

Abstract
Lyn, a member of the Src family of kinases, is a key factor in the dysregulation of survival and apoptotic pathways of malignant B cells in chronic lymphocytic leukemia. One of the effects of Lyn’s action is spatial and functional segregation of the tyrosine phosphatase SHP-1 into two pools, one beneath the plasma membrane in an active state promoting pro-survival signals, the other in the cytosol in an inhibited conformation and unable to counter the elevated level of cytosolic tyrosine phosphorylation. We herein show that SHP-1 activity can be elicited directly by nintedanib, an agent also known as a triple angiokinase inhibitor, circumventing the phospho-S591-dependent inhibition of the phosphatase, leading to the dephosphorylation of pro-apoptotic players such as procaspase-8 and serine/threonine phosphatase 2A, eventually triggering apoptosis. Furthermore, the activation of PP2A by using MP07-66, a novel FTY720 analog, stimulated SHP-1 activity via dephosphorylation of phospho-S591, which unveiled the existence of a positive feedback signaling loop involving the two phosphatases. In addition to providing further insights into the molecular basis of this disease, our findings indicate that the PP2A/SHP-1 axis may emerge as an attractive, novel target for the development of alternative strategies in the treatment of chronic lymphocytic leukemia.
Introduction
Reversible protein phosphorylation is the fundamental post-translational modification by which virtually all cellular events are regulated, enabling cells to respond properly to intra- and extra-cellular cues. The crucial players involved in this dynamic process are protein kinases and protein phosphatases, which are placed at the different levels of cellular signaling, and, albeit traditionally considered as functionally opposed to one another, not rarely cooperate to finely orchestrate and appropriately drive signal transduction.1–3 The significance of both classes of enzymes for a cell’s life and fate is mirrored by the effects of their dysregulation, whether related to altered expression or activity, which frequently underlies the onset and progression of a plethora of diseases.4–7 B-cell chronic lymphocytic leukemia (CLL), the most common leukemia in the western world,8–10 is no exception to this paradigm, with a high level of intracellular phosphorylation being mediated by the abnormal activity of several kinases downstream of the B-cell receptor, including Lyn, Syk, Btk, PI3K, and Akt.11 This condition, along with the aberrant expression of anti-apoptotic molecules12 and the supportive microenvironment,13 leads to the derangement of cell signaling and contributes to the growth and survival of leukemia cells.
Accumulating evidence suggests that the abnormal signal transduction is also supported by the lack of a proper counterbalance mediated by a number of phosphatases, whose expression or activity is altered in CLL cells. For instance, the expression of PTEN,14 PTPROt,15 PHLPP116,17 and SHIP118 is significantly decreased, leaving tonic pro-survival signaling intact, whereas PTPN22, which acts as a positive regulator of anti-apoptotic signals by hampering the negative regulation of B-cell receptor-dependent signaling pathways, is overexpressed.19 By contrast, protein phosphatase 2A (PP2A)20 and the Src homology 2 domain-containing phosphatase 1 (SHP-1)21 are expressed in CLL at levels comparable to those in normal B cells, but are functionally dysregulated by a variety of mechanisms, which are chiefly mediated by the Src family kinase, Lyn.22,23 In normal B cells, this tyrosine kinase is central to propagating signals initiated by engagement of the B-cell receptor through phosphorylation of the immunoreceptor tyrosine-based activation motifs of the B-cell receptor itself, promoting the formation of a multiprotein “signalosome”.24 It also possesses the unique ability to phosphorylate immunoreceptor tyrosine inhibitory motifs of inhibitory cell surface co-receptors, including FcγRIIb1, CD22, CD72 and CD5, and eventually to negatively regulate B-cell receptor signaling.25,26 In CLL cells, Lyn is overexpressed and constitutively active, distributed between the plasma membrane and an aberrant cytosolic multiprotein complex,27,28 exerting a crucial anti-apoptotic role by phosphorylating and thereby modifying the functional status of, a variety of protein targets.22,29
As to PP2A, the phosphorylation of its catalytic subunit (PP2Ac) by Lyn stabilizes the interaction of PP2Ac itself with SET, an endogenous PP2A inhibitor overexpressed in CLL cells,30 resulting in blockade of the phosphatase activity31,32 and a persistently high level of phosphorylation of factors implicated in CLL cell survival.22 On the other hand, Lyn induces the spatial segregation of SHP-1 into two pools, one being associated with the inhibitory co-receptor CD5 in an active form triggering membrane-derived anti-apoptotic signals, the other being located in the cytosol in an inactive conformation.30 Importantly, this latter condition is thought to be promoted by phosphorylation of the C-terminal tail at Ser591, whereas PP2A seems to play a role in the reactivation of SHP-1.22
The landscape of cell signaling inhibitors approved for the treatment of CLL has expanded rapidly and several agents with novel mechanisms of action (inhibitors of BTK, PI3K and Bcl2) have been introduced into routine clinical practice with promising results documented also in patients with relapsed/refractory disease.33–36 Because CLL is characterized by a high level of Lyn-dependent tyrosine phosphorylation in the cytosol,27–29 we wondered whether compounds capable of directly or indirectly driving the activation of SHP-1 could counter the pervasive action of Lyn and induce cell demise. Here we demonstrate that nintedanib, a small molecule tyrosine kinase inhibitor approved for the treatment of pulmonary fibrosis and lung adenocarcinoma,37 and MP07-66, a novel fingolimod analog designed and synthesized in our laboratories, generate a positive feedback signaling loop involving SHP-1 and PP2A, which dismantles the oncogenic machinery supported by the pro-survival action of the aberrant cytosolic form of Lyn. These molecules, which specifically target and activate molecular players other than those traditionally considered components of the CLL signalosome, may represent new weapons for the treatment of CLL patients.
Methods
Ethics statement
Written informed consent was obtained from all patients, prior to sample collection, according to the Declaration of Helsinki. The ethical approval for our study was granted by the local ethical committee of “Regione Veneto on Chronic Lymphocytic Leukemia” (3259/AO/14).
Patients, cell separation and culture conditions
B cells from 37 untreated patients with CLL were purified and cultured as previously described,27 and subjected to the treatments described throughout the text. The patients’ relevant features are reported in Online Supplementary Table S1.
Information concerning reagents, cell lysis and subcellular fractionation, SHP-1 activity assays immunoprecipitation of SHP-1, [32P]-Band 3 preparation, the PP2A activity assay, the Casp8 activity assay, cell transfection, co-culture conditions, apoptosis assays, western blotting, and the statistical analysis is detailed in the Online Supplementary Data.
