


Abstract
Methods for the direct C–H functionalization of aromatic compounds are in demand for a variety of applications, including the synthesis of agrochemicals, pharmaceuticals, and materials. Herein, we disclose the construction of aromatic nitriles via direct C–H functionalization using an acridinium photoredox catalyst and trimethylsilyl cyanide under an aerobic atmosphere. The reaction proceeds at room temperature under mild conditions and has proven to be compatible with a variety of electron-donating and -withdrawing groups, halogens, and nitrogen- and oxygen-containing heterocycles, as well as aromatic-containing pharmaceutical agents.
Nitrile-containing aromatic groups are well represented throughout agrochemicals and therapeutic molecules (Scheme 1A). In addition to being prevalent in a number of bioactive compounds, cyanoarenes provide an excellent functional group handle that can be transformed to give derivatives of benzoic acid through hydrolysis,1 aryl ketones or imines after treatment with a strong nucleophile,2 benzaldehydes or benzylamines by reduction,3 aryl-amides through hydration, and even tetrazoles through [3+2] cycloadditions with sodium azide.4
Scheme 1.

(A) Pharmaceuticals Containing Benzonitriles and (B) Photoredox-Catalyzed Generation of Benzonitriles
Classical approaches to cyanoarene synthesis include the Sandmeyer reaction from the corresponding diazonium salt5 or transition-metal-catalyzed cross-coupling with the requisite aryl halide.6–14 Few C–H functionalization methods exist for directly producing benzonitriles from unfunctionalized arenes, and they employ Rh or Co catalyst systems, often requiring elaborate directing groups and exotic reagents,15–20 harsh lithiation conditions,21 or electrophilic sources of cyanide that give poor selectivity or significant byproduct formation.22 While the aforementioned methods are successful at accessing arene cyanation, a mild and metal-free C–H cyanation of aromatic compounds employing a common and inexpensive cyanide source has yet to be reported.
Recent efforts from our laboratory have demonstrated the reactivity of arene cation radicals,23 generated via photoinduced electron transfer from the excited state of an acridinium catalyst,24 to accomplish direct C–H amination of a range of aromatics and heteroaromatics bearing a variety of functional groups.25 Importantly, this transformation does not require the presence of a directing group and is highly para-selective for monosubstituted benzenes.26–28 Recognizing the importance of cyanoaromatics, and previous literature citing the addition of cyanide to arene radical cations,29,30 we decided to investigate the use of cyanide nucleophiles in reactions with arene cation radicals to access this class of compounds. We proposed that the highly oxidizing 3,6-di-tert-butyl-9-mesityl-10-phenylacridinium tetrafluoroborate (1, E1/2red* = +2.15 V vs SCE)25 would be an effective photooxidant to generate reactive arene cation radicals as key electrophilic intermediates from a range of aromatics (Scheme 1B). Combining a nucleophilic source of cyanide and O2 as the terminal oxidant, we hoped to realize one of the only catalytic examples of direct C–H cyanation of substituted aromatic compounds.
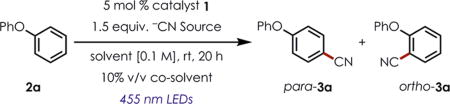
Reaction development began using diphenyl ether (2a) and the initial conditions from the aryl amination developed in our lab (Table 1, entry 1).25 Using catalytic TEMPO and either the potassium or tetrabutylammonium cyanide salts in 1,2-dichloroethane (DCE) did not result in any product formation (Table 1, entries 1 and 2). Switching to a more polar organic solvent and introducing an aqueous co-solvent yielded trace amounts of the para and ortho isomers of 3a, as detected by GC-MS (Table 1, entry 3). It was not until saturated aqueous NaHCO3 was added to the reaction mixture that more promising yields (12%) were observed (Table 1, entries 4–6). Both potassium and sodium cyanide afforded disappointing yields of the benzonitrile adduct (12% and 24%, respectively), as did the initial use of trimethylsilyl cyanide (TMSCN, 30% yield, entry 6). However, after we surveyed several different aqueous phosphate buffers as the co-solvent (Table 1, entries 7–9), pH 9 buffer, in conjunction with TMSCN, gave 3a in a combined 69% yield of para and ortho isomers (1.6:1 p:o) on a 0.5 mmol scale. The success of TMSCN as the optimal cyanation reagent can be rationalized by its ability to release cyanide slowly over the course of the reaction, allowing for a low effective concentration of the free cyanide anion during the reaction.31 Conducting the reaction in the absence of light or the acridinium photocatalyst did not afford any of the desired product (Table 1, entries 10 and 11).
