

Cardiac fibroblasts are essential for beneficial myocardial healing but also cause detrimental adverse remodeling following myocardial infarction. The mechanical properties of the infarcted myocardium and border regions display temporal and spatial characteristics that regulate different aspects of the profibrotic cardiac fibroblast phenotypes.
Abstract
Cardiac fibrosis is a serious condition currently lacking effective treatments. It occurs as a result of cardiac fibroblast (CFB) activation and differentiation into myofibroblasts, characterized by proliferation, extracellular matrix (ECM) production and stiffening, and contraction due to the expression of smooth muscle α-actin. The mechanical properties of myocardium change regionally and over time after myocardial infarction (MI). Although mechanical cues are known to activate CFBs, it is unclear which specific mechanical stimuli regulate which specific phenotypic trait; thus we investigated these relationships using three in vitro models of CFB mechanical activation and found that 1) paracrine signaling from stretched cardiomyocytes induces CFB proliferation under mechanical conditions similar to those of the infarct border region; 2) direct stretch of CFBs mimicking the mechanical environment of the infarct region induces a synthetic phenotype with elevated ECM production; and 3) progressive matrix stiffening, modeling the mechanical effects of infarct scar maturation, causes smooth muscle α-actin fiber formation, up-regulation of collagen I, and down-regulation of collagen III. These findings suggest that myocyte stretch, fibroblast stretch, and matrix stiffening following MI may separately regulate different profibrotic traits of activated CFBs.
INTRODUCTION
Cardiac fibroblasts (CFBs) are responsible for maintaining extracellular matrix (ECM) composition and organization in the heart wall. They are central mediators of the fibrosis that develops in many forms of cardiac disease, notably myocardial infarction (MI). In response to injury, the CFB becomes activated and acquires a profibrotic phenotype commonly referred to as a myofibroblast (Hinz, 2007), characterized by proliferative activity, excessive ECM production, and contractile function due to expression of smooth muscle α-actin (SMA). However, it is becoming increasingly clear that activated CFBs display multiple overlapping phenotypes depending on their spatial location and the stage of fibrosis (Tomasek et al., 2002; Hinz, 2007; van Putten et al., 2016). It has been established that the altered mechanical properties of the myocardium associated with cardiac diseases activate CFBs (van Putten et al., 2016). However, similar to the heterogeneity of CFB phenotypes, the mechanical alterations occurring in and adjacent to the infarct are regionally variable and change during the acute and chronic phases of infarction and postinfarct remodeling. Therefore teasing out which mechanical cues regulate specific phenotypic traits may identify novel targets that could prevent the adverse progression of cardiac fibrosis.
CFB mechanosensitivity is demonstrated by CFBs’ rapid transformation to a profibrotic myofibroblast phenotype when they are grown on plastic tissue culture dishes or polydimethylsiloxane (PDMS) membranes used to impose stretch; these materials have stiffnesses five and three orders of magnitude, respectively, greater than that of cardiac muscle (Berry et al., 2006; Engler et al., 2008). This may explain conflicting reports of both increased and decreased myofibroblast differentiation in response to mechanical stress (Wang et al., 2003; Papakrivopoulou et al., 2004; Husse et al., 2007; Waxman et al., 2012; Guo et al., 2013). Thus development of cell culture models that better imitate the in vivo mechanical environment and the changes postinfarction are needed (Spinale et al., 2016). The use of hydrogels to manipulate culture substrate stiffness has helped in vitro research on fibroblast function (Yeung et al., 2005; Goffin et al., 2006; Li et al., 2007; Kloxin et al., 2010; Olsen et al., 2011; Liu et al., 2015), but few studies have investigated the interplay of mechanical stimuli, especially in the context of CFB phenotypic plasticity. Thus all of the experiments here were performed on soft gels with physiological stiffness in an attempt to discriminate the differential effects of substrate stiffness and other applied mechanical stimuli, for example, biaxial stretch.
To formulate hypotheses concerning which mechanical forces drive specific CFB responses, it is important to understand the course of events triggered by MI, which roughly equates to acute scar formation and chronic scar remodeling. In the healthy heart, quiescent CFBs are probably at least partially protected against large changes in mechanical stress by their physical integration in the structurally stable ECM network. During acute MI, cardiomyocytes (CMs) in the ischemic myocardium rapidly cease generating active tension, while contraction of the surrounding functional myocardium gives rise to a functional border zone of perfused and viable tissue that is nonetheless dyskinetic or hypokinetic (Mazhari et al., 2000). The border zone is also associated with CFB proliferation and fibrosis (Virag and Murry, 2003), although the exact mechanism by which fibrosis occurs is unclear; CFBs may also be less protected from increased mechanical stress owing to rapid degradation and acute loss of native ECM stiffness. This scenario likely creates excessive stretch during systole, causing CFBs to acquire a synthetic phenotype producing matricellular proteins and structural ECM proteins and inducing collagen cross-linking that increases myocardial stiffness (Lopez et al., 2012). In later stages of scar remodeling, CFB differentiation into contractile myofibroblasts and ECM maturation cause scar shrinkage. In the normal adaptive response to scar healing, myofibroblasts undergo apoptosis as ECM tension of the scar is restored and takes over the mechanical load. In conditions in which mechanical load continues to be elevated, such as in the left ventricles of patients with chronic hypertension, persistent myofibroblast activity and progression of myocardial fibrosis is observed (Creemers and Pinto, 2011). The objective of this study was to investigate the distinct effects and relative contributions of different mechanical stimuli on CFB profibrotic phenotypes. For that, we developed three in vitro models that mimic the mechanical cues of MI. On the basis of known regional and temporal CFB responses, we hypothesized that 1) paracrine signaling from stretched CMs induces CFB proliferation; 2) direct stretch of CFBs induces ECM production; and 3) matrix stiffening promotes a contractile myofibroblast phenotype.
RESULTS
Matrix stiffness affects CFB phenotype and function
For determination of the matrix stiffness required for maintaining a quiescent phenotype in vitro, CFBs were grown on hyaluronic acid (HA) gels with stiffnesses corresponding to the range of intact myocardial tissue. CFBs grown on HA gels with a Young’s elastic modulus of 3 kPa had low or no expression of the myofibroblast marker SMA (Figure 1A). A substrate stiffness of 8 kPa, comparable to that of healthy myocardium (Berry et al., 2006; Engler et al., 2008), was sufficient for formation of SMA fibers. CFBs displayed prominent SMA fibers on 50 kPa HA gels corresponding to the myofibroblast phenotype observed in fibrotic myocardium, which has a stiffness of 20–100 kPa (Berry et al., 2006; Engler et al., 2008). Focal adhesion size and location, as determined from vinculin staining, were altered according to substrate stiffness, transitioning from small peripheral adhesions on 3 kPa substrates to large “supermature” focal adhesion structures that were evenly distributed throughout the cell membrane on 50 kPa substrates (Figure 1B). Although adhesion changes were more continuous, CFB area was markedly higher at and above 8 kPa than for 3 kPa HA gels (Figure 1C). The proliferation rates were similar for all stiffnesses (Figure 1D). We observed the lowest mRNA expression for ECM remodeling genes in CFBs on 8 kPa HA gels (Figure 1E), with the notable exception of thrombospondin 1 (THBS1). These data indicated that culturing CFBs on HA gels with stiffness of 3 and 8 kPa maintained the quiescent phenotype; these stiffnesses were therefore used in the subsequent in vitro assays. Because we also used polyacrylamide (PA) gels in this study, phenotyping experiments for SMA fiber incorporation and proliferation were repeated for CFBs on PA gels and showed no significant difference from experiments using HA gels (Supplemental Figure 1).
FIGURE 1:
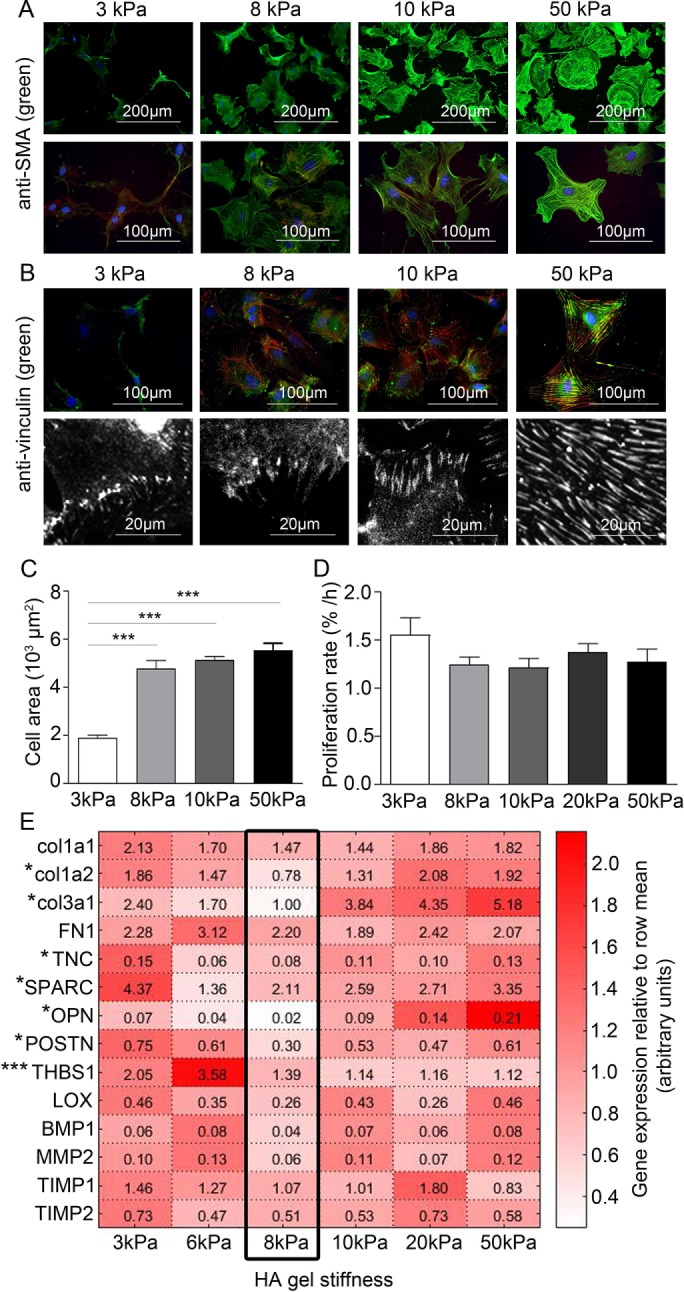
Maintaining a quiescent CFB phenotype by controlling substrate stiffness. Immunofluorescence staining for (A) SMA (green) and (B) vinculin. F-actin and nuclei were stained with phalloidin (red) and DAPI (blue), respectively. (C) Cell area and (D) proliferation rate (% of proliferating cells per hour) of CFBs plated on gels of 3, 8, 10, 20, and 50 kPa HA gels. One-way ANOVA with Tukey’s post hoc test was used in C and D. N = 6 and 4, respectively. (E) Heat map showing relative mRNA expression levels of collagen (col) 1a1, 1a2, and 3a1, fibronectin (FN1), TNC, SPARC, OPN, POSTN, THBS1, LOX, BMP1, matrix metalloproteinase (MMP) 2, TIMP1, and TIMP2 normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) in CFBs plated on HA gels with 3, 6, 8, 10, 20, and 50 kPa stiffness. Expression across genes was lowest at 8 kPa stiffness, as indicated by the black frame. Values in each box represent raw 2^(-delta Ct) values for comparison of expression levels among genes. *Denotes significant effect of stiffness as tested by one-way ANOVA and Kruskal-Wallis tests for parametric and nonparametric data, respectively. *P < 0.05, ***P < 0.001, N = 8–12.