Results
Nintedanib directly activates SHP-1 in the cytosol of chronic lymphocytic leukemia cells
We previously demonstrated that SHP-1 is present in CLL cells in two forms, one bound to the plasma membrane receptor CD5 in an active state, and the other in the cytosol in an inhibited conformation.23 As shown in Figure 1A, as well as in Online Supplementary Figure S1A, the plasma membrane-enriched fraction (particulate) and the cytosol of CLL cells were separated from total lysates and immunoblotted with anti-pY536-SHP-1 and anti-pS591-SHP-1 antibodies, a positive response to which is indicative of either activation or inhibition of SHP-1, respectively. As expected, the distribution of SHP-1 in the two cellular compartments paralleled the phosphorylation state, these characteristics proving independent of the diverse biological and clinical features of the single patients (Online Supplementary Table S1). Moreover, to establish how the phosphorylation state affected SHP-1 activity, SHP-1 was immunoprecipitated from the cytosolic and particulate fractions and its activity was measured by using [32P]-Band 3 as a substrate.38 The phosphatase activity of the cytosolic fraction of SHP-1 was negligible as compared to that of the particulate (Figure 1B and Online Supplementary Figure S1B), underscoring that the catalytic activity of SHP-1 can be profoundly changed by phosphorylation at different residues. This finding raised the issue of whether activating the cytosolic pool of SHP-1 might represent a means of countering the elevated level of tyrosine phosphorylation of CLL cells in this compartment, which was previously shown to promote anti-apoptotic mechanisms in this disease.22 We, therefore, first performed in vitro phosphatase activity assays on the cytosolic pool of SHP-1 in the presence of increasing concentrations of nintedanib, a receptor tyrosine kinase inhibitor recently shown to trigger SHP-1 activity39,40 despite the inhibitory S591 phosphorylation.41 First, SHP-1 was immunoprecipitated from the cytosolic fraction of CLL cells in the absence or presence of serine/threonine phosphatase inhibitors, so that the inhibitory phosphorylation at S591 could be removed or preserved, respectively (Figure 1C, inset), and the effect of nintedanib on the activity of the non-phosphorylated and phosphorylated forms of SHP-1 could be evaluated. Figure 1C shows that nintedanib was capable of activating the phosphorylated, and inhibited, form of SHP-1 (Ip2-SHP-1, right-hand panel), as expected,41 whereas the non-phosphorylated form was not influenced by this compound, the phosphatase being already active (Ip1-SHP-1, left-hand panel). These results are concordant with the hypothesis that this drug causes a change in the inhibited conformation of SHP-1 induced by phosphorylation at S591. Similarly, we analyzed the activity of SHP-1 immunoprecipitated from the cytosolic and particulate fractions of CLL cells treated with increasing concentrations of nintedanib, considering that SHP-1 in these compartments is differently phosphorylated and active. As shown in Figure 2A, SHP-1 reached full activation at a concentration as high as 15 μM nintedanib (left-hand panel), as determined by the dephosphorylation of [32P]-Band 3, whereas SHP-1 from the particulate was unaffected (right-hand panel). After incubating CLL cells with 15 μM nintedanib, we evaluated the phosphorylation status of SHP-1 from the cytosolic and particulate fractions by western blot analysis with anti-pS591-SHP-1 and anti-pY536-SHP-1 antibodies, respectively, and performed phosphatase activity assays on SHP-1 immunoprecipitates at each time interval. Nintedanib did not affect either the phosphorylation status (Figure 2B, right-hand panel) or the catalytic activity (Figure 2C, right-hand panel) of the SHP-1 pool of the particulate, whereas SHP-1 in the cytosol was readily activated by nintedanib, reaching maximal efficacy already at the earliest incubation times by circumventing the inhibitory phosphorylation at S591 (Figure 2B,C, left-hand panel). The data obtained from western blot analysis and phosphor imaging were quantitated as arbitrary units and are illustrated in Figure 2D for clarity’s sake.
Figure 1.
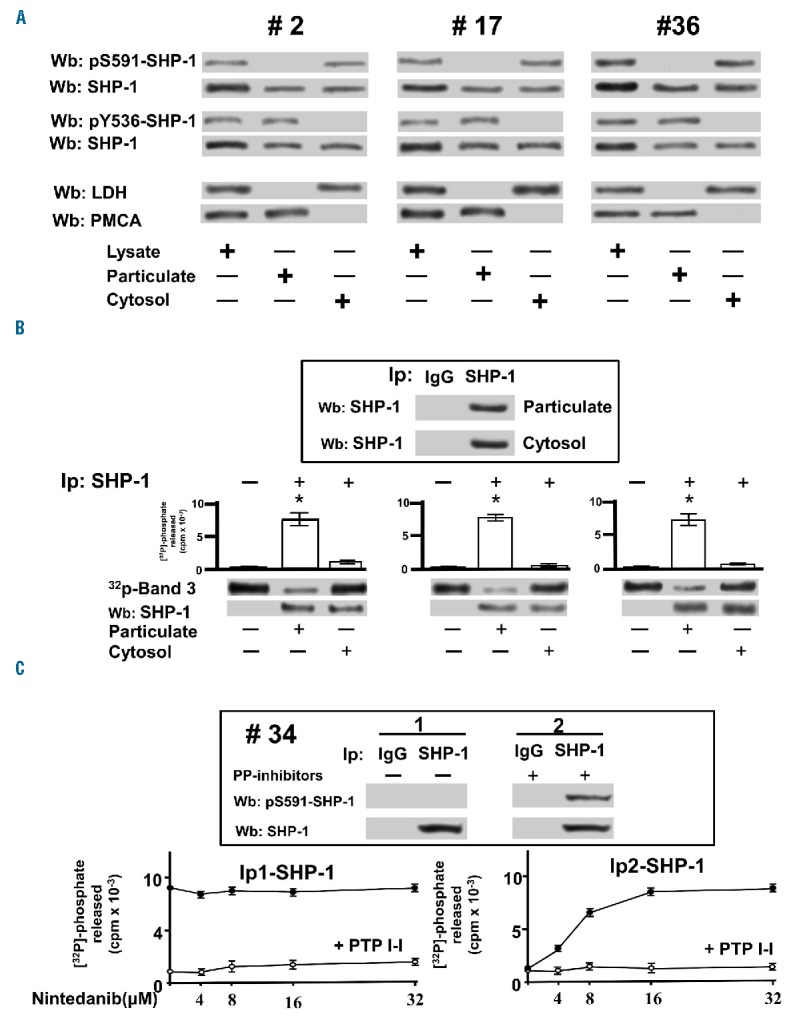
In vitro effect of nintedanib on the differently phosphorylated forms of SHP-1 pulled down from chronic lymphocytic leukemia cells. (A) Phosphorylation state of SHP-1 in total cell lysates, particulate and cytosol of CLL cells of patients belonging to various clinical and biological subtypes. Anti-LDH (cytosolic marker), anti-PMCA (plasma membrane marker). (B) Tyrosine phosphatase activity of SHP-1 immunoprecipitated from particulate and cytosol of CLL of patients #2, #17, and #36 measured as [32P] released from in vitro [32P]-Band 3. (C) Tyrosine phosphatase activity of SHP-1 immunoprecipitated in the absence (Ip1-SHP-1) and presence (Ip2-SHP-1) of serine/threonine phosphatase inhibitors from the cytosol of CLL cells of 15 patients as determined in vitro in the presence of increasing concentrations of nintedanib supplemented without (solid circles) or with 25 μM PTP I-I (open circles). Data are mean ± SD of three experiments performed in triplicate (*P≤0.01). LDH: lactate dehydrogenase; PMCA: plasma membrane Ca2+ ATPase; Wb: western blot; IP: immunoprecipitation.
