

Abstract
Chronic eosinophilic leukemia, not otherwise specified can be difficult to distinguish from idiopathic hypereosinophilic syndrome according to the current World Health Organization guideline. To examine whether the morphological features of bone marrow might aid in the differential diagnosis of these two entities, we studied a total of 139 patients with a diagnosis of chronic eosinophilic leukemia, not otherwise specified (n=17) or idiopathic hypereosinophilic syndrome (n=122). As a group, abnormal bone marrow morphological features, resembling myelodysplastic syndromes, myeloproliferative neoplasm or myelodysplastic/myeloproliferative neoplasm, were identified in 40/139 (27%) patients: 16 (94%) of those with chronic eosinophilic leukemia and 24 (20%) of those with hypereosinophilic syndrome. Abnormal bone marrow correlated with older age (P<0.001), constitutional symptoms (P<0.001), anemia (P=0.041), abnormal platelet count (P=0.002), organomegaly (P=0.008), elevated lactate dehydrogenase concentration (P=0.005), abnormal karyotype (P<0.001), as well as the presence of myeloid neoplasm-related mutations (P<0.001). Patients with abnormal bone marrow had shorter survival (48.1 months versus not reached, P<0.001), a finding which was independent of other confounding factors (P<0.001). The association between abnormal bone marrow and shorter survival was also observed in hypereosinophilic syndrome patients alone. In summary, most patients with chronic eosinophilic leukemia, not otherwise specified and a proportion of those with idiopathic hypereosinophilic syndrome show abnormal bone marrow features similar to the ones encountered in patients with myelodysplastic syndromes, myelodysplastic/myeloproliferative neoplasm or BCR-ABL1-negative myeloproliferative neoplasm. Among patients who are currently considered to have idiopathic hypereosinophilic syndrome, abnormal bone marrow is a strong indicator of clonal hematopoiesis. Similar to other myeloid neoplasms, bone marrow morphology should be one of the major criteria to distinguish patients with chronic eosinophilic leukemia, not otherwise specified or clonal hypereosinophilic syndrome from those with truly reactive idiopathic hypereosinophilic syndrome.
Introduction
Hypereosinophilia is defined by the presence of ≥1.5×109/L eosinophils in the peripheral blood (PB) and may be reactive, neoplastic or idiopathic.1–3 Chronic eosinophilic leukemia, not otherwise specified (CEL, NOS)4 is a myeloproliferative neoplasm (MPN), characterized by an expansion of eosinophils but lacking well-defined molecular genetic alterations such as BCR-ABL1 and rearrangements of PDGFRA, PDGFRB, FGFR1 and PCM1-JAK2. In idiopathic hypereosinophilic syndrome (HES), there is tissue/organ damage related to an eosinophilic infiltrate/activation, but the cause of the hypereosinophilia is unknown.
Due to substantial overlapping of their features, idiopathic HES was described alongside CEL, NOS in the World Health Organization (WHO) classification monograph;4 this categorization remains largely unmodified in the 2016 WHO revision.5 According to the current guidelines, CEL, NOS, can only be reliably separated from idiopathic HES by the presence of increased blasts in bone marrow (BM) and/or PB, or proof of clonality. Clonality was mainly determined by chromosomal analysis or testing for mutations well known to occur in MPN, such as JAK2, MPL, CALR and KIT. However, these mutations are uncommon in the eosinophilic diseases.6,7 More recently, next-generation sequencing (NGS) approaches have been applied to these eosinophilic disorders. Anderson and colleagues conducted whole-exome sequencing of nine patients with idiopathic HES8 and identified somatic missense mutations in three of them. The mutations they found involved the spliceosome gene PUF60 and the cadherin gene CDH17. More recently, using NGS with a gene panel targeted to somatic mutations commonly associated with myeloid neoplasms, we detected the presence of mutations at a relatively high (≥10%) variant allele frequency (VAF) in 25–30% of cases of idiopathic HES.7 These mutations mostly occurred in genes involved in DNA methylation and chromatin modification, such as AXSL1, TET2, EZH2, and DNMT3A. While such mutations would imply that some idiopathic HES are clonal stem cell neoplasms, they have also been reported in some aging individuals without evidence of a myeloid neoplasm,9,10 mandating caution in the use of mutations as definitive proof of a neoplastic myeloid process. On the other hand, the detection of mutations by NGS relies on the testing panel used, which may vary for the number of genes sequenced as well as the depth of sequencing.
Although some studies have suggested that abnormal eosinophil morphology is associated with clonal eosinophilia, it is generally felt that cytological abnormalities lack sufficient specificity to differentiate a neoplastic process from a reactive eosinophilia.4,11–14 As a result, BM morphology is not an integral part of the diagnosis and classification of hypereosinophilia. This is in apparent contrast to the situation in other myeloid neoplasms, in which abnormal BM features play a major role in establishing the diagnosis. In particular, BM morphology represents a “gold standard” in the diagnosis of myelodysplastic syndromes (MDS) and MDS/MPN. With regards to MPN, morphology has become one of the major criteria in the WHO classification (2016)5 of essential thrombocythemia, polycythemia vera, and primary myelofibrosis. In contrast, in the case of CEL, NOS or of HES with clonal eosinophilia, there is limited published information in relation to BM morphology. In our previous study,7 with molecular genetic information for patients, we observed some BM features that appeared to be preferentially present in cases with molecular genetic alterations. In this study, we conducted a thorough review of BM morphology of a large series of CEL, NOS and idiopathic HES cases collected from seven large medical centers in the USA using a defined set of morphological criteria, blinded to the original diagnosis and molecular genetic data. We used the morphological features to define an “abnormal” BM morphology, and correlated the morphological results with clinical and laboratory features, cytogenetics, mutation data, and patients’ outcomes. We sought to determine whether morphology can be utilized in the distinction of CEL, NOS and clonal HES from truly reactive idiopathic HES.