Paracrine signaling from stretched CMs induces proliferation of CFBs
CMs and CFBs were first cultured on separate substrates with common culture media (Figure 2A) to assess paracrine communication during CM stretch. CMs were subjected to nonequibiaxial biaxial (14% longitudinal, 3.6% transverse) static stretch using a custom device mimicking strains in the border region post-MI in vivo (Camelliti et al., 2006). Proliferation of the nonstretched CFBs markedly increased in the presence of CMs and was further enhanced by their stretch (Figure 2B). Whereas CM stretch was necessary to induce proliferation of grown on 8 kPa substrates, CFBs on 3 kPa substrates also responded to paracrine signaling from nonstretched CMs (Supplemental Figure 2A), indicating enhanced sensitivity of CFBs to paracrine signals. CFB proliferation rates were similar on 3 and 8 kPa substrates when cultured with stretched CMs, suggesting that responses to stretch-induced paracrine signaling are independent of substrate stiffness. The effect on CFB proliferation was not significant when CFBs were treated with conditioned media from stretched CMs, that is, without the coculture device, possibly indicating that paracrine mediators have short half-lives (Figure 2C). A coculture media screen indicated significant presence of a subset of cytokines (Supplemental Figure 2B), and among these, mRNA of colony-stimulating factor 1 (CSF-1) and platelet-derived growth factor B (PDGF-B), a growth factor known to induce proliferation of mesenchymal cells (Antoniades et al., 1979), were up-regulated in stretched CMs (Figure 2D). Although fibroblast growth factor 2 (FGF2) mRNA increased with 24 h stretch, it was reduced at the earlier time point, suggesting that this factor is not the initiator of CFB proliferation. Stimulation of CFBs on 8 kPa substrates with recombinant CSF-1 or PDGF-B induced CFB proliferation, and this response was enhanced by costimulation with both factors (Figure 2E). Finally, treatment with PDGF-B (AG-1295) and CSF-1 (GW-2580) receptor antagonists prevented the increase in CFB proliferation observed in cocultures with stretched CMs, suggesting PDGF-B and CSF-1 to be central to the paracrine signaling induced by stretch. Treatment with antagonists also reduced the proliferative response of CFBs on 8 kPa PA gels (Figure 2F), indicating a basic level of PDGF-B and CSF-1 receptor activity. This effect was abolished when CFBs were cocultured with nonstretched CMs. Despite effects on proliferation, paracrine signaling from stretched CMs had no effect on SMA fiber formation or fibronectin (FN) expression (Supplemental Figure 2, C–E). There was a slight reduction in cell area (Figure 2G), possibly as a result of the increased proliferation or morphological changes such as enhanced cell protrusions (Figure 2H), which may reflect cell responses to chemotactic agents in the media. Overall these data are suggestive of a direct effect of CM stretch on CFB proliferation via paracrine CSF-1 and PDGF-B signaling.
FIGURE 2:
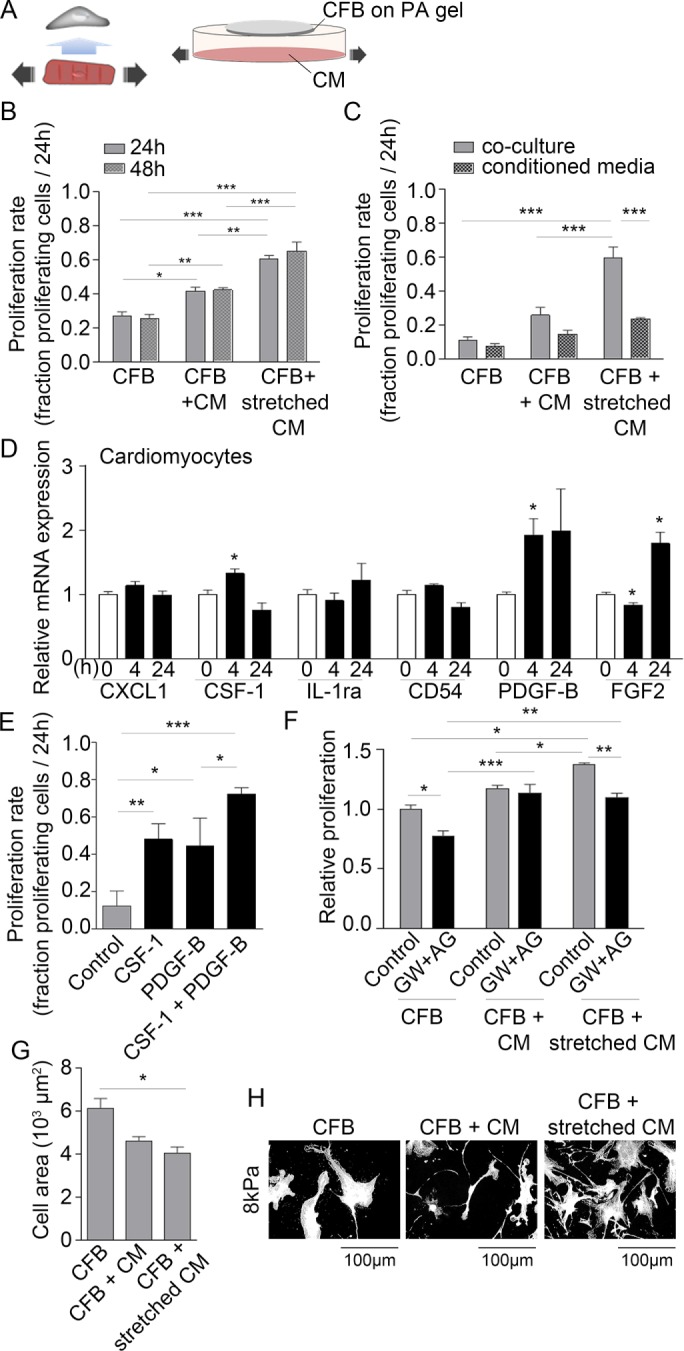
Paracrine signaling from stretched CMs induces CFB proliferation. (A) Schematic illustration of the principles of the paracrine signaling model. (B) Proliferation rate of CFB cultures alone or in coculture with nonstretched CMs or stretched CMs. CFBs were plated on PA gels with 8 kPa stiffness. Two-way ANOVA showed significant effect of culture type*** and Tukey’s post hoc test as indicated in figures. N = 3–6. (C) Proliferation rate of CFBs in CM coculture or treated with conditioned media from cocultures. Two-way ANOVA showed an effect of culture type***, conditioned media***, and a significant interaction**. Tukey’s post hoc test results are indicated in figures. N = 3. (D) Relative expression of chemokine (C-X-C motif) ligand 1 (CXCL1), CSF-1, interleukin-1 receptor antagonist (IL-1ra), cluster of differentiation 54 (CD54), PDGF-B, and FGF2 mRNA normalized to GAPDH mRNA in CMs stretched for 4 and 24 h. *Denotes significant difference from 0 h control as determined by Student’s t test. N = 9 (control) and 4 (stretch). (E) Proliferation rate following 24 h stimulation with recombinant CSF-1 and/or PDGF-B. One-way ANOVA showed significant effect of stimulation*** and Tukey’s post hoc test results as indicated. N = 3. (F) Relative proliferation rate of CFBs in cocultures in the presence of PDGF-B and CSF-1 receptor antagonists (AG and GW, respectively). Two-way ANOVA showed an effect of culture type*** and blockers*** as well as significant interaction* between the two. Tukey’s post hoc test results are indicated in the figure. N = 3. Cell area (G) and morphology (H) of CFBs on 8 kPa alone or in coculture with nonstretched and stretched CMs. One-way ANOVA showed significant effect of culture type* on cell area and Tukey’s post hoc test results as indicated in figure. *P < 0.05, **P < 0.01, ***P < 0.001.
Stretching CFBs on soft substrates induces SMA and ECM expression
To stretch CFBs grown on substrates of physiological relevant stiffness and thus maintained as inactivated quiescent fibroblasts, 3 and 8 kPa PA gels were attached to PDMS membranes (Figure 3A) using a previously published protocol (Simmons et al., 2013). Static 3% or 6% equibiaxial stretch (corresponding to a 10% and 20% cell area change, respectively; Lee et al., 1999) was applied using a custom circular stretch device (Camelliti et al., 2006). Although static stretch of the PA gels was sustained for 24 h, cells returned to their original size well within the 24 h of applied stretch (Supplemental Figure 3). Nevertheless, this static stretch induced massive up-regulation of mRNA for collagen 1a1, collagen 1a2, collagen 3a1, and FN (Figure 3B) and protein for FN (Figure 3C). The effect was more prominent in CFBs on 8 kPa PA gels and independent of stretch amplitude. Other ECM-remodeling genes were also altered by 3% stretch (Figure 3D), with tenascin C (TNC) being the only oppositely expressed gene between 3 and 8 kPa substrates. Stretch also induced opposing proliferation responses depending on substrate stiffness (Figure 3E).
FIGURE 3:

Stretch promotes ECM remodeling by CFBs. (A) Schematic illustration of the principles of the stretch model. (B) Collagen (col) 1a1, 1a2, and 3a1 and fibronectin (FN1) mRNA normalized to GAPDH mRNA in CFBs on 3 and 8 kPa gels subjected to 0%, 3%, and 6% stretch (x-axis). One-way ANOVA was used to test significant effect of stretch with Tukey’s post hoc tests for specific comparisons. Statistical results are indicated in the figure. N = 4 (control) and 12 (stretch). (C) Quantification and immunoblot of FN protein normalized to total protein determined by Ponceau staining. N = 4 (3 kPa) and 8 for (8 kPa). (D) Fold difference in LOX, BMP-1, OPN, POSTN, TNC, SPARC, THBS1, matrix metalloproteinase (MMP) 2, and TIMP 1 and 2 mRNA expression normalized to GAPDH mRNA. Significance was determined by multiple Student’s t tests corrected for multiple comparisons using Holm-Sidak’s post hoc test. p Values displayed in D are the uncorrected p values from the t test. N = 4 (control) and 12 (stretch). (E) Relative proliferation of CFB controls (C) or with 3% stretch (S). N = 3. (F) SMA mRNA normalized to GAPDH mRNA. N = 4 (control) and 12 (stretch). (G) Immunofluorescence staining for SMA (green) and DAPI staining for nuclei (blue). (H) Immunoblot for total and phophorylated focal adhesion kinase (FAK and pFAK, respectively) normalized to GAPDH. N = 4. Student’s t test was used to determine significant changes for C, E, F, and H. *P < 0.05, **P < 0.01, ***P < 0.001; n.s., nonsignificant.
