

Abstract
The Epithelial-to-mesenchymal transition (EMT) is a cell-biological program that confers mesenchymal traits on carcinoma cells and drives their metastatic dissemination. It is, unclear, however, whether activation of EMT in carcinoma cells can change their susceptibility to immune attack. We demonstrate here that mammary tumor cells arising from more epithelial carcinoma cell lines expressed high levels of MHC-I, low levels of PD-L1 and contained within their stroma CD8+ T cells and M1 (anti-tumor) macrophages. In contrast, tumors arising from more-mesenchymal carcinoma cell lines exhibiting EMT markers expressed low levels of MHC-I, high levels of PD-L1 and contained within their stroma regulatory T cells, M2 (pro-tumor) macrophages and exhausted CD8+ T cells. Moreover, the more mesenchymal carcinoma cells within a tumor retained the ability to protect their more epithelial counterparts from immune attack. Lastly, epithelial tumors were more susceptible to elimination by immunotherapy than corresponding mesenchymal tumors. Our results identify immune cells and immunomodulatory markers that can be potentially targeted to enhance the susceptibility of immunosuppressive tumors to various therapeutic regimens.
Keywords: EMT, tumor immunology, tumor microenvironment, breast carcinoma, immunosuppression
INTRODUCTION
The microenvironment of breast carcinomas is enriched with immunosuppressive cells that interact with carcinoma cells via a variety of heterotypic signaling mechanisms (1, 2). However, the factors that contribute to immunosuppression in the tumor microenvironment are poorly defined. The Epithelial to Mesenchymal Transition (EMT) is a cell-biological program that drives the metastatic dissemination of carcinomas including breast cancers (BrCa). After activation of an EMT, carcinoma cells begin to secrete a distinct cohort of cytokines such as TGF-β (3). TGF-β is required for the formation of immunosuppressive Tregs and reduces the cytotoxicity of natural killer (NK) cells (4, 5). It remains unclear, however, whether immunosuppression occurs as a direct consequence of the EMT program or develops through the mediation of additional, still-uncharacterized signaling channels. Moreover, carcinoma cells that have undergone an EMT are more refractory to various chemotherapeutic regimens (6). However, whether carcinoma cells residing in the epithelial versus mesenchymal state mount distinct responses to checkpoint blockade therapy is unknown. For these reasons, we undertook to understand the dynamics of the microenvironmental interactions of breast cancers cells that remain in a largely epithelial state or have acquired mesenchymal characteristics following activation of an EMT program.
MATERIALS AND METHODS
Mice
C57BL/6 mice were obtained from the Jackson laboratory. A colony of NOD-SCID mice was generated and maintained in-house. All animals used for experiments were 6–8 week old female mice. Animals were maintained in compliance with the guidelines and protocols approved by the Animal Care and Use Committees at the Massachusetts Institute of Technology.
Cell lines and cell culture
PyMT tumor cell lines from which epithelial (pB-2) and mesenchymal (pB-3) versions were derived, were kind gifts from the Harold L. Moses laboratory and were established and maintained as described previously (7). Cells were cultured in DMEM/F12 medium containing 5% adult bovine serum with 1X penicllin-streptomycin and 1X non-essential amino acids for the duration of this study. MCF7 and T47D cell lines were obtained from ATCC and established and maintained as previously described (8) in DMEM supplemented with 10% fetal bovine serum and 1X penicllin-streptomycin. Cells tested negative for mycoplasma (2014) and were not authenticated since first acquisition. All cell lines were obtained between 2009–2012 and used within 2–3 years since first acquisition. Cells were stably transfected with GFP, RAS or doxycycline-inducible EMT-TFs as previously described (8, 9). All cell lines containing doxycycline-inducible expression vectors were treated with 1.5 μg/ml (PyMT) or 1 μg/ml (MCF7RAS and T47DRAS) doxycycline hyclate (Sigma Aldrich).
In vivo models and tumor dissociation
For orthotopic tumor transplantations, sorted cell populations – EpCAMHI (epithelial PyMT cell line-pB-2), EpCAMLO(mesenchymal PyMT cell line-pB-3), EpCAMHISnail-YFPLO (Snail-lo) and EpCAMLOSnail-YFPHI (Snail-hi) were resuspended in 30μl media containing 20% Matrigel. 1 × 106 cells were implanted into the mammary fat pads of C57BL/6 or NOD/SCID mice. Animals were sacrificed once tumors reached 2cm in size. Tumors were excised and divided into two parts. One part was digested and used for flow cytometry analysis and the other part was fixed in formalin for tissue sections. For tumor digestions, tumors were minced with a razor blade and digested in RPMI containing 2mg/ml collagenase and 100 units/ml hyaluronidase (Roche) in a rotator at 37°C for 1hr. Dissociated tumors were washed two times in PBS and filtered through a 70 μm and 40 μm cell strainer to obtain single-cell suspensions. For immunotherapy experiments, mice bearing tumors arising from various cell lines were treated with anti-CTLA4, 200 ug, clone 9H10, every three days for 20 days.
