

Abstract
P27Kip1 (p27) can prevent cell proliferation by inactivating cyclin-dependent kinases. This function is impaired upon phosphorylation of p27 at tyrosine residue 88. We observed that FLT3 and FLT3-ITD can directly bind and selectively phosphorylate p27 on this residue. Inhibition of FLT3-ITD in cell lines strongly reduced p27 tyrosine 88 phosphorylation and resulted in increased p27 levels and cell cycle arrest. Subsequent analysis revealed the presence of tyrosine 88 phosphorylated p27 in primary patient samples. Inhibition of FLT3 kinase activity with AC220 significantly reduced p27 tyrosine 88 phosphorylation in cells isolated from FLT3 wild type expressing acute myeloid leukemia (AML) patients. In FLT3-ITD positive AML patients, p27 tyrosine 88 phosphorylation was reduced in 5 out of 9 subjects, but, surprisingly, was increased in 4 patients. This indicated that other tyrosine kinases such as Src family kinases might contribute to p27 tyrosine 88 phosphorylation in FLT3-ITD positive AML cells. In fact, incubation with the Src family kinase inhibitor dasatinib could decrease p27 tyrosine 88 phosphorylation in these patient samples, indicating that p27 phosphorylated on tyrosine 88 may be a therapeutic marker for the treatment of AML patients with tyrosine kinase inhibitors.
Introduction
Cell proliferation and cell cycle progression are tightly regulated by the sequential activation and inactivation of specific cyclin-dependent kinases (CDKs).2 Binding of the CDK inhibitor p27Kip1 (p27) can regulate CDK activity and thereby control cell cycle progression from G0/G1 phase to S phase. p27 regulates not only CDK activity, but also transcription and cell motility.2,3 p27 levels are elevated in non-proliferating cells and decline when cells progress towards S phase.4 Whereas p27 mRNA levels are frequently not altered during the cell cycle, protein levels of p27 can fluctuate dramatically.2,4 The rapid elimination of p27 at the G1/S transition is triggered through ubiquitin-dependent proteasomal degradation by the SCFSkp2 E3 ligase complex.5
Cyclin-dependent kinase inactivation by p27 involves the insertion of a 310-helix of the inhibitor into the catalytic cleft of the kinase, thereby blocking access of ATP.6 Interestingly, phosphorylation of p27 on residue tyrosine 88 (pY88) leads to the ejection of the inhibitory 310-helix from the catalytic cleft, permitting access of ATP7 and partial activation of p27-bound CDK complexes.7–11 The partially active cyclin-CDK2 can now phosphorylate substrates, including the bound p27 on T187.7 T187-phosphorylation is a prerequisite for p27 ubiquitination by SCFSkp2, initiating its proteasomal degradation.5 This mechanism directly couples mitogen-induced activation of tyrosine kinases to cell cycle control, but can also be used during oncogenic transformation of cancer cells.12 The non-receptor tyrosine kinases JAK2, Abl, BCR-Abl, Lyn, Yes, Src, and Brk can phosphorylate p27 on Y88 and likely employ this mechanism to inactivate p27 and to promote cell proliferation.7,8,11,13
The Fms-like tyrosine kinase 3 (FLT3) is a member of the class III subfamily of receptor tyrosine kinases and is activated by FLT3 ligand (FL).14 FLT3 is expressed in early hematopoietic progenitor cells in the bone marrow.14 High FLT3 levels have been detected in acute myeloid leukemia (AML),15,16 where activating FLT3 mutations can be found in approximately 30% of the patients.14,17 In fact, the most common mutation in AML is the internal tandem duplication (ITD) in the juxtamembrane domain of FLT3 with a 20–27% occurrence. FLT3-ITD serves as a prognostic marker since it positively correlates with higher blast counts, increased relapse rate, and worse overall survival.17–19 Several activating point mutations in the tyrosine kinase domain (TKD) have also been identified.14
Acute myeloid leukemia cells show increased proliferation and survival, as well as impaired hematopoietic differentiation.14 FLT3-ITD or FLT3 activation confers proliferative and survival advantages to cells14,20 by activating Src family tyrosine kinases (SFKs), the PI3K/Akt-, mitogen-activated protein kinase (MAPK) pathways, and, in the case of FLT3-ITD, also Stat5.20
Identifying the downstream targets of FLT3 and FLT3-ITD is essential to understanding the mechanisms through which they promote leukemia development. In the present study, we identified p27 as a novel direct substrate of FLT3 and FLT3-ITD. FLT3 inhibitor treatment efficiently reduced pY88-p27 in FLT3-ITD expressing cell lines and increased p27 protein levels. Analysis of cells from AML patients demonstrates for the first time that p27 is phosphorylated on Y88 in primary patient material. This uncovers a novel pathway with which FLT3 can promote hyperproliferation of AML cells.
Methods
Cell lines and primary cells
Cells were incubated at 37°C with 5% CO2 in DMEM (293T, U2OS) or RPMI (MV4;11, U937, Ba/F3, 32D) medium including 10% FCS. Primary blast cells were obtained from bone marrow aspirates or peripheral blood of AML patients. Written informed consent was obtained from all individuals in accordance with the Declaration of Helsinki. The use of human material was approved by the ethics committees of the Medical University of Innsbruck (AN2014-0362 344/4.22 345/4.4 346/4.1), Graz (27–372 14/15), and the Technical University of Munich (5689/13, 349/13, 276/15). Mononuclear cells were purified with Biocoll Separating Solution (Biochrom, Berlin, Germany), frozen in media containing 10% DMSO or immediately cultured in RPMI medium supplemented with 20% FCS for two hours (h) before treatment with the FLT3 inhibitor AC220.
Transient transfection, Western blot and immunoprecipitation
293T cells were analyzed 40 h after transfection using the calcium phosphate method.21 Protein extracts were prepared with 0.5% NP-40 containing lysis buffer and analyzed by Western blot as described.13 Bands were visualized by ECL- (GE-Healthcare) or Odyssey infrared fluorescence detection (LI-COR Biosciences) as indicated. Immunoprecipitation was performed as described13 using antibodies covalently coupled to protein A-agarose (Immunosorb A; Medicargo, Sweden). The antibodies are specified in the Online Supplementary Methods.
FACS
Cell cycle analyses were performed using bromodeoxyuridine (BrdU) and propidium iodide (PI) double staining, and analyzed as described previously.22
Protein interaction analysis
Pull down assays with recombinant isolated proteins were performed as described previously.13 Pull down experiments using His-p27 domains bound to AffiGel10 are described in the Online Supplementary Methods.
Kinase assay
In vitro phosphorylation was performed with Flag-tagged catalytically active cytoplasmic domain of FLT3-ITD (amino acids 564–1006) immunoprecipitated from transfected 293T cells or recombinant FLT3-ITD fused to GST-His6 (ProQinase) and His-p27, His-p21 and His-p57 as substrates, as described previously.13
Immunoflourescence
U2OS cells transfected with Flag-FLT3-ITD, Flag-FLT3, p27 or p27-Y88F were analyzed as described.13 Briefly, cells were fixed with 4% para-formaldehyde, permeabilized with 0.025% saponine, and blocked with 0.5% gelatine. A Leica DMi8 inverted widefield microscope was used for imaging.
Statistical analysis
P-values were determined by Wilcoxon test. ***P<0.001, **P<0.01, *P<0.05. n.s.: not significant.
Results
FLT3 and FLT3-ITD induce p27 phosphorylation, and inhibition of FLT3-ITD increases p27 expression
MV4;11 leukemia-derived cells express constitutively active FLT3-ITD.23 AC220 (Quizartinib) is a potent small molecule inhibitor of FLT3/FLT3-ITD24 currently under clinical phase III investigation (clinicaltrials.gov identifier: 02039726). Inhibition of FLT3-ITD with AC220 for 18 h resulted in a clear increase of p27 in MV4;11cells (Figure 1A and Online Supplementary Figure S1). As expected, AC220 also inhibited cell proliferation, induced an accumulation of cells in G0/G1 phase of the cell cycle, and increased the proportion of apoptotic cells (Figure 1B and C and Online Supplementary Figures S1–S3). To exclude potential cell-type specific effects of AC220, we also inhibited FLT3-ITD in a murine 32D cell line which had been stably transfected to express FLT3-ITD (32D-FLT3-ITD) and which proliferates independently of cytokines in the presence of this oncogene. Inhibition with AC220 led to a similar increase in p27, cell cycle arrest, and increased apoptosis (Figure 1D–F and Online Supplementary Figures S4 and S5). Inactivation of FLT3-ITD was confirmed by inhibited autophosphorylation on Y589/591 (Figure 1A and D, pY-FLT3). Since FLT3 undergoes glycosylation in the Golgi, immature (130 kDa) and mature forms (160 kDa) were detected.16,25
Figure 1.