Methods
Patients
Cases were collected from MD Anderson Cancer Center, Stanford University Medical Center, Cleveland Clinic, Massachusetts General Hospital, Weill-Cornell Medical College, the Hospital of the University of Pennsylvania and the University of New Mexico between 2005 and 2014. All included patients had persistent hypereosinophilia (≥1.5×109/L) and did not have acute leukemias, chronic myeloid leukemia, MDS, chronic myelomonocytic leukemia, systemic mastocytosis, or rearrangements of PDGFRA, PDGFRB, FGFR1 or PCM1-JAK2. For idiopathic HES, every patient had “end-organ damage” according to the definition by the working group on eosinophil disorders.15 Lymphocytic/T-cell variant HES1 was excluded based on the identification of aberrant T cells by flow cytometry with or without TCR gene rearrangement polymerase chain reaction studies. Clinical information was retrieved from the electronic medical records. This study was approved by the Institutional Review Boards of all participating institutions.
Bone marrow morphology and histology
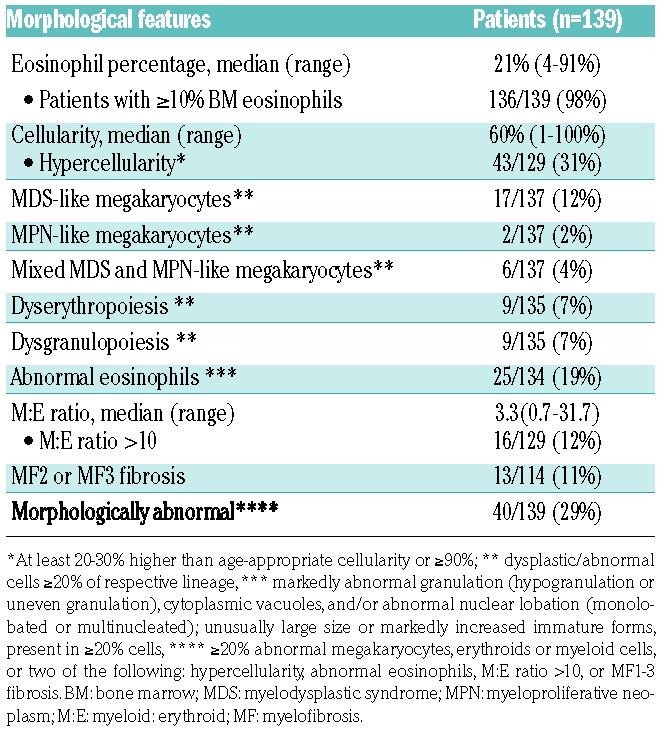
BM was assessed for the following morphological parameters (Table 1): cellularity; megakaryocyte numbers, morphology and distribution; fibrosis; dysgranulopoiesis; dyserythropoiesis; myeloid:erythroid (M:E) ratio; and eosinophil morphology. PB smears were also reviewed for eosinophil morphology and evidence of dysgranulopoiesis. Hypercellularity was defined by a cellularity at least 20% higher than the age-appropriate cellularity, and overall ≥70% in patients age 50–60 years; ≥60% in patients >60 years; and ≥90% in patients <30 years of age. Megakaryocyte morphology was recorded as predominantly MDS-like (small with hypolobated/non-lobated nuclei or separated nuclear lobes), MPN-like (medium-sized to large megakaryocytes with hyperlobulated, hyperchromatic, or bulbous nuclei, often with clustering and increase in numbers), mixed MDS and MPN-like, or within normal limits (WNL). In order to define dysgranulopoiesis and dyserythropoiesis strictly, the features had to be seen in ≥20% of cells of the assessed lineage. Myelofibrosis grade was assessed according to the European Bone Marrow Fibrosis Consensus criteria.16 For eosinophil morphology, abnormal features were markedly abnormal granulation (hypogranulation or uneven granulation), cytoplasmic vacuoles, abnormal nuclear lobation (non-lobated or multilobated), unusually large size or markedly increased immature forms. These features had to be observed in at least 20% of the eosinophils on the BM smears, since mild nuclear hypersegmentation and mild abnormal granulation in the PB can be seen with eosinophil activation17 or treatment with hydroxyurea.18 All cases were assessed by the members from the respective institution using the same set of criteria, which were developed by members of the Bone Marrow Pathology study group after having reviewed representative cases as a group. The features were reassessed by one observer (SAW); cases with borderline morphological abnormalities or discrepancy were again centrally reviewed by the group and scored by consensus. There was some disagreements on some of the parameters for approximately 10% of cases (n=13), but all members agreed on “abnormal” or “not abnormal” morphology for all cases. The disagreements mainly related to eosinophilic morphology, since the criteria were not previously defined; disagreements on scoring megakaryocyte morphology were present in a smaller subset of cases and centered on whether the features were MDS-like or mixed MDS/MPN-like. All morphology reviews were blinded to clinical features, molecular genetic data, original diagnoses and patients’ outcomes.
Table 1.
