

Abstract
Erdheim-Chester disease (ECD) is a rare form of systemic histiocytosis, typically presenting with striking osseous involvement characterized by bilateral osteosclerosis and involvement of organs such as the lung, pituitary gland, heart, and brain. Liver involvement with ECD is extremely uncommon. We report a 56-year-old woman presenting with newly diagnosed cirrhosis and signs concerning for intra-abdominal malignancy, including omental caking and peritoneal thickening. Liver biopsy demonstrated xanthogranulomatous infiltration from ECD. The patient showed initial improvement with interferon therapy, but she developed severe depression, which led to the discontinuation of the treatment. Shortly afterward, she died from progressive liver dysfunction resulting in hepatorenal syndrome.
Introduction
Erdheim-Chester disease (ECD) is a rare form of non-Langerhans cell histiocytosis that was described by Jacob Erdheim and William Chester in 1930.1 ECD is characterized by multifocal infiltration of foamy histiocytes in bone biopsies with or without involvement of extraskeletal organs. Sclerosis of the long bones is a hallmark of this disease. The clinical manifestations of ECD range from bone involvement to life-threatening multisystemic disease, with the most common presentations being bone pain, neurologic involvement, and diabetes insipidus.2 Patients with ECD rarely present with clinical features of liver and gastrointestinal (GI) involvement. Of approximately 500 cases in the literature, only 4 cases of hepatic involvement with ECD have been described. Mortality rates are high, and currently there is no standard treatment for patients with ECD. A broad range of therapies has been attempted for ECD, including corticosteroids, cytotoxic agents, interferon-α, IL-1 receptor antagonist, tyrosine kinase inhibitors, bisphosphonates, and autologous hematopoietic stem cell transplant. Recent studies have shown interferon-based therapy improves survival in patients with ECD.3,4
Case Report
A 56-year-old woman with no known prior medical history presented with acute onset of abdominal distension and leg swelling for the prior 2 weeks. She denied history of similar illness in the past. She denied fever, chills, nausea, vomiting, weight loss, jaundice, and bone pain. She denied smoking tobacco and intake of alcohol or any medication. She denied family history of liver disease. On presentation, her hemodynamics were stable. She was obese, and examination findings were suggestive of ascites with bilateral pitting pedal edema and muscle wasting but no other stigmata of chronic liver disease. She specifically had no asterixis or any sign of overt hepatic encephalopathy.
Complete blood count showed only mild thrombocytosis to 457,000/uL. Liver function tests were notable for elevated total bilirubin 2.2 mg/dL, direct bilirubin 1.2 mg/dL, alkaline phosphatase 1,157 U/L, alanine aminotransferase 128 U/L, protein 10.1 g/dL, albumin 3.2 g/dL, and normal aspartate aminotransferase 105 U/L. Diagnostic paracentesis showed scattered benign mesothelial cells and mixed inflammatory cells, as well as a serum-ascites albumin gradient of 2.4 g/dL, suggestive of ascites secondary to portal hypertension. Other lab results were normal.
Abdominal computed tomography (CT) imaging suggested peritoneal carcinomatosis (given the omental caking and peritoneal nodularity) and showed ascites and nodularity of the liver suggestive of cirrhosis (Figure 1). Her CA19-9 and CA-125 were elevated to 488 U/mL and 92.3 U/mL, respectively. A triphasic CT scan of the liver was negative for hepatic malignancy. A positron emission tomography (PET) scan prior to laparoscopic-guided biopsy showed centrally diffuse uptake in the liver (with a maximum serum uptake value 11.8), uptake in the anterior peritoneal wall (corresponding to omental caking; serum uptake value 5.5), multifocal mildly increased uptake in the right femur, and changes in the right maxillary sinus.
Figure 1.
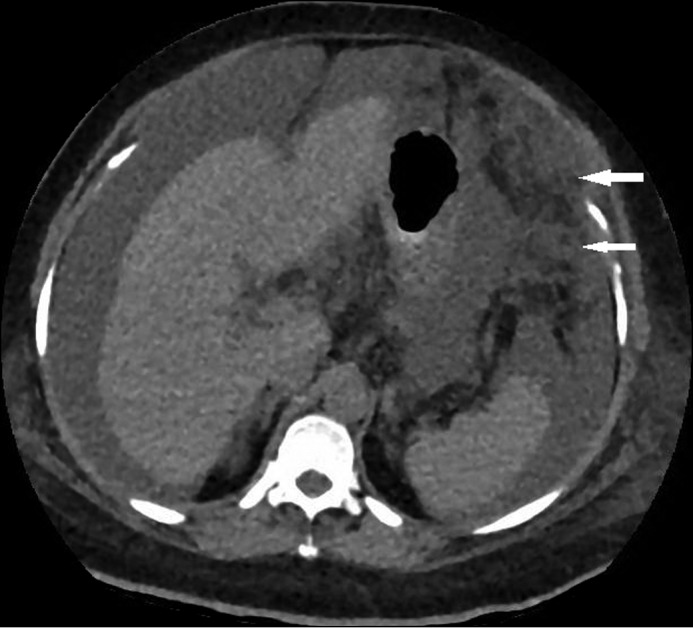
Abdominal computed tomography showing peritoneal carcinomatosis, including omental caking (arrows), scattered peritoneal nodularity, and ascites.