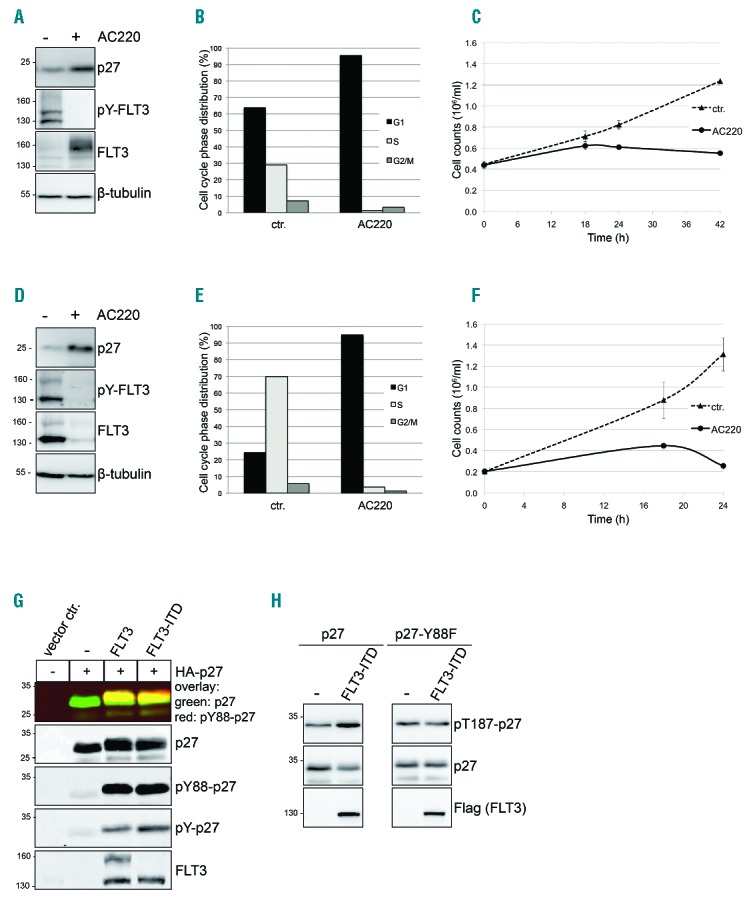
FLT3 and FLT3-ITD induce tyrosine phosphorylation of p27. Inhibition of FLT3-ITD with AC220 results in upregulation of p27 and cell cycle arrest in G1 phase of the cell cycle. Incubation of MV4;11 (A–C) and 32D-FLT3-ITD (D–F) cells in presence or absence of 100 nM AC220 for 18 hours (h). For Western blot and FACS analyses, one representative experiment out of three independent experiments is shown. (A and D) Western blot analysis: AC220 treatment increases p27 expression. pY589/Y591-FLT3 monitors kinase inhibition and β-tubulin serves as loading control. (B and E) Cell cycle phase distribution was determined by flow cytometry using propidium iodide (PI) and BrdU staining. (C and F) Cell proliferation is decreased in presence of AC220. Cells were seeded in a 12-well plate and cell numbers of viable (Trypan-blue negative) MV4;11 (C) and 32D-FLT3-ITD (F) cells were determined after 18, 24 and 42 h as indicated. (G) Expression of FLT3 or FLT3-ITD and HA-p27 leads to p27-Y88 phosphorylation. Transfected 293T cell lysates were heated [65°C, 10 minutes (min)] to precipitate the majority of proteins. The heat stable p27 and its tyrosine phosphorylation were simultaneously determined by Odyssey infrared imaging. The monoclonal 4G10 mouse antibody was used to detect overall p27 tyrosine-phosphorylation and a specific mouse monoclonal anti-pY88-p27 antibody to detect pY88-p27. Polyclonal rabbit anti-p27 antibodies were used to detect p27. FLT3 and FLT3-ITD were detected in non-heated lysates. (H) FLT3-ITD expression results in increased p27-T187 phosphorylation and decreased p27 levels. 293T cells were transfected with cyclin-E and CDK2 together with HA-p27, HA-p27-Y88F and Flag-FLT3-ITD as indicated. p27 phosphorylated on T187, p27 and FLT3 were detected with specific antibodies by Western blot analyses. ctr: control (vehicle-treated cells).
Whereas active FLT3-ITD predominantly accumulates in its underglycosylated form, inhibition of FLT3-ITD has been described to increase the ratio of the mature over the immature form, and to increase its abundance in MV4;11 cells25 (Figure 1A and Online Supplementary Figure S1).
To investigate if FLT3-ITD or FLT3 can induce tyrosine phosphorylation of p27, we expressed the proteins in 293T cells. Transfection of FLT3-ITD or FLT3 induced a strong phosphorylation of over-expressed p27 on tyrosine residues (Figure 1G, pY-p27). Using a monoclonal antibody specific for p27 phosphorylated at tyrosine residue 88 (pY88-p27), we observed that phosphorylation on this site was strongly induced by FLT3 and FLT3-ITD (Figure 1G, pY88-p27). Phosphorylation on Y88 converts p27 into a substrate of the bound CDK2/cyclin complex, leading to subsequent phosphorylation of p27 on T187 and its proteasomal degradation.7 Consistent with this observation, CDK2-cyclin E-dependent T187 phosphorylation of p27 was increased upon FLT3-ITD overexpression and resulted in decreased p27 levels (Figure 1H, left panel). Importantly, the p27-Y88F mutant failed to show the increase in p27-T187 phosphorylation as well as the decrease of p27 levels (Figure 1H, right panel), supporting the model that Y88 phosphorylation mediates the FLT3-ITD-induced decline of p27.
p27 is directly and selectively phosphorylated on tyrosine 88 by FLT3 and FLT3-ITD
To investigate the individual contribution of each of the three tyrosine residues present in p27 (Y74, Y88 and Y89), we expressed p27 proteins with one or several tyrosine residues mutated to phenylalanine together with FLT3-ITD. Tyrosine phosphorylation of all p27 mutant proteins lacking Y88 was dramatically reduced, and the mutant lacking only Y88 was not significantly phosphorylated on the remaining tyrosines (Figure 2A). This demonstrates that FLT3-ITD results in a highly selective phosphorylation of p27 on residue Y88 in vivo.
Figure 2.