Bone marrow morphological findings of patients with a diagnosis of chronic eosinopil leukemia, not otherwise specified or idiopathic hypereosinophilic syndrome
Cytogenetics, fluorescence in situ hybridization and molecular testing
Conventional cytogenetic analysis was performed on G-banded metaphase cells prepared from unstimulated BM aspirate cultures using standard techniques. Twenty metaphases were analyzed and the results were reported using the International System for Human Cytogenetic Nomenclature. Fluorescence in situ hybridization (FISH) and/or molecular genetic methods for detecting BCR-ABL1, PDGFRA, PDGFRB, and FGFR1 were performed at the respective institutions as part of the routine clinical work-up, if indicated.
Targeted next-generation sequencing
Targeted NGS had been performed on 57 patients previously7 and was performed in an additional 19 cases on DNA samples extracted from frozen unfractionated BM cells collected at the time of diagnosis, using the same method we described previously.7 The coding sequences of 44 genes (sequencing >90% gene coding regions), including ABL1, ASXL1, BCOR, BRAF, CALR, CBL, CEBPA, DNMT3A, ETV6, EZH2, FAM5C, FLT3 (ITD and TKD), GATA1, GATA2, HNRNPK, IDH1, IDH2, IKZF1, JAK1, JAK2, KDM6A, KIT, KRAS, MPL, NFE2, NOTCH1, NPM1, NRAS, PHF6, PTPN11, RAD21, RUNX1, SEPBP1, SF3B1, SH2B3, SMC1A, SMC3, STAG2, SUZ12, TET2, TP53, U2AF1, WT1, and ZRSR2, were investigated specifically for this study. Variant calling was performed with Illumina MiSeq Reporter software 1.3.17. using human genome build 19 (hg 19) as a reference.
Statistical analyses
Data for continuous variables were reported as medians and ranges. Data for nominal variables were reported as the number of patients unless otherwise specified. Survival was calculated from the date of diagnosis to the date of last follow-up or death not attributable to causes that were clearly not associated with disease (e.g., car accident, suicide). Patients who underwent hematopoietic stem cell transplantation were censored at the time of the procedure. Distribution of survival was estimated by Kaplan-Meier curves. Multivariable analysis was performed using a Cox regression model. Fisher exact and χ2 tests were used for categorical comparisons. All P values are two-tailed and considered statistically significant when <0.05. No adjustments for multiplicity were made.
Results
Patients, clinical data and molecular genetic data
A total of 139 patients were included in the study: these patients met the criteria for CEL, NOS (17 patients) or idiopathic HES (122 patients) after applying the exclusion and inclusion criteria and had sufficient material for morphological assessment. An abnormal karyotype was seen in 16 of the 17 CEL, NOS patients; detailed karyotype information on these cases was published previously.7 In brief, five patients had a complex karyotype, one had two cytogenetic abnormalities, nine had a single abnormality, and one was identified by FISH as having a del(9p) abnormality. Three patients had ≥5% BM blasts, including one patient with a normal BM karyotype who was also diagnosed as having CEL, NOS according to the WHO Classification criteria. The clinical and laboratory features of these patients as a group are shown in Table 2.
Table 2.
Clinical, molecular genetic features and survival comparison of patients with morphologically abnormal bone marrow or bone marrow within normal limits.

NGS was performed in 76 patients. In total, mutations were found in 21/76 patients (27.6%). The mutation data and frequency are shown in Figure 1. In brief, the mutations, in decreasing frequency, were: ASXL1 (7/76, 9.2%); TET2 (5/76, 6.6%); EZH2 (5/76, 6.6%), DNMT3A (5/76, 6.6%), NOTCH1 (4/76, 5.3%), SETBP1 (3/76, 4.0%); CBL (2/76, 2.6%); U2AF1 (2/76, 2.6%), and one each (1.3%) of TP53, JAK2 exon 13, NRAS, BCOR, GATA2, CSF3R and ETV6. Two or more mutations were found in 8/76 (11%) patients. Overall, mutations were identified in 18/70 (25.7%) tested cases of idiopathic HES and 3/6 tested cases of CEL, NOS (50%).
Figure 1.
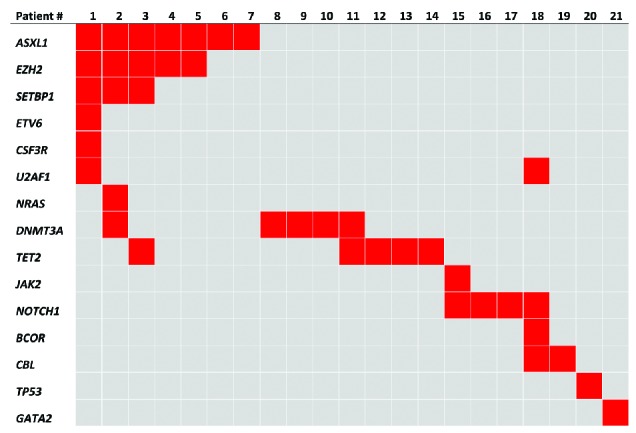
Mutations detected in 21 patients with a diagnosis of chronic eosinophilic leukemia, not otherwise specified/idiopathic hypereosinophilic syndrome.
Bone marrow morphology
BM morphology was evaluated in conjunction with PB smears, blinded to all clinical, laboratory, molecular genetic data and patients’ outcome. Increased BM eosinophils were seen in the majority of the cases, accounting for a median of 21% (range, 4–91%) of BM cells; only three patients had <10% eosinophils in the BM. In two-thirds of the cases (71%), BM was unremarkable except for increased eosinophils (Figure 2). In contrast, in one-third of cases (29%), besides the increased eosinophils, a number of other changes in BM were observed; these are shown in Table 1. The most common abnormalities (Figure 3) were BM hypercellularity, abnormal eosinophils, abnormal megakaryocytes, a markedly elevated M:E ratio ≥10; moderate to marked fibrosis, dysgranulopoiesis, and dyserythropoiesis.