Figure 2.
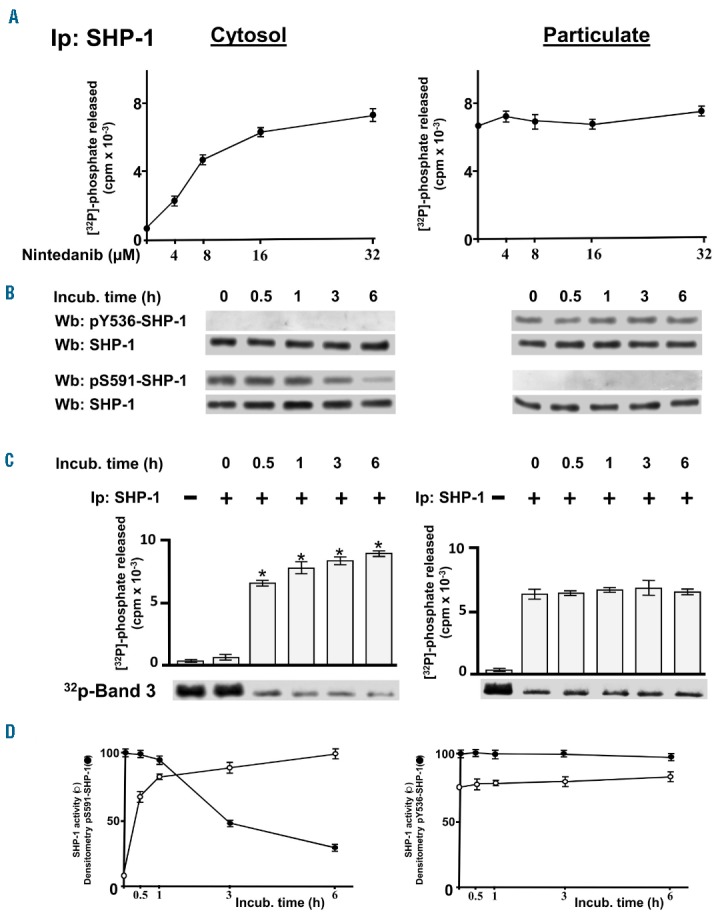
Effect of nintedanib on the activity of the differently phosphorylated forms of SHP-1 inside chronic lymphocytic leukemia cells. (A) Phosphatase activity of SHP-1 immunoprecipitated from the cytosol (left) and particulate (right) of CLL cells cultured in the absence or presence of increasing concentrations of nintedanib for 1 h measured as [32P] released from [32P]-Band 3. Data are mean ± SD of three experiments performed in triplicate. (B) Expression and phosphorylation state of SHP-1 of CLL cells cultured in the presence of 15 μM nintedanib over time. (C) Phosphatase activity of SHP-1 immunoprecipitated from the cytosol (left) or particulate (right) of the CLL cells described in (B) measured as [32P] released from [32P]-Band 3. (D) Densitometric analysis of western blots probed with anti-pS591 or anti-pY536 antibody (arbitrary units, open circles, left- and right-hand panel, respectively) and phosphatase activity from (C) normalized as percentage (solid circles, left and right panel, respectively) and plotted as line graphs. Data are mean ± SD of three experiments performed in triplicate (*P≤ 0.01), n=16. Wb: western blot; IP: immunoprecipitation; incub: incubation.
Nintedanib induces activation of caspase-8 and PP2A by decreasing tyrosine phosphorylation in chronic lymphocytic leukemia cells
To verify whether the inhibition of SHP-1 contributed to the elevated tyrosine phosphorylation of CLL cells, which we earlier demonstrated to be dependent on the delocalized and constitutively active HSP90-bound form of Lyn27,28 (Online Supplementary Figure S2), freshly isolated CLL cells underwent SHP-1 knockdown (Figure 3A) and then incubated for 1 h in the presence of increasing concentrations of nintedanib (0–30 μM). After cell lysis, western blotting with anti-phosphotyrosine antibody showed that nintedanib caused a dramatic reduction in tyrosine phosphorylation at a concentration of 10 μM (Figure 3B and Online Supplementary S3B, left-hand panel), which is significantly higher than the nanomolar range reported to inhibit the receptor tyrosine kinases that nintedanib is known to target.39,40 Importantly, the effect of nintedanib was abrogated by the genetic inhibition of the phosphatase itself (Figure 3B and Online Supplementary S3B, right-hand panel). On the other hand, tyrosine kinase activities were only affected to a limited extent by nintedanib, as assessed by the level of phosphorylation of poly-Glu-Tyr or cdc2[6-20], peptide substrates used to determine global (including that of receptor tyrosine kinases) or Src family kinase-specific tyrosine kinase activity, respectively.42,43 In fact, as shown by the histograms in Figure 3C and Online Supplementary Figure S3C, both types of activities were affected at most by 30% of the control only at high concentrations of nintedanib (over 10 μM). Notably, the slight decrease in tyrosine kinase activity paralleled the activation state of Lyn, as monitored by western blot analysis with anti-pTyr-396-Lyn antibody (Figure 3D and Online Supplementary Figure S3D), whereas the phosphorylation of CD5, to which the pool of SHP-1 associated with the plasma membrane is bound,23 was unchanged (Figure 3E and Online Supplementary Figure S3E). Moreover, stimulation with 20 μg/mL anti-IgM antibody at different time points, which only weakly increased the level of phosphorylation of CLL cells compared to that in the resting state,23,27 did not affect the decrease in tyrosine phosphorylation caused by pre-incubation with 15 μM nintedanib for 1 h (Online Supplementary Figure S4), again underscoring the importance of the activation of the cytosolic pool of SHP-1 in dampening phospho-tyrosine-mediated signals.
Figure 3.

Effect of nintedanib on the tyrosine phosphorylation of chronic lymphocytic leukemia cells. (A) Expression of SHP-1 in CLL cells of patient #18 transfected with scrambled and SHP-1 siRNA. (B) Phosphorylation pattern of CLL cells transfected with scrambled or SHP-1 siRNA and cultured in the absence or presence of increasing concentrations of nintedanib for 1 h. (C) Global tyrosine kinase activity and specific Src activity in CLL cells cultured as in (B) determined by [32P] incorporation into the nonspecific random polymer polyGlu4Tyr (top panel) or the specific peptide substrate cdc2(6–20) (bottom panel). (D) Activation state of Lyn in CLL cells cultured as in (B) determined by western blotting with anti-pTyr396-Lyn antibody. (E) Phosphorylation state of CD5 as assessed by western blotting with anti-pTyr antibody of CD5 immunoprecipitates from total cell lysates of CLL cells treated as in (B). Data are mean ± SD of three experiments performed in triplicate (*P≤0.05). Wb: western blotting; IP: immunoprecipitation.