SMA mRNA levels were markedly substrate dependent, with 300- and 8-fold increases for the 3 and 8 kPa PA gels, respectively (Figure 3F). This was also reflected in immunostaining for SMA, wherein SMA intensity was clearly increased in 3 kPa CFB cultures after stretch, albeit not incorporated into fibers (Figure 3G). Although development of SMA fibers was not induced by stretch, cell morphology of 8 kPa CFB cultures was altered by stretch, displaying more elongated cells, which suggests that cells remodel and readjust cell size during the 24 h static stretch.
Because vinculin staining (Figure 1) showed differences in focal adhesion size for CFBs on 3 and 8 kPa substrates, we measured activation (by phosphorylation) of focal adhesion kinase (FAK). FAK was phosphorylated by 24 h static stretch only in CFBs on 8 kPa, suggesting active mechanotransduction by integrins (Figure 3H).
Transforming growth factor β (TGFβ) is the most well-known driver of myofibroblast differentiation (Biernacka et al., 2011), and mRNA levels were increased by stretch only in CFBs on 3 kPa PA gels (Supplemental Figure 4A). However, this was not accompanied by TGFβ activity 24 h after static stretch (Supplemental Figure 4B), and TGFβ activity was reduced by stretch for CFBs on 8 kPa PA gels. TGFβ is known to regulate expression of the myofibroblast marker gene FN extradomain A (EDA), which also was increased in CFBs on 3 kPa but reduced in CFBs on 8 kPa PA gels following stretch (Supplemental Figure 4C), supporting that EDA may be downstream of TGFβ signaling and not independently regulated by stretch. Accompanying higher FN protein staining (Supplemental Figure 4D), mRNA levels of αv, a FN receptor and TGFβ-activating integrin, were higher in CFBs on 3 kPa compared with 8 kPa substrates (Supplemental Figure 4E), suggesting that these CFBs are more capable of activating TGFβ. Collectively these data suggest an interplay between substrate stiffness and stretch to induce a synthetic phenotype, but in a limited set of niche conditions.
Matrix stiffening from physiological to pathological stiffness induces up-regulation of collagen I, down-regulation of collagen III, and SMA fiber formation
Because stretch effects depend on local stiffness, it is important to note that stiffness itself depends on ECM production and cross-linking, and this can increase during scar formation postinfarct. For determination of how dynamic changes in stiffness could affect CFBs, cells were grown on soft (3 or 8 kPa) HA gels for 5 d before substrates were stiffened to 30 kPa (Figure 4A); Although this treatment is UV based, we did not observe p53 immunostaining and viability changes suggestive of cell damage (Supplemental Figure 5). HA gels were stiffened from 3 kPa (where CFBs have no SMA fibers) to 30 kPa to detect changes in SMA expression and fiber formation. Stiffening caused massive cell spreading, increased SMA intensity, and SMA fiber formation (Figure 4B). Similar to CFBs on 10 kPa HA gels (see Figure 1A), SMA fibers were present at 8 kPa (Supplemental Figure 6A), and stiffening from 8 to 30 kPa did not induce additional presence of SMA fibers or cell spreading. These data suggest that the threshold for SMA fiber formation in two-dimensional cultures is somewhere in the range of >3 kPa to <8 kPa. TGFβ and EDA mRNAs were unchanged after stiffening from 3 to 30 kPa and 8 to 30 kPa (Supplemental Figure 6B), indicating that the effect on SMA was a direct effect of stiffening and not downstream of TGFβ signaling. Stiffening from 8 to 30 kPa increased collagen I but reduced collagen III mRNA as measured 24 h after stiffening (Figure 4C), suggesting differential transcriptional regulation of these two genes. Although FN mRNA expression was not was up-regulated in CFBs 24 h after stiffening, immunofluorescence staining intensity of FN protein increased 48 h after stiffening from 8 to 30 kPa (Figure 4D). No change was observed for lysyl oxidase (LOX) mRNA (Figure 4C) or activity (Supplemental Figure 6C), nor was there any change in other ECM proteins after stiffening from 8 to 30 kPa (Supplemental Figure 6D). Interestingly, osteopontin (OPN) mRNA was reduced from 8 to 30 kPa following substrate stiffening (Figure 4C), even though this gene has previously been found to be up-regulated in response to mechanical stress (Herum et al., 2015). Proliferation was unchanged by stiffening from 8 to 30 kPa (Supplemental Figure 6E). Taken together, these data specifically imply that SMA expression and its assembly are affected by temporal changes in passive niche properties, for example, stiffness.
FIGURE 4:
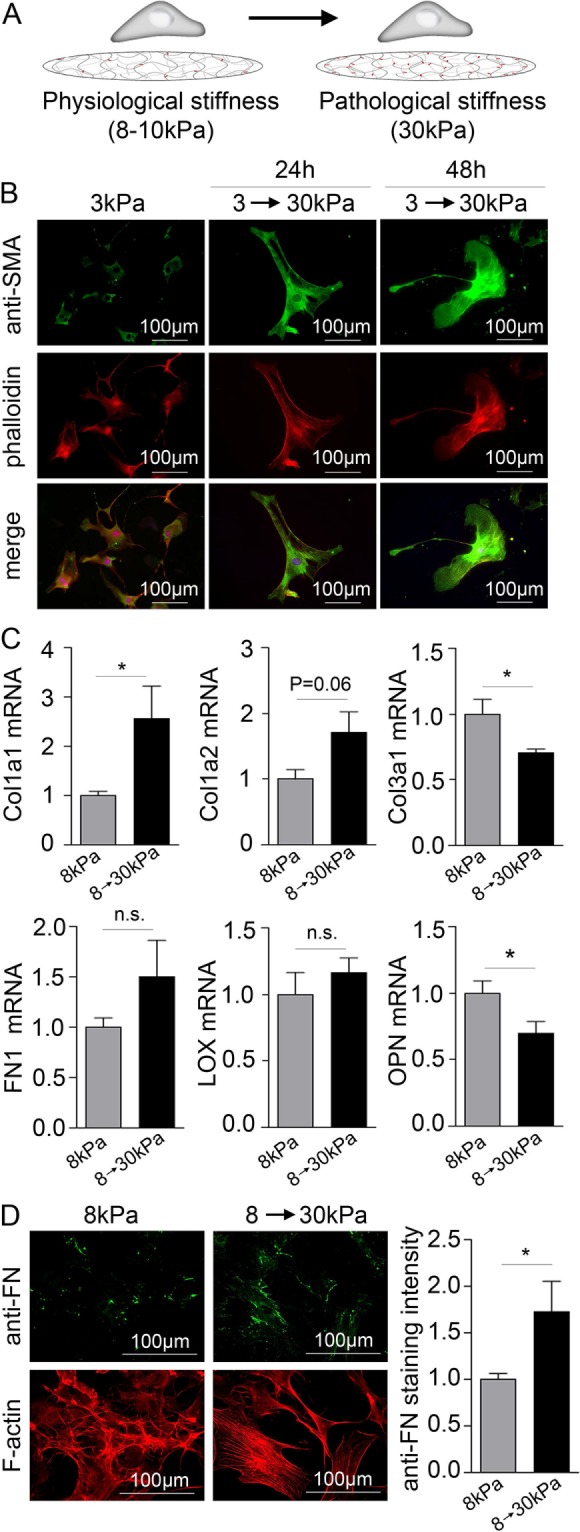
Matrix stiffening causes cell spreading, SMA fiber formation, up-regulation of collagen I, and down-regulation of collagen III. (A) Schematic illustration of the principles of the stiffening model. (B) immunofluorescence staining for SMA (green). F-actin and nuclei were stained with phalloidin (red) and DAPI (blue), respectively. (C) Collagen (col) 1a1, 1a2, and 3a1, fibronectin (FN1), LOX, and OPN mRNA normalized to GAPDH mRNA in CFBs on 8 kPa gels stiffened to 30 kPa. N = 4 (control) and 12 (stiffened). (D) Immunofluorescence staining for FN (green) and quantification of staining intensity 48 h after gels stiffening. N = 8. F-actin was stained with phalloidin (red). Student’s t tests were applied to determine significant changes. *P < 0.05; n.s., nonsignificant.
DISCUSSION
In these studies we found the following: 1) Paracrine signaling from stretched CMs induces proliferation of CFBs, consistent with the high proliferative activity of fibroblasts in the functional border region after acute MI (Figure 5, point 1). 2) ECM production by CFBs in response to stretch is dependent upon matrix stiffness. This suggests a mechanism for differential regulation of ECM across the infarct and peri-infarct regions during the acute phase when ECM stiffness is decreased due to rapid degradation of ECM (Figure 5, point 2a) and the subsequent remodeling phase when ECM stiffness increases with the accumulation of newly synthesized ECM (Figure 5, point 2b). 3) Matrix stiffening induces massive cell spreading and SMA fiber formation as hypothesized. We also report the novel finding that substrate stiffening promotes collagen I expression but inhibits collagen III expression (Figure 5, point 3). This response imitates the late stages of infarct healing when ECM stiffening dominates the mechanical environment of the infarct scar. Figure 5 summarizes the results in the context of the mechanics of myocardium following MI.
FIGURE 5:

Proposed model of mechanical regulation of CFBs following MI. 1) Paracrine signaling involving PDGF-B and CSF-1 from stretched CMs in the border region leads to proliferation of CFBs. 2a) Stretch of the infarct region during the acute phase after MI (ECM stiffness ∼3 kPa) promotes collagen I, LOX, BMP-1, POSTN, SPARC, THBS1, TIMP1 and 2, while inhibiting TNC expression by CFBs. Proliferation is increased during this phase. 2b) Stretch of the infarct region during the remodeling phase (ECM stiffness ∼8 kPa) also up-regulates collagen I in addition to collagen III, FN, and TNC, while it down-regulates the matricellular protein OPN. Proliferation is decreased during this phase. Stretch causes up-regulation of SMA mRNA during both the acute and remodeling phases; however, incorporation into stress fibers requires an accompanying increase in matrix stiffness. 3) At late stages of infarct healing, ECM stiffening to pathological stiffnesses (∼30 kPa) dominates the mechanical environment. This promotes a contractile CFB phenotype due to SMA fiber formation that has elevated, albeit dampened, collagen I production compared with that of stretched CFBs. Collagen III and OPN expression is reduced, supporting scar maturation and healing. However, continuous stretch of CFBs at this stage will cause persistent ECM production and thus development of pathological fibrosis.
