

Summary
Mitochondrial p53 is involved in apoptosis and tumor suppression. However, its regulation is not well studied. Here, we show that TRAF6 E3 ligase is a crucial factor to restrict mitochondrial translocation of p53 and spontaneous apoptosis by promoting K63-linked ubiquitination of p53 at K24 in cytosol, and such ubiquitination limits the interaction between p53 and MCL-1/BAK. Genotoxic stress reduces this ubiquitination in cytosol by S13/T330 phosphorylation-dependent translocation of TRAF6 from cytosol to nucleus, where TRAF6 also facilitates the K63-linked ubiquitination of nuclear p53 and its transactivation by recruiting p300 for p53 acetylation. Functionally, K63-linked ubiquitination of p53 compromised p53-mediated apoptosis and tumor suppression. Colorectal cancer samples with WT p53 reveals that TRAF6 overexpression negatively correlates with apoptosis and predicts poor response to chemo/radiotherapy. Together, our study identifies TRAF6 as a critical gatekeeper to restrict p53 mitochondrial translocation and such mechanism may contribute to tumor development and drug resistance.
eTOC Blurb
Zhang et al discovered that TRAF6 prevents the mitochondrial translocation of p53 and spontaneous apoptosis by promoting K63-linked ubiquitination of p53 in cytosol. Genotoxic stress overrides this protection mechanism by translocating TRAF6 into nucleus. Deregulation of this mechanism may contribute to cancer development and resistance to chemotherapy and radiotherapy.
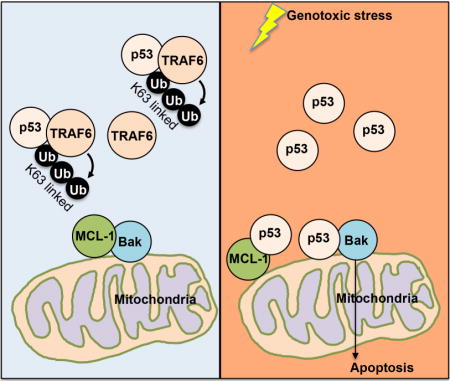
Introduction
Cell fate determination is a tightly regulated process that is involved in human development and diseases. A key factor responsible for cell fate determination is p53, which is widely involved in diverse biological processes, including cell cycle arrest, apoptosis, cellular senescence and energy homeostasis (Brady et al., 2011; Vousden and Prives, 2009; Vousden and Ryan, 2009). Since p53 is frequently mutated in a wide variety of human cancers, it is considered as the most important tumor suppressor. Therefore, it is critical to understand the molecular mechanism by which p53 activity is regulated.
p53 is a short-lived protein whose stability is tightly controlled by Mdm2 E3 ligase (Brooks and Gu, 2006; Haupt et al., 1997; Kubbutat et al., 1997). Upon genotoxic stresses, such as ionizing radiation (IR) and DNA damage agents, p53 protein is stabilized and accumulated in the cells presumably due to the loss of its interaction with Mdm2 and subsequent reduction in p53 ubiquitination. Majority of p53 resides in the nucleus where it can bind to the promoter and/or enhancer of its target genes to either induce or repress gene expression. Induction of p21 and Gadd45 is responsible for p53-mediated cell cycle arrest (el-Deiry et al., 1993; Kastan et al., 1992), whereas upregulation of Puma, Bax and Noxa are thought to mediate p53-dependent apoptosis (Miyashita et al., 1994; Nakano and Vousden, 2001; Oda et al., 2000). Loss of p53 impairs both genotoxic stress-induced cell cycle arrest and apoptosis, suggesting that p53 is a key mediator in the cell fate decision upon DNA damage. Thus, it is traditionally accepted that p53 exerts its biological functions primarily through its transcriptional activity. In addition to ubiquitination dependent degradation, p53 activity is also regulated by other posttranslational modifications, such as acetylation (Kruse and Gu, 2009). Acetylation of p53 is generally required for its transcriptional activity (Gu and Roeder, 1997; Tang et al., 2008). p53 acetylation is induced upon exposure to genotoxic stresses, which is the result of enhanced interaction of p53 with HAT domain-containing proteins, such as CBP/p300. However, how genotoxic stresses trigger the association of p53 and p300/CBP remains largely unanswered.