Additionally, we wanted to evaluate whether nintedanib could affect CLL cell viability, considering that compounds decreasing the tyrosine phosphorylation in CLL cells also induced apoptosis.27,28 Freshly isolated CLL cells incubated with increasing concentrations of nintedanib (0–24 μM) for 24 and 48 h exhibited a marked level of apoptosis, as determined by annexin V–propidium iodide flow cytometry and pooled densitometric analysis (Figure 4A, left-hand panel). Moreover, the degree of variability observed among the different subsets of patients as spontaneous apoptosis was attenuated after treatment with nintedanib (Online Supplementary Figure S5). Notably, the concentrations of nintedanib able to induce apoptosis in CLL cells did not alter the survival of normal B cells (Online Supplementary Figure S6). Mechanistically, PARP cleavage suggested that activation of the apoptotic pathway was the major route leading to cell death (Figure 4A, right-hand panel), which was further confirmed by the use of the pan-caspase inhibitor z-VADfmk44 (Online Supplementary Figure S7A) and by experiments exploring alternative mechanisms possibly contributing to cell demise, such as necroptosis or autophagy. In fact, survival of CLL cells did not vary if treatment with nintedanib followed pre-incubation with necrostatin-1, an inhibitor of necroptosis (Online Supplementary Figure S7B), and the protein levels of p62/SQSTM1, the degradation of which is indicative of autophagy,45,46 remained stable for the entire duration of the experiments performed in the presence of nintedanib (Online Supplementary Figure S7C). In the latter case, the positive control was provided by supplementing the incubation medium of CLL cells with the mTOR inhibitor everolimus, which caused a decrease in the protein level of p62/SQSTM1 (Online Supplementary Figure S7D).47 Similar results were obtained by co-culturing CLL cells with bone marrow mesenchymal stromal cells, which had been cultured in monolayer and grown to confluence (approximately 1×105 mesenchymal stromal cells per well), in a 20:1 ratio, to mimic the tissue microenviroment in which CLL cells proliferate48 (Online Supplementary Figure S8).
Figure 4.

Effect of nintedanib on the viability of chronic lymphocytic leukemia cells. Apoptosis of CLL cells from ten patients belonging to the various clinical and biological subtypes cultured (A) in the presence of increasing concentrations of nintedanib for 24 and 48 and assessed h by annexin V–propidium iodide flow cytometry, (B) in the absence (bars 1–2, 5–6) or presence (bars 3–4, 7–8) of 15 μM nintedanib for 0 or 24 h after transfection with either scrambled siRNA or SHP-1-siRNA (white and gray bars, respectively, left-hand panel) analyzed as in (A), and (C) pre-incubated without (1–4) or with 25 μM PTP-I-I (5–8) for 1 h (white and gray bars, respectively, left-hand panel) and then cultured in the absence (bars 1–2, 5–6) or presence (bars 3–4, 7–8) of 15 μM nintedanib for 0 or 24 h analyzed as in (A). Expression of SHP-1 in CLL cells of patient #34 after transfection with either scrambled siRNA or SHP-1-siRNA by western blotting analysis and pooled densitometric analysis (arbitrary units) of the Wb bands of ten patients is represented by the histograms in the inset of (B). Data are mean percentage of early and late apoptosis ± SD from three separate experiments performed in triplicate (left-hand panel, *P≤0.01). Western blotting analysis with anti-caspase 3 and anti-PARP antibodies monitored caspase-dependent apoptosis; anti-β-actin antibody was used as a loading control (right-hand panels). Wb: western blot.
To further investigate the contribution of SHP-1 to nintedanib-induced apoptosis, we used genetic and pharmacological inhibition of SHP-1 itself. Figure 4B (left-hand panel) shows that the rate of apoptosis after 24 h of treatment with nintedanib (bar 4) was dramatically reduced by the silencing of SHP-1 (bar 8) to levels comparable to those reached by knocking down the phosphatase in the absence of nintedanib (bar 6). At the molecular level, nintedanib induced caspase-dependent apoptosis, as witnessed by the cleavage of caspase-3 and PARP (Figure 4B, right-hand panel, lane 4), the latter event being negligible when silencing SHP-1, irrespective of the presence of nintedanib (lanes 6 and 8). These findings were consistent with our previous data demonstrating that SHP-1 knockdown brings about caspase-independent apoptosis by targeting the plasma membrane pool of the phosphatase, which is catalytically active and orchestrates anti-apoptotic signals.21,23 The pharmacological inhibition of SHP-1 mediated by PTP-I-I provided results overlapping those obtained by using SHP-1 short interfering RNA (Figure 4C). These observations led us to hypothesize that the dephosphorylation of specific SHP-1 substrates induced apoptosis following treatment with nintedanib in CLL cells. We, therefore, focused on two factors that we had previously explored and the activity of which we found to be inhibited in CLL via phosphorylation by the aberrant cytosolic form of Lyn, procaspase-8 (procasp8)29 and PP2A.22 After incubating freshly isolated CLL cells with increasing concentrations of nintedanib, we performed western blot analysis with antibodies directed against the phosphorylated form of specific inhibitory residues of these two proteins, Y380 of procasp8 and Y307 of PP2Ac. Both tyrosines were phosphorylated when SHP-1 was not silenced and nintedanib was not added to the incubation medium (Figure 5A,C, left-hand panels, 0 μM), the level of phosphorylation gradually declining as nintedanib concentration increased (1–20 μM). Moreover, total lysates from the same samples were assayed for the activity of the two enzymes in vitro (see the Online Supplementary Data for details on the commercial kits employed), which allowed us to conclude that the activation of both enzymes depended on the action of nintedanib exerted on SHP-1 (Figure 5B,D, left-hand panels). Importantly, dephosphorylation and activation of these factors were blocked, albeit in the presence of nintedanib, by silencing SHP-1 in CLL cells (Figure 5, right-hand panels), indicating that procasp8 and PP2Ac were substrates for SHP-1 and effectors of a SHP-1- dependent pro-apoptotic pathway.
Figure 5.