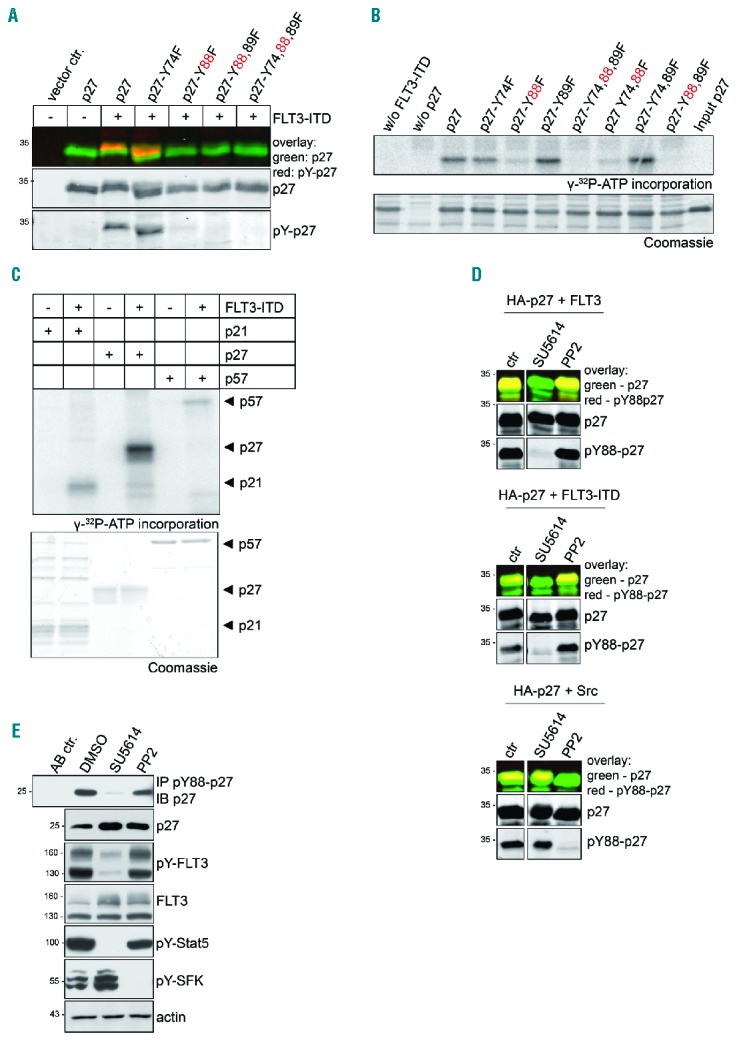
FLT3 phosphorylates p27 selectively on Y88 in vivo and in vitro and Y88-phosphorylation of p27 is Src family tyrosine kinase (SFK) independent. (A) FLT3-ITD phosphorylates p27 on Y88. In a similar approach as described in Figure 1G, HA-p27 and HA-p27 mutants where tyrosines were replaced by phenylalanine were expressed together with FLT3-ITD in 293T cells. p27 was detected simultaneously with overall tyrosine phosphorylation in heat-treated extracts. Co-migration indicates that the tyrosine-phosphorylated protein is p27 (pY-p27). (B) FLT3-ITD phosphorylates p27 on Y88 in vitro. Kinase assays were performed with recombinant His-p27 or His-p27 mutant proteins as indicated. The Flag-tagged cytoplasmic domain of FLT3-ITD was expressed in 293T cells and precipitated with anti-Flag antibodies. p27 protein or mutants were incubated with the sepharose-A bound kinase and γ-32P-ATP. Incorporation of γ-32P-ATP in p27 was detected by autoradiography (top panel) following SDS-PAGE and Coomassie brilliant blue staining (bottom panel). (C) FLT3-ITD selectively phosphorylates p27. In vitro kinase assay was performed as described above (B) using the cytoplasmic domain of recombinant FLT3-ITD isolated from insect cells as kinase and recombinant His-p21, His-p27 and His-p57 as substrates. (D) The receptor tyrosine kinase inhibitor SU5614, but not the SFK inhibitor PP2, decreases pY88-p27 after FLT3 or FLT3-ITD expression. 293T cells were treated with SU5614 (1 μM), PP2 (10 μM), or vehicle (DMSO) for 3 hours (h) following transient tansfection of HA-p27 together with FLT3 (top panel), FLT3-ITD (middle panel), or Src (bottom panel). Western blot analysis was performed as described in Figure 1G. (E) p27-Y88 phosphorylation occurs independently of activated SFK members. 32D-FLT3-ITD cells were treated with SU5614 (1 μM), PP2 (10 μM), or vehicle (DMSO) for 2 h. pY88-p27 was immunoprecipitated with pY88-p27-specific antibody and analyzed by Western blot with HRP-coupled anti-p27 antibodies. pY694-Stat5 serves as a control for SU5614 and pY416-SFK for PP2 treatment. Actin was used as loading control. One representative experiment of three independent experiments is shown. ctr: control (vehicle-treated cells).
p27 can be a direct substrate of FLT3/FLT3-ITD or its phosphorylation might be indirect, e.g. through Src family kinases (SFKs) activated by FLT3 kinases. To investigate if FLT3-ITD can phosphorylate recombinant purified p27 in vitro, p27 was incubated with the catalytically active cytoplasmic domain of FLT3-ITD. While wild-type p27 was efficiently phosphorylated, mutation of Y88 of p27 to phenylalanine strongly decreased the phosphorylation (Figure 2B and Online Supplementary Figure S6). No reduction of phosphorylation was observed when Y89 and/or Y74 were mutated. A mutant preserving Y88 as single tyrosine residue (p27-Y74,89F) was as efficiently phosphorylated as wild-type p27 (Figure 2B and Online Supplementary Figure S6), supporting the hypothesis that Y88 is selectively and directly phosphorylated by FLT3 in vitro. When p27 phosphorylation by recombinant FLT3-ITD was compared to p21 and p57, an efficient phosphorylation was only observed for p27 (Figure 2C), suggesting that p27 is the principal FLT3 target of the CIP/KIP family members.
FLT3/FLT3-ITD activates SFK members such as Src, Lyn and Fyn,26–28 and Src and Lyn can phosphorylate p27.7,8 Phosphorylation of p27 by Src, however, also includes Y74,8 but phosphorylation at this tyrosine was not detected in transfection assays (Figure 2A, p27-Y88,89F), suggesting that Src is not the main tyrosine kinase that contributes to FLT3-ITD-induced p27 phosphorylation. To assess the contribution of SFKs to p27 phosphorylation in cell lines, we used SU5614 to inactivate FLT3,29 and PP2 as SFK inhibitor. In 293T cells over-expressing p27 and FLT3 or FLT3-ITD, p27-Y88 phosphorylation was inhibited by SU5614, and remained unchanged by PP2 treatment (Figure 2D, top and middle panels), indicating that SFK activity is not required for FLT3-induced p27 phosphorylation. As expected, PP2 treatment abolished p27-Y88 phosphorylation in cells transfected with p27 and Src (Figure 2D, lower panel). Importantly, Y88 phosphorylation of endogenous p27 in 32D-FLT3-ITD cells was also strongly reduced by SU5614, whereas treatment with the SFK inhibitor PP2 did not inhibit p27 phosphorylation (Figure 2E). Inhibition of FLT3-ITD by SU5614 as well as inhibition of SFK by PP2 was confirmed by the phosphorylation status of Y694-Stat5 and Y416-SFK, respectively, demonstrating that SFKs remain active in SU5614-treated cells (Figure 2E). These observations are consistent with the hypothesis that p27 is a direct substrate of FLT3 and FLT3-ITD.
p27 is in a complex with FLT3 and FLT3-ITD
If p27 is a substrate of FLT3/FLT3-ITD, both proteins might associate in a stable complex. To investigate if p27 can bind to FLT3/FLT3-ITD, we initially performed pull down experiments with the GST-tagged cytoplasmic domain of FLT3-ITD (cytFLT3-ITD) and His-tagged p27. To exclude the presence of potential bridging factors, the proteins were expressed in E.coli. GST-cytFLT3-ITD, but not GST, co-precipitated p27 (Figure 3A), demonstrating a stable and direct interaction in vitro. The interaction of p27 with FLT3-ITD was confirmed in 293T cells in which HA-p27 was co-immunoprecipitated with Flag-FLT3-ITD, and vice versa (Figure 3B). HA-p27 co-precipitated also with Flag-FLT3 (Online Supplementary Figure S7). Importantly, a stable complex of endogenous p27 and endogenous FLT3-ITD was precipitated from MV4;11 cells (Figure 3D). Similarly, endogenous p27 co-precipitated with FLT3-ITD in 32D-FLT3-ITD cells (Figure 3C). These data demonstrate that p27 can form a stable complex with FLT3 and FLT3-ITD, and support the hypothesis that p27 is a direct substrate of these kinases.
Figure 3.