Matrix stiffness regulates fiber formation, while stretch regulates expression of SMA
For the first time, we show here the effect of matrix stiffening on CFB phenotype, causing massive cell spreading and incorporation of SMA into stress fibers. This is in agreement with reports from other fibroblast-like cells, including hepatic stellate cells (Guvendiren et al., 2014; Caliari et al., 2016), mesenchymal stem cells (Guvendiren and Burdick, 2012), and the NiH3T3 fibroblast cell line (Ondeck and Engler, 2016). Although equibiaxial stretch also caused a dramatic increase in SMA mRNA, it did not promote SMA stress fiber formation, suggesting that substrate stiffness is the dominant determining mechanical factor for the assembly of SMA into fibers and subsequent contraction of the infarct scar. From a mechanical standpoint, this is consistent with the cell tensegrity theory (Ingber, 2008). Interestingly, Cui and colleagues showed that cyclic stretch of CFBs on soft gels did induce F-actin stress fibers even in the absence of matrix stiffening (Cui et al., 2015). Whether this also applied to SMA fiber formation was not studied. The massive up-regulation of SMA mRNA in stretched CFBs on 3 kPa PA gels might partly be due to increased TGFβ signaling, because TGFβ and its target gene EDA also exhibited increased expression following stretch. This was not the case for CFBs on 8 kPa substrates, suggesting a mechanotransduction pathway independent of TGFβ to account for this change, for example, mechanotransduction via the transmembrane proteoglycan syndecan-4 has been shown to have such an effect (Herum et al., 2013).
Mechanical cues regulate collagen differentially
Enhanced production of structural ECM proteins is essential for development of fibrosis, with the amount and composition of collagen being of particular importance due to collagen’s central role in myocardial stiffening. Stretch (3%, 8 kPa substrates) caused massive up-regulation of collagen III and FN, thus confirming our previous results from CFBs stretched on PDMS substrates (with an elastic modulus in the MPa range) (Lee et al., 1999). However, responses by CFBs on soft substrates were 5- and 2.5-fold higher for collagen III and FN, respectively, suggesting a more mechanosensitive CFB phenotype in this setting. Also, increasing stretch to 6% did not reduce FN mRNA as previously reported for standard PDMS membranes (Lee et al., 1999), again supporting different responses to stretch depending on substrate stiffness. Both stretch and substrate stiffening promoted collagen I expression, although the response was 10 times higher for stretch in the case of col1a1. Interestingly, and in contrast to stretch, collagen III was down-regulated in response to stiffening. Mass spectrometry has provided insight about the ECM composition of infarct regions (Sullivan et al., 2014), showing elevated collagen I and reduced collagen III levels in the mature scar compared with earlier stages of remodeling. Along these lines, the collagen I and III ratio has been suggested to influence myocardial stiffness, with a high collagen I:III ratio being associated with a less compliant ventricle (Mukherjee and Sen, 1990). Other subtypes of collagens, including nonstructural collagens, have also been associated with cardiovascular disease (Rasi et al., 2010; Luther et al., 2012; Skrbic et al., 2015). Determining the mechanoregulation and role of these less-studied collagens may provide valuable information for evaluating their importance in different stages of cardiac fibrosis development.
Collagen maturation is promoted by stretch
Collagen cross-linking is emerging as an essential player in myocardial stiffening and diastolic dysfunction (Kasner et al., 2011; Lopez et al., 2012). mRNAs for the collagen cross-linking enzyme LOX and its activator bone morphogenetic protein 1 (BMP1) were increased by stretch, but no change in LOX mRNA and activity were observed after matrix stiffening, suggesting the absence of a feed-forward loop involving further stiffening by enzymatic collagen cross-linking. In agreement, OPN and periostin (POSTN), which are known to induce LOX expression (Lopez et al., 2013; Herum et al., 2015) and increase collagen cross-linking (Maruhashi et al., 2010), were not up-regulated by substrate stiffening. Thus stretch seems to be the mechanical trigger for enzymatic collagen cross-linking. Indeed LOX has been shown to become rapidly up-regulated before myocardial stiffening in an experimental mouse model of left ventricular pressure overload (Herum et al., 2015). Cross-linked collagen is less susceptible to degradation by the matrix metalloproteinase (MMP) enzymes. This, together with the increase in MMP endogenous inhibitors tissue inhibitor of metalloproteinase (TIMP) 1 and 2 by CFBs on 3 kPa PA gels, supports that stretch promotes collagen maturation and ECM stabilization.
The effect of stretch on matricellular gene expression is dependent on matrix stiffness
Matricellular proteins such as OPN, POSTN, secreted protein acidic and rich in cysteine (SPARC), and THBS1 play important roles in post-MI remodeling and were up-regulated by stretch when cells were cultured on 3 kPa but not 8 kPa PA gels. A stiffness of 3 kPa likely reflects the ECM stiffness in the early phase after an infarct, when a sudden increase in MMP activity (Takahashi et al., 1990) leads to degradation of collagen and thus decreases myocardial stiffness. Indeed, results from experimental animal models of MI and left-ventricular pressure overload generally show a rapid increase of OPN, POSTN, and SPARC at early stages and somewhat of a decline at later stages of ECM remodeling (Schellings et al., 2009; Sullivan et al., 2014; Herum et al., 2015). As new ECM accumulates and remodeling of the infarct progresses, myocardial stiffness will gradually increase. Reflecting the mechanical environment of this stage of remodeling, OPN mRNA was reduced with stretch of CFBs on 8 kPa PA gels. Interestingly, cardiac MMPs have recently been found to cleave OPN, resulting in OPN fragments with distinct biological functions that are associated with fibrosis (Lindsey et al., 2015). Thus a stiffness resembling the healthy myocardium or later stages of post-MI remodeling may exert a negative feedback effect on fibrosis development.
Possible mechanisms for stiffness-specific stretch responses
Although mechanical activation of FAK has been studied extensively (Parsons, 2003; Wong et al., 2012), we here show that stretch-induced activation is stiffness-dependent and could therefore be involved in the stiffness-specific response of CFBs to stretch. Indeed, stiffness has been shown to be a regulator of FAK activity (Carraher and Schwarzbauer, 2013) and required for nuclear translocation of the transcriptional cofactor four-and-a-half LIM domains 2, which controls cell proliferation by modulating p21 gene expression (Nakazawa et al., 2016). This is in agreement with the stiffness-dependent inhibition of proliferation observed along with FAK activation. FAK is activated by integrins in response to mechanical stress, and signals downstream to activate mitogen-activated protein kinase signaling (Hynes, 2002), known to induce myofibroblast differentiation and ECM production (Davis and Molkentin, 2014; Lighthouse and Small, 2016; Molkentin et al., 2017), and inhibition of FAK was recently successfully used to prevent fibrosis after MI (Zhang et al., 2017). Thus inhibition of FAK may hold therapeutic potential for treatment of cardiac fibrosis, and it is therefore important to understand which mechanical cues regulate its activity in CFBs. Although we did not see increased TGFβ activity in CFBs on 3 kPa PA gels 24 h after static stretch, there were other indications that TGFβ signaling was enriched only for CFBs on 3 kPa substrates. Collectively our data indicate that the stiffness of the ECM directly surrounding the CFBs seems to influence which profibrotic signaling mechanisms are activated by stretch.
Matrix stiffness dictates the effect of stretch on TNC expression
The matricellular protein TNC distinguished itself from the other matricellular proteins in that it exhibited regulation in opposite directions according to matrix stiffness, being down-regulated on 3 kPa PA gels and up-regulated on 8 kPa PA gels in response to stretch. Interestingly, TNC expression post-MI is seen in regions expected to have mechanical properties resembling the stiffer 8 kPa PA gels, including the viable border zone during acute MI and the central infarct zone during the chronic stages of remodeling, where it is coexpressed and colocalized with FN (Willems et al., 1996). The different domains of TNC have distinct functions that manifest themselves according to the environment. TNC can disrupt CM adhesions at the CM costameres, thereby facilitating tissue reorganization in the border region after MI (Imanaka-Yoshida et al., 2001) while promoting myofibroblast differentiation in the infarct region during the remodeling phase (Duerr et al., 2011). The differential TNC gene expression demonstrated here suggests a critical importance of matrix stiffness in determining the specific mechanosignaling pathways are engaged in response to stretch.
Paracrine signaling from stretched CMs induces CFB proliferation via PDGF and CSF-1
The main effect of paracrine signaling from stretched CMs was enhanced proliferation of CFBs, and this response was independent of substrate stiffness. This resembles the intense proliferative activity of fibroblasts present in the border zone and infiltrating the infarct region (Virag and Murry, 2003). Results from immunohistochemical studies and in situ hybridization experiments suggest that stretched CMs in the infarct border zone produce a variety of cytokines (Gwechenberger et al., 1999) and chemokines (Tarzami et al., 2002). However, the identity and relative contribution of CM-derived inflammatory mediators for the profibrotic response of CFBs is poorly understood. We here identify PDGF-B and CSF-1 as factors up-regulated in CMs by stretch and capable of inducing the observed response on CFB proliferation. Whereas PDGF has been shown to be rapidly up-regulated in the border zone (Zhao et al., 2011) and to promote proliferation of rat atrial fibroblasts (Jiang et al., 2016), studies on the effect of CSF-1 on CFBs are limited, although it has been shown to regulate the survival, proliferation, and differentiation of mononuclear phagocytes (Chitu and Stanley, 2006) and can be expressed by cardiac cells (Hohensinner et al., 2007). Antagonists for PDGF-B and CSF-1 receptors could prevent the increase in proliferation of CFBs induced by paracrine signaling from stretched CMs, supporting a role for PDGF-B and CSF-1 in CM-induced proliferation.
Limitations of the in vitro CFB models
The static stretch is a model of a chronic hemodynamic overload such as an increase in ventricular volume, rather than the effects of phasic cardiac strains. Although cyclic stretching does more closely reflect the contraction cycle of the heart, in vitro models do not recapitulate the frequencies of cyclic strain of the mouse heart (≈10 Hz). Previously published work shows that the low-frequency (0.1 Hz) cyclic strain of CFBs on soft substrates induces greater SMA fiber formation (Cui et al., 2015). This would suggest either the sustainment of a mechanical memory, as was recently demonstrated for microRNA-21 in mesenchymal stem cells (Li et al., 2017), or that the continuous cyclic stretch prevents cells from remodeling back to their original size, as we observed for 24 h static stretch. The fact that we saw dramatic responses to static stretch that mimic the CFB responses observed after MI in vivo supports pathological remodeling being mainly driven by an increase in baseline stretch.