Table 1.
Optimization of Reaction Conditionsa
| |||||
|---|---|---|---|---|---|
|
| |||||
| entry | notes | −CN source | solvent | co-solvent | yield, % (p:o) |
| 1 | 20 mol% TEMPO | KCN | DCE | ||
| 2 | 20 mol% TEMPO | Bu4N+CN− | DCE | ||
| 3 | KCN | MeCN | H2O | trace | |
| 4b | KCN | MeCN | NaHCO3 (aq) | 12 (3:1) | |
| 5b | NaCN | MeCN | NaHCO3 (aq) | 24 (2:1) | |
| 6b | Me3SiCN | MeCN | NaHCO3 (aq) | 30 (2:1) | |
| 7c | Me3SiCN | MeCN | pH 8 buffer | 28 (2.5:1) | |
| 8c,d | Me3SiCN | MeCN | pH 9 buffer | 69 (1.6:1) | |
| 9c | Me3SiCN | MeCN | pH 10 buffer | 33 (2.6:1) | |
| 10c | no LEDs | Me3SiCN | MeCN | pH 9 buffer | |
| 11c | no catalyst | Me3SiCN | MeCN | pH 9 buffer | |
Reactions were carried out at 0.2 mmol scale, in O2-sparged MeCN [0.1 M] under two LED lamps (λmax = 450 nm) for 24 h. Yields were obtained from the crude reaction mixture by GC-MS relative to 3-bromotoluene internal standard.
NaHCO3 was prepared as a saturated aqueous solution.
Buffer prepared from KH2PO4 and K2HPO4 at [4 M] with respect to phosphate ion.
0.5 mmol scale.
With the optimal conditions identified, we explored the scope of this transformation, beginning with simple monosubstituted aromatic systems (Chart 1). Biphenyl was functionalized to form the corresponding phenylbenzonitrile (3b) in good yield (80%), showing a preference for the para-substituted product (3:1). Benzyl-protected phenol (2c) was converted to the desired product 3c in 52% yield, albeit with 1.2:1 regioselectivity. Silyl-protected phenols (2d, 2e) were also tested under these reaction conditions; both tert-butyldiphenylsilyl- (TBDPS) and triisopropylsilyl (TIPS)-protected phenols were converted to 4-cyanophenol as the only product after being subjected to the reaction conditions for 72 h. It is likely that, after addition of cyanide to the silyl-protected radical cation and formation of the desired benzonitrile, the silyl group is cleaved by the mildly basic aqueous conditions.32 Cyanation of naphthalene (2f) and 2-methoxynaphthalene (2g) proved successful, in each case giving C–H cyanation solely at the 1-position. 1,3,5-Trisubstituted aromatic compounds were screened with mesitylene (2h), furnishing 2,4,6-trimethylbenzonitrile (3h) in 35% yield; however, this was the only product detected in the reaction, as no benzylic oxidation products were observed. Subjection of 3,5-dimethylanisole (2i) to the C–C bond-forming conditions gave a mixture of benzonitrile products 3i in excellent yield, forming the C–CN bonds on 2- and 4-positions indiscriminately.
Chart 1.

Scope of Arene Cyanation Reaction
aSingle regioisomer. b72 h reaction time. c4.0 equiv of Me3SiCN used. dProduct prone to decomposition; see SI for details.