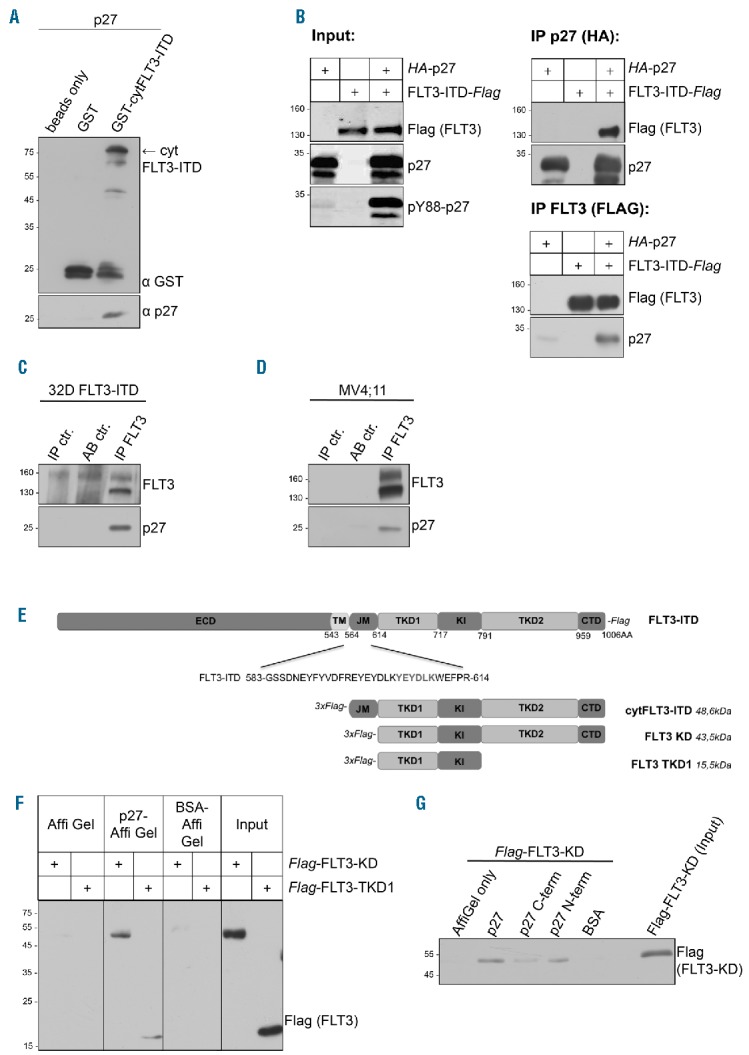
FLT3 and FLT3-ITD directly bind to p27. (A) FLT3-ITD interacts directly with p27 in vitro. GST and the GST-tagged cytoplasmic domain of FLT3-ITD were isolated from E.coli, bound to glutathione-sepharose, and incubated with recombinant p27. Eluates were analyzed by Western blot using anti-GST and anti-p27 antibodies. (B) HA-p27 co-precipitates with Flag-FLT3-ITD, and vice versa after expression in 293T cells. Protein expression and Y88-phosphorylation of p27 (Input, left panel) as well as p27-co-immunoprecipitates (right panel) and FLT3-co-immunoprecipitates (lower panel) were analyzed by Western blot. (C and D) Endogenous p27 co-precipitates with FLT3-ITD in 32D-FLT3-ITD cells (C) and with endogenous FLT3-ITD in MV4;11 cells (D). Immunoprecipitates were detected with anti-FLT3 antibodies and co-immunoprecipitates with anti-p27 antibodies in Western blots. Normal rabbit IgG was used in control IPs (AB ctr). Coupled pY88-p27 antibodies were loaded to exclude unspecific signals of the antibody (IP ctr.). (E) Schematic representation of FLT3-ITD and its domains used in (A, F and G). (F) Recombinant His-p27 was bound to Affi-Gel10 and incubated with cell lysates from 293T cells transfected with the respective Flag-FLT3 domains. Eluates were analyzed by Western blot using anti-Flag-antibodies. Bovine serum albumin (BSA) coupled to Affi-Gel as well as Affi-Gel alone incubated with Flag-FLT3-ITD served as negative controls. (G) Recombinant full-length His-p27 and its C-terminal (C-term) and N-terminal (N-term) domains were bound to Affi-Gel 10 and incubated with cell lysates from 293T cells transfected with the Flag-FLT3 tyrosine kinase domain (Flag-FLT3-KD). Eluates were analyzed by Western blot using Flag-antibody. BSA coupled to Affi-Gel as well as Affi-Gel alone, incubated with Flag-FLT3-ITD, served as negative controls. One representative experiment of three independent experiments is shown. IP: immunoprecipitation; cytFLT3-ITD: cytoplasmic domain; KD: kinase domain; TKD1: tyrosine kinase domain 1.
To identify essential domains for the p27-FLT3 interaction, we performed pull down experiments using fragments of both proteins. FLT3 can be subdivided in an extracellular domain, a transmembrane portion, and a cytoplasmic region containing the split kinase domain30 (Figure 3E). Cell lysates expressing different regions of the cytoplasmic domain were incubated with recombinant His-tagged p27. p27 bound to the FLT3 kinase domain (KD) and to its N-terminal lobe (TKD1) (Figure 3F). Co-immunoprecipitation experiments with transfected proteins confirmed binding of the kinase domain and the TKD1 domain to p27 (Online Supplementary Figure S8). On the other hand, full-length p27 and its N-terminal kinase-inhibitory domain precipitated FLT3-KD efficiently (Figure 3G). Interestingly, this mapping analysis indicates that the kinase domain of FLT3 interacts with the CDK-inhibitory domain of p27.
In order to be a significant substrate of FLT3/FLT3-ITD, p27 and the kinase should at least partially co-localize. FLT3 localizes to the plasma membrane and can be restrained in ER membranes upon incomplete glycosylation.25 p27 is predominantly a nuclear protein that shuttles between nucleus and cytoplasm.31 To interact with the cytoplasmic domain of the FLT3 kinases, p27 needs to be, at least partly, exported to the cytoplasm. To determine the subcellular localization of both proteins, U2OS cells were transfected with p27 and FLT3-ITD (Figure 4, panels 1–8) or p27 and FLT3 (Figure 4, panels 9–16). Immunoflourescence analyses revealed that FLT3-ITD and FLT3 are mainly localized in extended structures surrounding the nucleus, potentially representing ER membranes (Figure 4, panels 2,6 and 10,14, respectively). With p27 accumulated in the nucleus, however, a portion of the protein was detected in the cytoplasm (Figure 4, panels 5,13). This extranuclear fraction of p27 partially co-localized with FLT3-ITD/FLT3 (Figure 4, panels 7,15). Interestingly, when Y88-phosphorylated p27 was analyzed, the degree of co-localization with FLT3-ITD and FLT3 was significantly enhanced (Figure 4, panels 3,11). These observations support the hypothesis that p27 interacts with FLT3-ITD and FLT3.
Figure 4.
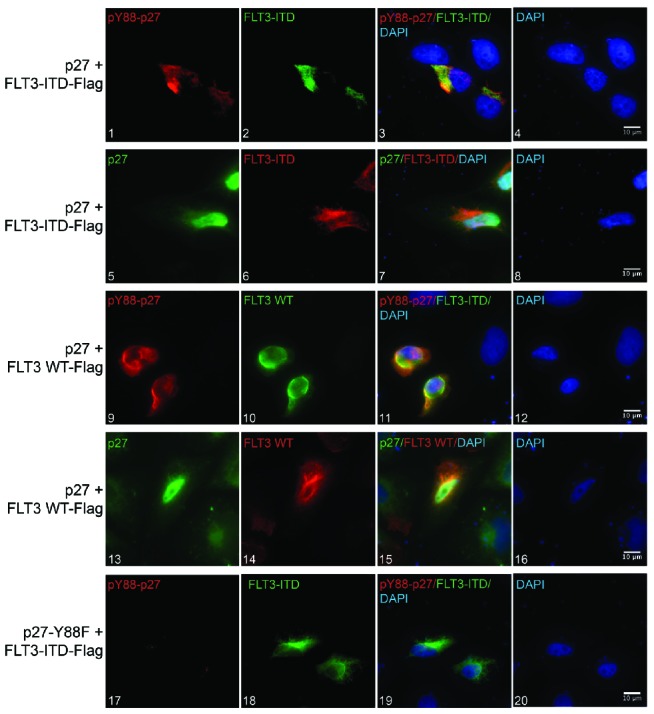
p27 partially co-localizes with FLT3 and FLT3-ITD. Immunofluorescence analysis of U2OS cells transfected with p27 and Flag-FLT3-ITD (panels 1–8) or Flag-FLT3 WT (panels 9–16) with a Leica DMi8 inverted widefield microscope. Panels 1–3 and 9–11 show the localization of pY88-p27 (red) and FLT3-ITD or FLT3 WT (green), respectively. Panels 5–7 and 13–15 show the localization of p27 (green) and FLT3-ITD or FLT3 WT (red). As a specificity control for pY88-p27 antibodies, U2OS cells were transfected with a phosphodeficient p27 mutant (p27-Y88F) and FLT3-ITD (panels 17–20). DNA was stained with DAPI (blue). Scale bar represents 10 μm.