Liver biopsy revealed extensive bridging fibrosis and early nodule formation (stage 4/4), attributed to nonalcoholic steatohepatitis, along with prominent bile ductular proliferation and histiocytic infiltration of the sinusoids (Figure 2). Diagnostic laparoscopy with omental biopsy showed infiltration of fibroadipose tissue by numerous foamy histiocytes admixed with Touton-type giant cells and a chronic inflammatory cell infiltrate. Immunohistochemistry revealed histiocytes positive for CD163 and CD68, while factor XIIIa and S-100 were weakly positive, confirming ECD (Figure 3). BRAFV600E mutational analyses performed on the omental biopsy specimen and bone marrow biopsy specimen were negative.
Figure 2.

Erdheim-Chester disease histology. (A) Hematoxylin and eosin (H&E) stain of the liver biopsy showing hepatic parenchyma infiltrated by pale histiocytes (100x). (B) High magnification of H&E stain showing foamy histiocytes (arrow) (400x). (C) Trichrome stain showing cirrhosis characterized by extensive bridging fibrosis and nodular formation (100x). (D) H&E section of peritoneal biopsy showing similar foamy histiocytes (black arrow) and a Touton giant cell (blue arrow) (400x).
Figure 3.
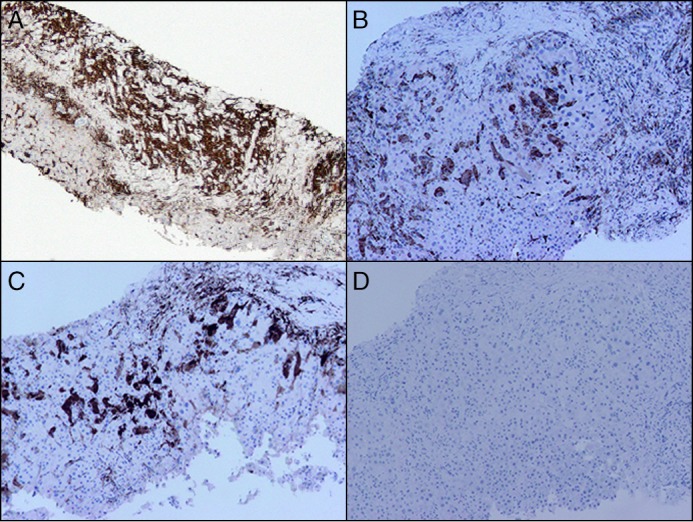
Immunohistochemical stains on liver biopsy. (A) CD163 showing numerous positive histiocytes filling the liver sinusoids (100x). (B) CD68 showing positivity in the histiocytes (100x). (C) Factor XIIIa showing weak to strong positivity in the histiocytes (100x). (D) CD1a is negative in the histiocytes (100x).
The patient was initiated on pegylated interferon-α2a(c) 135 μg weekly and was discharged home. During follow-up, there were mild improvements in her ascites and liver function tests. A restaging PET scan showed evidence of mild disease response. However, the patient developed severe depression with suicidal intent, leading to temporary discontinuation of interferon-based therapy. She developed progressive liver dysfunction with worsening liver enzymes and total bilirubin, along with hepatic encephalopathy and subsequent hepatorenal syndrome requiring hemodialysis. Mutational testing from the omental biopsy confirmed a mitogen-activated protein kinase 2 (MAP2K) mutation. While awaiting insurance approval for Trametinib (MEK inhibitor), the patient was restarted on lower-dose pegylated interferon-α 70 μg weekly. She did not tolerate this therapy well and developed worsening encephalopathy. She opted for comfort care, and hemodialysis was discontinued. She experienced rapid clinical deterioration with worsening encephalopathy, treatment-refractory hypotension, and bradycardia. She passed away shortly thereafter.
Discussion
ECD is a rare form of non-Langerhans cell histiocytosis characterized by multifocal infiltration of foamy histiocytes in bone biopsies with or without involvement of extraskeletal organs.2 Universally, almost all patients with ECD have bilateral symmetrical osteosclerosis of the long bones, with only 4% of patients lacking osteosclerosis at this site.3,5 Only rarely do these patients present with clinical features of liver and GI involvement, as discussed in our case report. Mortality rates are high. Recent studies have shown that interferon-based therapy improves survival in these patients.4
The pathogenesis of this rare disorder is still unclear, but it is characterized by clonal expansion of histiocytes marked by hyperactivation of MAPK signaling. BRAF V600E mutation, activation of proto-oncogene of the serine-threonine kinase family has been found in 38–69% of patients with ECD.3 Immunohistochemistry testing in our patient, however, was negative for BRAF V600E mutations.