Beyond its mere role in the nucleus for gene expression, transcription-independent role of p53 in inducing mitochondrial apoptosis has been recently suggested (Moll et al., 2005; Vaseva and Moll, 2009). Several studies have shown that the p53 mutants with defects in transcription activity are still able to induce apoptosis (Chen et al., 1996; Haupt et al., 1995; Kokontis et al., 2001; Mihara et al., 2003). Later studies unraveled that p53 moves to the mitochondrial outer membrane in response to DNA damage where it binds to BAK/Bax to promote BAK/Bax oligomerization (Chipuk et al., 2004; Mihara et al., 2003). This event leads to mitochondrial outer membrane permeabilization (MOMP), cytochrome C release, caspase-3 activation and consequent apoptosis (Chipuk and Green, 2008; David, 2012). Consistent with this notion, recombinant p53 proteins can bind to the purified intact mitochondria and robustly trigger cytochrome C release in the in vitro assay (Chipuk et al., 2004; Mihara et al., 2003). These studies therefore underscore the direct function of p53 in the mitochondria to induce apoptosis. While there is increasing appreciation about the role of p53 in the mitochondria, it is also critical to elucidate the mechanism by which the complexity of p53 activation and localization in the mitochondria is regulated.
Here we provide evidence that TRAF6 is the critical factor to control the p53 mitochondria translocation. TRAF6 triggers K63-linked ubiquitination of p53 at K24 in the cytoplasm, which reduces the interaction between p53 and MCL-1/BAK, thus keeping p53 away from mitochondria and preventing p53 mediated activation of BAK. Genotoxic stress rapidly removes this K63-linked ubiquitination to trigger p53-mediated apoptosis pathway in the mitochondria by increasing the free p53 to TRAF6 ratio. In contrast, genotoxic stress triggers the localization of TRAF6 to nucleus, where it also mediates the K63-linked ubiquitination of p53 at K24, which promotes the interaction of p53 with p300, thereby facilitating p53 acetylation and downstream gene expression, including p21 and GADD45, for the cell survival under stress condition. Our results provide great insights into how genotoxic stress coordinates transcription-dependent and transcription-independent role of p53 in apoptosis by modulating the dynamic status of K63-linked ubiquitination of p53 in distinct cellular compartments.
Results
p53 translocates from cytosol to mitochondria to trigger cytochrome C release and subsequent apoptosis in response to genotoxic stresses. Although p53 protein is drastically induced by genotoxic stress, both nuclear and cytosolic p53 protein are still detectable in various mice tissues and cancer cell lines without exposing to genotoxic agents (Figure S1A and S1B). Since unmodified recombinant p53 readily binds to and efficiently triggers apoptosis pathway in mitochondria in vitro(Chipuk et al., 2004; Mihara et al., 2003), we hypothesized that there may be a mechanism to restrict p53 in cytosol and prevent its mitochondria translocation to avoid undesired apoptosis under physiological conditions. While BCL-2, BCL-xL and MCL-1 are known direct sequesters for cytosolic p53, it is also reported that knockout of liver specific gene IGFBP1 accumulates p53 in mitochondria and induces apoptosis in liver(Leu and George, 2007), suggesting that the whole machinery to antagonize cytoplasmic p53 is more complicated than just the traditional sequesters. Since K63-linked ubiquitination has been linked to protein trafficking, we examined whether p53 is modified through K63-linked ubiquitination in distinct cellular compartments. Surprisingly, we found that cytosolic pool of p53 was ubiquitinated through the K63-linkage, but such modification was not detected for the mitochondrial p53 (Figure S1C). Also, a profound decrease in K63-linked ubiquitination of cytosolic p53 was correlated with the increase in p53 mitochondrial translocation upon genotoxic stress (Figure S1C), indicating that K63-linked ubiquitination of p53 could be a potential negative signal to restrict p53 mitochondria translocation.
To study whether K63-linked ubiquitination of p53 plays a suppressive role in the mitochondrial translocation of p53, we aimed to identify E3 ligases that interact with p53 and trigger K63-linked ubiquitination of p53. We screened a panel of E3 ligases and found that ectopic expression of TRAF6 E3 ligase readily induced K63-linked ubiquitination of p53 (Figure 1A, S1D and S1E). p53 is a short-lived protein whose stability is regulated by K48-linked ubiquitination and proteasomal degradation(Dornan et al., 2004; Haupt et al., 1997; Leng et al., 2003). Our data showed that TRAF6 only promoted K63-linked, but not K48-linked ubiquitination of p53 (Figure 1B). We further demonstrated that TRAF6, but not TRAF6-C70A E3 ligase dead mutant, induced in vivo and in vitro p53 ubiquitination (Figure 1C and 1D). Reciprocal co-immunoprecipitation assay revealed that TRAF6 interacted with p53 endogenously (Figure 1E). Endogenous ubiquitination assay further demonstrated that K63-linked ubiquitination of p53 in cytosol was reduced in Traf6−/− MEFs or upon genotoxic agent treatment (Figure 1F). Together, these data suggest that TRAF6 is a direct E3 ligase for p53.