Stimulation of FLT3 with its ligand FL induces p27 phosphorylation. Inhibition of FLT3-ITD in AML cell lines reduces p27-Y88 phosphorylation and restores p27 protein levels
To investigate the potential physiological role of p27-Y88 phosphorylation by FLT3, we investigated if endogenous p27 tyrosine phosphorylation follows FL-induced activation of endogenous FLT3 in U937 cells. pY88-p27 was low in asynchronously proliferating cells and remained low upon serum starvation (Figure 5A). FLT3 autophosphorylation is known to occur 5–15 min after FL addition.32 Interestingly, 5 min after activation by FL p27-Y88 phosphorylation was already strongly induced (Figure 5A), suggesting that p27 phosphorylation is an immediate event following FLT3 activation. p27-Y88 phosphorylation slowly declined within 90 min (Figure 5A). FLT3 protein started to decline 60 min after activation (Figure 5A), probably following internalization.33
Figure 5.
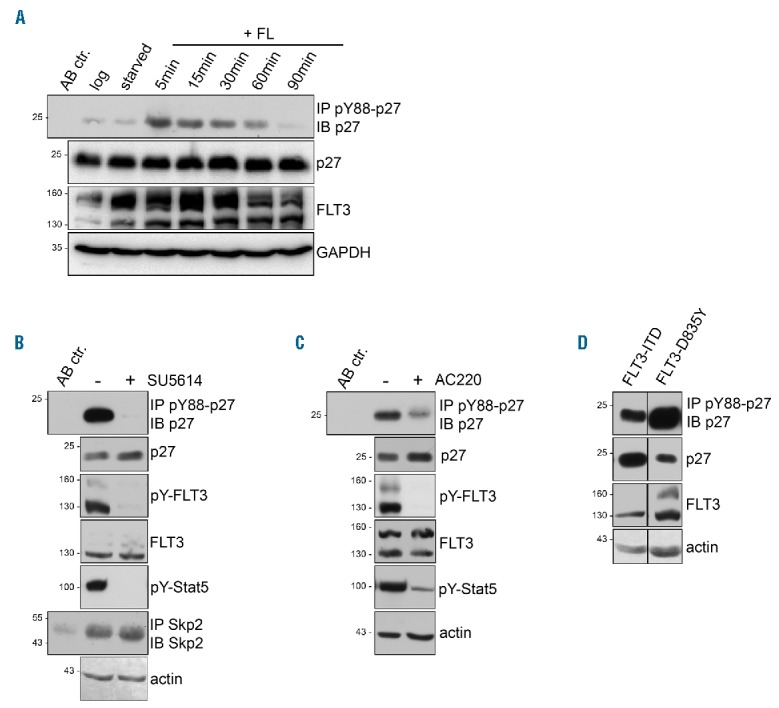
FLT3 ligand induces p27-Y88 phosphorylation in U937 cells. pY88-p27 is decreased by FLT3 inhibitors in FLT3-ITD positive acute myeloid leukemia (AML) cell lines. (A) FLT3 ligand (FL) induces p27-Y88-phosphorylation in U937 cells. U937 were serum-starved for 6 hours (H) (starved) before stimulation with 50 ng/ml rhFL for the indicated time points. pY88-p27 was immunoprecipitated with specific antibodies and detected in Western blots using HRP-coupled anti-p27 antibodies. Further, anti-FLT3, p27 and GAPDH antibodies were used. Mouse IgG served as isoptype control for immunoprecipitation (AB ctr.). (B and C) pY88-p27 is decreased and p27 levels are increased by FLT3 inhibitors. Western blot analyses are shown. (B) MV4;11 cells were incubated with SU5614 (1 μM) or vehicle for 1 h. (C) Ba/F3 cells were incubated with AC220 (50 nM) or vehicle for 1 h. p27, FLT3 and pY88-p27 were detected as described above. pY589/Y591-FLT3 and pY694-Stat5 served as controls for inhibitor treatment and actin as loading control. Skp2 immunoprecipitates revealed no change in Skp2 expression. Mouse IgG was used as isoptype control for immunoprecipitation (AB ctr.). (D) p27 is phosphorylated on Y88 in FLT3-D835Y cells. Total p27 correlates inversely with pY88-p27 in FLT3-D835Y and FLT3-ITD cells. Analyses were performed as described in (B and C). One representative experiment of three independent experiments is shown.
In asynchronous FLT3-ITD-positive MV4;11 cells, p27 was strongly phosphorylated on Y88 (Figure 5B). The FLT3 inhibitor SU5614 abolished p27-Y88 phosphorylation to undetectable levels, suggesting that this phosphorylation depends on FLT3-ITD kinase activity (Figure 5B). The efficiency of SU5614 was monitored by reduction of pY589/Y591-FLT3 and pY694-Stat5 (Figure 5B). Similar results were observed in Ba/F3-FLT3-ITD cells treated with AC220 (Figure 5C). Y88 phosphorylation of p27 can initiate its SCFSkp2-mediated proteasomal degradation.7,8,13 Consistent with our earlier findings,7,13 long-term inhibition of FLT3-ITD led to a robust accumulation of p27 (Figure 1A and F) which could be the cause or consequence of the cell cycle arrest. To exclude cell cycle positioning effects, we analyzed p27 after very short periods of inhibition. In MV4;11 or Ba/F3-FLT3-ITD cells, an initial increase in p27 protein could already be detected 60 min after FLT3-ITD inhibition (Figure 5B and C). When p27 phosphorylation was compared in 32D cells stably expressing either FLT3-ITD or FLT3-TKD (D835Y), higher p27-Y88 phosphorylation and lower p27 levels were found in FLT3-D835Y expressing cells (Figure 5D). These observations are consistent with the previous finding that pY88-p27 can initiate its SCFSkp2-triggered degradation, a mechanism that leads to CDK activation and cell proliferation.7,8,13
p27 is phosphorylated on Y88 in primary AML cells
The p27-Y88 phosphorylation and its physiological consequences have been studied in vitro and in cell lines.7–11,13,34,35 However, the presence, abundance or regulation of this modification have not yet been established in primary patient material. We went on to investigate the detectability of pY88-p27 in primary blast cells obtained from AML patients. Immunoprecipitation analysis of cell extracts from AML patients positive for FLT3 wild type (WT) revealed the presence of pY88-p27 and p27 at levels comparable to those in FLT3-ITD positive MV4;11 cells (Figure 6A and Online Supplementary Figure S9).
Figure 6.
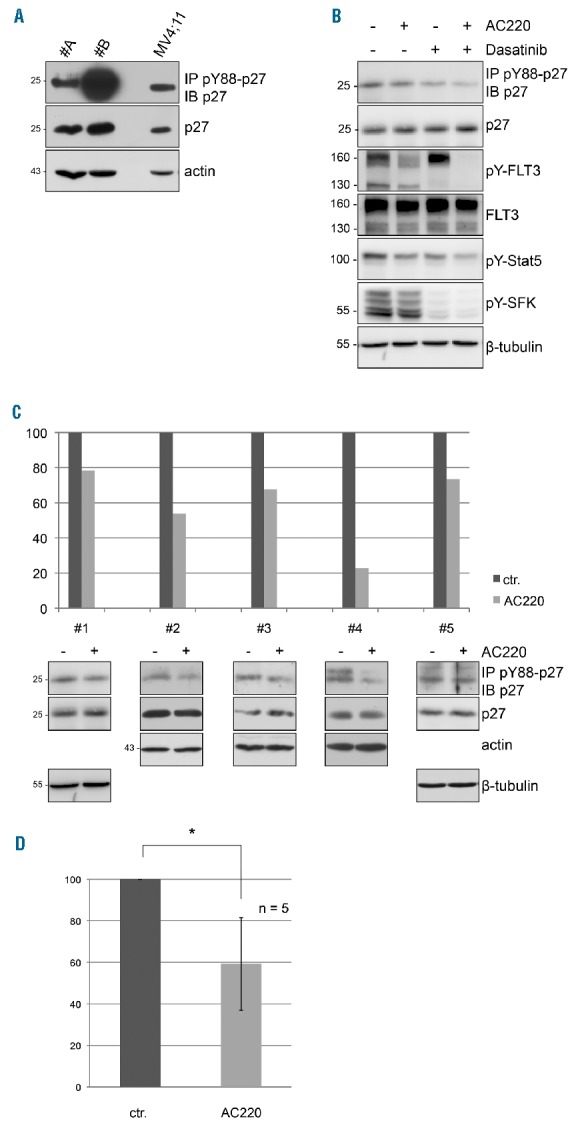
pY88-p27 is detected in primary FLT3 wild-type (WT) acute myeloid leukemia (AML) cells and reduced upon FLT3 inhibition. (A) Western blot of pY88-p27 immunoprecipitates, p27 and actin in FLT3 primary AML cells. FLT3 WT expressing primary cells were cultured overnight in RPMI medium containing 20% FBS and equal amounts of protein extracts were analyzed by immunoblotting. Cells from patient #B were stimulated with FL (100 ng/mL) for 20 minutes (min). (B-D) AC220 treatment of FLT3 expressing primary AML cells down-regulates pY88-p27. Primary AML cells were cultured overnight (frozen samples) or for 2 hours (h) (fresh samples) in medium containing 20% FBS before incubation with AC220 (100 nM), dasatinib (100 nM) or vehicle for 2 h. Equal total protein amounts as determined by DC protein quantification assay were used for pY88-p27 immunoprecipitation with specific antibodies and subjected to Western blot analysis using HRP-coupled anti-p27 antibodies. (B) Immunoblot analysis of one representative patient (#1) expressing FLT3 WT. p27, FLT3, and the loading control actin were detected. pY589/Y591-FLT3, pY694-Stat5, and pT202/Y204-Erk served as controls for FLT3 inhibition. (C) Quantitative analysis (top panel) of Western blots (bottom panel) determining pY88-p27 levels in all 5 FLT3 WT AML patient cells that were treated with or without AC220. Sample #1 is also shown in Figure 6B. Western blots were quantified by densitometry using ImageJ software and normalized for actin. (D) Mean decrease of pY88-p27 levels of 5 FLT3 WT AML patients upon AC220 treatment (2 h). Wilcoxon test, *P=0.043.