Figure 2.
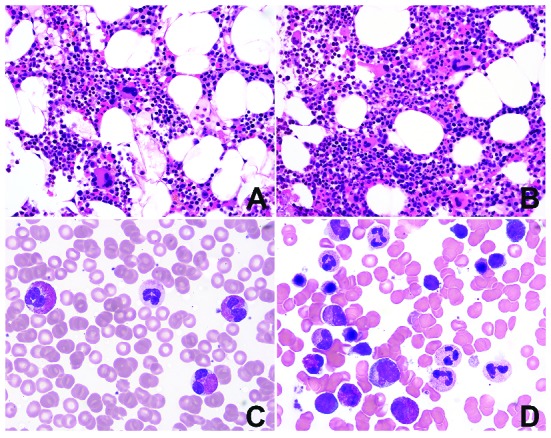
Many cases with idiopathic hypereosinophilic syndrome. show unremarkable bone marrow morphology. Bone marrow (BM) cellularity is either age-appropriate [(A) patient aged 48 years] or only slightly increased [(B) patient aged 45 years], with increased BM eosinophils and normal-appearing megakaryocytes. (C) Eosinophils in peripheral blood (PB) may show mild uneven granulation (D) but are unremarkable on BM smear. No dysgranulopoiesis or dyserythropoiesis (BM biopsy, hematoxylin & eosin, original magnification ×400; PB and BM smears Wright-Giemsa, original magnification ×1000).
Figure 3.
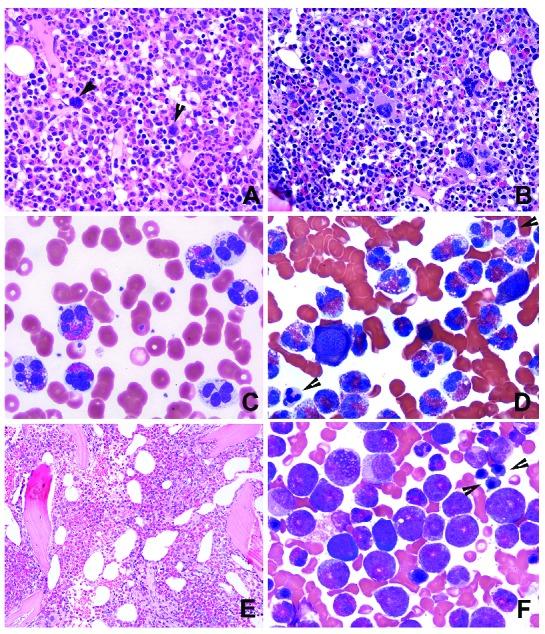
Morphologically abnormal bone marrow. (A, B) Bone marrow (BM) hypercellularity with increased eosinophils and neutrophilic granulocytic elements; frequent small hypolobated MDS-like megakaryocytes (A, arrows) or mixed MDS- and MPN-like megakaryocytes (B). (C) Peripheral blood (PB) shows abnormal eosinophils with multiple lobes and marked hypogranulation or agranulation. (D) The same changes are also observed in the BM from the same case. In addition, dysplastic erythroids and granulocytes (arrows) are evident. (E, F) A case with decreased megakaryocytes, hypercellularity with disrupted BM topography (E) and a BM smear showing markedly increased immature eosinophils and dyserythropoiesis (F, arrows). (BM biopsy: hematoxylin & eosin, original magnification ×400; PB and BM smears: Wright-Giemsa, original maginification ×1000)
Cases were considered to be morphologically abnormal if they showed overtly abnormal megakaryocytes (resembling those in MDS or MPN), significant dysgranulopoiesis or dyserythropoiesis, or increased (≥5%) BM blasts. These included 25 cases with abnormal megakaryocytes, most of which showed MDS-like morphology (Online Supplementary Table S1). Of these 25 cases, 15 also had abnormal eosinophils; three had ≥5% BM blasts, six had MF2 or MF3 myelofibrosis; 19 had hypercellularity, six had dysgranulopoiesis, and eight showed dyserythropoiesis. Of patients whose BM did not show abnormal megakaryocytes or had insufficient megakaryocytes for assessment, in four cases the BM was concluded to be abnormal, including three with marked dysgranulopoiesis and one with marked dyserythropoiesis (2 of these also showed abnormal eosinophils; 2 with hypercellularity and 1 with MF2 fibrosis). An additional 11 cases were scored as “abnormal” because of the presence of at least two of the following changes: BM hypercellularity (n=10); MF2 or MF3 fibrosis (n=6); abnormal eosinophils (n=4); M:E ratio >10 (n=1); and markedly decreased/near absence of megakaryocytes (n=2), of which one with a subset of MDS-like megakaryocytes (see Online Supplementary Table S1). There were also increased macrophages/histiocytes, stromal cells, vessels, and a disarrayed distribution of the BM cellular elements in some of these cases. These 11 cases were centrally reviewed, and one example is shown in Figure 3E, F.
Thus, 40/139 cases (29%) were considered to have abnormal BM morphology and 99 had either normal morphology or only one morphological abnormality that did not include significant dysplasia, abnormal megakaryocytes, or excess blasts. In total, 16 of the 17 (94%) cases of CEL, NOS and 24 of 122 (22%) of the HES cases were morphologically abnormal. If the current WHO definitions of CEL, NOS and HES were used to anchor the reviewed cases as “true positives” for each diagnosis, abnormal morphology would have a sensitivity of 94.1% (95% confidence interval: 71.3–99.8%) and a specificity of 84.7% (95% confidence interval: 77.8–90.2%) for CEL, NOS.