Flow cytometry
Dissociated tumors were resuspended in wash buffer (PBS containing 0.1% BSA) and stained for surface markers using CD45 PECy7 (30F-11; Affymetrix), CD45 FITC (30F-11; Affymetrix), CD4 PE (RM4-5; Affymetrix), CD8a APC (53-6.7; Affymetrix), CD25 PercpCy5.5 (pc61.5; Affymetrix), CD44 PercpCy5.5 (IM7; Affymetrix), PD-1 FITC (J43; Affymetrix), CTLA4 PE (UC10-4B9; Affymetrix), CD107a PercpCy5.5 (1D4B; Affymetrix), CD11B PercpCy5.5 (M1/70; Affymetrix), F480 PECY7 (BM8; Affymetrix), LY6C E450 (HK1.4; Affymetrix), LY6G APC (1A8; Affymetrix), CD206 APC (MR6F3; Affymetrix), CD3 PercpCy5.5 (17A2; Affymetrix), NK1.1 PE (PK136; Affymetrix), MHC I (H-2Kb) PE (AF6-88.5.5.3; Affymetrix), CD274 BV605 (10F.9G2; Biolegend), HLA-ABC APC (W6/32; Affymetrix), PDL1 FITC (MIH1; BD Biosciences). CD8+ T-cells were sorted from digested tumor samples and co-cultured with the respective PyMT cells (1 × 106 cells/ ml) for 5 hrs in the presence of Monensin Golgi Stop (BD Biosciences). Intracellular cytokine staining was performed using the Intracellular Fixation and Permeabilization Buffer Set (Affymetrix). Intra-cellular staining for FOXP3 was performed using the FOXP3/Transcription Factor Staining Buffer Set (Affymetrix) using FOXP3 Alexa488 (FJK-16s; Affymetrix), NOS2 FITC (CXNFT; Affymetrix), IL-12 PE (C15.6; Biolegend), IFNγ PECY7 (XMG1.2; Affymetrix). Flow cytometry data was acquired on a BD LSRFortessa and data was analyzed using the FlowJo (TreeStar) software.
Western Blot
Whole cell lysates were made in RIPA Buffer (150mM NaCl, 1% IgeCal-CA 360, 0.1% SDS, 50mM Tris, pH-8.0, 0.5% Sodium deoxycholate) and resolved on a gradient gel. Protein was transferred on a nitro-cellulose membrane and blocked in 5% milk powder and 0.2% Tween-20 in PBS. Membranes were probed over-night with primary antibody, washed and incubated with horseradish peroxidase (HRP) labeled secondary antibody and developed using ECL substrate (ThermoFisher). Primary antibodies used were E-cadherin, Vimentin, Zeb1, Snail, GAPDH, β-tubulin, β2-microglobulin (Cell Signaling Technology), Fibronectin (BD Biosciences), Twist (Abcam).
Immunofluorescence Staining
Tumors were fixed in 10% neutral buffered formalin for12–24 hrs and transferred to 70% ethanol, followed by embedding and sectioning. Tumor sections were washed two times in Histoclear II, followed by one wash each in 100%, 95%, 75% ethanol, PBS and 1X wash buffer (Dako). Antigen retrieval was done in 1X Target Retrieval Solution, pH 6.1 (Dako) in a microwave. Sections were blocked in PBS containing 0.3% Triton-X100 and 1% normal donkey serum (Jackson ImmunoResearch Laboratories) for 1hr at room temperature. Sections were incubated with primary antibody at 4°C, overnight. Sections were washed two times in 1X wash buffer followed by incubation with secondary antibody (Biotium) for 2 hrs. Sections were washed three times with 1X wash buffer and incubated with DAPI for 10 mins, followed by 1 wash in PBS. Sections were mounted using Prolong gold antifade reagent (Invitrogen). Tumor cell lines were fixed in 2.5% neutral buffered formalin on ice for 15 mins, followed by three washes in PBS+. Cells were fixed in Triton-X100 for 3 mins and blocked in PBS- containing 3% normal donkey serum. Cells were incubated with primary antibody at 4°C, overnight. Cells were washed three times with PBS- followed by incubation with secondary antibody 2 hrs. Cells were washed three times with PBS- and incubated with DAPI for 10 mins, followed by 1 wash in PBS. Cells were mounted using Prolong gold antifade reagent. Primary antibodies used were E-cadherin (BD Biosciences), Vimentin (Cell Signaling Technology), Zeb1 (Santa Cruz Biotechnology), CD3 (AbCam), CD8 (Abbiotec), F480 (ThermoFisher), Arginase1 (Santa Cruz Biotechnology). Images were acquired using the Zeiss confocal microscope and analyzed using the Zen software.