To gain more insight into the p27-Y88 phosphorylation in primary AML patient samples, we investigated the response to FLT3 inhibition by AC220. (We did not test the effect of other FLT3 inhibitors.) Primary blast cells from 14 AML patients, including 5 FLT3 WT and 9 FLT3-ITD positive patients, were analyzed (Table 1). All samples included in this analysis were positive for FLT3 or FLT3-ITD expression and FLT3 autophosphorylation, and responded to AC220 as monitored by reduced pY589/Y591-FLT3, pY694-Stat5, or pT202/Y204-Erk (Figures 6B, 7A and D, Online Supplementary Figures S10 and S11, and data not shown). Inhibitor treatment of FLT3 WT positive AML cells for 2 h resulted in a significant reduction of p27-Y88 phosphorylation to an average 59% (Figure 6C and D). All individual samples of this group responded to AC220 with a decline in p27-Y88 phosphorylation (Figure 6C). In FLT3-ITD-expressing AML cells, AC220 treatment decreased pY88-p27 levels in 5 out of 9 patient samples (Figure 7B). Surprisingly, in 4 patients, pY88-p27 levels were even increased in the presence of FLT3 inhibitor (Figure 7C), although FLT3-ITD kinase activity was inhibited, as monitored by marker protein phosphorylation (reduction of pY589/Y591-FLT3, pY694-Stat5, or pT202/Y204-Erk) (Figure 7D, Online Supplementary Figure S11, and data not shown). The impaired ability of AC220 to prevent p27-Y88 phosphorylation in FLT3-ITD expressing cells could be attributed to other activated tyrosine kinases present in these cells. Members of the SFK family can be activated in AML cells.26–28,36,37 Therefore, we investigated the activity of SFKs in the patient with the strongest increase of pY88-p27 upon AC220 incubation (#13, 63% increase). In this case, the SFK autophosphorylation did not decrease upon AC220 incubation (Figure 7D). SFKs can also phosphorylate p27 on Y88.7,8 To investigate if increased SFK activity might contribute to p27-Y88 phosphorylation, we treated these cells with AC220, dasatinib or both inhibitors (Figure 7D). pY88-p27 was increased 1.7-fold by AC220 alone, despite efficient inhibition of FLT3 autophosphorylation (Figure 7D). Interestingly, dasatinib treatment alone reduced pY88-p27 to 38% of vehicle-treated cells and the combination of dasatinib and AC220 further reduced pY88-p27 to 23%. Phosphorylation of marker proteins Stat5, ERK and SFK was also efficiently reduced following dasatinib treatment (Figure 7D), suggesting that, in this patient, increased activity of SFKs (or other desatinib-sensitive kinases) significantly contribute to p27-Y88 phosphorylation. Subsequent analyses of patient samples #11 and #14, where AC220 treatment also increased pY88-p27 levels, revealed that dasatinib treatment decreased p27-Y88 phosphorylation status of patient sample #14 but had no effect on sample #11 (Online Supplementary Figure S11).
Table 1.
Characteristics of acute myeloid leukemia patients.

Figure 7.

AC220 regulates pY88-p27 in primary FLT3-ITD acute myeloid leukemia (AML) cells. Experiments with FLT3-ITD positive AML cells were performed as described in Figure 6. (A) Western blot analysis of a representative FLT3-ITD patient (#6) of the group of patients, where pY88-p27 decreased upon AC220 treatment. pY589/Y591-FLT3 and pY694-Stat5, which is a direct substrate of FLT3-ITD, serve as controls for FLT3 inhibition. p27, FLT3 and β-tubulin were monitored as loading control. (B) Quantitative analysis (top panel) of Western blots (bottom panel) determining pY88-p27 levels in 5 FLT3-ITD AML patients where AC220 treatment for 2 hours (h) decreased pY88-p27 levels. (C) Western blots (bottom panel) and its quantification (top panel) of pY88-p27 levels from 4 FLT3-ITD patients where AC220 treatment increased pY88-p27 levels. (D) Dasatinib treatment reduces pY88-p27 in an FLT3-ITD positive patient sample where AC220 increased pY88-p27. FLT3-ITD AML cells (#13) were treated for 2 h with AC220, dasatinib, the combination of both inhibitors, or vehicle. p27, FLT3, and the loading control β-tubulin were detected. pY589/Y591-FLT3 (pY-FLT3), pY694-Stat5 (pY-Stat5), and pT202/Y204-Erk (pY-Erk) serve as controls for FLT3 inhibition. pY416-SFK (pY-SFK) is a control for dasatinib-mediated Src inhibition.
These data demonstrate that pY88-p27 can be detected in primary human patient material and that p27 is targeted by FLT3 and FLT3-ITD in primary AML cells. Some AML patients express additional p27-phosphorylating tyrosine kinases that can be inhibited by dasatinib or combinations of tyrosine kinase inhibitors.
Discussion
p27 is a substrate of several non-receptor tyrosine kinases.7–11,13 Here we report that the receptor tyrosine kinase FLT3 and its constitutively active counterpart FLT3-ITD can bind and selectively phosphorylate p27on Y88.
p27 can shuttle between the nucleus and cytoplasm.31 Increased cytoplasmic localization of p27 results from mitogen stimulation and involves Akt dependent phosphorylation events.2,38 Since FLT3/FLT3-ITD signaling activates the PI3K/Akt pathway, it is likely that enhanced Akt activity reinforces the nuclear export of p27.
Binding of p27 to FLT3 involves the N-terminal domain of p27 and TKD1 of the tyrosine kinase domain of FLT3. The N-terminal domain of p27 uses a folding-on-binding mechanism for cyclin/CDK binding, and inserts a 310-helix surrounding Y88 into the catalytic cleft of the kinase.6,39 Since the kinase domain and especially TKD1 of FLT3 bind to the CDK-inhibitory domain of p27, it will be interesting to determine if this binding follows a similar folding-on-binding mechanism of p27 on FLT3. This may explain the enhanced binding by the entire kinase domain and might also position Y88 for selective phosphorylation. The precise mechanism and dynamics of p27 binding to the FLT3 tyrosine kinase domain still need to be determined.
