at the C1′ position, which may not be as accurately mimicked by the DADMe-ImmA scaffold. The EPS maps for the Rv0091 TS structure (Figure 5a) and MT-DADMe-ImmA (Figure 5b) reveal greater positive charge is localized at the 1′ position of the inhibitor. Accordingly, differences in TS geometry and electrostatics between E. coli MTAN and Rv0091 may also contribute to the lower potency observed for DADMe-ImmA inhibitors with the Rv0091 protein.
The TDR M. tuberculosis gene function library classifies Rv0091 as a nonessential gene.45,46 This is consistent with the assignment of Rv0091 as a putative MTAN with roles in quorum sensing and SAM recycling. With the function of Rv0091 now defined as a 5′-dAdo hydrolase, the in vivo activity of Rv0091 is more likely coupled to radical SAM reactions that act to generate radical reaction centers, with 5′-dAdo and methionine as products. Radical SAM enzymes are recognized by the CX3CX2C motif,47 and 21 of these proteins have been annotated in the M. tuberculosis genome.48 Preliminary growth experiments with M. tuberculosis indicate that MT-DADMe-ImmA (3) and butylthio-DADMe-immucillinA (4) have no effect on growth of cultured cells.
CONCLUSION
The Rv0091 protein from M. tuberculosis, previously annotated as a putative MTAN, exists as a dimer in solution and displays substrate specificity for 5′-dAdo relative to MTA and SAH. Thus, the protein is not crucial in quorum sensing pathways but more likely plays a role in reactions involving SAM radical enzymes. Intrinsic KIE measurements and quantum chemical calculations were used to establish a model for the TS structure of Rv0091. The TS for Rv0091 differs from the transition states of previously reported MTANs. The Rv0091 TS is characterized by a significant decrease in C1′–N9 bond order, substantial oxocarbenium character of the ribosyl ring, and weak participation of the water nucleophile. DADMe-ImmA TS analogues resemble the Rv0091 TS structure and inhibit Rv0091 activity with Ki values in the range of 10−9 to 10−11 M. The most potent inhibitor of Rv0091 activity identified in this study was hexyl-thio-DADMe-immucillinA, which exhibited a Ki of 87 pM. Studies to investigate the in vivo role of Rv0091 in mycobacteria, as well as the potential for DADMe-ImmA TS analogues to function as modulators of virulence in M. tuberculosis, are ongoing.
METHODS
Full details for all experimental methods are provided in the Supporting Information.
Supplementary Material
Acknowledgments
We thank M. Poulin (Einstein), Z. Wang (Einstein), and A. Gizzi (Einstein) for helpful discussions. The inhibitor molecules were a generous gift of P. C. Tyler and G. B. Evans from the Ferrier Research Institute, Victoria University of Wellington, New Zealand. This work was supported by research grant GM041916 and training grant T32AI070117 from the National Institutes of Health.
Footnotes
ASSOCIATED CONTENT
- Supporting Information Figure S1–S5 and Supporting Information Tables S1–S2; complete experimental procedures for expression and purification of Rv0091; kinetic assays and oligomeric state determination of Rv0091; synthesis of isotopically labeled MTA substrates; measurement of KIEs and forward commitment; computational analyses; and determination of inhibition constants (PDF)
The authors declare no competing financial interest.