More diverse functionality on the arene proved to be compatible under the acridinium photoredox conditions. Electron-rich systems such as 2-allyloxyanisole (2j) and 1,3-dimethoxybenzene (2k) were suitable substrates, forging the benzonitrile products in fair yields while retaining the allyl group in the case of 2j, which is typically unstable to Pd-catalyzed conditions.33–35 Benzyl-protected 3-allylphenol (2l) afforded the anticipated cyano adduct with complete regiocontrol, demonstrating the compatibility of unsaturated systems under these conditions. Both cyclopropylmethyl- and cyclopropyl-substituted compounds 2m and 2n, respectively, successfully underwent aryl cyanation under these conditions to give the corresponding benzonitrile without undergoing ring opening. Electron-rich aryl halides such as 3-fluoroanisole (2o) gave a mixture of C4 and C6 substitution products 3o as a 1:2 mixture in good yield, while 2-chloroanisole (2p) led to a single regioisomer of product 3p in 48% yield. Electron-withdrawing groups were also investigated under these conditions, demonstrated by benzophenone (2q), which was converted to the benzonitrile product 3q, giving a single regioisomer with complete site selectivity for the more electron-rich aromatic ring. Methyl ester 2r produced the corresponding benzonitrile exclusively para to the methoxy group (3r). Interestingly, 1,2-dialkoxybenzenes are recalcitrant to the reaction conditions, possibly due to insufficient charge density at the other four sites on the ring.36 However, we discovered that changing one of the alkoxy groups to a more electron-deficient tosylate, as in 2s, rendered this 1,2-dialkoxybenzene reactive, giving a single regioisomer of the benzonitrile 3s.
We next investigated the applicability of the cyanation reaction conditions to heteroaromatic systems. When N-Boc-5-methoxyindole (2t) was subjected to cyanation conditions, the corresponding cyanated product 3t was produced exclusively at the C4 position. Typically, this is a difficult site to functionalize, often requiring harsh conditions with low substrate compatibility.37,38 Similarly, 2,6-dimethoxypyridine (2u) was successfully cyanated at the C3 position exclusively to give the pyridinecarbonitrile product 3u in modest yield, while N-methylindazole (2v) afforded the cyanation products 3v at the C7 and C3 positions in a 1.3:1 ratio, respectively, in 51% yield. The oxygen-containing heterocycle chromane (2w) was a suitable substrate in this reaction, giving the cyanated product 3w para to the oxygen substituent as a single product.
The reaction conditions were also suitable for more structurally complex bioactive substrates. Naproxen methyl ester (2x) was successfully cyanated adjacent to the methoxy group, similar to what was observed in the naphthalene derivatives discussed previously (3f, 3g). Gemfibrozil methyl ester (2y) was cyanated exclusively para to the alkoxy-donating group to give 3y. Indole-containing N-Boc-melatonin (2z) was functionalized exclusively at the C4 position, and last, fenoprofen methyl ester (2aa) was well tolerated under these conditions, affording cyanide addition solely to the more electron-rich ring, and giving a single regioisomer (3aa), demonstrating selectivity for substrates containing multiple arene rings.
In general, the substrates investigated gave clean reactivity, either leading to the desired product or returning unreacted starting material. Additionally, in the cases where 4.0 equiv of TMSCN was required for optimal reactivity, only the monocyanated product was observed, with no dicyanation products detected. Substrates that led to a mixture of product isomers exhibit substitution patterns consistent with previously developed calculations outlining the electrophilic sites of electron-rich arene radical cations.39
While the mechanism of this reaction still requires further investigation, we hypothesize that the reaction proceeds through the pathway outlined in Scheme 2. The acridinium photocatalyst 1 is promoted to its highly oxidizing excited state, Mes-Acr+* (E1/2red* = +2.15 V vs SCE) using blue light. The arene 2 is oxidized to the corresponding radical cation 5, giving the reduced acridinium, Mes-Acr• (6). Cyanide then engages 5 either ortho or para to the electron-donating substituent. Fukuzumi proposed that cyclohexadienyl radical 7 can be oxidized by molecular oxygen to give the desired benzonitrile product 3.26,27 In order to complete the catalytic cycle, it is likely that either the hydroperoxy radical or molecular oxygen can oxidize Mes-Acr• (6) to regenerate the ground-state catalyst 1. It is worth noting that the oxidation potential of the free cyanide anion was found to be +0.60 V vs SCE, opening the possibility that Mes-Acr+* is able to oxidize cyanide to the corresponding radical. However, it is unlikely that this reaction proceeds through this pathway, as it has been shown that cyanide radical adds to arenes in an unselective manner, giving nearly statistical mixtures of the ortho-, meta-, and para-substituted products.40 Additionally, it has been shown that the oxidation of cyanide in the presence of water quickly generates cyanate anions.41 It is unlikely that cyanate anions would act as reactive species to produce the desired benzonitriles in this system. Finally, the observed ortho–para regioselectivity is consistent with prior work and can be best explained by the location of partial charges on intermediate 5.25,27
Scheme 2.