Figure 1. p53 is ubiquitinated by TRAF6 through K63-linkage.
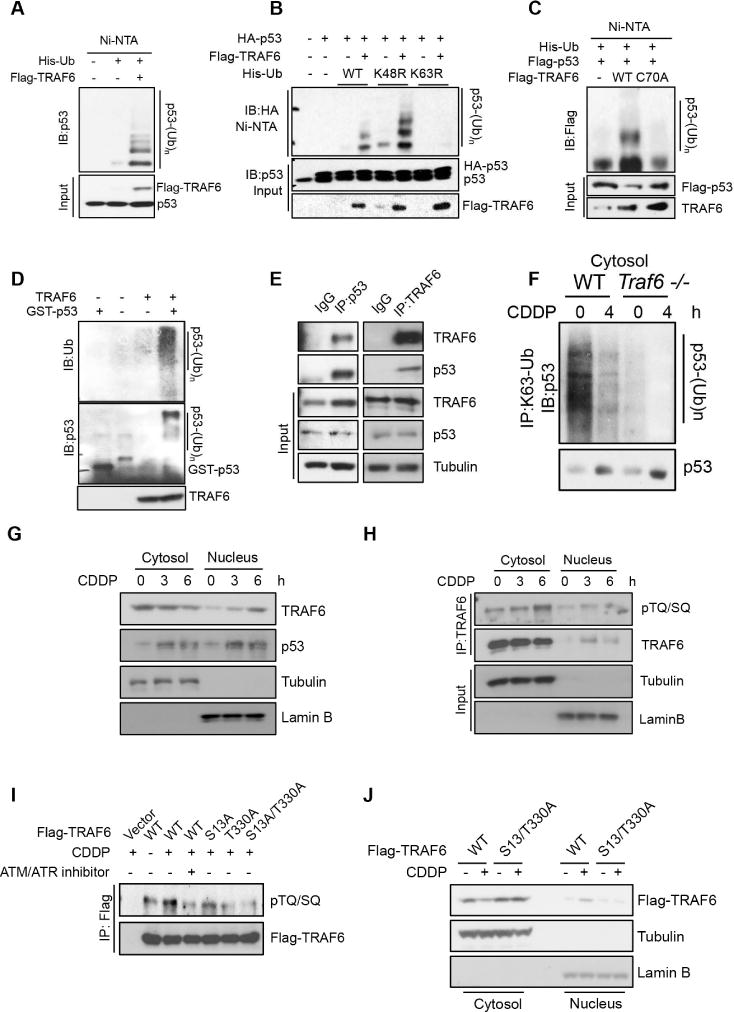
(A) U2OS cells were transfected with indicated plasmids for in vivo ubiquitination assay. See also experimental procedures.
(B) In vivo ubiquitination assay was performed for U2OS cells transfected with indicated plasmids.
(C) U2OS cells were transfected with indicated plasmids for in vivo ubiquitination.
(D) TRAF6 ubiquitinates p53 in vitro. See also experimental procedures.
(E) Immunoprecipitation assay was performed for U2OS cells.
(F) Immunoprecipitation assay was performed for the cytosol fraction of WT and Traf6 −/− primary MEFs.
(G) Cellular fractionation assay was performed for U2OS cells treated with CDDP for indicated times.
(H) Immunoprecipitation assay was performed for U2OS cells treated with CDDP for indicated times. Phosphorylation was detected by the anti-phosphor-TQ/SQ motif, which is the consensus phosphorylation site for ATM and ATR kinase.
(I) Immunoprecipitation assay was performed for 293T cells expressing WT and mutant TRAF6 as indicated. CDDP and/or ATM inhibitor was also treated as indicated.
(J) Cellular fractionation was performed for 293T cells expressing WT and mutant TRAF6 with or without CDDP treatment.