Although there was only unilateral bone involvement on the bone scan, our patient had typical histopathologic findings that confirmed the diagnosis of ECD.5 The presence of CD68- and CD163-positive cells on immunohistiochemistry indicates a histiocytic origin. Histiocytes can be Langerhans cells that are of dendritic cell origin, are positive for CD68, CD1a, and S100 protein, and show Birbeck granules in their cytoplasm, or they can be non-Langerhans cells that are of monocytic-macrophagic origin and are characterized by the absence of Birbeck granules on histology and the presence of factor XIIIa, as seen in our patient.6 Non-Langerhans cell histiocytosis could be categorized into those involving cutaneous sites, cutaneous sites with systemic manifestations, and those only with systemic involvement. The systemic form of non-Langerhans cell histiocytosis could be either Rosai-Dorfman disease or ECD. Rosai-Dorfman disease usually involves lymph nodes, and the histology shows intracytoplasmic lymphocytes and immunohistiochemistry strongly positive for S-100.7 Our patient did not have any lymph node involvement; histology showed histiocytosis with xanthomatous features with fibrosis, and immunohistiochemistry was only weakly positive for S-100, confirming the diagnosis of ECD.3
Liver involvement in these patients is rarely reported. To date, only 4 cases of liver involvement in patients with ECD have been reported.8-11 In the prior literature, liver involvement presented as nodular lesions, tumor-like lesions of the biliary tract and diffuse infiltration.8,9,11 These patients had either mild transaminase elevation or cholestasis from extrinsic biliary obstruction or normal liver function.9-11 Of these 4 case reports, only one died from progressive respiratory failure in spite of steroid therapy.
This is the first case report showing liver fibrosis with histiocytic infiltration leading to progressive liver decompensation. Although our patient did have fatty change and her cirrhosis was likely due to nonalcoholic steatohepatitis, her progressive liver decompensation was likely caused by xanthogranulomatous infiltration from ECD. The pathogenesis of ECD has implicated T helper-1 cell activation leading to recruitment of proinflammatory cells and histiocytes leading to fibrosis.5 High-dose interferon therapy is the standard of care for the management of ECD; however, our patient did not respond well, perhaps because her limited hepatic reserve was unable to sustain the immunologic response to the treatment. Patients with such liver dysfunction may benefit from treatment directed by mutation analysis rather than a trial of interferon-based therapy. Overall prognosis is modest, with 68% survival after 3 years of diagnosis.4
Disclosures
Author contributions: G. Balasubramanian wrote the manuscript. A. Modiri, M. Affi, CE Hagen, B. Batdorf, K. Oshima, and L. Michaelis edited the manuscript. K. Saeian edited the manuscript and is the article guarantor.
Financial disclosure: None to report.
Informed consent could not be obtained, as the patient is deceased. All identifying information has been removed to protect patient privacy.
References
- 1.Chester W. Uber lipoidgranulomatose. Virchows Arch Pathol Anat. 1930;279:561–602. [Google Scholar]
- 2.Cavalli G, Guglielmi B, Berti A, et al. The multifaceted clinical presentations and manifestations of Erdheim-Chester disease: Comprehensive review of the literature and of 10 new cases. Ann Rheum Dis. 2013;72:1691–5. [DOI] [PubMed] [Google Scholar]
- 3.Diamond EL, Dagna L, Hyman DM, et al. Consensus guidelines for the diagnosis and clinical management of Erdheim-Chester disease. Blood. 2014;124:483–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arnaud L, Hervier B, Néel A, et al. CNS involvement and treatment with interferon-alpha are independent prognostic factors in Erdheim-Chester disease: A multicenter survival analysis of 53 patients. Blood. 2011;117:2778–82. [DOI] [PubMed] [Google Scholar]
- 5.Haroche J, Arnaud L, Cohen-Aubart F, et al. Erdheim-Chester disease. Rheum Dis Clin North Am. 2013;39:299–311. [DOI] [PubMed] [Google Scholar]
- 6.Wilejto M, Abla O. Langerhans cell histiocytosis and Erdheim-Chester disease. Curr Opin Rheumatol. 2012;24:90–6. [DOI] [PubMed] [Google Scholar]
- 7.Weitzman S, Jaffe R. Uncommon histiocytic disorders: The non-Langerhans cell histiocytoses. Pediatr Blood Cancer. 2005;45:256–264. [DOI] [PubMed] [Google Scholar]
- 8.Gundling F, Nerlich A, Heitland WU, et al. Biliary manifestation of Erdheim-Chester disease mimicking Klatskin's carcinoma. Am J Gastroenterol. 2007;102:452–4. [DOI] [PubMed] [Google Scholar]
- 9.Pan A, Doyle T, Schlup M, et al. Unusual manifestation of Erdheim-Chester disease. BMC Gastroenterol. 2011;11:77.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ivan D, Neto A, Lemos L, et al. Erdheim-Chester disease: A unique presentation with liver involvement and vertebral osteolytic lesions. Arch Pathol Lab Med. 2003;127:e337–e339. [DOI] [PubMed] [Google Scholar]
- 11.Gupta A, Aman K, Al-Babtain M, et al. Multisystem Erdheim-Chester disease: A unique presentation with liver and axial skeletal involvement. Br J Haematol. 2007;138:280.. [DOI] [PubMed] [Google Scholar]