Possible Mechanism for Photoredox-Mediated Generation of Benzonitriles
In conclusion, we have developed a photoredox-catalyzed system taking advantage of an acridinium photooxidant to achieve direct C–H functionalization to furnish aromatic and heteroaromatic nitriles. The conditions developed are mild and proved to be compatible with sensitive or reactive functionalities. The reaction also avoids undesired reactivity at the benzylic positions of the substrates, allowing for exclusive aromatic ring C–H functionalization. Future studies are aimed at elucidating the origins of the regioselectivity in more complex substrates.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (NIGMS) Award No. R01 GM120186 and an Eli Lilly Grantee Award (D.A.N.).
Footnotes
ASSOCIATED CONTENT
- Experimental procedures and supporting data 1H and 13C NMR spectra (PDF)
The authors declare no competing financial interest.
References
- 1.Hyland J, O’Connor C. J. Chem. Soc., Perkin Trans. 1973;2:223–227. [Google Scholar]
- 2.Swain C. J. Am. Chem. Soc. 1947;69:2306–2309. [Google Scholar]
- 3.Finholt AE, Jacobson EC, Ogard AE, Thompson P. J. Am. Chem. Soc. 1955;77:4163–4163. [Google Scholar]
- 4.Das B, Reddy C, Kumar D, Krishnaiah M, Narender R. Synlett. 2010;2010:391–394. [Google Scholar]
- 5.Sandmeyer T. Ber. Dtsch. Chem. Ges. 1884;17:1633–1635. [Google Scholar]
- 6.Sekiya A, Ishikawa N. Chem. Lett. 1975;4:277–278. [Google Scholar]
- 7.Takagi K, Okamoto T, Sakakibara Y, Ohno A, Oka S, Hayama N. Bull. Chem. Soc. Jpn. 1975;48:3298–3301. [Google Scholar]
- 8.Dalton JR, Regen SL. J. Org. Chem. 1979;44:4443–4444. [Google Scholar]
- 9.Akita Y, Shimazaki M, Ohta Synthesis. 1981;1981:974–975. [Google Scholar]
- 10.Chatani N, Hanafusa T. J. Org. Chem. 1986;51:4714–4716. [Google Scholar]
- 11.Sato N, Suzuki M. J. Heterocycl. Chem. 1987;24:1371–1372. [Google Scholar]
- 12.Nair V, Purdy DF, Sells TB. J. Chem. Soc., Chem. Commun. 1989:878–879. [Google Scholar]
- 13.Takagi K, Sasaki K, Sakakibara Y. Bull. Chem. Soc. Jpn. 1991;64:1118–1121. [Google Scholar]
- 14.Tschaen DM, Desmond R, King AO, Fortin MC, Pipik B, King S, Verhoeven TR. Synth. Commun. 1994;24:887–890. [Google Scholar]
- 15.Liu W, Ackermann L. Chem. Commun. 2014;50:1878. doi: 10.1039/c3cc49502g. [DOI] [PubMed] [Google Scholar]
- 16.Han J, Pan C, Jia X, Zhu C. Org. Biomol. Chem. 2014;12:8603–8606. doi: 10.1039/c4ob01736f. [DOI] [PubMed] [Google Scholar]
- 17.Gong T-J, Xiao B, Cheng W-M, Su W, Xu J, Liu Z-J, Liu L, Fu Y. J. Am. Chem. Soc. 2013;135:10630–10633. doi: 10.1021/ja405742y. [DOI] [PubMed] [Google Scholar]
- 18.Gu L-J, Jin C, Wang R, Ding H-Y. Chem Cat Chem. 2014;6:1225–1228. [Google Scholar]