We next determined how genotoxic stress reduced K63-linked ubiquitination of p53 in cytosol, which was also observed in multiple other cell lines with CDDP or etoposide treatment (Figure S1F to S1J). Besides the stabilization of p53 protein in cytosol upon genotoxic stress, we also observed a decreased localization of TRAF6 protein in cytosol (Figure 1G and S1K–M), which resulted in an increase of free p53 to TRAF6 ratio in cytosol and may lead to the reduction of K63-linked ubiquitination of cytosolic p53 upon genotoxic stress. To study how TRAF6 localization in cytosol is regulated by genotoxic stress, we examined whether genetoxic stress-induced DNA damage signaling is involved. Inhibition of ATM/ATR, which are the major upstream kinases activated by DNA damage, prevented the reduction of TRAF6 level in cytosol (Figure S1N). Immunoprecipitation assay further demonstrated that genotoxic agents induced the phosphorylation of TRAF6 at TQ/SQ motifs (Figure 1H and S1O), which are consensus ATM/ATR substrate sites. Mutations on TQ/SQ sites S13 and T330 to alanine on TRAF6 reduced genotoxic stress induced phosphorylation of TRAF6 (Figure 1I). Such mutant also displayed resistance to the genotoxic agents mediated reduction of TRAF6 level in cytosol (Figure 1J and S1P), while its affinity to p53 is similar to TRAF6 WT (Figure S1Q). Together, our data suggest that genotoxic stress promotes TRAF6 phosphorylation, which reduces the level of TRAF6 in cytosol and may contribute to the reduction of K63-linked ubiquitination of p53 in cytosol.
If K63-linked ubiquitination of cytosolic p53 acts as a suppressive signal for p53 mitochondrial localization, TRAF6 would serve as a suppressor for p53 mitochondrial localization and apoptosis, whereas its loss will favor p53 mitochondria accumulation and spontaneous apoptosis. Using Traf6−/− genetic mouse model, we found that Traf6−/− thymus, spleen and lung displayed higher spontaneous apoptosis rate than their WT counterparts (Figure 2A, 2B and S2A). The spontaneous apoptosis level in thymus cells from Traf6−/− mice was also comparable to that in thymus cells from WT mice upon IR treatment (Figure 2A). Notably, treating Traf6−/− mice with pifithrin-μ, a potent inhibitor that specifically inhibits p53 mitochondrial translocation, reversed the spontaneous apoptosis in Traf6−/− thymus, while pifithrin-α, the specific inhibitor for p53 transcriptional activity failed to do so (Figure 2C and 2D), suggesting that mitochondrial p53 contributes to spontaneous apoptosis upon TRAF6 deficiency. It should be noted that a recent study suggests that in addition to preventing p53 mitochondria accumulation, pifithrin-μ can also disrupt the interaction between Hsp70 and its chaperones, thus leading to apoptosis(Leu et al., 2009). Therefore, it is highly unlikely that pifithrin-μ acts through disrupting Hsp70 activity to reverse spontaneous apoptosis in Traf6−/− thymus. Consistently, knockdown of TRAF6 in U2OS cells also triggered spontaneous apoptosis (Figure 2E and S2B). Like in mice thymus, such spontaneous apoptosis upon TRAF6 deficiency acts through p53, as knockdown of p53 reversed the elevated spontaneous apoptosis in TRAF6 deficient U2OS cells (Figure 2E and S2B).
Figure 2. TRAF6 inhibits p53 translocation to mitochondria and apoptosis.
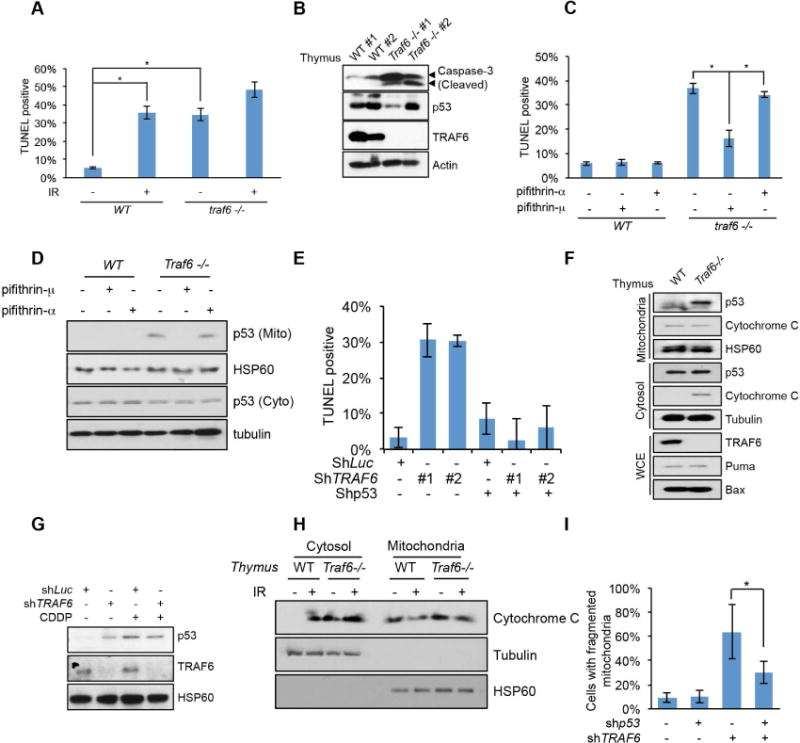
(A) WT and Traf6−/− total thymus cells were analyzed by TUNEL assay. See also experimental procedures.