References
- 1.World Health Organization. WHO global tuberculosis report. World Health Organization; Geneva, Switzerland: 2015. [Google Scholar]
- 2.CDC - Tuberculosis (TB) http://www.cdc.gov/tb/
- 3.Parveen N, Cornell KA. Methylthioadenosine/S-adenosylhomocysteine nucleosidase, a critical enzyme for bacterial metabolism. Mol. Microbiol. 2011;79:7–20. doi: 10.1111/j.1365-2958.2010.07455.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schramm VL. Enzymatic transition states, transition-state analogs, dynamics, thermodynamics, and lifetimes. Annu. Rev. Biochem. 2011;80:703–732. doi: 10.1146/annurev-biochem-061809-100742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Singh V, Lee JE, Nunez S, Howell PL, Schramm VL. Transition state structure of 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidase from Escherichia coli and its similarity to transition state analogues. Biochemistry. 2005;44:11647–11659. doi: 10.1021/bi050863a. [DOI] [PubMed] [Google Scholar]
- 6.Singh V, Schramm VL. Transition-state analysis of S. pneumoniae 5′-methylthioadenosine nucleosidase. J. Am. Chem. Soc. 2007;129:2783–2795. doi: 10.1021/ja065082r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Singh V, Luo M, Brown RL, Norris GE, Schramm VL. Transition-state structure of neisseria meningitides 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidase. J. Chem. Soc. 2007;129:13831–13833. doi: 10.1021/ja0754204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schramm VL. Enzymatic transition state theory and transition state analogue design. J. Biol. Chem. 2007;282:28297–28300. doi: 10.1074/jbc.R700018200. [DOI] [PubMed] [Google Scholar]
- 9.Schramm VL. Enzymatic transition states and transition state analog design. Annu. Rev. Biochem. 1998;67:693–720. doi: 10.1146/annurev.biochem.67.1.693. [DOI] [PubMed] [Google Scholar]
- 10.Schramm VL. Enzymatic transition-state analysis and transition-state analogs. Methods Enzymol. 1999;308:301–355. doi: 10.1016/s0076-6879(99)08015-5. [DOI] [PubMed] [Google Scholar]
- 11.Singh V, Shi W, Almo SC, Evans GB, Furneaux RH, Tyler PC, Painter GF, Lenz DH, Mee S, Zheng R, Schramm VL. Structure and inhibition of a quorum sensing target from Streptococcus pneumoniae. Biochemistry. 2006;45:12929–12941. doi: 10.1021/bi061184i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gutierrez JA, Luo M, Singh V, Li L, Brown RL, Norris GE, Evans GB, Furneaux RH, Tyler PC, Painter GF, Lenz DH, Schramm VL. Picomolar inhibitors as transition-state probes of 5′-methylthioadenosine nucleosidases. ACS Chem. Biol. 2007;2:725–734. doi: 10.1021/cb700166z. [DOI] [PubMed] [Google Scholar]
- 13.Schramm VL. Enzymatic transition states and transition state analogues. Curr. Opin. Struct. Biol. 2005;15:604–613. doi: 10.1016/j.sbi.2005.10.017. [DOI] [PubMed] [Google Scholar]
- 14.Gutierrez JA, Crowder T, Rinaldo-Matthis A, Ho MC, Almo SC, Schramm VL. Transition state analogs of 5′-methylthioadenosine nucleosidase disrupt quorum sensing. Nat. Chem. Biol. 2009;5:251–257. doi: 10.1038/nchembio.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang S, Cameron SA, Clinch K, Evans GB, Wu Z, Schramm VL, Tyler PC. New Antibiotic Candidates against Helicobacter pylori. J. Am. Chem. Soc. 2015;137:14275–14280. doi: 10.1021/jacs.5b06110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Souza GA, Leversen NA, Malen H, Wiker HG. Bacterial proteins with cleaved or uncleaved signal peptides of the general secretory pathway. J. Proteomics. 2011;75:502–510. doi: 10.1016/j.jprot.2011.08.016. [DOI] [PubMed] [Google Scholar]