Conclusion and future perspectives
Antifibrotic therapies have failed owing to the pleiotropy of targets and difficulties of regional and temporal targeting of the postinfarcted myocardium. For instance, while blocking well-known fibrotic pathways inhibits excessive fibrosis and diastolic dysfunction, cardiac rupture becomes more prevalent (Daskalopoulos et al., 2012; Hermans et al., 2016). Hence it may be necessary to promote myofibroblast transformation in some regions and certain times (e.g., to stabilize ECM in the infarct zone at early stages of post-MI remodeling), while inhibiting myofibroblast actions in other regions and times (e.g., preventing fibrosis in remote and border regions during the chronic stages of post-MI remodeling). Although many of our observations have been made in other cell types, this study provides new information about profibrotic responses of CFBs to specific and distinct mechanical cues that occur in vivo in the complex mechanical milieu of post-MI remodeling. Further, our results suggest that the engaged mechanotransduction pathways are specific to particular mechanical cues. With these novel experimental tools and established CFB phenotypes in place, differential targeting of mechanosignaling pathways present at different stages and regions of fibrosis development is an obtainable goal. These targets include components of the ECM itself, such as the family of proteoglycans for which accumulating data support a central role in cardiac mechanosignaling and differential expression patterns at different stages of fibrosis development (Melchior-Becker et al., 2011; Engebretsen et al., 2013; Lunde et al., 2016; Melleby et al., 2016). Another approach that holds great promise is the combination of high-sensitivity analyses of entire CFB transcriptomes and advanced systems biology computational models (McCulloch, 2016; Zeigler et al., 2016) to draw connections between changes at the transcriptional level to upstream signaling pathways as well as downstream phenotypic changes. In this way, established signaling pathways may prove to play previously unknown roles in mechanotransduction, and novel profibrotic mechanosensitive signaling pathways may be identified.
MATERIALS AND METHODS
Isolation of adult CFBs
CFBs were isolated from adult CD1 male and female mice (Charles River Labs, San Diego, CA), and all animal procedures followed the Guide for the Care and Use of Laboratory Animals (8th ed.) and were approved by the University of California−San Diego Animal Subjects Committee (protocol no. S01013M). Mice were killed by cervical dislocation after initial anesthesia with “open drop” isoflurane. Hearts were excised and rinsed in Hank’s balanced salt solution (HBSS, cat. no. 14170-120; Life Technologies, Grand Island, NY), and the left ventricles were cut into 8 pieces. These were subjected to predigestion in 0.6 mg/ml trypsin (cat. no. 22715; USB, Cleveland, OH) overnight at 4°C. Hearts were further digested with 1 mg/ml (330 U/ml) collagenase type 2 (cat. no. LS004176; Worthington Biochem, Lakewood, NJ) for 10 min at 37°C and mechanically dissolved by repeated pipetting. Cell solution was passed through a 100 µm cell strainer, after which the collagenase was neutralized by adding fibroblast media consisting of DMEM (cat. no. 11965-092; Life Technologies) supplemented with 10% fetal bovine serum (FBS; cat. no. 16000-044; Life Technologies) and 1% antibacterial/antimicrobial (Sigma, St. Louis, MO). The cell solution was centrifuged at 200 × g for 4 min, after which the supernatant was aspirated and the pellet resuspended in fibroblast media. The solution was then incubated in a T75 tissue culture flask for 30 min at 37°C and 5% CO2, allowing fibroblasts to attach to the flask so that the supernatant containing CMs and ECM debris could be removed. CFBs were immediately trypsinized in 0.05% trypsin-EDTA (cat. no. 25300-054; Life Technologies), counted, and seeded onto PA or HA gels for further experiments.
Isolation of neonatal CMs
Neonatal CD1 mouse pups (protocol no. S01013M; Charles River Labs) were killed by decapitation, and the hearts were removed and rinsed in HBSS. Predigestion was performed with 0.46 mg/ml trypsin overnight and digestion with 0.7 mg/ml collagenase type 2 for 7 min at 37°C. Further digestion was done by repeated pipetting, after which the cell solution was passed through a 100 µm cell strainer. Collagenase was neutralized by adding Dark media consisting of 3:4 DMEM, 1:4 M-199 (cat. no. 11150-059; Life Technologies) and supplemented with 1% HEPES (1 M), 10% horse serum (cat. no. DH-05; Omega Scientific, Tarzana, CA), 5% FBS, and 1% penicillin/streptomycin (cat. no. 30-00261; Corning, Manassas, VA). The cell solution was centrifuged at 200 × g for 4 min. After the pellet was resuspended in Dark medium, preplating was performed by letting non-CM cells attach to T75 flasks for 1.5 h at 37°C and 5% CO2. The supernatant containing CMs was collected, centrifuged for 4 min at 200 × g, and resuspended in 2 ml Dark media. Cell number was determined using a Bürcher cell counter chamber by counting live cells after the addition of Trypan blue (cat. no. K940; VWR Amresco, Solon, OH). CMs (700,000 ) were added to each cell stretcher membrane and incubated at 37°C and 5% CO2 for 3 d before stretch experiments were performed. For some control experiments, 10 μM AraC (cytosine-B-d-arabino-furanoside hydrochloride; cat. no. C6645; Sigma) was added on day 2 after isolation to prevent survival of “contaminating” CFBs (Ehler et al., 2013).
Preparation of PA gels
PA gels were fabricated with elastic moduli E of 3.13 (3 kPa) and 8.7 kPa (8 kPa) by adjusting the relative acrylamide and bis-acrylamide concentrations (Tse and Engler, 2010). Acrylic acid (0.5% ) was added to enable subsequent collagen attachment. The photoinitiated cross-linker Irgacure 2959 (Sigma) was dissolved in 100% ethanol to obtain a 10% solution that was then diluted to 0.05% in the PA solution. PA solution was then sandwiched between a 12-mm-diameter coverslip that was UV-ozone activated and methacrylated to permit hydrogel binding and a glass slide that was treated with dichlorodimethylsilane (DCDMS, cat. no. 430881000; Acros Organics, Geel, Belgium) to avoid adherence to the gel. For initiation of gel polymerization, the hydrogel was subjected to UV light with a wavelength of 350 nm for 5 min using a transilluminator (4 mW/cm2; UVP, Upland, CA)
PDMS membrane
Two-part PDMS (cat. no. 30097358-1004, Sylgard 186; Dow Corning, Midland, MI) was mixed at a 10:1 ratio of base to curing agent according to the manufacturer’s instructions. The mixture was then loaded into 10 ml syringes with Luer-Lok stopcocks, and large air pockets were removed in an IEC Centra CL2 centrifuge at 2500 rpm for 3 min. Seven milliliters of elastomer mix was then extruded onto either an unpatterned or a patterned (microgrooves of 10 µm width, 5 µm depth, and spaced 10 µm apart) silicon wafer in a spin coater. The spin coater was run at 650 rpm for 30 s for each wafer. All wafers were then placed in a vacuum chamber to degas in short cycles for 20–40 min, or until all visible bubbles were gone. The wafers were then oven-cured for 30 min at 70°C and cooled at room temperature overnight. The membranes could then be peeled off the wafer and stored in a clean environment for up to 1 yr (Camelliti et al., 2006).
Treatment of PDMS membranes with benzophenone in to allow PA adherence
To allow PA gel adherence to the PDMS surface, we used a previously described protocol (Simmons et al., 2013). The silicone elastomer PDMS membranes were immersed in a solution of 10% benzophenone (cat. no. A10739; Alfa Aesar) dissolved in a water/acetone mixture (35:65 wt/wt) for 1 min. The membranes were then immediately rinsed with methanol and dried with a stream of nitrogen. Following benzophenone treatment, PA gel solution was prepared as described above, pipetted onto the surface of the membrane, covered with a DCDMS-treated 25 mm coverslip, and exposed to UV light (350 nm) for 25 min. Coverslips were removed, and gels that successfully adhered to PDMS membranes were equilibrated in phosphate-buffered saline (PBS) overnight.
Assembly and stretch of circular and elliptical stretch device
Before being coated with protein, PDMS membranes were assembled in circular (for CFBs on PA gels on unpatterned PDMS stretch membranes) or elliptical (for CMs on patterned PDMS stretch membranes) stretch devices built in house (Camelliti et al., 2006). A perimeter of silicone grease was placed around the area intended for cell attachment and coated with either collagen I for CFB cultures or laminin (10 µg/ml, 10 min UV exposure, cat. no. L2020; Sigma) for CM cultures. After 3 d of cell culture, static stretch was applied for 24 and 48 h by turning the screw top of each device to the extent corresponding to the desired amount of stretch. Stretch has previously been confirmed to transfer to cells cultured on PA gels attached to PDMS stretch membranes (Simmons et al., 2013). For determining whether cells return to their original size after stretch, the cell area of 8 CFBs on PDMS stretch membranes was measured before stretch; immediately after stretch; and 10, 20, 30, and 60 min after applying static stretch. Cell areas for nonstretched and stretched CFBs on 3 and 8 kPa PA gels were measured 24 h after applying the static stretch.
Interface coculture device for studying paracrine signaling in combination with stretch
To study the effect on paracrine signaling from stretched CMs on CFB phenotype, we designed a coculture device that allowed CFBs on PA gels attached to coverslips to be inverted onto CMs in the elliptical stretch device while preventing physical contact between cell cultures. The design allowed sufficient gas exchange, close proximity (1 mm) between cell cultures to ensure maximum effect of paracrine signals, easy access to media, and easy collection of CFB cultures after completion of the experiments and was made of a biocompatible inert material (polylactic acid [PLA]). The device was manufactured by 3D-PLA printing. CMs were stretched by rotating the screw top 720°, corresponding to a 14% longitudinal and 3.6% transverse stretch of the CM. CFBs and media were collected after 24 or 48 h coculture.