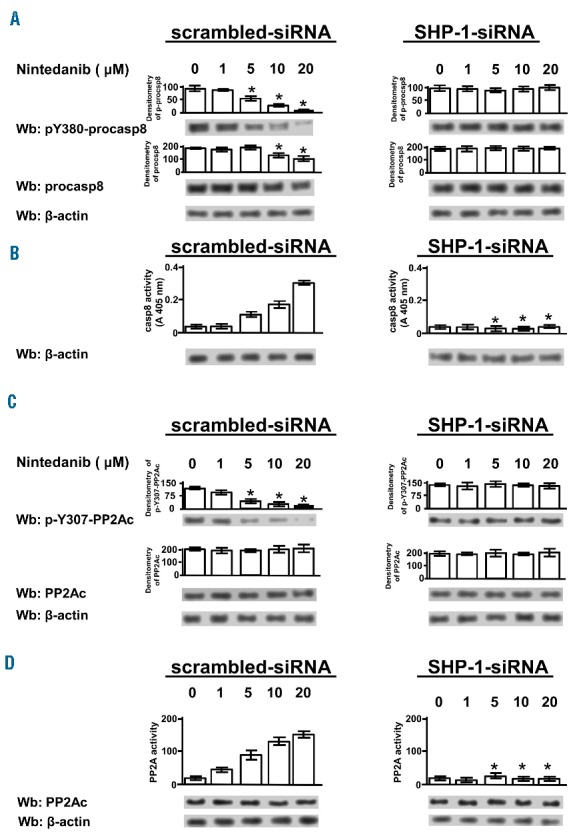
Effect of nintedanib on the phosphorylation state and activity of procaspase 8 and PP2Ac. (A) Western blotting (Wb) analysis with anti-pY380-procasp8 antibody, stripped and reprobed with anti-procasp8 antibody and with anti-β-actin antibody as a loading control, of total cell lysates of CLL cells from ten patients belonging to the various clinical and biological subtypes transfected by nucleofection with either scrambled siRNA (left-hand panels) or SHP-1-siRNA (right-hand panels) and cultured in the presence of increasing concentrations of nintedanib for 1 h. Densitometric analysis (arbitrary units) of the pY380-procasp8 and procasp8 bands is represented as histograms. (B) In vitro casp8 activity on cell lysates from CLL cells treated as in (A) as described in the Methods section. Compared with the effect of nintedanib, changes due to siRNA were statistically significant (*P≤0.01). (C) Wb analysis with anti-pY307-PP2Ac antibody, stripped and reprobed with anti-PP2Ac antibody and with anti-β-actin antibody as a loading control, of total cell lysates of CLL cells from ten patients treated as in (A). Densitometric analysis (arbitrary units) of the pY307-PP2Ac and PP2Ac bands is represented as histograms. Data are mean ± SD from four experiments performed in triplicate. (D) In vitro PP2A activity on cell lysates from CLL cells treated as in (A) by using a specific phosphopeptide as a substrate, as described in the Methods section of the Online Supplementary Data. Compared with the effect of nintedanib, changes due to siRNA were statistically significant (*P≤0.01).
Apoptosis of chronic lymphocytic leukemia cells can be induced by indirect activators of SHP-1
The data collected thus far confirmed that nintedanib could trigger SHP-1 by circumventing the phosphorylation at the inhibitory residue S591. Interestingly, we recently demonstrated that pS591 could be dephosphorylated by PP2A, the activity of which was impaired by phosphorylation at Y307 as well as by the interaction with its endogenous inhibitor SET.22 In this scenario, the restoration of PP2A activity by a fingolimod analog devoid of immunosuppressive action, the so-called MP07-66 [(2,2-diethoxyethyl<4-(hexyloxy)phenyl]methyl})amine], and the subsequent dephosphorylation of PP2A substrates, was shown to trigger apoptosis.22 These findings led us to conjecture that MP07-66 could be used as an indirect activator of SHP-1 via PP2A, and exploited to potentiate the action of nintedanib on SHP-1 itself to reinforce the apoptotic response of CLL cells. As shown in Figure 6A (left-hand panel), incubation with increasing concentrations of MP07-66 (0–24 μM) for 24 and 48 h brought about a marked level of apoptosis in CLL cells, as determined by annexin V–propidium iodide flow cytometry. As in the case of nintedanib (Figure 4), MP07-66 evoked apoptosis in a caspase-dependent manner, as shown by the use of the pan-caspase inhibitor zVADfmk44 (Figure 6A, Online Supplementary Figure S9A,B), with no evidence of necroptosis, the number of viable cells remaining unchanged following pre-incubation with necrostatin-1 prior to MP07-66 treatment, or autophagy, as indicated by the stable level of expression of p62/SQSTM145 (Online Supplementary Figure S9C) for the duration of the experiments in the presence of PP2A activator. Under these conditions, the survival of normal B cells was not altered (Online Supplementary Figure S10). To evaluate whether SHP-1 was implicated in these events, aliquots from the cytosol for each time interval were immunoblotted with anti-pS591-SHP-1 antibody and tested for phosphatase activity in the presence of [32P]-Band 3 as a substrate. The elevation of activity of SHP-1 was concomitant with the dephosphorylation thereof as the concentration of MP07-66 increased (Figure 6B,C) and the results were highly similar for all the samples tested (Online Supplementary Figure S11A,B). Further evidence for the role of SHP-1 in mediating apoptosis of CLL cells upon treatment with MP07-66 was provided by western blotting with antibodies against pY380-procasp8 and pY307-PP2Ac, which revealed the dephosphorylation of the inhibitory residues (Figure 6D, lane 6) at 6 h. As expected, this event was blocked by the specific SHP-1 inhibitor PTP-I-I (Figure 6D, lane 8) as well as by okadaic acid, a phosphatase inhibitor that is highly selective toward PP2A in the low nanomolar range49 (lane 7), which confirmed that the activation of PP2A drove the dephosphorylation of SHP-1 S591, thereby promoting global tyrosine dephosphorylation and cell death. Again, these results were similar for all the samples tested, as reported in the pooled densitometric analysis shown in Online Supplementary Figure S11C.
Figure 6.