(B) WT and Traf6−/− total thymus cells were analyzed by Western blot.
(C) WT and Traf6−/− mice were treated with pifithrin-μ or pifithrin-α and apoptosis of total thymus cells was analyzed by TUNEL assay.
(D) Mitochondria fractionation was performed for WT and Traf6−/− thymus treated with pifithrin-μ or pifithrin-α See also experimental procedures.
(E) TUNEL assay was performed for U2OS cells with indicated knockdown and examined by fluorescence microscopy.
(F and G) Mitochondria were isolated from total thymus cells (E) or U2OS cells (F). U2OS cells were treated with or without CDDP for 6 hours.
(H) Mitochondria fractionation was performed for the WT and Traf6−/− mice thymus treated with or without IR.
(I) TRAF6 deficiency induced mitochondria fragmentation was p53 dependent. Immunofluorescence assay was performed for U2OS cells with indicated knockdown.
All data are represented as mean +/− SEM. * indicates p<0.01 in student T test.
By isolating mitochondria, we found a spontaneous accumulation of p53 in mitochondria in Traf6−/− thymus, MEFs and TRAF6 knockdown U2OS cells, reaching to the level similar to that observed in control cells treated with CDDP (Figure 2F, 2G and S2C). However, CDDP treatment did not further enhance the level of p53 in mitochondria in these TRAF6 deficient cells (Figure 2G and S2C). Consistent with the mitochondrial translocation of p53 and apoptosis, cytochrome C, the effector of mitochondrial apoptosis pathway, was released from mitochondria to cytosol in Traf6−/− thymus before IR, while cytochrome C was only released upon IR in WT thymus (Figure 2H). Since mitochondria fragmentation and reduced mitochondria potential are also important readouts for the activation of mitochondrial apoptosis pathways(Arnoult, 2007; Brooks et al., 2007; Ly et al., 2003), we examined whether TRAF6 deficient cells also exhibited such phenotypes. Remarkably, TRAF6 deficiency readily induced mitochondrial fragmentation and reduced mitochondria potential even before genotoxic stress (Figure S2D, S2E and S2F). Knockdown of p53 partially reversed mitochondria fragmentation phenotype in TRAF6 knockdown cells (Figure 2I). Accordingly, these results suggest that TRAF6 serves as a key player to restrict p53 mitochondria translocation and spontaneous apoptosis in unstressed cells both in vivo and in vitro.
To directly support the functional role of K63-linked ubiquitination of p53 in these processes, we then determined TRAF6 dependent ubiquitination site(s) on p53. We mutated every conserved lysine (K) residue on p53 and found that TRAF6 preferentially ubiquitinates p53 at K24 in the in vivo ubiquitination assay (Figure 3A, S3A, S3B and S3C). Mutation of K24R on p53 also reduced in vitro ubiquitination of p53 by TRAF6 (Figure S3D). We then determined whether K63-linked ubiquitination of p53 affects p53 mitochondrial localization in a manner similar to TRAF6 does. Ectopic expression of p53 K24R mutant showed increased localization to mitochondria (Figure 3B), phenocopying TRAF6 deficiency. Hence, TRAF6-mediated K63-linked ubiquitination acts as a suppressive signal for p53 mitochondrial localization.
Figure 3. K63-linked ubiquitination of p53 inhibited the mitochondria translocation of p53.