Whereas p27 levels were low in cells expressing active FLT3-ITD, inhibition of FLT3-ITD led to increased p27 levels and cell cycle arrest in G0/G1-phase. To avoid cell cycle position effects, we analyzed p27 after very short inhibition of FLT3. Already one hour following FLT3 inhibition, we observed a reproducible increase in p27. Degradation induced by Y88 phosphorylation requires CDK-dependent phosphorylation of the inhibitor and expression of the cell cycle-regulated ubiquitin E3 ligase SCFSkp2.5,7 Since proliferating cells express the SCFSkp2 E3 ligase, it is likely that FLT3-ITD-mediated phosphorylation of p27 contributes to the reduction of p27 levels in MV4;11 and proliferating AML cells. SCFSkp2-dependent proteasomal degradation of T187-phosphorylated p27 may occur in the cytoplasm or in the nucleus. Since Skp2 translocates to the cytoplasm upon Akt phosphorylation, it may also support cytoplasmic p27 ubiquitination. On the other hand, p27 was phosphorylated on Y88 but did not decline upon FL stimulation of serum starved U937 cells. Lack of p27 degradation is likely caused by low Skp2 expression in starved cells.40
Interestingly, decreased p27 levels have been associated with poor prognostic outcome in AML patients.41–43 In contrast to these observations, Haferlach et al. reported that low CDKN1B levels correlates with a better prognosis in AML patients.44 However, Haferlach and co-workers analyzed only CDKN1B mRNA expression and p27 mRNA frequently does not correlate with its protein expression.8 In addition, Y88 phosphorylation and cytoplasmic localization can inactivate the CDK inhibitor function and convert the p27 tumor suppressor into a cytoplasmic oncogene.45 The molecular consequences of p27-Y88 phosphorylation have been extensively described.7–11,13,34,35 In contrast to data in cell culture systems, confirmation of the presence of pY88-p27 in primary patient material has been lacking. We found that pY88-p27 could readily be detected in primary AML blast cells from patients expressing either FLT3 WT or FLT3-ITD. Levels of pY88-p27 are comparable to levels detected in MV4;11 cells (Figure 6A and Online Supplementary Figures S9 and S10). This is the first time that pY88-p27 has been detected in primary patient material.
FLT3 inhibition with AC220 significantly reduced pY88-p27 in all FLT3 WT patient samples. These data indicate that p27 is a substrate of FLT3 in primary AML cells. In contrast to experiments with leukemic cell lines, p27 protein levels were not increased following AC220 treatment. This is most likely caused by the cell culture conditions, which do not support proliferation of the primary cells.42 This would prevent Skp2 expression, and the E3 ligase is required for p27 degradation following p27 tyrosine phosphorylation. The reduction of pY88-p27 by AC220 in FLT3 WT AML cells reflects an increased CDK inhibitory activity of p27 and possibly increased p27 levels in proliferating cells. These findings support the suggestion that FLT3 WT AML patients may benefit from FLT3 inhibitor treatment.46 Of note, a phase II study is currently investigating the effect of AC220 monotherapy in FLT3 WT AML patients (clinicaltrials.gov identifier: 00989261). Interestingly, we observed that pY88-p27 was also decreased by dasatinib treatment in the single FLT3 WT AML patient investigated (Figure 6B), indicating that FLT3 WT patients might also benefit from multikinase inhibitor therapy.
AC220 is under investigation in a phase III trial for patients expressing FLT3-ITD (clinicaltrials.gov identifier: 02039726). Approximately 30% of FLT3-ITD patients do not respond to FLT3 inhibitor treatment.47 Surprisingly, we observed both up- and downregulation of pY88-p27 following AC220 treatment of individual FLT3-ITD patient samples. The increase of pY88-p27 upon AC220 treatment in some patient samples raised the question as to whether other tyrosine kinases were responsible for p27-Y88 phosphorylation in these patients. We found that SFKs might contribute to this phosphorylation since dasatinib treatment efficiently reduced pY88-p27 in 2 out of 3 FLT3-ITD positive patients with increased pY88-p27 levels after AC220 treatment (Figure 7D and Online Supplementary Figure S11, patient #14). Dasatinib treatment further reduced pY88-p27 in one out of 2 FLT3-ITD patients where AC220 already reduced pY88-p27 levels (Online Supplementary Figure S10, patient #10). SFKs can be activated in primary AML cells36,37 and Src, Yes and Lyn can phosphorylate p27.7,8 Of note, dasatinib can also inhibit other tyrosine kinases including Abl, which might cause p27 phosphorylation. It will be important to explore if p27-Y88 phosphorylation can serve as a therapeutic marker in AML and act as an indicator for treatment decisions involving tyrosine kinase inhibitors. Similarly, it should be determined if inhibition of p27-Y88 phosphorylation correlates with therapeutic outcome in AML patients undergoing therapy with tyrosine kinase inhibitors.
Interestingly, small molecule CDK inhibitors are currently being investigated for use in AML therapy48 (clinicaltrials.gov identifiers: 02310243 and 00278330), and recent investigations indicate a synergistic effect of CDK4/6 kinase inhibitor and FLT3 inhibitors to impair the survival of leukemic FLT3-ITD positive cells.49 Preventing p27-Y88 phosphorylation by combinations of tyrosine kinase inhibitors might be an alternative approach to restore CDK inactivation in AML.
Supplementary Material
Acknowledgments
The authors would like to thank Caroline Linhart for performing statistical analyses of the ethics proposal, Jonathan Vosper for proofreading and all members of the Hengst lab for support and stimulating discussions.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/102/8/1378
Funding
This work has been supported by the Austrian Science Fund (FWF) Grant P24031 (LH) and the Österreichische Krebshilfe Tirol (ÖKH-KG) (IP).
References
- 1.Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9(3):153–166. [DOI] [PubMed] [Google Scholar]
- 2.Chu IM, Hengst L, Slingerland JM. The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nat Rev Cancer. 2008;8(4):253–267. [DOI] [PubMed] [Google Scholar]
- 3.Sharma SS, Pledger WJ. The non-canonical functions of p27(Kip1) in normal and tumor biology. Cell Cycle. 2016;15(9):1189–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hengst L, Reed SI. Translational control of p27Kip1 accumulation during the cell cycle. Science. 1996;271(5257):1861–1864. [DOI] [PubMed] [Google Scholar]
- 5.Carrano AC, Eytan E, Hershko A, Pagano M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat Cell Biol. 1999;1(4):193–199. [DOI] [PubMed] [Google Scholar]
- 6.Russo AA, Jeffrey PD, Patten AK, Massague J, Pavletich NP. Crystal structure of the p27Kip1 cyclin-dependent-kinase inhibitor bound to the cyclin A-Cdk2 complex. Nature. 1996;382(6589):325–331. [DOI] [PubMed] [Google Scholar]
- 7.Grimmler M, Wang Y, Mund T, et al. Cdk-inhibitory activity and stability of p27Kip1 are directly regulated by oncogenic tyrosine kinases. Cell. 2007;128(2):269–280. [DOI] [PubMed] [Google Scholar]
- 8.Chu I, Sun J, Arnaout A, et al. p27 phosphorylation by Src regulates inhibition of cyclin E-Cdk2. Cell. 2007;128(2):281–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.James MK, Ray A, Leznova D, Blain SW. Differential modification of p27Kip1 controls its cyclin D-cdk4 inhibitory activity. Mol Cell Biol. 2008;28(1):498–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Larrea MD, Liang J, Da Silva T, et al. Phosphorylation of p27Kip1 regulates assembly and activation of cyclin D1-Cdk4. Mol Cell Biol. 2008;28(20):6462–6472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patel P, Asbach B, Shteyn E, et al. Brk/Protein tyrosine kinase 6 phosphorylates p27KIP1, regulating the activity of cyclin D-cyclin-dependent kinase 4. Mol Cell Biol. 2015;35(9):1506–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jakel H, Peschel I, Kunze C, Weinl C, Hengst L. Regulation of p27 (Kip1) by mitogen-induced tyrosine phosphorylation. Cell Cycle. 2012;11(10):1910–1917. [DOI] [PubMed] [Google Scholar]