Statistical Analysis
All data are represented as mean +/− SEM. Statistical Analysis was performed using the GraphPad Prism software. P values were calculated using an unpaired two-tailed student’s t test. * p<0.05, ** p<0.01, *** p<0.001, ns – not significant.
RESULTS
To determine whether the immune responses to epithelial and mesenchymal carcinoma cells were fundamentally different, we used cell lines that were established from tumors arising in the transgenic MMTV-PyMT mouse model of breast adenocarcinoma development (7). Some of these cell lines were more epithelial and expressed high levels of E-cadherin and EpCAM, whereas others were more mesenchymal and in contrast expressed vimentin, low levels of EpCAM, and significant levels of the Zeb1, Twist and Snail EMT-inducing transcription factors (EMT-TFs) (Fig 1A, Supplementary Fig 1A, B).
Fig 1. Immunosuppressive cells in tumors arising from mesenchymal cell lines.
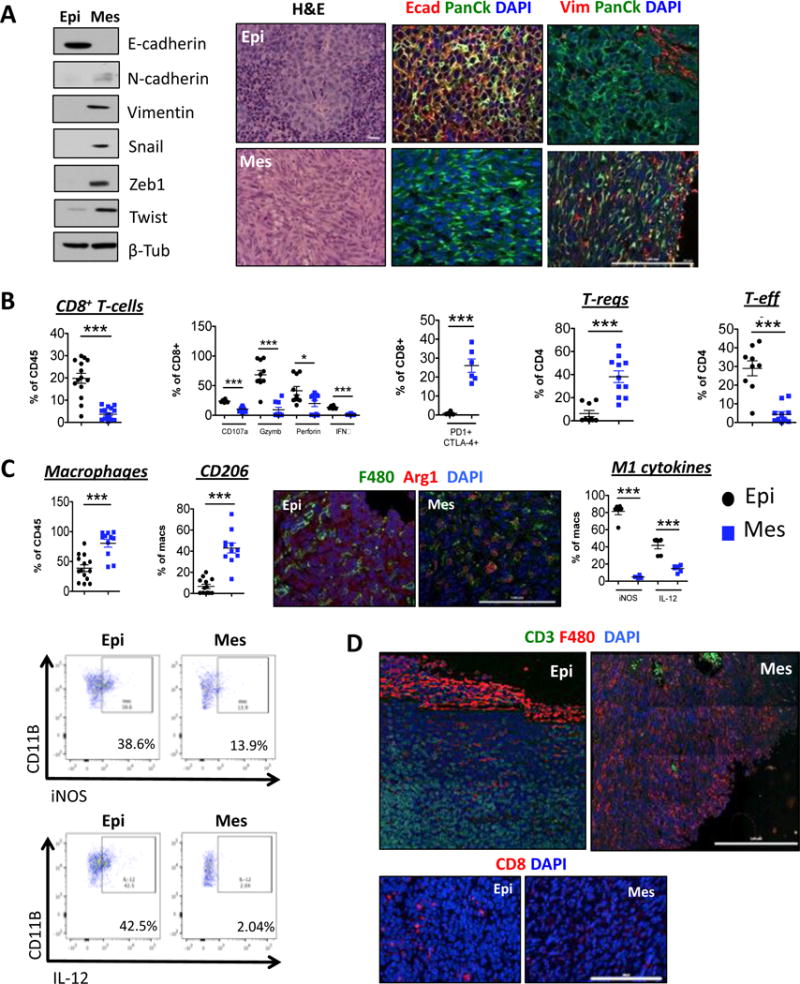
(A) Western blot for EMT markers expressed by cell lines. Haemotoxylin and Eosin and (B, D) Immunofluorescent staining of tumor sections for the indicated markers (N=5). Scale bar, 100 μm. (B, C) Digested tumors were analyzed for the indicated immune populations by flow cytometry after gating on CD45+ cells. Data represent three independent experiments
To determine whether tumors formed from the epithelial and mesenchymal cell lines were differentially susceptible to immune attack, we implanted these cells orthotopically into syngeneic immunocompetent hosts. Tumors arising from the more-epithelial cell line were well-differentiated adenocarcinomas and expressed E-cadherin, while tumors arising from the more mesenchymal cells were poorly differentiated, exhibited a sarcomatoid histomorphology, and expressed instead the mesenchymal marker vimentin (Fig 1A).
Most strikingly, the stroma of primary tumors arising from the more mesenchymal cell line, relative to those spawned by the more epithelial carcinoma cells, contained significantly higher numbers of CD25+ FOXP3+ Tregs and lower numbers of CD25+ FOXP3− effector CD4+ T-cells (Fig 1B, Supplementary Fig 1D). Conversely, tumors arising from the epithelial cell line contained higher numbers of CD8+ cytotoxic T-cells instead (Fig 1B, Supplementary Fig 1C). Furthermore, a higher percentage of CD8+ T-cells present in the stroma of epithelial tumors expressed CD107a, Perforin, Granzyme b and IFN-γ compared to those present in mesenchymal tumors, indicative of effector function (Fig 1B). Finally, the few CD8+ T-cells present in the stroma of the more mesenchymal tumors co-expressed markers associated with functional exhaustion, PD-1 and CTLA4 (Fig 1B).