- 19.Yu D-G, Gensch T, de Azambuja F, Vásquez-Céspedes S, Glorius F. J. Am. Chem. Soc. 2014;136:17722–17725. doi: 10.1021/ja511011m. [DOI] [PubMed] [Google Scholar]
- 20.Pawar AB, Chang S. Org. Lett. 2015;17:660–663. doi: 10.1021/ol503680d. [DOI] [PubMed] [Google Scholar]
- 21.Sato N, Yue Q. Tetrahedron. 2003;59:5831–5836. [Google Scholar]
- 22.Okamoto K, Watanabe M, Murai M, Hatano R, Ohe K. Chem. Commun. 2012;48:3127. doi: 10.1039/c2cc18008a. [DOI] [PubMed] [Google Scholar]
- 23.Schmittel M, Burghart A. Angew. Chem., Int. Ed. Engl. 1997;36:2550–2589. [Google Scholar]
- 24.Romero NA, Nicewicz DA. Chem. Rev. 2016;116:10075–10166. doi: 10.1021/acs.chemrev.6b00057. [DOI] [PubMed] [Google Scholar]
- 25.Romero NA, Margrey KA, Tay NE, Nicewicz DA. Science. 2015;349:1326–1330. doi: 10.1126/science.aac9895. [DOI] [PubMed] [Google Scholar]
- 26.Ohkubo K, Fujimoto A, Fukuzumi S. J. Phys. Chem. A. 2013;117:10719–10725. doi: 10.1021/jp408315a. [DOI] [PubMed] [Google Scholar]
- 27.Ohkubo K, Mizushima K, Iwata R, Fukuzumi S. Chem. Sci. 2011;2:715. [Google Scholar]
- 28.Morofuji T, Shimizu A, Yoshida J. J. Am. Chem. Soc. 2014;136:4496–4499. doi: 10.1021/ja501093m. [DOI] [PubMed] [Google Scholar]
- 29.Shu Z, Ji W, Wang X, Zhou Y, Zhang Y, Wang J. Angew. Chem., Int. Ed. 2014;53:2186–2189. doi: 10.1002/anie.201309791. [DOI] [PubMed] [Google Scholar]
- 30.Dohi T, Morimoto K, Kiyono Y, Tohma H, Kita Y. Org. Lett. 2005;7:537–540. doi: 10.1021/ol0476826. [DOI] [PubMed] [Google Scholar]
- 31.Sundermeier M, Mutyala S, Zapf A, Spannenberg A, Beller M. J. Organomet. Chem. 2003;684:50–55. [Google Scholar]
- 32.Ankala S, Fenteany G. Tetrahedron Lett. 2002;43:4729–4732. [Google Scholar]
- 33.Tsukamoto H, Suzuki T, Kondo Y. Synlett. 2007;2007:3131–3136. [Google Scholar]
- 34.Vutukuri DR, Bharathi P, Yu Z, Rajasekaran K, Tran M-H, Thayumanavan S. J. Org. Chem. 2003;68:1146–1149. doi: 10.1021/jo026469p. [DOI] [PubMed] [Google Scholar]
- 35.Chandrasekhar S, Reddy CR, Rao RJ. Tetrahedron. 2001;57:3435–3438. [Google Scholar]
- 36.Riener M, Nicewicz DA. Chem. Sci. 2013;4:2625. doi: 10.1039/C3SC50643F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dong Y, Busacca CA. J. Org. Chem. 1997;62:6464–6465. [Google Scholar]
- 38.Bronner SM, Goetz AE, Garg NK. J. Am. Chem. Soc. 2011;133:3832–3835. doi: 10.1021/ja200437g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zweig A, Hodgson W, Jura W. J. Am. Chem. Soc. 1964;86:4124–4129. [Google Scholar]
- 40.Spagnolo P, Testaferri L, Tiecco M. J. Chem. Soc. B. 1971:2006–2008. [Google Scholar]
- 41.Tamura H, Arikado T, Yoneyama H, Matsuda Y. Electrochim. Acta. 1974;19:273–277. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.