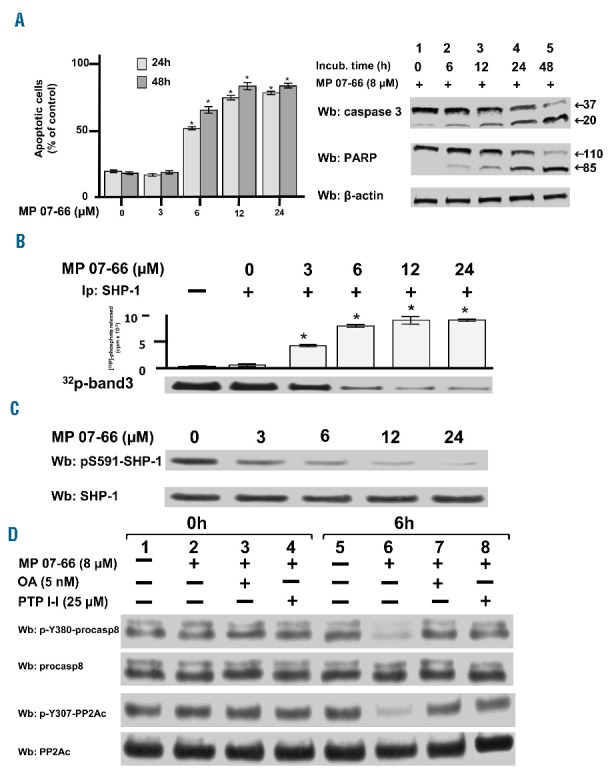
Effect of MP 07-66 on the chronic lymphocytic leukemia cell survival. (A) Apoptosis of CLL cells from ten patients belonging to the various clinical and biological subtypes cultured in the presence of increasing concentrations of MP07-66 for 24 and 48 h analyzed by annexin V– propidium iodide flow cytometry (left-hand panel). Data are mean percentages of early and late apoptosis ± SD from three separate experiments performed in triplicate (*P≤0.01). Western blotting (Wb) analysis of total cell lysate of CLL cells with anti-caspase 3 and anti-PARP antibodies monitored caspase-dependent apoptosis; anti-β-actin antibody was used as a loading control (right-hand panel). (B) Tyrosine phosphatase activity of SHP-1 immunoprecipitated from the cytosol of CLL cells of patient #28 cultured in the presence of increasing concentrations of MP07-66 for 1 h and measured as [32P] released from in vitro [32P]-Band 3. Data are expressed as mean ± SD from one experiment performed in triplicate (*P≤ 0.01). (C) Expression of SHP-1 and phosphorylation state of pS591 of CLL cells of patient #28 cultured in the presence of increasing concentrations of MP07-66. Data are expressed as mean ± SD from one experiment performed in triplicate (*P≤0.01). (D) Expression and phosphorylation state of procasp8 and PP2Ac in the CLL cells of patient #28 cultured in the absence (lanes 1 and 5) and presence of 8 μM MP07-66 supplemented with 5 nM okadaic acid (OA) (lanes 3 and 7) or 25 μM PTP-I-I (lanes 4 and 8) for 0 and 6 h.
All these observations were consistent with the hypothesis that SHP-1 and PP2A form a signaling axis wherein the single phosphatases, when stimulated, can activate one another, and that such a process can be amplified by a combination of molecules activating both simultaneously.
MP07-66 potentiates the pro-apoptotic effect of nintedanib
Since our data suggest that the activation of either PP2A or SHP-1 triggered by specific small molecules caused stimulation of each other’s activity, thereby evoking a positive feedback signaling loop promoting apoptosis, we wondered whether the combination of nintedanib and MP07-66 could result in a more robust apoptotic response of CLL cells. We, therefore, incubated freshly isolated CLL cells with 15 μM nintedanib and 8 μM MP07-66 at different times and monitored apoptosis by annexin V–propidium iodide flow cytometry (Figure 7A). Nintedanib proved moderately effective at inducing apoptosis of CLL cells after 6 and 12 h of incubation, its efficacy being large ly improved by the concomitant presence of MP07-66, which itself exhibited a pro-apoptotic activity overlapping that of nintedanib when used as a single agent. Furthermore, the variability exhibited by the different subsets of patients as spontaneous apoptosis was attenuated after treatment with the combination of the two agents (Online Supplementary Figure S12). Similarly, co-cultures of CLL cells with bone marrow mesenchymal stromal cells were treated as above with overlapping results (Online Supplementary Figure S13). Moreover, to explore how PP2A activation took part in the apoptotic process induced by MP07-66, CLL cells were incubated with 15 μM nintedanib for 6 h in the absence or presence of okadaic acid. As expected, 5 nM okadaic acid drastically reduced the rate of apoptosis of CLL cells treated with MP07-66 or the combination with nintedanib, but only to a lesser extent with nintedanib alone (Figure 7B, left-hand panel). Moreover, the PP2A activity assay performed using a commercial PP2A assay kit on cell lysates of CLL cells treated as above showed a trend similar to that observed for the apoptotic rate (right-hand panel). Overall, these data corroborate the hypothesis that the inhibition of PP2A is central to CLL cell viability and that its activation is facilitated by the supportive action of SHP-1, as demonstrated by the effect produced by the simultaneous use of the respective activators.
Figure 7.

Effect of the combined action of nintedanib and MP07-66 on chronic lymphocytic leukemia cell survival. (A) Apoptosis of CLL cells from ten patients belonging to the various clinical and biological subtypes cultured in the absence and presence of 15 μM nintedanib, 8 μM MP07-66 or both over time analyzed by annexin V–propidium iodide flow cytometry. (B) Apoptosis of CLL cells from ten patients as described in (A) supplemented without or with 5 nM okadaic acid (OA) (right panels) for 6 h. Analysis by annexin V– propidium iodide flow cytometry is expressed as mean percentages of early and late apoptosis ± SD from three separate experiments performed in triplicate (*P≤0.01). (C) PP2A phosphatase activity performed on the total lysates of CLL cells treated as in (B). (D) Working model of the positive feedback signling loop triggered by the combination of nintedanib and MP07-66, which counters the high level of cytosolic tyrosine phosphorylation, the crucial factor sustaining the oncogenic machinery in CLL cells, mediated by the aberrant form of HSP90-bound Lyn.
Discussion
In this study, we show that nintedanib induces caspase-dependent apoptosis in CLL cells via dephosphorylation of pro-apoptotic key players such as procasp8 and PP2A by directly activating the cytosolic pool of the tyrosine phosphatase SHP-1.