Tumors arising from the more mesenchymal cell lines also contained a significantly higher number of macrophages that expressed the mannose receptor CD206 and stained positive for Arginase1, indicating a pro-tumor M2 phenotype, relative to the macrophages present in tumors arising from the more epithelial cells, which by contrast did not express either of these two markers but instead stained positively for iNOS and IL-12, markers of M1, anti-tumor macrophages (Fig 1C, Supplementary Fig 1E–F).
Immunofluorescence analysis of tumor sections revealed that CD3+ and CD8+ T-cells had infiltrated the tumors arising from the epithelial cell line, while the macrophages remained largely at the periphery. This contrasted with tumors arising from the mesenchymal cell line, which were infiltrated by macrophages (Fig 1D). We did not however, observe any significant differences in the numbers of monocytic or granulocytic MDSCs infiltrating these two classes of tumors (Supplementary Fig 1G, H). These results indicated that, in contrast to the stroma assembled by the more-epithelial tumors, the stroma of tumors arising from the more-mesenchymal cells, was largely immunosuppressive.
Using MMTV-PyMT reporter mice that bear knock-in IRES-YFP constructs reporting the activity of the endogenous Snail and Slug promoters, we recently demonstrated that, when expressed at physiologic levels, Snail is associated with a CSC-like phenotype, whereas Slug choreographs the normal mammary epithelial stem cell state (7). To determine whether the Snail-expressing carcinoma cells would also be immunosuppressive, we sorted cells that differed in their expression of the Snail-YFP reporter and thus endogenous Snail protein, from tumors arising in MMTV-PyMT-Snail-YFP mice using YFP expression in conjunction with EpCAM, as previously described (Supplementary Fig 2A and (7)). Snail-hi-YFP cells were mesenchymal and did not express any epithelial markers, but expressed instead N-cadherin, vimentin, fibronectin and the Zeb1 EMT-TF. In contrast, Snail-lo-YFP cells were less mesenchymal and expressed high levels of both EpCAM and E-cadherin and low levels of Zeb-1 (Fig 2A, Supplementary Fig 2B). This approach allowed us to compare the immune response to related cell lines, which were established from the same tumor and differed in their levels of Snail expression.
Fig 2. Immunosuppressive cells in tumors arising from Snail+ cells.
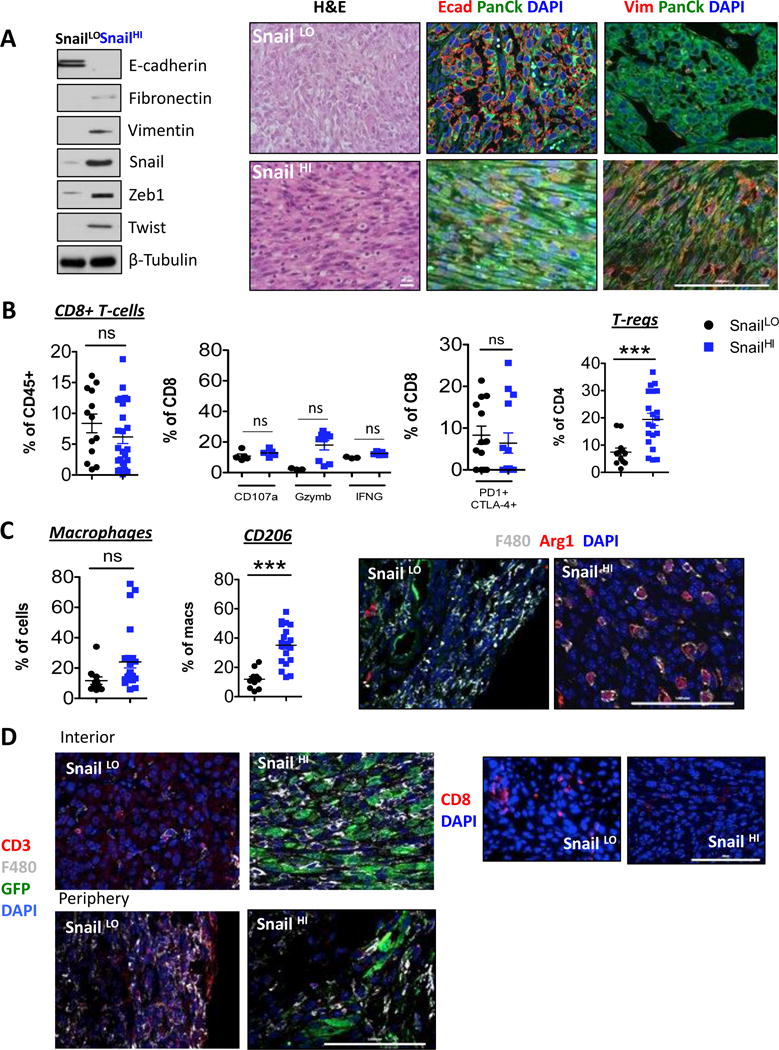
(A) Western blot for EMT markers. Haemotoxylin and Eosin and (C, D) Immunofluorescent staining of tumor sections for the indicated markers (N=5). Scale bar, 100 μm. (B, C) Digested tumors were analyzed for the indicated immune populations by flow cytometry after gating on CD45+ cells. Data represent three independent experiments.