- 13.Jakel H, Weinl C, Hengst L. Phosphorylation of p27Kip1 by JAK2 directly links cytokine receptor signaling to cell cycle control. Oncogene. 30(32):3502–3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gilliland DG, Griffin JD. The roles of FLT3 in hematopoiesis and leukemia. Blood. 2002;100(5):1532–1542. [DOI] [PubMed] [Google Scholar]
- 15.Birg F, Courcoul M, Rosnet O, et al. Expression of the FMS/KIT-like gene FLT3 in human acute leukemias of the myeloid and lymphoid lineages. Blood. 1992; 80(10):2584–2593. [PubMed] [Google Scholar]
- 16.Carow CE, Levenstein M, Kaufmann SH, et al. Expression of the hematopoietic growth factor receptor FLT3 (STK-1/Flk2) in human leukemias. Blood. 1996; 87(3):1089–1096. [PubMed] [Google Scholar]
- 17.Meshinchi S, Appelbaum FR. Structural and functional alterations of FLT3 in acute myeloid leukemia. Clin Cancer Res. 2009; 15(13):4263–4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schnittger S, Schoch C, Dugas M, et al. Analysis of FLT3 length mutations in 1003 patients with acute myeloid leukemia: correlation to cytogenetics, FAB subtype, and prognosis in the AMLCG study and usefulness as a marker for the detection of minimal residual disease. Blood. 2002;100(1):59–66. [DOI] [PubMed] [Google Scholar]
- 19.Thiede C, Steudel C, Mohr B, et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood. 2002;99(12):4326–4335. [DOI] [PubMed] [Google Scholar]
- 20.Takahashi S. Downstream molecular pathways of FLT3 in the pathogenesis of acute myeloid leukemia: biology and therapeutic implications. J Hematol Oncol. 2011;4:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Graham FL, van der Eb AJ. A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology. 1973; 52(2):456–467. [DOI] [PubMed] [Google Scholar]
- 22.Vosper J, Masuccio A, Kullmann M, Ploner C, Geley S, Hengst L. Statin-induced depletion of geranylgeranyl pyrophosphate inhibits cell proliferation by a novel pathway of Skp2 degradation. Oncotarget. 2015;6(5):2889–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Quentmeier H, Reinhardt J, Zaborski M, Drexler HG. FLT3 mutations in acute myeloid leukemia cell lines. Leukemia. 2003;17(1):120–124. [DOI] [PubMed] [Google Scholar]
- 24.Zarrinkar PP, Gunawardane RN, Cramer MD, et al. AC220 is a uniquely potent and selective inhibitor of FLT3 for the treatment of acute myeloid leukemia (AML). Blood. 2009;114(14):2984–2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schmidt-Arras D, Bohmer SA, Koch S, et al. Anchoring of FLT3 in the endoplasmic reticulum alters signaling quality. Blood. 2009;113(15):3568–3576. [DOI] [PubMed] [Google Scholar]
- 26.Chougule RA, Kazi JU, Ronnstrand L. FYN expression potentiates FLT3-ITD induced STAT5 signaling in acute myeloid leukemia. Oncotarget. 2016;7(9):9964–9974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leischner H, Albers C, Grundler R, et al. SRC is a signaling mediator in FLT3-ITD-but not in FLT3-TKD-positive AML. Blood. 119(17):4026–4033. [DOI] [PubMed] [Google Scholar]
- 28.Robinson LJ, Xue J, Corey SJ. Src family tyrosine kinases are activated by Flt3 and are involved in the proliferative effects of leukemia-associated Flt3 mutations. Exp Hematol. 2005;33(4):469–479. [DOI] [PubMed] [Google Scholar]
- 29.Spiekermann K, Dirschinger RJ, Schwab R, et al. The protein tyrosine kinase inhibitor SU5614 inhibits FLT3 and induces growth arrest and apoptosis in AML-derived cell lines expressing a constitutively activated FLT3. Blood. 2003;101(4):1494–1504. [DOI] [PubMed] [Google Scholar]
- 30.Rosnet O, Marchetto S, deLapeyriere O, Birnbaum D. Murine Flt3, a gene encoding a novel tyrosine kinase receptor of the PDGFR/CSF1R family. Oncogene. 1991;6(9):1641–1650. [PubMed] [Google Scholar]
- 31.Connor MK, Kotchetkov R, Cariou S, et al. CRM1/Ran-mediated nuclear export of p27(Kip1) involves a nuclear export signal and links p27 export and proteolysis. Mol Biol Cell. 2003;14(1):201–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grafone T, Palmisano M, Nicci C, Storti S. An overview on the role of FLT3-tyrosine kinase receptor in acute myeloid leukemia: biology and treatment. Oncol Rev. 2012;6(1):e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Turner AM, Lin NL, Issarachai S, Lyman SD, Broudy VC. FLT3 receptor expression on the surface of normal and malignant human hematopoietic cells. Blood. 1996; 88(9):3383–3390. [PubMed] [Google Scholar]
- 34.Kardinal C, Dangers M, Kardinal A, et al. Tyrosine phosphorylation modulates binding preference to cyclin-dependent kinases and subcellular localization of p27Kip1 in the acute promyelocytic leukemia cell line NB4. Blood. 2006;107(3):1133–1140. [DOI] [PubMed] [Google Scholar]
- 35.Ou L, Ferreira AM, Otieno S, Xiao L, Bashford D, Kriwacki RW. Incomplete folding upon binding mediates Cdk4/cyclin D complex activation by tyrosine phosphorylation of inhibitor p27 protein. J Biol Chem. 2011;286(34):30142–30151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dos Santos C, Demur C, Bardet V, Prade-Houdellier N, Payrastre B, Recher C. A critical role for Lyn in acute myeloid leukemia. Blood. 2008;111(4):2269–2279. [DOI] [PubMed] [Google Scholar]
- 37.Ozawa Y, Williams AH, Estes ML, et al. Src family kinases promote AML cell survival through activation of signal transducers and activators of transcription (STAT). Leuk Res. 2008;32(6):893–903. [DOI] [PubMed] [Google Scholar]
- 38.Shin I, Yakes FM, Rojo F, et al. PKB/Akt mediates cell-cycle progression by phosphorylation of p27(Kip1) at threonine 157 and modulation of its cellular localization. Nat Med. 2002;8(10):1145–1152. [DOI] [PubMed] [Google Scholar]
- 39.Lacy ER, Filippov I, Lewis WS, et al. p27 binds cyclin-CDK complexes through a sequential mechanism involving binding-induced protein folding. Nat Struct Mol Biol. 2004;11(4):358–364. [DOI] [PubMed] [Google Scholar]
- 40.Wei W, Ayad NG, Wan Y, Zhang GJ, Kirschner MW, Kaelin WG., Jr Degradation of the SCF component Skp2 in cell-cycle phase G1 by the anaphase-promoting complex. Nature. 2004;428(6979):194–198. [DOI] [PubMed] [Google Scholar]
- 41.Abdel Alim A, Youssef SR, Farouk HM. The prognostic potential of bone marrow cyclin E and P27, in Egyptian patients with acute myeloid leukemia. Egypt J Immunol. 2010;17(1):9–18. [PubMed] [Google Scholar]
- 42.Radosevic N, Delmer A, Tang R, Marie JP, Ajchenbaum-Cymbalista F. Cell cycle regulatory protein expression in fresh acute myeloid leukemia cells and after drug exposure. Leukemia. 2001;15(4):559–566. [DOI] [PubMed] [Google Scholar]
- 43.Yokozawa T, Towatari M, Iida H, et al. Prognostic significance of the cell cycle inhibitor p27Kip1 in acute myeloid leukemia. Leukemia. 2000;14(1):28–33. [DOI] [PubMed] [Google Scholar]
- 44.Haferlach C, Bacher U, Kohlmann A, et al. CDKN1B, encoding the cyclin-dependent kinase inhibitor 1B (p27), is located in the minimally deleted region of 12p abnormalities in myeloid malignancies and its low expression is a favorable prognostic marker in acute myeloid leukemia. Haematologica. 2011;96(6):829–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Besson A, Dowdy SF, Roberts JM. CDK inhibitors: cell cycle regulators and beyond. Dev Cell. 2008;14(2):159–169. [DOI] [PubMed] [Google Scholar]
- 46.Ozeki K, Kiyoi H, Hirose Y, et al. Biologic and clinical significance of the FLT3 transcript level in acute myeloid leukemia. Blood. 2004;103(5):1901–1908. [DOI] [PubMed] [Google Scholar]
- 47.Kindler T, Lipka DB, Fischer T. FLT3 as a therapeutic target in AML: still challenging after all these years. Blood. 2010;116(24): 5089–5102. [DOI] [PubMed] [Google Scholar]
- 48.Bose P, Grant S. Rational Combinations of Targeted Agents in AML. J Clin Med. 2015;4(4):634–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Uras IZ, Walter GJ, Scheicher R, et al. Palbociclib treatment of FLT3-ITD+ AML cells uncovers a kinase-dependent transcriptional regulation of FLT3 and PIM1 by CDK6. Blood. 2016;127(23):2890–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.