SHP-1 is a tyrosine phosphatase that negatively regulates signaling in cells of hematopoietic lineage, having a key role in modulating the response to antigens and contributing to the development of tolerance to self-antigens. In CLL, SHP-1 undergoes multiple regulatory mechanisms leading to both spatial and functional segregation, which seem to be crucial in supporting the cancer phenotype.23 Phosphorylation of different residues, especially at the C-terminus, significantly changes the activation status and localization of SHP-1, phospho-Y536 being typical of the activated pool bound to the plasma membrane co-receptor CD5, and phospho-S591 characterizing the inhibited pool of SHP-1 in the cytosol. This latter form appears to be one of the factors that sustains the aberrant Lyn-dependent tyrosine phosphorylation of countless proteins in the cytosol of CLL cells, which is ultimately key to the anti-apoptotic signaling network in this disease.22,27,29 Here, we demonstrate that nintedanib, a small molecule known to act as a triple angiokinase inhibitor in the low nanomolar range,39,40 activates the cytosolic fraction of SHP-1 by circumventing pS591-dependent inhibition, as recently described in another cancer model.41 Significantly, in addition to leaving SHP-1 at the plasma membrane unaffected, nintedanib only marginally modifies tyrosine kinase activities in CLL cells even at micromolar concentrations. On the other hand, a dramatic drop in tyrosine phosphorylation occurs as a result of SHP-1 activation in the cytosol with consequent caspase-dependent apoptosis, suggesting that the massive tyrosine phosphorylation in CLL cells directly affects the function of factors that counteract the oncogenic machinery. Notably, although genetic or pharmacological inhibition of SHP-1 can prevent the caspase-dependent apoptosis evoked by nintedanib, again supporting the hypothesis that the action of this drug is mediated by SHP-1, such treatments can still induce caspase-independent apoptosis, which is in line with the role of the CD5-bound form of SHP-1 as a pro-survival agent in CLL.23
Our findings indicate that the distribution of the two forms of SHP-1 is central to their differentiated function, at the plasma membrane in an active form taking part in a signalosome that orchestrates survival signals, and in the cytosol in an inhibited conformation. This condition renders SHP-1 unable to dephosphorylate cytosolic Lyn targets endowed with pro-apoptotic potential, such as procasp8 and PP2A. Procasp8 occurs as an inactive homodimer in CLL cells, the trigger for dimerization being the phosphorylation of Y380 mediated by Lyn.29 Here, nintedanib, via direct activation of SHP-1 in the cytosol, induces dephosphorylation, autocatalysis and activation of procasp8, which explains the caspase-dependent apoptosis observed. As to PP2A, Lyn-mediated phosphorylation at Y307 of the catalytic subunit stabilizes its interaction with its physiological inhibitor SET, hampering the activity of the phosphatase.22 This results in the persistent serine/threonine phosphorylation of PP2A substrates, including the cytosolic pool of SHP-1, and propagates pro-survival and anti-apoptotic signals.22 Conversely, nintedanib-activated SHP-1 dephosphorylates PP2A, facilitating the disruption of the PP2A/SET complex, with activation of PP2A itself and dephosphorylation of SHP-1 (Figure 7D), eventually triggering the apoptotic response. This latter event is magnified by the combination of nintedanib with the FTY720 analog MP07-66 in that this latter compound interferes directly with the interaction between PP2A and SET,22 further aiding in the reactivation of PP2A. In this scenario, nintedanib and MP07-66, direct activators of SHP-1 and PP2A, respectively, appear to initiate a positive feedback signaling loop which opposes Lyn-mediated oncogenic signaling, thus promoting the dephosphorylation of crucial players in the deranged signaling architecture of CLL, switching off anti-apoptotic signals and unleashing cell death.
In conclusion, our findings indicate that phosphatase activators may represent a new weapon against this form of leukemia, especially in the light of the heterogeneity and the unavoidable progression of the disease as well as the resistance to the front-line drugs currently in use, not to mention the adverse effects recently reported for the most promising second-line drugs.50
Supplementary Material
Acknowledgments
This work was supported by grants from A.I.R.C. (Milan) to GS and LT by MIUR with funds to LT; by Regione Veneto on Chronic Lymphocytic Leukemia; by AIRC Regional Project with Fondazione CARIPARO and CARIVERONA and by FIRB (Rome).
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/102/8/1401
References
- 1.Johnson LN. The regulation of protein phosphorylation. Biochem Soc Trans. 2009;37(4): 627–641. [DOI] [PubMed] [Google Scholar]
- 2.Lim WA, Pawson T. Phosphotyrosine signalling: evolving a new cellular communication system. Cell. 2010;142(5):661–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tonks NK. Protein tyrosine phosphatases. from house keeping enzymes to master regulators of signal transduction. FEBS J. 280(2):346–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cohen P. Protein kinases - the major drug targets of the twenty-first century¿ Nat Rev Drug Discov. 2002;1(4):309–315. [DOI] [PubMed] [Google Scholar]
- 5.Lahiry P, Torkamani A, Schork NJ, Hegele RA. Kinase mutations in human diseases: interpreting genotype-phenotype relationships. Nat Rev Genet. 2010;11(1):60–74. [DOI] [PubMed] [Google Scholar]
- 6.Julien SG, Dube N, Hardy S, Tremblay ML. Inside the human cancer tyrosine phos-phatome. Nat Rev Cancer. 2011;1(1):35–49. [DOI] [PubMed] [Google Scholar]
- 7.Khatri A, Wang J, Pendergast AM. Multifunctional Abl kinases in health and disease. J Cell Sci. 2016;129(1): 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dighiero G, Hamblin TJ. Chronic lymphocytic leukaemia. Lancet. 2008;371(9617): 1017–1029. [DOI] [PubMed] [Google Scholar]
- 9.Burger JA, Chiorazzi N. B cell receptor signalling in chronic lymphocytic leukemia. Trends Immunol. 2013;34(12):592–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hallek M. Chronic lymphocytic leukemia: 2015 Update on diagnosis, risk stratification, and treatment. Am J Hematol. 2015;90(5):446–460. [DOI] [PubMed] [Google Scholar]
- 11.Zhang S, Kipps TJ. The pathogenesis of chronic lymphocytic leukemia. Annu Rev Pathol. 2014;9:103–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Del Gaizo Moore V, Brown JR, Certo M, Love TM, Novina CD, Letai A. Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT-737. J Clin Invest. 2007;117(1):112–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ten Hacken E, Burger JA. Microenvironment interactions and B-cell receptor signalling in chronic lymphocytic leukemia: implications for disease pathogenesis and treatment. Biochim Biophys Acta. 2016;1863(3):401–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zou ZJ, Fan L, Wang L, et al. miR-26a and miR-214 down-regulate expression of the PTEN gene in chronic lymphocytic leukemia, but not PTEN mutation or promoter methylation. Oncotarget. 2015;6(2): 1276–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Motiwala T, Majumder S, Kutay H, et al. Methylation and silencing of protein tyrosine phosphatase receptor type O in chronic lymphocytic leukemia. Clin Cancer Res. 2007;13(11):3174–3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Suljagic M, Laurenti L, Tarnani M, Alam M, Malek SN, Efremov DG. Reduced expression of the tumor suppressor PHLPP1 enhances the antiapoptotic B-cell receptor signal in chronic lymphocytic leukemia B-cells. Leukemia. 2010;24(12):2063–2071. [DOI] [PubMed] [Google Scholar]