Tumors arising from orthotopic implantation of the Snail-lo cells were heterogeneous and contained some sectors of E-cadherin-expressing cells. In contrast, Snail-hi tumors were poorly differentiated and lacked E-cadherin expression (Fig 2A). Although we did not observe any difference in the numbers and function of CD8+ T-cells (Fig 2B, Supplementary Fig 2C), Snail-hi tumors exhibited a significantly higher percentage of infiltrating Tregs (Fig 2B, Supplementary Fig 2D), and an increase in CD206-expressing M2-macrophages (Fig 2C, Supplementary Fig 2E–F), relative to Snail-lo tumors. These macrophages also expressed Arginase1, in contrast to Snail-lo tumors, which lacked Arginase1-expressing macrophages (Fig 2C). Furthermore, these macrophages had infiltrated Snail-hi tumors, which was in sharp contrast to Snail-lo tumors, where the macrophages were confined largely at the tumor periphery (Fig 2D). Conversely, Snail-lo tumors were infiltrated by CD3+ and CD8+ T-cells (Fig 2D). Additionally, adenocarcinomas arising from another epithelial cell line established from Slug reporter mice (7), (Supplementary Fig 3A–B) were also infiltrated by significantly higher numbers of CD8+ T-cells and significantly fewer numbers of Tregs and M2 macrophages relative to Snail-hi cells (Supplementary Fig 3C–H). Once again these observations demonstrated that more mesenchymal Snail-hi cells were capable of inducing a tumor microenvironment rich in immunosuppressive Tregs and M2 macrophages in contrast to the less mesenchymal Snail-lo cells.
We also observed that the more mesenchymal cell lines, which clearly recruited immunosuppressive cells to the tumor-associated stroma, showed a striking reduction in the levels of surface MHC-I compared to the more epithelial cell lines (Supplementary Fig 4A–D). Furthermore, tumors arising from the more mesenchymal carcinoma cell lines in both immune-competent and immune-deficient mice, exhibited significantly fewer MHC-I+ cells compared to those arising from the epithelial cell lines. In contrast, we found that tumors arising from the more mesenchymal carcinoma cell lines contained instead a higher percentage of PD-L1- positive cells (Supplementary Fig 4E–H). Of note, activation of an EMT in epithelial cell lines by forced expression of various doxycycline-inducible EMT-TFs (Supplementary Fig 5A–C) resulted in a decrease in protein levels of MHC-I expression, which was also accompanied by decreased expression of β2-microglobulin (β2M) (Supplementary Fig 5D, F, H) and increased expression of PD-L1 (Supplementary Fig 5E, G). Thus, the immunoevasive and immunosuppressive maneuvers assembled by the carcinoma cells resulted directly from the cell-autonomous actions of the EMT program.
Carcinomas of the type studied here are comprised of heterogeneous mixtures of both epithelial and mesenchymal cells. However, it was unclear whether the minority subpopulations of immunosuppressive mesenchymal carcinoma cells are able to protect the entire tumor including its epithelial cells from immune attack or whether, their immunosuppressive effects were limited to their immediate vicinity. To address this question, we injected mixtures of unlabeled epithelial cells with two different GFP-labelled, mesenchymal cell lines described earlier.
We observed that a decrease in the proportion of epithelial cells (10:1, 4:1 and 1:1 ratio of E:M cells) was accompanied by a concomitant decrease in the number of infiltrating CD8+ T-cells (Fig 3A and Supplementary Fig 6A, C). CD8+ T-cells present in these various co-mixed tumors did not show any reduction in the expression of markers associated with effector function compared to those present in tumors arising exclusively from epithelial cells. Nonetheless, the CD8+ T-cells that were present in all three types of co-mixed tumors co-expressed PD1 and CTLA4, indicative of partial exhaustion, relative to CD8+ T-cells present in tumors arising only from epithelial cells (Fig 3A and Supplementary Fig 6A). In sharp contrast, a decrease in the proportion of epithelial cells in all co-mixed tumors was accompanied by an increase in the number of suppressive Tregs (CD25+ FOXP3+) (Fig 3A and Supplementary Fig 6A, C) and a decrease in the number of CD4+ effector T-cells (CD25+ FOXP3− and CD44+ FOXP3−) (Fig 3B and Supplementary Fig 6B).