- 17.Payne JW. Polymerization of proteins with glutaraldehyde. Soluble molecular-weight markers. Biochem. J. 1973;135:867–873. doi: 10.1042/bj1350867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haapalainen AM, Thomas K, Tyler PC, Evans GB, Almo SC, Schramm VL. Salmonella enterica MTAN at 1.36 A resolution: a structure-based design of tailored transition state analogs. Structure. 2013;21:963–974. doi: 10.1016/j.str.2013.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ronning DR, Iacopelli NM, Mishra V. Enzyme-ligand interactions that drive active site rearrangements in the Helicobacter pylori 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidase. Protein Sci. 2010;19:2498–2510. doi: 10.1002/pro.524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee JE, Cornell KA, Riscoe MK, Howell PL. Structure of Escherichia coli 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidase inhibitor complexes provide insight into the conformational changes required for substrate binding and catalysis. J. Biol. Chem. 2003;278:8761–8770. doi: 10.1074/jbc.M210836200. [DOI] [PubMed] [Google Scholar]
- 21.Buckoreelall K, Sun Y, Hobrath JV, Wilson L, Parker WB. Identification of Rv0535 as methylthioadenosine phosphorylase from Mycobacterium tuberculosis. Tuberculosis (Oxford, U. K.) 2012;92:139–147. doi: 10.1016/j.tube.2011.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buckoreelall K, Wilson L, Parker WB. Identification and characterization of two adenosine phosphorylase activities in Mycobacterium smegmatis. J. Bacteriol. 2011;193:5668–5674. doi: 10.1128/JB.05394-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thomas K, Cameron SA, Almo SC, Burgos ES, Gulab SA, Schramm VL. Active site and remote contributions to catalysis in methylthioadenosine nucleosidases. Biochemistry. 2015;54:2520–2529. doi: 10.1021/bi501487w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Banerjee S, Agrawal MJ, Mishra D, Sharan S, Balaram H, Savithri HS, Murthy MR. Structural and kinetic studies on adenylosuccinate lyase from Mycobacterium smegmatis and Mycobacterium tuberculosis provide new insights on the catalytic residues of the enzyme. FEBS J. 2014;281:1642–1658. doi: 10.1111/febs.12730. [DOI] [PubMed] [Google Scholar]
- 25.Northrop DB. The expression of isotope effects on enzyme-catalyzed reactions. Annu. Rev. Biochem. 1981;50:103–131. doi: 10.1146/annurev.bi.50.070181.000535. [DOI] [PubMed] [Google Scholar]
- 26.Cleland WW. The use of isotope effects to determine transition-state structure for enzymic reactions. Methods Enzymol. 1982;87:625–641. doi: 10.1016/s0076-6879(82)87033-x. [DOI] [PubMed] [Google Scholar]
- 27.Rose IA. The isotope trapping method: desorption rates of productive E.S complexes. Methods Enzymol. 1980;64:47–59. doi: 10.1016/s0076-6879(80)64004-x. [DOI] [PubMed] [Google Scholar]
- 28.Berti PJ, Tanaka KSE. Transition State Analysis Using Multiple Kinetic Isotope Effects: Mechanisms of Enzymatic and Non-enzymatic Glycoside Hydrolysis and Transfer. Adv. Phys. Org. Chem. 2002;37:239–314. [Google Scholar]
- 29.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima TE, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09. Gaussian, Inc; Wallingford, CT: 2009. [Google Scholar]
- 30.Lee JE, Singh V, Evans GB, Tyler PC, Furneaux RH, Cornell KA, Riscoe MK, Schramm VL, Howell PL. Structural rationale for the affinity of pico- and femtomolar transition state analogues of Escherichia coli 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidase. J. Biol. Chem. 2005;280:18274–18282. doi: 10.1074/jbc.M414471200. [DOI] [PubMed] [Google Scholar]
- 31.Singh V, Evans GB, Lenz DH, Mason JM, Clinch K, Mee S, Painter GF, Tyler PC, Furneaux RH, Lee JE, Howell PL, Schramm VL. Femtomolar transition state analogue inhibitors of 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidase from Escherichia coli. J. Biol. Chem. 2005;280:18265–18273. doi: 10.1074/jbc.M414472200. [DOI] [PubMed] [Google Scholar]