- 17.O’Hayre M, Niederst M, Fecteau JF, et al. Mechanisms and consequences of the loss of PHLPP1 phosphatase in chronic lymphocytic leukemia (CLL). Leukemia. 2012;26(7):1689–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cui B, Chen L, Zhang S, et al. MicroRNA-155 influences B-cell receptor signalling and associates with aggressive disease in chronic lymphocytic leukemia. Blood. 2014;124(4):546–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Negro R, Gobessi S, Longo PG, et al. Overexpression of the autoimmunity-associated phosphatase PTPN22 promotes survival of antigen-stimulated CLL cells by selectively activating AKT. Blood. 2012;119(26):6278–6287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perrotti D, Neviani P. Protein phosphatase 2A (PP2A), a druggable tumor suppressor in Ph1(+) leukemias. Cancer Metastasis Rev. 2008;27(2):159–168. [DOI] [PubMed] [Google Scholar]
- 21.Tsui FW, Martin A, Wang J, Tsui HW. Investigations into the regulation and function of the SH2 domain-containing protein-tyrosine phosphatase, SHP-1. Immunol Res. 2006;35(1–2):127–136. [DOI] [PubMed] [Google Scholar]
- 22.Zonta F, Pagano MA, Trentin L, et al. Lyn sustains oncogenic signalling in chronic lymphocytic leukemia by strengthening SET-mediated inhibition of PP2A. Blood. 2015;125(24):3747–3755. [DOI] [PubMed] [Google Scholar]
- 23.Tibaldi E, Brunati AM, Zonta F, et al. Lyn-mediated SHP-1 recruitment to CD5 contributes to resistance to apoptosis of B-cell chronic lymphocytic leukemia cells. Leukemia. 2011;25(11):1768–1781. [DOI] [PubMed] [Google Scholar]
- 24.Pierce SK, Liu W. The tipping points in the initiation of B cell signalling: how small changes make big differences. Nat Rev Immunol. 2010;10(11):767–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DeFranco AL, Chan VW, Lowell CA. Positive and negative roles of the tyrosine kinase Lyn in B cell function. Semin Immunol. 1998; 10(4):299–307. [DOI] [PubMed] [Google Scholar]
- 26.Xu Y, Harder KW, Huntington ND, Hibbs ML, Tarlinton DM. Lyn tyrosine kinase: accentuating the positive and the negative. Immunity. 2005;22(1):9–18. [DOI] [PubMed] [Google Scholar]
- 27.Contri A, Brunati AM, Trentin L, et al. Chronic lymphocytic leukemia B cells contain anomalous Lyn tyrosine kinase, a putative contribution to defective apoptosis. J Clin Invest. 2005;115(2):369–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trentin L, Frasson M, Donella-Deana A, et al. Geldanamycin-induced Lyn dissociation from aberrant Hsp90-stabilized cytosolic complex is an early event in apoptotic mechanisms in B-chronic lymphocytic leukemia. Blood. 2008;112(12):4665–4674. [DOI] [PubMed] [Google Scholar]
- 29.Zonta F, Pagano MA, Trentin L, et al. Lyn-mediated procaspase 8 dimerization blocks apoptotic signalling in B-cell chronic lymphocytic leukemia. Blood. 2014;123(6): 875–883. [DOI] [PubMed] [Google Scholar]
- 30.Christensen DJ, Chen Y, Oddo J, et al. SET oncoprotein overexpression in B-cell chronic lymphocytic leukemia and non-Hodgkin lymphoma: a predictor of aggressive disease and a new treatment target. Blood. 2011;118(15):4150–4158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oaks J, Ogretmen B. Regulation of PP2A by sphingolipid metabolism and signalling. Front Oncol. 2015;4:388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saddoughi SA, Gencer S, Peterson YK, et al. Sphingosine analogue drug FTY720 targets I2PP2A/SET and mediates lung tumour suppression via activation of PP2A-RIPK1-dependent necroptosis. EMBO Mol Med. 2013(1);5:105–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Woyach JA, Furman RR, Liu TM, et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N Engl J Med. 2014;370(24):2286–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Furman RR, Sharman JP, Coutre SE, et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med. 2014;370(11):997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Burger JA, Tedeschi A, Barr PM, et al. Ibrutinib as initial therapy for patients with chronic lymphocytic leukemia. N Engl J Med. 2015;373(25):2425–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roberts AW, Davids MS, Pagel JM, et al. Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia. N Engl J Med. 2016;374(4):311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McCormack PL. Nintedanib: first global approval. Drugs. 2015;75(1):129–139. [DOI] [PubMed] [Google Scholar]
- 38.Tibaldi E, Zonta F, Bordin L, et al. The tyrosine phosphatase SHP-1 inhibits proliferation of activated hepatic stellate cells by impairing PDGF receptor signalling. Biochim Biophys Acta. 2014;1843(2):288–298. [DOI] [PubMed] [Google Scholar]
- 39.Hilberg F, Roth GJ, Krssak M, et al. BIBF 1120: triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy. Cancer Res. 2008;68(12):4774–4782. [DOI] [PubMed] [Google Scholar]
- 40.Roth GJ, Heckel A, Colbatzky F, et al. Design, synthesis, and evaluation of indolinones as triple angiokinase inhibitors and the discovery of a highly specific 6-methoxycarbonyl-substituted indolinone (BIBF 1120). J Med Chem. 2009;52(14):4466–4480. [DOI] [PubMed] [Google Scholar]
- 41.Tai WT, Shiau CW, Li YS, et al. Nintedanib (BIBF-1120) inhibits hepatocellular carcinoma growth independent of angiokinase activity. J Hepatol. 2014;61(1):89–97. [DOI] [PubMed] [Google Scholar]
- 42.Gringeri E, Carraro A, Tibaldi E, et al. Lyn-mediated mitochondrial tyrosine phosphorylation is required to preserve mitochondrial integrity in early liver regeneration. Biochem J. 2009;425(2):401–412. [DOI] [PubMed] [Google Scholar]
- 43.Tibaldi E, Venerando A, Zonta F, et al. Interaction between the SH3 domain of Src family kinases and the proline-rich motif of HTLV-1 p13: a novel mechanism underlying delivery of Src family kinases to mitochondria. Biochem J. 2011;439(3):505–516. [DOI] [PubMed] [Google Scholar]
- 44.Pietkiewicz S, Schmidt JH, Lavrik IN. Quantification of apoptosis and necroptosis at the single cell level by a combination of imaging flow cytometry with classical annexin V/propidium iodide staining. J Immunol Methods. 2015;423:99–103. [DOI] [PubMed] [Google Scholar]
- 45.Degterev A, Hitomi J, Germscheid M, et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4(5):313–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Katsuragi Y, Ichimura Y, Komatsu M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 2015;282(24):4672–4678. [DOI] [PubMed] [Google Scholar]
- 47.Calimeri T, Ferreri AJ. m-TOR inhibitors and their potential role in haematological malignancies. Br J Haematol. 2017;177(5): 684–702. [DOI] [PubMed] [Google Scholar]
- 48.Kurtova AV, Balakrishnan K, Chen R, et al. Diverse marrow stromal cells protect CLL cells from spontaneous and drug-induced apoptosis: development of a reliable and reproducible system to assess stromal cell adhesion-mediated drug resistance. Blood. 2009;114(20):4441–4450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Swingle M, Ni L, Honkanen RE. Small-molecule inhibitors of ser/thr protein phosphatases: specificity, use and common forms of abuse. Methods Mol Biol. 2007;365:23–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brown JR. Idelalisib has CLL on the run! Blood. 2015;126(25):2656–2657. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.