Correlation of bone marrow morphology with clinical features
The clinical presentations of the 40 patients with abnormal BM differed from those of the 99 patients who lacked significantly abnormal BM findings (Table 2). The patients with abnormal BM were older and presented with a higher white blood cell count and a higher absolute eosinophil count. These patients also had lower hemoglobin levels and more commonly had abnormal platelet counts (thrombocytopenia or thrombocytosis) (P=0.002). Clinically, more patients with abnormal BM morphology presented with constitutional symptoms (19/40 versus 17/97, P<0.001), but fewer with allergy/hypersensitivity (2/40 versus 30/97, P<0.001); cough, bronchitis, or pneumonitis (1/40 versus 24/97, P<0.001); gastrointestinal symptoms (5/40 versus 28/97, P=0.049); and heart failure, myocardial infarction or pericardial effusion (2/40 versus 20/97, P=0.012). Skin rashes and various forms of dermatitis were common in both groups of patients. An abnormal BM also correlated with more frequent organomegaly and elevated lactate dehydrogenase levels (Table 2).
Correlation of bone marrow morphology with molecular genetic data
Of 17 patients with abnormal cytogenetics and/or increased BM blasts (CEL, NOS by the current WHO criteria), 16 had an abnormal BM. The one patient with CEL-NOS without abnormal BM morphology was a 64-year old female who presented with chest pain, and was found to have elevated troponin, and pericardial and pleural effusions. This patient had del(16)(q23q24) and her BM showed 40% eosinophils, but otherwise had normal morphology. FISH was negative for both CBFB and CHIC2. The patient was alive at 24 months of follow-up.
Mutations detected by NGS were more frequent in patients with abnormal BM (12/20 versus 9/56, P<0.001). Mutations involving two or more genes were significantly more common in patients with abnormal BM (7/20 versus 1/56, P<0.001). Moreover, TP53, EZH2, SETBP1, NRAS, JAK2 exon 13 and CSF3R were only seen in patients with abnormal BM. Of patients who lacked abnormal BM findings, mutations included single gene mutations in TET2 (n=2), DNMT3A (n=3, 2 with 5–10% VAF); ASXL1 (n=1), CBL (n=1), or NOTCH1 (n=1). The only patient who had two mutations (TET2, VAF 25% and DNMT3A, VAF 15%) but no abnormal BM findings, was a 24-year old man who presented with fever and chest pain, dizziness and sensory abnormalities in both hands. The patient had abnormal magnetic resonance imaging findings in the brain and lungs, likely due to eosinophilic infiltrates. He showed some response to corticosteroids, but did not tolerate imatinib. He was alive at 28 months of follow-up.
Correlation of bone marrow morphology with outcome data
These patients were treated with various agents recommended for people with idiopathic HES/CEL, NOS, including corticosteroids with or without interferon, hydroxyurea for cytoreduction, cyclosporine, methotrexate, and alemtuzumab. Tyrosine kinase inhibitors, mostly imatinib and dasatinib in some cases, were used in 54/126 (43%) patients over the course of the disease. Hypomethylating agents, single-agent chemotherapy, and high-dose chemotherapy were also used in some patients when their disease progressed or was refractory to other treatment modalities. A total of seven patients underwent hematopoietic stem cell transplantation.
The median follow-up for all 139 patients was 38.9 months (range, 0 – 405.3 months). Three unrelated deaths (1 due to suicide, 1 due to a car accident and 1 due to diffuse large B-cell lymphoma), were censored at the time of death. Of 40 patients with abnormal BM, there were 18 deaths, including three due to progression of acute myeloid leukemia. The other causes of death included infection, bleeding and organ failure. Among the five patients in this group who received a hematopoietic stem cell transplant, four were alive and one died of disease recurrence. In contrast, of 99 patients without abnormal BM morphology, none experienced acute myeloid leukemia progression. There were eight deaths in this group, including four due to myocardial infarction or heart failure, two due to chronic obstructive lung disease, one due to the complication of a bone fracture as a result of long-term steroid use, and another of unknown cause. Both patients who received a hematopoietic stem cell transplant were alive at the last follow-up. The median overall survival for patients with an abnormal BM was 48.1 months (range, 1–120.1 months), which was significantly inferior to that of patients with a normal BM (not reached; range, 0–277.2) (Kaplan-Meier log rank, P<0.001) (Figure 4A). Survival was also compared in patients with a normal karyotype and <5% BM blasts, who would be considered as having idiopathic HES by the current WHO criteria. Within this group of patients, abnormal BM morphology remained a predictor of an inferior survival (median 125.5 months versus not reached, Kaplan-Meier log rank, P<0.001) (Figure 4B).
Figure 4.