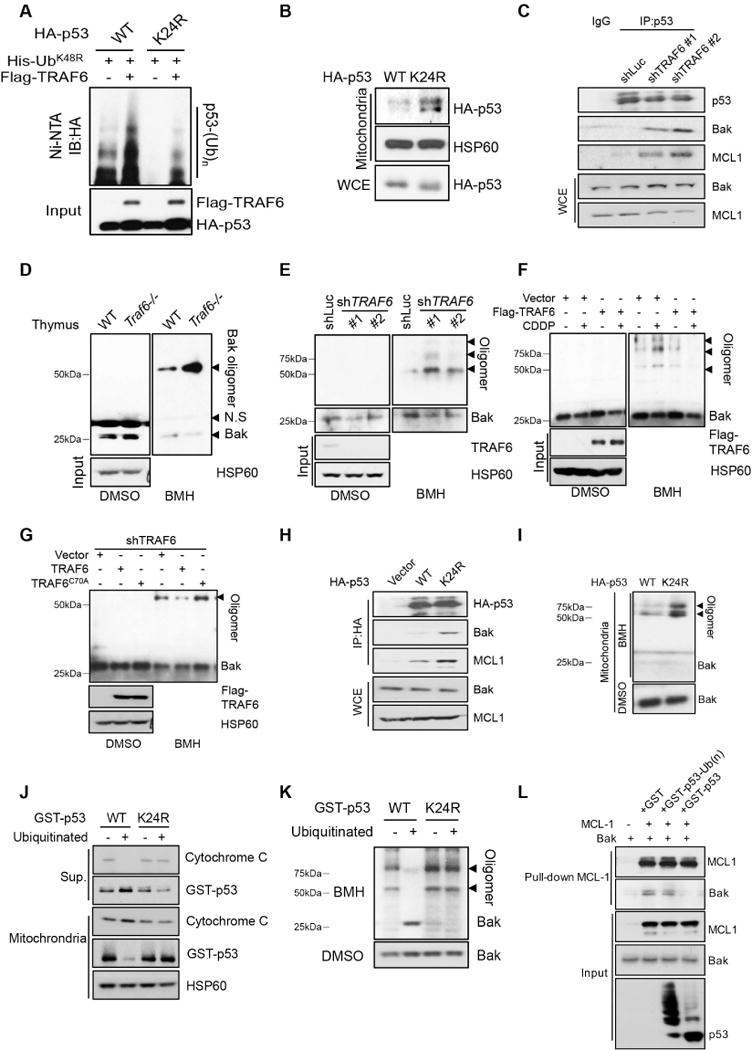
(A) In vivo ubiquitination assay was performed for U2OS cells transfected with indicated plasmids.
(B) Mitochondria were isolated from H1299 cells transfected with p53 WT or K24R.
(C) Immunoprecipitation assay was performed for control and TRAF6 knockdown U2OS.
(D and E) Mitochondria were isolated from total thymus cells (D) or U2OS cells (E) and cross-linked as described in experimental procedures.
(F) U2OS cells were treated with or without CDDP for 6 hours before BAK oligomerization assay.
(G) Mitochondria were isolated from TRAF6 knockdown U2OS cells restored with TRAF6 WT or C70A and cross-linked by BMH.
(H) Immunoprecipitation assay was performed for H1299 cells transfected with p53 WT and K24R.
(I) Mitochondria were isolated from H1299 cells transfected with p53 WT and K24R and cross-linked by BMH.
(J and K) Ubiquitinated p53 failed to interact with mitochondria and trigger BAK oligomerization in vitro. See also figure S3H and experimental procedures.
(L) In vitro binding assay was performed for recombinant MCL-1, BAK and GST-p53 derived from in vitro ubiquitination assay.
As previous report and our data showed that BAK, but not Bax is required to recruit p53 to mitochondria upon genotoxic stress and BAK is the principal target for p53 in mitochondria (figure S3E)(Leu et al., 2004; Pietsch et al., 2007), we then rationalized that TRAF6 mediated K63-linked ubiquitination of p53 may inhibit p53 mitochondrial translocation by disrupting the interaction between p53 and BAK. In support of this notion, TRAF6 knockdown increased the binding between p53 and BAK without genotoxic agent treatment (Figure 3C). As a consequence, TRAF6 deficiency readily promoted BAK oligomerization in mouse thymus and multiple cell lines (Figure 3D, 3E and S3F), while p53 deficiency inhibited such effect (Figure S3G). Upon CDDP treatment, TRAF6 overexpression also attenuated genotoxic stress induced BAK oligomerization (Figure 3F). Restoration of TRAF6, but not TRAF6 E3 ligase dead mutant (C70A) inhibited BAK oligomerization in TRAF6 deficient cells (Figure 3G). Similarly, p53 K24R mutant displayed higher binding affinity to BAK and stronger activity to induce BAK oligomerization in comparison with p53 WT (Figure 3H and 3I). Of note, TRAF6 knockdown or p53 K24R mutation also increased the binding between p53 and MCL-1 (Figure 3C and 3H), which is the major sequester of BAK in mitochondria. Since p53 is also shown to interact with MCL-1 and antagonize the sequestering effect of MCL-1 on BAK (Leu et al., 2004), the increased interaction between p53 and MCL-1 triggered by the deficiency in K63-linked ubiquitination of p53 may also contribute to the release of BAK from inhibition, BAK oligomerization and subsequent apoptosis.