Production and stiffening of HA hydrogels
HA was methacrylated and HA hydrogels synthesized according to the protocols described by the Burdick (Guvendiren et al., 2014) and Engler (Ondeck and Engler, 2016) groups. Sodium hyaluronate (50 kDa; Lifecore Biomedical, Chaska, MN) was dissolved in deionized water overnight, after which 20 M methacrylate anhydride was added for an additional 12 h, dialyzed against deionized water at 4°C for 3 d, and lyophilized for an additional 3 d. Hydrogels were synthesized by dissolving methacrylated HA at 1% in PBS containing 0.2 M triethanolamine (Sigma). HA hydrogel solution containing 0.02% Irgacure 2959 was sandwiched between a 12-mm-diameter coverslip that was UV-ozone activated and methacrylated, and a DCDMS-treated glass slide. The hydrogel was subjected to UV light with a wavelength of 350 nm for 0.5–2 min using a transilluminator to induce gel polymerization. HA hydrogels were stiffened by incubation for 30 min with 0.05% vol/vol Irgacure 2959 per 1 ml cell media consisting of DMEM without phenol red (cat. no. SH30284.01; GE Life Sciences, Logan, UT) supplemented with 10% FBS before 2 min exposure to UV light (350 nm). Cell cultures were immediately washed three times with cell media. Atomic force microscopy (MFP-3D-Bio atomic force microscope; Asylum Research, Santa Barbara, CA) was used to determine hydrogel elastic modulus as previously described (Ondeck and Engler, 2016). To ensure that UV exposure was not detrimental to cells, we performed live–dead stain (cat. no. R37609; Molecular Probes, Eugene, OR) and immunostaining for p53.
Collagen attachment to PA and HA gels
Collagen I from rat tail was attached by incubating PA gels with 20 mM 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (ProteoChem, Hurricane, UT), 50 mM N-hydroxysuccinimide (Alfa Aesar, Heysham, UK), and 100 µg/ml type I rat tail collagen (cat. no. 354249; BD Biosciences, Bedford, MA) dissolved in PBS overnight at 37°C. For fluorescence labeling of collagen on the surface of the hydrogel to compare amount of rat tail type I collagen binding to HA and PA hydrogel, a polyclonal rabbit type 1 collagen antibody (1:200; Novus Biological NBP1-30054) and 568 conjugated goat anti-mouse secondary antibody (1:1000; Thermo A-10042) were used. Confocal cross-sections of hydrogels were taken, and intensities were obtained using ImageJ (National Institutes of Health) and plotted using Matlab. Collagen staining intensity was normalized to background, and the maximum intensity was used to quantify relative collagen amount. Values were normalized to the amount found on HA gels.
Proliferation assay
Proliferation was measured with the Click-iT EdU imaging kit (cat. no. C10337; Life Technologies, Grand Island, NY), according to the manufacturer’s instructions and imaged and analyzed with the EVOS FL Auto fluorescence microscope (cat. no. AMAFD1000; Life Technologies). Ten pictures were taken per gel at 10× magnification, and EdU- and 4′,6-diamidino-2-phenylindole (DAPI)-positive nuclei were counted and represented as the fraction of proliferating cells per 24 h. CFBs were stimulated with 10 ng/ml recombinant PDGF-B (cat. no. 558802; BioLegend, San Diego, CA), 10 ng/ml CSF-1 (cat. no. 14-8983-62; Affymetrix, San Diego, CA), and their respective receptor blockers AG1295 (10 µM, cat. no. ab142375; Abcam, Cambridge, MA) and GW2580 (5 µM, cat. no. SML1047; Sigma).
Activity assay
LOX and TGFβ activity was measured in the media of CFB cultures 24 h after stiffening using a LOX activity assay (cat. no. ab112139; Abcam) and TGFβ activity assay (cat. no. 437707; BioLegend, San Diego, CA) according to the manufacturers’ instructions.
Immunocytochemistry
CFBs were fixed in 4% paraformaldehyde, permeabilized in 0.1% Triton X (Sigma), quenched with 25 mM glycin (cat. no. 646500; Fisher Scientific, Fair Lawn, NJ), blocked in 5% goat serum (cat. no. G6767; Sigma) in PBS for 20 min, and incubated at 4°C overnight with primary antibodies toward SMA (1:200, mouse anti-mouse α-SMA, cat. no. A5228; Sigma), FN (1:200, rabbit anti-mouse FN, cat. no. ab2413; Abcam), vinculin (1:200, mouse anti-vinculin, cat. no. V9131; Sigma), and p53 (1:200, rabbit anti-p53, cat. no. sc6243; Santa Cruz Biotechnology, Santa Cruz, CA) in 2% goat serum in PBS. After three 10-min washes of CFB with PBS, secondary antibodies were added (1:1000 in 2% goat serum, Alexa Fluor 488 goat anti-mouse, cat. no. A11029; Alexa Fluor 700, goat anti-rabbit, cat. no. A21038; Molecular Probes). We used 633-phalloidin (1: 200 in PBS, 20 min at room temperature, cat. no. A22284; Molecular Probes) to stain F-actin and DAPI to stain nuclei. Coverslips with cells were mounted on microscope slides using SlowFade Gold antifade reagent (cat. no. S36940; Molecular Probes) and imaged and analyzed with an EVOS FL Auto fluorescence microscope. FN was imaged with 20× magnification, and the staining intensity was analyzed using ImageJ (1.50 f). Settings were kept constant for the same experiment (from a same date), time point, and stiffness. The presence of SMA fibers was evaluated for at least 10 micrographs per gel at 20× magnification. For some micrographs, the EdU proliferation assay and SMA staining were performed simultaneously at the same excitation and emission wavelength. Because EdU was restricted to the nucleus and SMA is located in the cytosol, these were easily distinguished during analysis. Cell area was measured by using brightness and contrast thresholds that clearly displayed the autofluorescence of the cell cytoplasm and thus enabled automatic quantification of the area of the micrograph covered with cells. This area was then divided by the number of cell nuclei as determined by DAPI staining.
RNA isolation, cDNA synthesis, and real-time PCR
RNA isolation was performed using the TRIzol method. First, 1 ml of TRIzol (cat. no. 15596026; Ambion, Carlsbad, CA) was added to each of the gels to lyse CFBs. The cell lysate was then added to prespun Phase Lock Gel-Heavy tubes (cat. no. 2302830; 5 Prime GmbH, Hamburg, Germany), spun at 1500 × g for 30 s, and incubated for 5 min at 22°C. Chloroform (0.2 ml) was then added to each gel and shaken vigorously for 15 s. The lysate/chloroform mixture was then centrifuged at 12,000 × g for 10 min at 4°C. Following this spin, there was a visible separation between the layers of solution, with a clear aqueous layer entirely above the Phase Lock Gel. This aqueous layer, containing RNA, was then removed and placed into a fresh 1.7 ml tube. To each of the RNA solutions, 0.5 ml of isopropyl alcohol was added and mixed by inversion. Samples were then allowed to incubate at 22°C for 10 min followed by centrifugation for 10 min at 12,000 × g (4°C). The supernatant was then decanted, and 1 ml of freshly prepared 75% ethanol was added to each sample. One microliter of glycogen was added to each sample to visualize the pellet. Samples were then centrifuged for 5 min at 7500 × g (4°C). Following this centrifugation, the supernatant was decanted from each sample, and the RNA pellet was dried. Finally, each RNA pellet was resuspended in 15 μl of molecular biology grade water and stored for later quantification and analysis. For CFBs on stretchers, RNA isolation was performed with the RNeasy Mini Kit (cat. no. 74104; Qiagen, Hilden, Germany) according to the manufacturer’s protocol. RNA quantification was performed with the Qubit RNA BR assay kit (cat. no. Q33211; Molecular Probes), and cDNA synthesis was performed with the iScript kit (cat. no. 04896866001; Roche, Indianapolis, IN) according to the manufacturer’s protocol. Quantitative real-time PCR was performed using StepOnePlus Real-time PCR machine (Applied Biosystems, Foster City, CA) and KAPA SYBR Fast Universal qPCR kit (cat. no. 07959397001; Kapa Biosystems, Cape Town, South Africa) and primers targeting the genes of interest (Integrated DNA Technologies, Indianapolis, IN). Primer sequences are available in Supplemental Table 1.
Statistics
Data are presented as mean ± SEM. N represents biological replicates, and all experiments were replicated in at least two independent cell isolations. Changes in gene expression presented in Figure 3C were calculated using log2-transformed values, where fold changes were calculated by taking the normalized (meanstretch − meancontrol) and an SEM being √((SDcontrol2/ncontrol) + (SDstretch2/ nstretch)). Statistical analysis was performed using GraphPad Prism 6. Normal distribution of data was determined by D’Agostino and Pearson omnibus normality test and assisted in the choice of subsequent parametric or nonparametric statistical tests. For comparison of two groups, two-sided a Student’s t test or Mann-Whitney test was used. If multiple t tests were performed, these were corrected for multiple comparisons using the Holm-Sidak post hoc test. For comparison between three or more groups, one-way analysis of variance (ANOVA) with Tukey’s post hoc testing (parametric) and Kruskal-Wallis with Dunn’s post hoc testing (nonparametric) was used. For data sets comparing two interventions (e.g., substrate stiffness and stretch), two-way ANOVA with the Tukey’s post hoc test was used. For correlation of parametric data, Pearson’s test was used. ***, P < 0.001; **, P < 0.01; *, P < 0.05.
Supplementary Material
Acknowledgments
We thank Jennifer Stowe, Vandya Juneja, and Joey Ochoa for excellent technical assistance and Jeffrey H. Omens for valuable scientific input. We are grateful for funding from the Research Council of Norway (K.M.H.), the European Commission program (K.M.H.), the National Science Foundation Graduate Research Fellowship Program (A.K.), and National Institutes of Health grants R01CA206880 and DP2OD006460 (A.J.E.). This work was supported in part by National Institutes of Health grants (GM094503, GM103426, HL105242, HL122199, and HL105242) and by a California Institute of Regenerative Medicine grant (CIRM RT3-07899).
Abbreviations used:
- ANOVA
analysis of variance
- BMP1
bone morphogenetic protein 1
- CFB
cardiac fibroblast
- CM
cardiomyocyte
- CSF-1
colony-stimulating factor 1
- DCDMS
dichlorodimethylsilane
- ECM
extracellular matrix
- FAK
focal adhesion kinase
- FBS
fetal bovine serum
- FGF2
fibroblast growth factor 2
- FN
fibronectin
- GADPH
glyceraldehyde 3-phosphate dehydrogenase
- HA
hyaluronic acid
- LOX
lysyl oxidase
- MI
myocardial infarction
- MMP
matrix metalloproteinase
- OPN
osteopontin
- PA
polyacrylamide
- PBS
phosphate-buffered saline
- PDGF-B
platelet-derived growth factor B
- PDMS
polydimethylsiloxane
- PLA
polylactic acid
- SMA
smooth muscle α-actin
- SPARC
secreted protein acidic and rich in cysteine
- TGFβ
transforming growth factor β
- THBS1
thrombospondin 1
- TIMP
tissue inhibitor of metalloproteinase
- TNC
tenascin C.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E17-01-0014) on May 3, 2017.