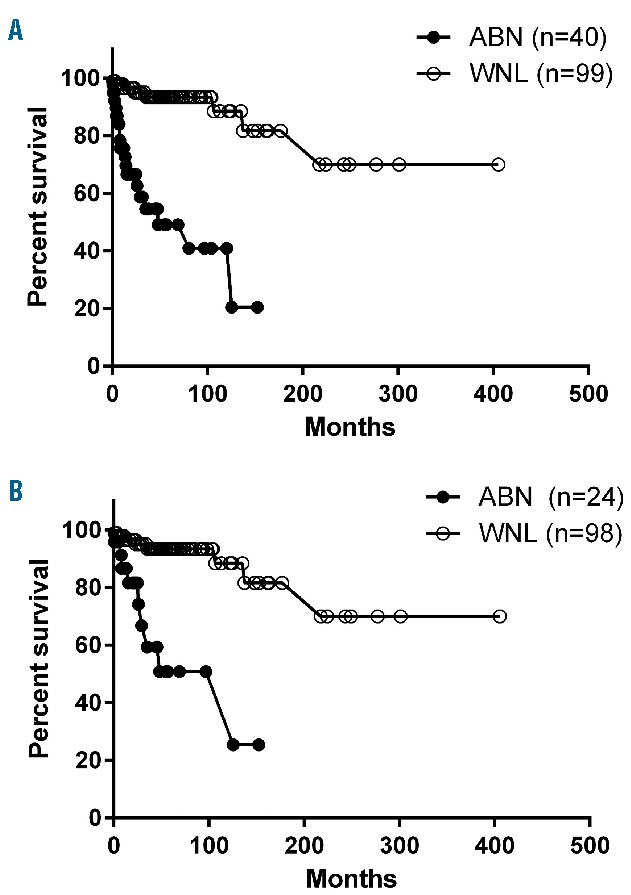
Comparison of survival of patients with chronic eosinophilic leukemia, not otherwise specified/idiopathic hypereosinophilic syndrome. (A) All patients (n=139): patients with morphologically abnormal bone marrow (ABN) had a median survival of 48.1 months, which is significantly inferior to that of patients with a normal BM (WNL) (unreached, P<0.001). (B) For patients who would otherwise be classified as having idiopathic HES (a normal karyotype and/or <5% blasts, n=122), an abnormal BM was also significantly associated with a shorter survival (P<0.001).
The prognostic significance of abnormal morphology was tested in multivariable analysis. The variables included age, gender, white blood cell count, absolute eosinophil count, hemoglobin level, lactate dehydrogenase concentration, organomegaly, constitutional symptoms, heart or brain involvement, cytogenetics, mutations, and the presence of two or more mutations. In the final multivariable Cox regression model, only age, thrombocytopenia, heart and/or brain involvement and abnormal BM morphology emerged as significant prognostic factors (Table 3); abnormal karyotype, mutations, and other clinical and laboratory parameters were not independently significant. Multivariable analysis was also performed in the subset of 122 patients with a normal karyotype and <5% BM blasts, who would be classified as having idiopathic HES by the current WHO criteria; abnormal morphology remained an independent predictor for inferior survival (Table 3).
Table 3.
Factors independently predicting an inferior survival of patients with chronic eosinophilic leukemia, not otherwise specified/idiopathic hypereosinophilic syndrome (n=139) as well as of patients with idiopathic hypereosinophilic syndrome (n=122) only in multivariable analysis*

Discussion
In this study, we reviewed the BM of 139 patients who presented with hypereosinophilia without recurrent molecular genetic alterations or a known reactive cause. According to the current WHO classification criteria, 17 (12%) of these patients would be classified as having CEL, NOS, either due to the presence of an abnormal karyotype and/or increased BM blasts. However, abnormal BM morphology, with features resembling MDS, MDS/MPN or MPN, was observed in 40 of these patients, including 16 of 17 (94%) patients who were classified as having CEL, NOS and 24 of the 122 (20%) patients who were classified as having idiopathic HES.
The criteria used to assess BM morphology were in part derived from what we had observed previously7 by comparing cases with molecular genetic abnormalities versus no identifiable abnormalities. These included increased blasts, hypercellularity, abnormal megakaryocytes, dyserythropoiesis and dysgranulopoiesis, markedly elevated M:E ratio and fibrosis, and abnormal eosinophils. The definitions of “abnormal” BM findings were similar to those characteristically found in other myeloid neoplasms, including MDS, MPN and MDS/MPN, except for the inclusion of eosinophil morphology. In these patients, a BM eosinophilic infiltrate was invariably present, with only three patients having <10% eosinophils in the BM. However, in two-thirds of the patients, an increase in BM eosinophils either did not significantly alter or only led to a slight increase in BM cellularity. In patients with significant BM hypercellularity, this was frequently due to increased neutrophils and their precursors, megakaryocytes, and in some, erythroid precursors, or less commonly to an increased number of macrophages. Additional changes included increased stromal cells, histiocytes, vessels, and disarrayed cellular distribution, which are often referred by others as altered BM topography.19 Recognizing that cytological eosinophil atypia may be seen in reactive eosinophilia,4,11–14 we arbitrarily considered eosinophil morphology abnormal only if at least 20% of the eosinophils were involved. Interestingly, we found that mild atypical changes in reactive eosinophils were more frequently observed in PB than in BM (Figure 2), suggesting that BM smears may be more reliable for the assessment of eosinophil morphology. Nevertheless, of the 25 patients who showed significant numbers of abnormal eosinophils, 22 also had other BM abnormalities and only three patients had it as the sole alteration. Of these latter three patients, two had a long-standing history of allergy and gastrointestinal symptoms and one patient had deep venous thrombosis, endocardial fibrosis and Budd-Chiari syndrome. All three of these patients had a normal karyotype, and two tested by NGS were negative for mutations. Our findings suggest that alterations in eosinophil morphology can be used in conjunction with other BM findings in morphological assessment, but, if it is the sole alteration, may be unreliable to differentiate a neoplastic process from reactive eosinophilia.4,11–14
Prior to this study, there have been very few published studies with descriptions of megakaryocytes in patients with hypereosinophilia,20 even in well-defined entities such as PDGFRA- and PDGFRB-rearranged myeloid/lymphoid neoplasms.21–23 In our series, abnormal megakaryocyte morphology was frequently observed, with cytological features mostly resembling those of MDS-type megakaryocytes or mixed small and large megakaryocytes, with only a few cases showing megakaryocyte morphology similar to that present in BCR/ABL1-negative MPN. Some patients showed dysgranulopoiesis and/or dyserythropoiesis. These findings in a patient with eosinophilia suggest a clonal neoplastic process. On the other hand, the presence of dysplastic changes in association with thrombocytopenia and anemia, seen in some of these patients, may raise the question of whether such cases should be considered more closely related to a MDS/MPN rather than to a true MPN, the nosological attribution of CEL, NOS and HES in the current WHO classification scheme.