To validate our notion further, we incubated recombinant p53 or recombinant p53 that was ubiquitinated by TRAF6 in vitro with the mitochondria isolated from p53-deficient H1299 cells (figure S3D and S3H). Strikingly, p53 that was ubiquitinated by TRAF6 in vitro not only failed to interact with mitochondria, but also lost its ability to trigger BAK oligomerization and cytochrome C release in vitro compared with non-ubiquitinated p53 and p53 K24R mutant (Figure 3J and 3K), providing strong evidence that TRAF6-mediated K63-linked ubiquitination of p53 prevents p53 from binding to mitochondria and inducing BAK oligomerization. We also performed in vitro assay to test whether K63-linked ubiquitination of p53 affects its capability to antagonize MCL-1. While non-ubiquitinated p53 WT or p53 K24R mutant could readily disrupt the binding between BAK and MCL-1 in vitro, ubiquitinated p53 failed to do so (Figure 3L). This was also supported by the in vitro binding assay showing that ubiquitinated p53 failed to interact with MCL-1 (Figure S3I). In addition, ubiquitinated p53 also showed reduced affinity to BAK (Figure S3J), suggesting that K63-linked ubiquitination of p53 may inhibit its binding to both BAK and MCL-1, and these two mechanisms may contribute to the suppression of BAK oligomerization and activation at the same time.
Our finding that TRAF6 protect cells from spontaneous apoptosis by inhibiting mitochondrial localization of p53 in multiple cell types prompted us to examine whether TRAF6-mediated p53 ubiquitination at K24 regulates p53-mediated tumor suppression. We found that introducing p53 WT or p53 K24R to H1299 cells could inhibit in vitro colony formation and in vivo tumor growth in xenograft models (Figure 4A, 4B and 4C). Notably, while TRAF6 overexpression could block p53-mediated cell killing and tumor suppression, it failed to do so in cancer cells expressing p53-K24R mutant (Figure 4A, 4B and 4C), suggesting that TRAF6-mediated p53 ubiquitination at K24 generally protects cancer cells from p53-mediated tumor suppression. As p53 plays a critical role in chemotherapy- and radiotherapy-induced apoptosis of cancer cells, we examined whether TRAF6 affect cancer cell survival under the treatment of genotoxic agents. We found that TRAF6 knockdown or C70A mutation sensitize the cells to genotoxic stress (Figure S4A–D), confirming the protective role of TRAF6 against genotoxic agents. We further studied the clinical relevance of TRAF6 and chemo- or IR-induced apoptosis. Strikingly, higher TRAF6 protein levels negatively correlated with the level of cleaved caspase-3 and TUNEL-positive staining, two readouts for apoptosis, and predicted the poor survival outcome of colorectal cancer in response to chemotherapy and IR treatment (Figure 4D–H, S4E and Table). Thus, clinical data indicate that TRAF6 overexpression is correlated with less apoptosis and predicts poor response of colorectal cancer patients with WT p53 to chemotherapy and IR treatment. Our finding may provide a mechanism to explain how TRAF6 regulates apoptosis in the p53 WT context.
Figure 4. TRAF6 inhibited p53 WT, but not p53 K24R induced tumor suppression.
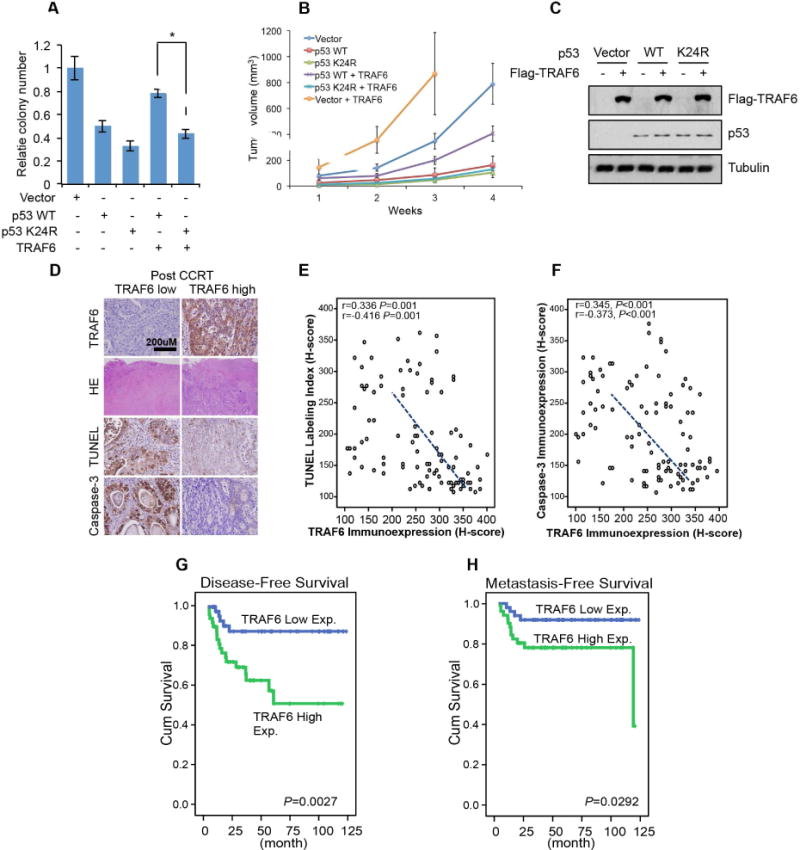
(A–C) Colony forming assay (A), Xenograft tumor growth assay (B) and Western blot assay (C) were performed for H1299 cells expressing indicated exogenous proteins.