Fig 3. Ability of mesenchymal cells to protect epithelial cells from immune attack.
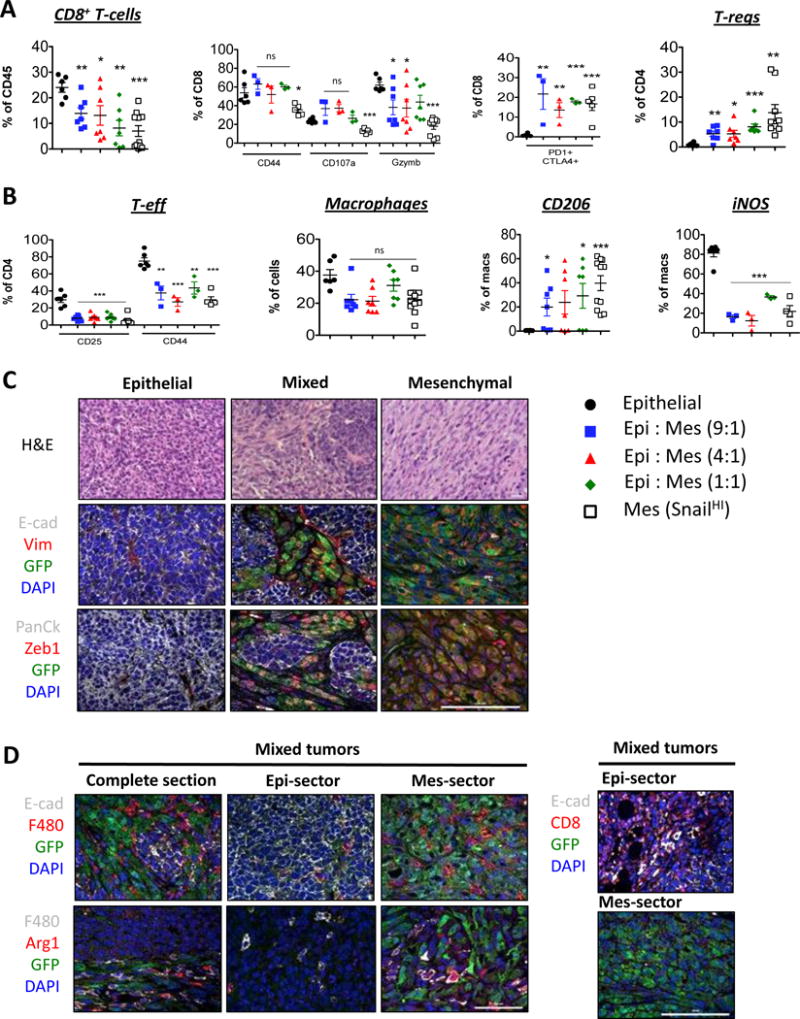
(A, B) Digested tumors were analyzed for the indicated immune populations by flow cytometry after gating on CD45+ cells. (C) Haemotoxylin and Eosin and (C, D) Immunofluorescent staining of tumor sections for the indicated markers. (N=5). Scale bar, 100 μm. Data represent three independent experiments.
Similarly, macrophages present in all types of co-mixed tumors expressed pro-tumor M2 markers CD206 and Arginase1 and low levels of the iNOS anti-tumor, M1 marker, relative to macrophages present in tumors arising from the epithelial cells alone (Fig 3B, Supplementary Fig 6B, C). Hence even when the mesenchymal cells were present as small minority subpopulations (~10%), they were nonetheless able to influence the numbers of suppressive Tregs and M2 macrophages in the tumors as a whole.
Both immunofluorescent and H&E staining of tumors arising from the mixtures of cells injected in different ratios revealed that the epithelial and mesenchymal cells became segregated to distinct sectors of the tumor and thus did not physically intermingle (Fig 3C– top, Supplementary Fig 6D). Additionally, mesenchymal sectors within the co-mixed tumors continued to express features associated with the EMT such as vimentin and nuclear Zeb1, in contrast to their epithelial counterparts, which expressed E-cadherin instead (Fig 3C- middle, bottom, Supplementary Fig 6D).
This clear segregation of the two types of tumor cells to distinct sectors of tumors allowed us to examine the topological relationships between recruited immune cells and readily identifiable epithelial and mesenchymal sectors of the tumors. As we found, CD8+ T-cells were found near the epithelial sectors of mixed tumors. Conversely, numerous Arginase1+ F480+ M2-macrophages were interspersed only among the GFP-positive, mesenchymal cells of the tumor, while only a few scattered macrophages were found at the periphery of epithelial sectors within the same tumor (Fig 3D – top, Supplementary Fig 6E). Most importantly, the latter macrophages did not stain positive for Arginase1 (Fig 3D – middle), indicating that the juxtaposition of macrophages with mesenchymal but not epithelial cells within a tumor was likely to be crucial for regulating their immunosuppressive function. These data suggest that the proportion of mesenchymal cells dictates the numbers and types of suppressive immune cells recruited to the tumor.