Clinically, patients with abnormal BM often showed features suggestive of a myeloid neoplasm, more frequently having constitutional symptoms, organomegaly, higher lactate dehydrogenase concentration, higher white blood cell and absolute eosinophil counts, abnormal platelet count and anemia. In contrast, they had fewer symptoms related to eosinophil activation, such as allergy, hypersensitivity, arthritis, muscle aches, and gastrointestinal, pulmonary, and cardiac-related symptoms. There were 18 (45%) disease-associated deaths in patients with abnormal BM morphology, including three which occurred due to progression to acute myeloid leukemia; many deaths were due to complications of BM failure. In contrast, there were only eight (8%) deaths in patients with normal BM morphology, and the causes of deaths were mainly cardiac or pulmonary complications. Patients with abnormal BM had a significantly inferior median survival compared to that of patients without significantly abnormal BM morphology. The median survival of 48.1 months appeared to be better than the previously reported 15–22 months7,20 in patients with CEL, NOS. However, the previous studies only included cases with an abnormal karyotype and/or increased blasts. A similar significance of abnormal BM morphology for survival was also observed in HES patients with a normal karyotype and no increased BM or PB blasts, who otherwise would be diagnosed as having idiopathic HES. These findings were underscored in the multivariable analysis, which showed that abnormal BM morphology, but not abnormal BM karyotype, was an independent prognostic marker when other factors were co-analyzed.
In this study, we were also able to correlate mutational data with morphology and clinical data. Of 76 patients studied, 21 (27.6%) were found to have mutations. Similarly to what we found previously,7 mutations frequently involved genes involved in DNA methylation and chromatin modification, such as ASXL1, TET2, and DNMT3A. Although, most of these mutations are also frequently reported to occur in normal aging individuals,9,10 making it challenging to apply mutation data in the establishment of a clonal hematopoietic stem cell neoplasm, mutations involving at least one gene (60% versus 16%) as well as two or more genes were significantly more frequent in patients with abnormal BM morphology. The caveat was that the patients with abnormal BM morphology were significantly older (64.0 versus 47.7 years). Interestingly, it has been shown recently that in chronic myelomonocytic leukemia, age-related somatic mutations through successive acquisition convert a myelomonocytic biased hematopoiesis into overt leukemia.24 We noted that in our patients mutations more specific for a myeloid neoplasm (TP53, EZH2, SETBP1, NRAS, CSF3R, JAK2) were only found in subjects with abnormal BM morphology. Similar findings have been reported in MDS, in that certain specific mutations, the number of mutations and VAF, are predictive of MDS evolvement in cytopenic patients with clonal hematopoiesis of undetermined potential.9,25 Based on our findings of differences in mutation frequency and affected genes associated with BM morphology, we recommend that abnormal BM morphology should prompt NGS study using a myeloid mutation panel to try to establish evidence of clonality, and identify a neoplastic hematopoietic disease.
In summary, we found that most patients with CEL, NOS and about 20% of those with idiopathic HES have abnormal BM morphology, while the remainder have unremarkable BM morphology with the exception of increased eosinophils. The abnormal BM findings in these cases are similar to those seen in MDS, MDS/MPN and/or BCR-ABL1-negative MPN. Isolated cytological abnormality of eosinophils is not entirely specific for a neoplastic process, but its presence should prompt careful assessment of BM morphology and appropriate molecular genetic testing. We found that abnormal BM morphology correlates with clinical presentations typically associated with a myeloid neoplasm, such as constitutional symptoms, splenomegaly, high lactate dehydrogenase concentration, anemia and abnormal platelet counts and less commonly with symptoms associated with an eosinophil activation syndrome, such as allergy, respiratory or gastrointestinal symptoms, or cardiac involvement. Abnormal BM morphology is significantly correlated with abnormal karyotype and the presence of myeloid neoplasm-related mutations, and is highly associated with inferior patients’ outcome. The prognostic significance is independent of the effect of abnormal karyotypes, mutations or other risk factors. We conclude that abnormal BM morphology should be regarded as a critical parameter useful for identifying a neoplastic subset of patients considered to have idiopathic HES. The presence of abnormal BM findings should be added to the abnormal karyotype and excess PB and/or BM blasts as one of the defining criteria for diagnosing CEL, NOS.