REFERENCES
- Antoniades HN, Scher CD, Stiles CD. Purification of human platelet-derived growth factor. Proc Natl Acad Sci USA. 1979;76:1809–1813. doi: 10.1073/pnas.76.4.1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry MF, Engler AJ, Woo YJ, Pirolli TJ, Bish LT, Jayasankar V, Morine KJ, Gardner TJ, Discher DE, Sweeney HL. Mesenchymal stem cell injection after myocardial infarction improves myocardial compliance. Am J Physiol Heart Circ Physiol. 2006;290:H2196–H2203. doi: 10.1152/ajpheart.01017.2005. [DOI] [PubMed] [Google Scholar]
- Biernacka A, Dobaczewski M, Frangogiannis NG. TGF-β signaling in fibrosis. Growth Factors. 2011;29:196–202. doi: 10.3109/08977194.2011.595714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caliari SR, Perepelyuk M, Cosgrove BD, Tsai SJ, Lee GY, Mauck RL, Wells RG, Burdick JA. Stiffening hydrogels for investigating the dynamics of hepatic stellate cell mechanotransduction during myofibroblast activation. Sci Rep. 2016;6:21387. doi: 10.1038/srep21387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camelliti P, Gallagher JO, Kohl P, McCulloch AD. Micropatterned cell cultures on elastic membranes as an in vitro model of myocardium. Nat Protoc. 2006;1:1379–1391. doi: 10.1038/nprot.2006.203. [DOI] [PubMed] [Google Scholar]
- Carraher CL, Schwarzbauer JE. Regulation of matrix assembly through rigidity-dependent fibronectin conformational changes. J Biol Chem. 2013;288:14805–14814. doi: 10.1074/jbc.M112.435271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitu V, Stanley ER. Colony-stimulating factor-1 in immunity and inflammation. Curr Opin Immunol. 2006;18:39–48. doi: 10.1016/j.coi.2005.11.006. [DOI] [PubMed] [Google Scholar]
- Creemers EE, Pinto YM. Molecular mechanisms that control interstitial fibrosis in the pressure-overloaded heart. Cardiovasc Res. 2011;89:265–272. doi: 10.1093/cvr/cvq308. [DOI] [PubMed] [Google Scholar]
- Cui Y, Hameed FM, Yang B, Lee K, Pan CQ, Park S, Sheetz M. Cyclic stretching of soft substrates induces spreading and growth. Nat Commun. 2015;6:6333. doi: 10.1038/ncomms7333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daskalopoulos EP, Janssen BJ, Blankesteijn WM. Myofibroblasts in the infarct area: concepts and challenges. Microsc Microanal. 2012;18:35–49. doi: 10.1017/S143192761101227X. [DOI] [PubMed] [Google Scholar]
- Davis J, Molkentin JD. Myofibroblasts: trust your heart and let fate decide. J Mol Cell Cardiol. 2014;70:9–18. doi: 10.1016/j.yjmcc.2013.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duerr GD, Elhafi N, Bostani T, Ellinger J, Swieny L, Kolobara E, Welz A, Dewald O. Comparison of myocardial remodeling between cryoinfarction and reperfused infarction in mice. J Biomed Biotechnol. 2011;2011:961298. doi: 10.1155/2011/961298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehler E, Moore-Morris T, Lange S. Isolation and culture of neonatal mouse cardiomyocytes. J Vis Exp. 2013;2013:50154. doi: 10.3791/50154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engebretsen KV, Lunde IG, Strand ME, Waehre A, Sjaastad I, Marstein HS, Skrbic B, Dahl CP, Askevold ET, Christensen G, et al. Lumican is increased in experimental and clinical heart failure, and its production by cardiac fibroblasts is induced by mechanical and proinflammatory stimuli. FEBS J. 2013;280:2382–2398. doi: 10.1111/febs.12235. [DOI] [PubMed] [Google Scholar]
- Engler AJ, Carag-Krieger C, Johnson CP, Raab M, Tang HY, Speicher DW, Sanger JW, Sanger JM, Discher DE. Embryonic cardiomyocytes beat best on a matrix with heart-like elasticity: scar-like rigidity inhibits beating. J Cell Sci. 2008;121:3794–3802. doi: 10.1242/jcs.029678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goffin JM, Pittet P, Csucs G, Lussi JW, Meister JJ, Hinz B. Focal adhesion size controls tension-dependent recruitment of α-smooth muscle actin to stress fibers. J Cell Biol. 2006;172:259–268. doi: 10.1083/jcb.200506179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Zeng QC, Zhang CQ, Zhang XZ, Li RX, Wu JM, Guan J, Liu L, Zhang XC, Li JY, Wan ZM. Extracellular matrix of mechanically stretched cardiac fibroblasts improves viability and metabolic activity of ventricular cells. Int J Med Sci. 2013;10:1837–1845. doi: 10.7150/ijms.6786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guvendiren M, Burdick JA. Stiffening hydrogels to probe short- and long-term cellular responses to dynamic mechanics. Nat Commun. 2012;3:792. doi: 10.1038/ncomms1792. [DOI] [PubMed] [Google Scholar]
- Guvendiren M, Perepelyuk M, Wells RG, Burdick JA. Hydrogels with differential and patterned mechanics to study stiffness-mediated myofibroblastic differentiation of hepatic stellate cells. J Mech Behav Biomed Mater. 2014;38:198–208. doi: 10.1016/j.jmbbm.2013.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwechenberger M, Mendoza LH, Youker KA, Frangogiannis NG, Smith CW, Michael LH, Entman ML. Cardiac myocytes produce interleukin-6 in culture and in viable border zone of reperfused infarctions. Circulation. 1999;99:546–551. doi: 10.1161/01.cir.99.4.546. [DOI] [PubMed] [Google Scholar]
- Hermans KC, Daskalopoulos EP, Blankesteijn WM. The Janus face of myofibroblasts in the remodeling heart. J Mol Cell Cardiol. 2016;91:35–41. doi: 10.1016/j.yjmcc.2015.11.017. [DOI] [PubMed] [Google Scholar]
- Herum KM, Lunde IG, Skrbic B, Florholmen G, Behmen D, Sjaastad I, Carlson CR, Gomez MF, Christensen G. Syndecan-4 signaling via NFAT regulates extracellular matrix production and cardiac myofibroblast differentiation in response to mechanical stress. J Mol Cell Cardiol. 2013;54:73–81. doi: 10.1016/j.yjmcc.2012.11.006. [DOI] [PubMed] [Google Scholar]
- Herum KM, Lunde IG, Skrbic B, Louch WE, Hasic A, Boye S, Unger A, Brorson SH, Sjaastad I, et al. Syndecan-4 is a key determinant of collagen cross-linking and passive myocardial stiffness in the pressure-overloaded heart. Cardiovasc Res. 2015;106:217–226. doi: 10.1093/cvr/cvv002. [DOI] [PubMed] [Google Scholar]
- Hinz B. Formation and function of the myofibroblast during tissue repair. J Invest Dermatol. 2007;127:526–537. doi: 10.1038/sj.jid.5700613. [DOI] [PubMed] [Google Scholar]
- Hohensinner PJ, Kaun C, Rychli K, Niessner A, Pfaffenberger S, Rega G, de Martin R, Maurer G, Ullrich R, Huber K, Wojta J. Macrophage colony stimulating factor expression in human cardiac cells is upregulated by tumor necrosis factor-α via an NF-κB dependent mechanism. J Thromb Haemost. 2007;5:2520–2528. doi: 10.1111/j.1538-7836.2007.02784.x. [DOI] [PubMed] [Google Scholar]
- Husse B, Briest W, Homagk L, Isenberg G, Gekle M. Cyclical mechanical stretch modulates expression of collagen I and collagen III by PKC and tyrosine kinase in cardiac fibroblasts. Am J Physiol Regul Integr Comp Physiol. 2007;293:R1898–R1907. doi: 10.1152/ajpregu.00804.2006. [DOI] [PubMed] [Google Scholar]
- Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- Imanaka-Yoshida K, Hiroe M, Nishikawa T, Ishiyama S, Shimojo T, Ohta Y, Sakakura T, Yoshida T. Tenascin-C modulates adhesion of cardiomyocytes to extracellular matrix during tissue remodeling after myocardial infarction. Lab Invest. 2001;81:1015–1024. doi: 10.1038/labinvest.3780313. [DOI] [PubMed] [Google Scholar]
- Ingber DE. Tensegrity and mechanotransduction. J Bodyw Mov Ther. 2008;12:198–200. doi: 10.1016/j.jbmt.2008.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Z, Zhong G, Wen L, Hong Y, Fang S, Sun P, Li S, Li S, Feng G. The role of platelet-derived growth factor-B/platelet-derived growth factor receptor-β signaling in chronic atrial fibrillation. Cardiology. 2016;133:242–256. doi: 10.1159/000442940. [DOI] [PubMed] [Google Scholar]
- Kasner M, Westermann D, Lopez B, Gaub R, Escher F, Kuhl U, Schultheiss HP, Tschope C. Diastolic tissue Doppler indexes correlate with the degree of collagen expression and cross-linking in heart failure and normal ejection fraction. J Am Coll Cardiol. 2011;57:977–985. doi: 10.1016/j.jacc.2010.10.024. [DOI] [PubMed] [Google Scholar]
- Kloxin AM, Benton JA, Anseth KS. In situ elasticity modulation with dynamic substrates to direct cell phenotype. Biomaterials. 2010;31:1–8. doi: 10.1016/j.biomaterials.2009.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AA, Delhaas T, McCulloch AD, Villarreal FJ. Differential responses of adult cardiac fibroblasts to in vitro biaxial strain patterns. J Mol Cell Cardiol. 1999;31:1833–1843. doi: 10.1006/jmcc.1999.1017. [DOI] [PubMed] [Google Scholar]
- Li CX, Talele NP, Boo S, Koehler A, Knee-Walden E, Balestrini JL, Speight P, Kapus A, Hinz B. MicroRNA-21 preserves the fibrotic mechanical memory of mesenchymal stem cells. Nat Mater. 2017;16:379–389. doi: 10.1038/nmat4780. [DOI] [PubMed] [Google Scholar]
- Li Z, Dranoff JA, Chan EP, Uemura M, Sevigny J, Wells RG. Transforming growth factor-β and substrate stiffness regulate portal fibroblast activation in culture. Hepatology. 2007;46:1246–1256. doi: 10.1002/hep.21792. [DOI] [PubMed] [Google Scholar]
- Lighthouse JK, Small EM. Transcriptional control of cardiac fibroblast plasticity. J Mol Cell Cardiol. 2016;91:52–60. doi: 10.1016/j.yjmcc.2015.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsey ML, Zouein FA, Tian Y, Padmanabhan Iyer R, de Castro Bras LE. Osteopontin is proteolytically processed by matrix metalloproteinase 9. Can J Physiol Pharmacol. 2015;93:879–886. doi: 10.1139/cjpp-2015-0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Lagares D, Choi KM, Stopfer L, Marinkovic A, Vrbanac V, Probst CK, Hiemer SE, Sisson TH, Horowitz JC, et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am J Physiol Lung Cell Mol Physiol. 2015;308:L344–L357. doi: 10.1152/ajplung.00300.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez B, Gonzalez A, Lindner D, Westermann D, Ravassa S, Beaumont J, Gallego I, Zudaire A, Brugnolaro C, Querejeta R, et al. Osteopontin-mediated myocardial fibrosis in heart failure: a role for lysyl oxidase? Cardiovasc Res. 2013;99:111–120. doi: 10.1093/cvr/cvt100. [DOI] [PubMed] [Google Scholar]