Having observed substantial differences in T-cells infiltrating the primary tumors arising from implanted epithelial versus mesenchymal carcinoma cells, we asked whether these tumors would differ in their susceptibility to checkpoint blockade therapy (10, 11). While epithelial tumor bearing mice showed a complete response to anti-CTLA4 immunotherapy, mesenchymal-tumor bearing mice were refractory to treatment (Fig 4A, B). Interestingly, tumors arising from 1:1 or 9:1 mixtures of epithelial and mesenchymal cells respectively, also did not respond to anti-CTLA4 treatment (Fig 4C). Additionally, anti-CTLA4 treatment did not alter the numbers or EMT-features of epithelial and mesenchymal cells within mixed tumors (Supplementary Fig 7A, B). Hence, these data suggest that the immunosuppressive tumor microenvironment of mesenchymal tumors made them resistant to elimination via check-point blockade therapy (12, 13). Moreover, the presence of even a small minority of mesenchymal carcinoma cells resulted in the loss of responsiveness of tumors to checkpoint blockade therapy.
Fig 4. Response of Epithelial and Mesenchymal tumors to immunotherapy.
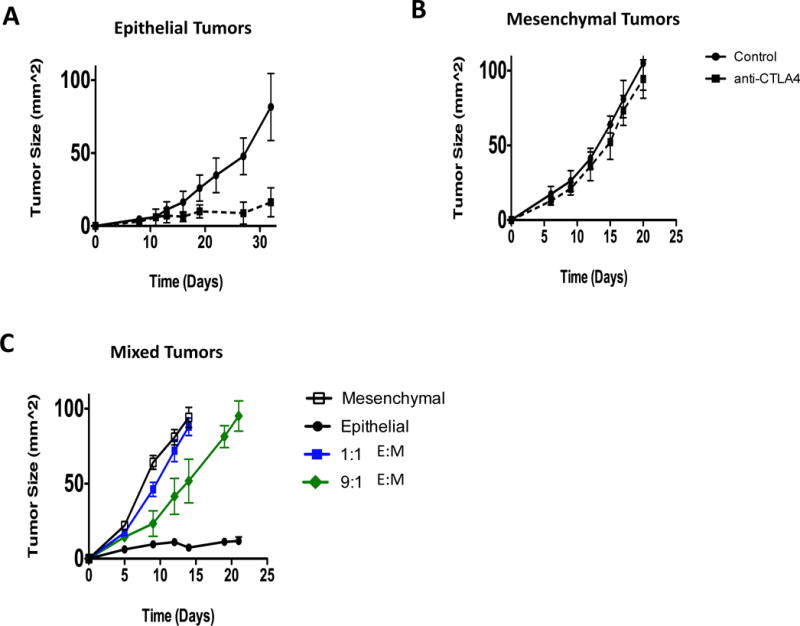
(A, B, C) Average tumor growth of mice that did or did not receive anti-CTLA4 therapy. Data represent three independent experiments.
DISCUSSION
Several studies have demonstrated an association between the EMT and immunosuppression in other cancer types following forced expression of EMT-TFs (12). However, high-level expression of EMT-TFs elicits functional changes in carcinoma cells that do not recapitulate the changes occurring when these EMT-TFs are expressed under cell-physiologic control. (7). This explains why we undertook the presently described work to determine whether an association between the EMT and immunosuppression does indeed occur when EMT-TFs are expressed under physiologic control using cell lines that were established directly from tumors arising in MMTV-PyMT mice.
Mesenchymal cell lines and the tumors arising from them expressed markedly lower levels of cell-surface MHC-I compared to epithelial cell lines and their corresponding tumors, raising the question of whether such MHC-I-low expression levels render tumor cells vulnerable to NK-mediated killing (14). Indeed, we did observe a higher percentage of NK cells infiltrating MHC-I-low tumors arising from mesenchymal cell lines relative to MHC-I-high tumors arising from epithelial cell lines (Supp Fig 1E and 2E). Importantly, however, these tumors were not eliminated despite the presence of such NK cells. We attribute this outcome to the actions of other immunosuppressive subsets, such as Tregs, which were present abundantly in mesenchymal tumors and are known to dampen NK function (4, 15).
The presence of cytotoxic lymphocytes in MHC-I expressing epithelial tumors is likely to make them more susceptible to elimination via checkpoint blockade therapy. Indeed, patients with tumor-infiltrating lymphocytes (TILs) generally have a relatively good prognosis and are thought to generate favorable responses to checkpoint blockade treatment regimes (10, 16). In contrast, the strongly immunosuppressive stroma of mesenchymal tumors may render them refractory to such therapy (13).