Supplementary Material
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/102/8/1352
References
- 1.Gotlib J. World Health Organization-defined eosinophilic disorders: 2015 update on diagnosis, risk stratification, and management. Am J Hematol. 2015; 90(11):1077–1089. [DOI] [PubMed] [Google Scholar]
- 2.Bain BJ. Eosinophilic leukaemias and the idiopathic hypereosinophilic syndrome. Br J Haematol. 1996;95(1):2–9. [PubMed] [Google Scholar]
- 3.Butt NM, Lambert J, Ali S, et al. Guideline for the investigation and management of eosinophilia. Br J Haematol. 2017;176(4): 553–572. [DOI] [PubMed] [Google Scholar]
- 4.Bain BJ, G D, Vardiman JW, Horny H-P. Chronic eosinophilic leukemia, not otherwise specified. In: Swerdlow SH, Campo E, Harris NL, et al., ed. WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues: Lyon, France: IARC Press, 2008:51–53. [Google Scholar]
- 5.Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–2405. [DOI] [PubMed] [Google Scholar]
- 6.Schwaab J, Umbach R, Metzgeroth G, et al. KIT D816V and JAK2 V617F mutations are seen recurrently in hypereosinophilia of unknown significance. Am J Hematol. 2015;90(9):774–777. [DOI] [PubMed] [Google Scholar]
- 7.Wang SA, Tam W, Tsai AG, et al. Targeted next-generation sequencing identifies a subset of idiopathic hypereosinophilic syndrome with features similar to chronic eosinophilic leukemia, not otherwise specified. Mod Pathol. 2016;29(8):854–864. [DOI] [PubMed] [Google Scholar]
- 8.Andersen CL, Nielsen HM, Kristensen LS, et al. Whole-exome sequencing and genome-wide methylation analyses identify novel disease associated mutations and methylation patterns in idiopathic hypereosinophilic syndrome. Oncotarget. 2015;6(38):40588–40597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cargo CA, Rowbotham N, Evans PA, et al. Targeted sequencing identifies patients with preclinical MDS at high risk of disease progression. Blood. 2015;126(21):2362–2365. [DOI] [PubMed] [Google Scholar]
- 10.Steensma DP, Bejar R, Jaiswal S, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. 2015; 126(1):9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chusid MJ, Dale DC, West BC, Wolff SM. The hypereosinophilic syndrome: analysis of fourteen cases with review of the literature. Medicine (Baltimore). 1975;54(1):1–27. [PubMed] [Google Scholar]
- 12.Flaum MA, Schooley RT, Fauci AS, Gralnick HR. A clinicopathologic correlation of the idiopathic hypereosinophilic syndrome. I. Hematologic manifestations. Blood. 1981;58(5):1012–1020. [PubMed] [Google Scholar]
- 13.Kueck BD, Smith RE, Parkin J, Peterson LC, Hanson CA. Eosinophilic leukemia: a myeloproliferative disorder distinct from the hypereosinophilic syndrome. Hematol Pathol. 1991;5(4):195–205. [PubMed] [Google Scholar]
- 14.Weller PF, Bubley GJ. The idiopathic hypereosinophilic syndrome. Blood. 1994; 83(10):2759–2779. [PubMed] [Google Scholar]
- 15.Valent P, Klion AD, Horny HP, et al. Contemporary consensus proposal on criteria and classification of eosinophilic disorders and related syndromes. J Allergy Clin Immunol. 2012;130(3):607–612.e609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thiele J, Kvasnicka HM, Facchetti F, Franco V, van der Walt J, Orazi A. European consensus on grading bone marrow fibrosis and assessment of cellularity. Haematologica. 2005;90(8):1128–1132. [PubMed] [Google Scholar]
- 17.Yamamoto A, Kojima T, Aoki T, Sasai M, Taniuchi S, Kobayashi Y. Eosinophil hypersegmentation is a possible marker to monitor the disease activity of atopic dermatitis. Allergology International. 2001;50(4): 325–330. [Google Scholar]
- 18.Xu X. Nuclear hypersegmentation of neutrophils, eosinophils, and basophils due to hydroxycarbamide (hydroxyurea). Blood. 2014;124(9):1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Orazi A. Histopathology in the diagnosis and classification of acute myeloid leukemia, myelodysplastic syndromes, and myelodysplastic/myeloproliferative diseases. Pathobiology. 2007;74(2):97–114. [DOI] [PubMed] [Google Scholar]
- 20.Helbig G, Soja A, Bartkowska-Chrobok A, Kyrcz-Krzemien S. Chronic eosinophilic leukemia-not otherwise specified has a poor prognosis with unresponsiveness to conventional treatment and high risk of acute transformation. Am J Hematol. 2012;87(6):643–645. [DOI] [PubMed] [Google Scholar]
- 21.Pardanani A, Brockman SR, Paternoster SF, et al. FIP1L1-PDGFRA fusion: prevalence and clinicopathologic correlates in 89 consecutive patients with moderate to severe eosinophilia. Blood. 2004;104(10):3038–3045. [DOI] [PubMed] [Google Scholar]
- 22.Walz C, Metzgeroth G, Haferlach C, et al. Characterization of three new imatinib-responsive fusion genes in chronic myeloproliferative disorders generated by disruption of the platelet-derived growth factor receptor beta gene. Haematologica. 2007;92(2):163–169. [DOI] [PubMed] [Google Scholar]
- 23.Schwaab J, Jawhar M, Naumann N, et al. Diagnostic challenges in the work up of hypereosinophilia: pitfalls in bone marrow core biopsy interpretation. Ann Hematol. 2016;95(4):557–562. [DOI] [PubMed] [Google Scholar]
- 24.Mason CC, Khorashad JS, Tantravahi SK, et al. Age-related mutations and chronic myelomonocytic leukemia. Leukemia. 2016;30(4):906–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fernandez-Pol S, Ma L, Ohgami RS, Arber DA. Significance of myelodysplastic syndrome-associated somatic variants in the evaluation of patients with pancytopenia and idiopathic cytopenias of undetermined significance. Mod Pathol. 2016;29(9):996–1003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.