- Lopez B, Querejeta R, Gonzalez A, Larman M, Diez J. Collagen cross-linking but not collagen amount associates with elevated filling pressures in hypertensive patients with stage C heart failure: potential role of lysyl oxidase. Hypertension. 2012;60:677–683. doi: 10.1161/HYPERTENSIONAHA.112.196113. [DOI] [PubMed] [Google Scholar]
- Lunde IG, Herum KM, Carlson CC, Christensen G. Syndecans in heart fibrosis. Cell Tissue Res. 2016;365:539–552. doi: 10.1007/s00441-016-2454-2. [DOI] [PubMed] [Google Scholar]
- Luther DJ, Thodeti CK, Shamhart PE, Adapala RK, Hodnichak C, Weihrauch D, Bonaldo P, Chilian WM, Meszaros JG. Absence of type VI collagen paradoxically improves cardiac function, structure, and remodeling after myocardial infarction. Circ Res. 2012;110:851–856. doi: 10.1161/CIRCRESAHA.111.252734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruhashi T, Kii I, Saito M, Kudo A. Interaction between periostin and BMP-1 promotes proteolytic activation of lysyl oxidase. J Biol Chem. 2010;285:13294–13303. doi: 10.1074/jbc.M109.088864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazhari R, Omens JH, Covell JW, McCulloch AD. Structural basis of regional dysfunction in acutely ischemic myocardium. Cardiovasc Res. 2000;47:284–293. doi: 10.1016/s0008-6363(00)00089-4. [DOI] [PubMed] [Google Scholar]
- McCulloch AD. Systems biophysics: multiscale biophysical modeling of organ systems. Biophys J. 2016;110:1023–1027. doi: 10.1016/j.bpj.2016.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melchior-Becker A, Dai G, Ding Z, Schafer L, Schrader J, Young MF, Fischer JW. Deficiency of biglycan causes cardiac fibroblasts to differentiate into a myofibroblast phenotype. J Biol Chem. 2011;286:17365–17375. doi: 10.1074/jbc.M110.192682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melleby AO, Strand ME, Romaine A, Herum KM, Skrbic B, Dahl CP, Sjaastad I, Fiane AE, Filmus J, Christensen G, Lunde IG. The heparan sulfate proteoglycan glypican-6 is upregulated in the failing heart, and regulates cardiomyocyte growth through ERK1/2 signaling. PLoS One. 2016;11:e0165079. doi: 10.1371/journal.pone.0165079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molkentin JD, Bugg D, Ghearing N, Dorn LE, Kim P, Sargent MA, Gunaje J, Otsu K, Davis JM. Fibroblast-specific genetic manipulation of p38 MAPK in vivo reveals its central regulatory role in fibrosis. Circulation. 2017 doi: 10.1161/CIRCULATIONAHA.116.026238. DOI:10.1161/CIRCULATIONAHA.116.026238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee D, Sen S. Collagen phenotypes during development and regression of myocardial hypertrophy in spontaneously hypertensive rats. Circ Res. 1990;67:1474–1480. doi: 10.1161/01.res.67.6.1474. [DOI] [PubMed] [Google Scholar]
- Nakazawa N, Sathe AR, Shivashankar GV, Sheetz MP. Matrix mechanics controls FHL2 movement to the nucleus to activate p21 expression. Proc Natl Acad Sci USA. 2016;113:E6813–E6822. doi: 10.1073/pnas.1608210113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen AL, Bloomer SA, Chan EP, Gaca MD, Georges PC, Sackey B, Uemura M, Janmey PA, Wells RG. Hepatic stellate cells require a stiff environment for myofibroblastic differentiation. Am J Physiol Gastrointest Liver Physiol. 2011;301:G110–G118. doi: 10.1152/ajpgi.00412.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ondeck MG, Engler AJ. Mechanical characterization of a dynamic and tunable methacrylated hyaluronic acid hydrogel. J Biomech Eng. 2016;138:021003. doi: 10.1115/1.4032429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papakrivopoulou J, Lindahl GE, Bishop JE, Laurent GJ. Differential roles of extracellular signal-regulated kinase 1/2 and p38MAPK in mechanical load-induced procollagen α1(I) gene expression in cardiac fibroblasts. Cardiovasc Res. 2004;61:736–744. doi: 10.1016/j.cardiores.2003.12.018. [DOI] [PubMed] [Google Scholar]
- Parsons JT. Focal adhesion kinase: the first ten years. J Cell Sci. 2003;116:1409–1416. doi: 10.1242/jcs.00373. [DOI] [PubMed] [Google Scholar]
- Rasi K, Piuhola J, Czabanka M, Sormunen R, Ilves M, Leskinen H, Rysa J, Kerkela R, Janmey P, Heljasvaara R, et al. Collagen XV is necessary for modeling of the extracellular matrix and its deficiency predisposes to cardiomyopathy. Circ Res. 2010;107:1241–1252. doi: 10.1161/CIRCRESAHA.110.222133. [DOI] [PubMed] [Google Scholar]
- Schellings MW, Vanhoutte D, Swinnen M, Cleutjens JP, Debets J, van Leeuwen RE, d’Hooge J, Van de Werf F, Carmeliet P, Pinto YM, et al. Absence of SPARC results in increased cardiac rupture and dysfunction after acute myocardial infarction. J Exp Med. 2009;206:113–123. doi: 10.1084/jem.20081244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons CS, Ribeiro AJ, Pruitt BL. Formation of composite polyacrylamide and silicone substrates for independent control of stiffness and strain. Lab Chip. 2013;13:646–649. doi: 10.1039/c2lc41110e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skrbic B, Engebretsen KV, Strand ME, Lunde IG, Herum KM, Marstein HS, Sjaastad I, Lunde PK, Carlson CR, Christensen G, et al. Lack of collagen VIII reduces fibrosis and promotes early mortality and cardiac dilatation in pressure overload in mice. Cardiovasc Res. 2015;106:32–42. doi: 10.1093/cvr/cvv041. [DOI] [PubMed] [Google Scholar]
- Spinale FG, Frangogiannis NG, Hinz B, Holmes JW, Kassiri Z, Lindsey ML. Crossing into the next frontier of cardiac extracellular matrix research. Circ Res. 2016;119:1040–1045. doi: 10.1161/CIRCRESAHA.116.309916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan KE, Quinn KP, Tang KM, Georgakoudi I, Black LD., III Extracellular matrix remodeling following myocardial infarction influences the therapeutic potential of mesenchymal stem cells. Stem Cell Res Ther. 2014;5:14. doi: 10.1186/scrt403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi S, Barry AC, Factor SM. Collagen degradation in ischaemic rat hearts. Biochem J. 1990;265:233–241. doi: 10.1042/bj2650233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarzami ST, Cheng R, Miao W, Kitsis RN, Berman JW. Chemokine expression in myocardial ischemia: MIP-2 dependent MCP-1 expression protects cardiomyocytes from cell death. J Mol Cell Cardiol. 2002;34:209–221. doi: 10.1006/jmcc.2001.1503. [DOI] [PubMed] [Google Scholar]
- Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002;3:349–363. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- Tse JR, Engler AJ. Preparation of hydrogel substrates with tunable mechanical properties. Curr Protoc Cell Biol. 2010:10.10.16. doi: 10.1002/0471143030.cb1016s47. [DOI] [PubMed] [Google Scholar]
- van Putten S, Shafieyan Y, Hinz B. Mechanical control of cardiac myofibroblasts. J Mol Cell Cardiol. 2016;93:133–142. doi: 10.1016/j.yjmcc.2015.11.025. [DOI] [PubMed] [Google Scholar]
- Virag JI, Murry CE. Myofibroblast and endothelial cell proliferation during murine myocardial infarct repair. Am J Pathol. 2003;163:2433–2440. doi: 10.1016/S0002-9440(10)63598-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Chen H, Seth A, McCulloch CA. Mechanical force regulation of myofibroblast differentiation in cardiac fibroblasts. Am J Physiol Heart Circ Physiol. 2003;285:H1871–H1881. doi: 10.1152/ajpheart.00387.2003. [DOI] [PubMed] [Google Scholar]
- Waxman AS, Kornreich BG, Gould RA, Moise NS, Butcher JT. Interactions between TGFβ1 and cyclic strain in modulation of myofibroblastic differentiation of canine mitral valve interstitial cells in 3D culture. J Vet Cardiol. 2012;14:211–221. doi: 10.1016/j.jvc.2012.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willems IE, Arends JW, Daemen MJ. Tenascin and fibronectin expression in healing human myocardial scars. J Pathol. 1996;179:321–325. doi: 10.1002/(SICI)1096-9896(199607)179:3<321::AID-PATH555>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Wong VW, Longaker MT, Gurtner GC. Soft tissue mechanotransduction in wound healing and fibrosis. Semin Cell Dev Biol. 2012;23:981–986. doi: 10.1016/j.semcdb.2012.09.010. [DOI] [PubMed] [Google Scholar]
- Yeung T, Georges PC, Flanagan LA, Marg B, Ortiz M, Funaki M, Zahir N, Ming W, Weaver V, Janmey PA. Effects of substrate stiffness on cell morphology, cytoskeletal structure, and adhesion. Cell Motil Cytoskeleton. 2005;60:24–34. doi: 10.1002/cm.20041. [DOI] [PubMed] [Google Scholar]
- Zeigler AC, Richardson WJ, Holmes JW, Saucerman JJ. A computational model of cardiac fibroblast signaling predicts context-dependent drivers of myofibroblast differentiation. J Mol Cell Cardiol. 2016;94:72–81. doi: 10.1016/j.yjmcc.2016.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Fan G, Zhao H, Wang Z, Li F, Zhang P, Zhang J, Wang X, Wang W. Targeted inhibition of focal adhesion kinase attenuates cardiac fibrosis and preserves heart function in adverse cardiac remodeling. Sci Rep. 2017;7:43146. doi: 10.1038/srep43146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Zhao T, Huang V, Chen Y, Ahokas RA, Sun Y. Platelet-derived growth factor involvement in myocardial remodeling following infarction. J Mol Cell Cardiol. 2011;51:830–838. doi: 10.1016/j.yjmcc.2011.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.