Our studies demonstrate that epithelial and mesenchymal carcinomas differ in their susceptibility to immune attack. Although we have yet to determine the precise molecular mechanism(s) by which EMT-induced immunosuppression takes place, we suggest that this may likely occur by several mechanisms. Among these are dysregulated expression of immunomodulatory markers associated with immune-evasion and alterations in cytokine and chemokine secretion patterns, which may act as the first step that triggers a cascade of events that ultimately culminate in immunosuppression. All of these immunoevasive maneuvers utilized by carcinoma cells could act in concerted fashions to (i) recruit specific immunosuppressive cells to the tumor microenvironment and/or (ii) reprogram pre-existing cells to adopt an immunosuppressive phenotype.
Supplementary Material
Acknowledgments
We thank Richard A. Goldsby and Barbara A. Osborne for critical reading of the manuscript. We thank Brian Bierie and Xin Ye for cells and all other members of the Weinberg Lab for helpful discussion. We thank the Flow Cytometry Core facility at the Whitehead Institute for assistance with cell sorting and flow cytometry analysis; the Keck Imaging facility at the Whitehead Institute for assistance with microscopy. We thank Roderick Bronson for histopathological analysis of tumor sections and the Histology Facility at the Koch Institute/MIT for tissue sectioning.
Financial support: R.A. Weinberg received support from the NIH (P01 CA080111), Breast Cancer Research Foundation, Samuel Waxman Cancer Research Foundation and Ludwig Center for Molecular Oncology. H. L. Ploegh received support from NIH (R01-AI087879-06, R01-GM100518-04), and the Lustgarten Foundation. A.Dongre was supported by the Ludwig Fund for Cancer Research. M. Rashidian was supported by the Cancer Research Institute.
References
- 1.Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21(3):309–22. doi: 10.1016/j.ccr.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 2.Kerkar SP, Restifo NP. Cellular Constituents of Immune Escape within the Tumor Microenvironment. Cancer Res. 2012;72(13):3125–30. doi: 10.1158/0008-5472.CAN-11-4094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scheel C, Eaton EN, Li SH, Chaffer CL, Reinhardt F, Kah KJ, et al. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell. 2011;145(6):926–40. doi: 10.1016/j.cell.2011.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crane CA, Han SJ, Barry JJ, Ahn BJ, Lanier LL, Parsa AT. TGF-beta downregulates the activating receptor NKG2D on NK cells and CD8+ T cells in glioma patients. Neuro Oncol. 2010;12(1):7–13. doi: 10.1093/neuonc/nop009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Viel S, Marcais A, Guimaraes FS, Loftus R, Rabilloud J, Grau M, et al. TGF-beta inhibits the activation and functions of NK cells by repressing the mTOR pathway. Sci Signal. 2016;9(415):ra19. doi: 10.1126/scisignal.aad1884. [DOI] [PubMed] [Google Scholar]
- 6.Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29(34):4741–51. doi: 10.1038/onc.2010.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ye X, Tam WL, Shibue T, Kaygusuz Y, Reinhardt F, Eaton EN, et al. Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nature. 2015;525(7568):256+. doi: 10.1038/nature14897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guo W, Keckesova Z, Donaher JL, Shibue T, Tischler V, Reinhardt F, et al. Slug and Sox9 cooperatively determine the mammary stem cell state. Cell. 2012;148(5):1015–28. doi: 10.1016/j.cell.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Cock JM, Shibue T, Dongre A, Keckesova Z, Reinhardt F, Weinberg RA. Inflammation triggers Zeb1-dependent escape from tumor latency. Cancer Res. 2016 doi: 10.1158/0008-5472.CAN-16-0608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–64. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348(6230):56–61. doi: 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- 12.Kudo-Saito C, Shirako H, Takeuchi T, Kawakami Y. Cancer Metastasis Is Accelerated through Immunosuppression during Snail-induced EMT of Cancer Cells. Cancer Cell. 2009;15(3):195–206. doi: 10.1016/j.ccr.2009.01.023. [DOI] [PubMed] [Google Scholar]
- 13.De Henau O, Rausch M, Winkler D, Campesato LF, Liu C, Cymerman DH, et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kgamma in myeloid cells. Nature. 2016;539(7629):443–7. doi: 10.1038/nature20554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Waldhauer I, Steinle A. NK cells and cancer immunosurveillance. Oncogene. 2008;27(45):5932–43. doi: 10.1038/onc.2008.267. [DOI] [PubMed] [Google Scholar]
- 15.Rook AH, Kehrl JH, Wakefield LM, Roberts AB, Sporn MB, Burlington DB, et al. Effects of transforming growth factor beta on the functions of natural killer cells: depressed cytolytic activity and blunting of interferon responsiveness. J Immunol. 1986;136(10):3916–20. [PubMed] [Google Scholar]
- 16.Callahan MK, Wolchok JD, Allison JP. Anti-CTLA-4 antibody therapy: immune monitoring during clinical development of a novel immunotherapy. Seminars in oncology. 2010;37(5):473–84. doi: 10.1053/j.seminoncol.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.