- 32.Bates C, Kendrick Z, McDonald N, Kline PC. Transition state analysis of adenosine nucleosidase from yellow lupin (Lupinus luteus) Phytochemistry. 2006;67:5–12. doi: 10.1016/j.phytochem.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 33.Berti PJ, McCann JA. Toward a detailed understanding of base excision repair enzymes: transition state and mechanistic analyses of N-glycoside hydrolysis and N-glycoside transfer. Chem. Rev. 2006;106:506–555. doi: 10.1021/cr040461t. [DOI] [PubMed] [Google Scholar]
- 34.Kline PC, Schramm VL. Purine nucleoside phosphorylase. Catalytic mechanism and transition-state analysis of the arsenolysis reaction. Biochemistry. 1993;32:13212–13219. doi: 10.1021/bi00211a033. [DOI] [PubMed] [Google Scholar]
- 35.Schwartz PA, Vetticatt MJ, Schramm VL. Transition state analysis of the arsenolytic depyrimidination of thymidine by human thymidine phosphorylase. Biochemistry. 2011;50:1412–1420. doi: 10.1021/bi101900b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gawlita E, Szylhabel-Godala A, Paneth P. Kinetic Isotope Effects on the Menshutkin Reaction: Theory Versus Experiment. J. Phys. Org. Chem. 1996;9:41–49. [Google Scholar]
- 37.Wang S, Lim J, Thomas K, Yan F, Angeletti RH, Schramm VL. A complex of methylthioadenosine/S-adenosylhomocysteine nucleosidase, transition state analogue, and nucleophilic water identified by mass spectrometry. J. Am. Chem. Soc. 2012;134:1468–1470. doi: 10.1021/ja211176q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Evans GB, Furneaux RH, Lewandowicz A, Schramm VL, Tyler PC. Synthesis of second-generation transition state analogues of human purine nucleoside phosphorylase. J. Med. Chem. 2003;46:5271–5276. doi: 10.1021/jm030305z. [DOI] [PubMed] [Google Scholar]
- 39.Furneaux RH, Limberg G, Tyler PC, Schramm VL. Synthesis of transition state inhibitors for N-riboside hydrolases and transferases. Tetrahedron. 1997;53:2915–2930. [Google Scholar]
- 40.Evans GB, Furneaux RH, Gainsford GJ, Schramm VL, Tyler PC. Synthesis of transition state analogue inhibitors for purine nucleoside phosphorylase and N-riboside hydrolases. Tetrahedron. 2000;56:3053–3062. [Google Scholar]
- 41.Wolfenden R. Transition state analogues for enzyme catalysis. Nature. 1969;223:704–705. doi: 10.1038/223704a0. [DOI] [PubMed] [Google Scholar]
- 42.Wolfenden R, Snider MJ. The depth of chemical time and the power of enzymes as catalysts. Acc. Chem. Res. 2001;34:938–945. doi: 10.1021/ar000058i. [DOI] [PubMed] [Google Scholar]
- 43.Thomas K, Haapalainen AM, Burgos ES, Evans GB, Tyler PC, Gulab S, Guan R, Schramm VL. Femtomolar inhibitors bind to 5′-methylthioadenosine nucleosidases with favorable enthalpy and entropy. Biochemistry. 2012;51:7541–7550. doi: 10.1021/bi3009938. [DOI] [PubMed] [Google Scholar]
- 44.Copeland RA. Enzymes. 2. Wiley; Canada: 2000. [Google Scholar]
- 45.TDR Targets. http://tdrtargets.org.
- 46.Sassetti CM, Rubin EJ. Genetic requirements for mycobacterial survival during infection. Proc. Natl. Acad. Sci. U. S. A. 2003;100:12989–12994. doi: 10.1073/pnas.2134250100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sofia HJ, Chen G, Hetzler BG, Reyes-Spindola JF, Miller NE. Radical SAM, a novel protein superfamily linking unresolved steps in familiar biosynthetic pathways with radical mechanisms: functional characterization using new analysis and information visualization methods. Nucleic Acids Res. 2001;29:1097–1106. doi: 10.1093/nar/29.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Akiva E, Brown S, Almonacid DE, Barber AE, 2nd, Custer AF, Hicks MA, Huang CC, Lauck F, Mashiyama ST, Meng EC, Mischel D, Morris JH, Ojha S, Schnoes AM, Stryke D, Yunes JM, Ferrin TE, Holliday GL, Babbitt PC. The Structure-Function Linkage Database. Nucleic Acids Res. 2014;42:D521–530. doi: 10.1093/nar/gkt1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.