(D) Representative rectal cancers after concurrent chemoradiotherapy (CCRT) showing low (left panel) and high (right panel) TRAF6 immunoreactivity were significantly associated with a higher tumor regression grade (TRG) and increased TUNEL and Caspase-3 expression (left panel) and vice versa (right panel). (E and F) In post-CCRT rectal cancers, TRAF6 expression is significantly and negatively related to cell death as determined by TUNEL and active Caspase-3 staining.
(G and H) Survival analysis plotted by using Kaplan-Meier methods discloses TRAF6 expression in Post-CCRT biopsy specimens is significantly predictive for disease-free and metastasis-free survival.
All data are represented as mean +/− SEM. * indicates p<0.01 in student T test.
As another important role of p53 is the stress sensing transcription factor, which modulates cell survival upon stress, we tested whether TRAF6 also regulates the transcriptional activity of p53. Hence, we applied gene expression microarray using the mouse thymus isolated from WT and Traf6−/− mice treated with or without IR. Interestingly, the IR induced expression pattern of p53 target genes was deregulated in Traf6−/− thymus (Figure 5A and S5A). Importantly, p21 and Gadd45, which are important downstream mediators of p53 for the cell survival upon genotoxic stresses (Figure S5B) (Gartel and Tyner, 2002; Lu et al., 2008), are not induced in Traf6−/− thymus as confirmed by both real-time PCR and Western blot analysis (Figure 5B and 5C). The impairment of IR-induced p21 and Gadd45 expression upon Traf6 deficiency could be extended to MEFs and various human cell lines (Figure S5C–E). Again, by analyzing human colorectal cancer samples with WT p53, we found that TRAF6 protein expression was significantly correlated with the expression of p21 (Figure 5D, 5E and table S1). Collectively, our data suggest that TRAF6 regulates the genotoxic stress induced expression of p53 downstream targets, p21 and Gadd45 in diverse cell types both in mouse and human, which may account for the role of TRAF6 in cell survival upon genotoxic stress and poor response to CCRT in colorectal cancer patients with WT p53 (Figure 4D–H).
Figure 5. TRAF6 is required for genotoxic stress induced p53 target gene expression.
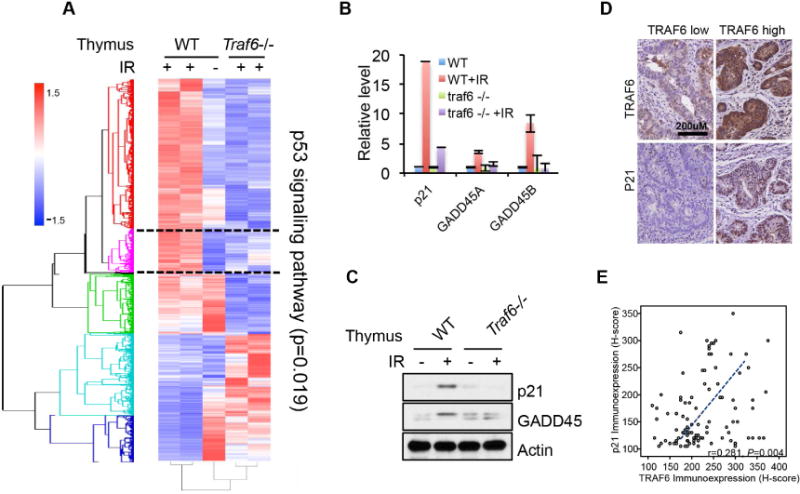
(A) 2.5-week-old WT and Traf6−/− mice were IR (15 Gy) treated for 10 hours before thymus were isolated and total RNA or total protein was extracted. Genes with greater than 1.5 fold change in mRNA expression were used to generate the heat map.
(B and C) The expression of p53 target genes, p21 and GADD45 were validated by qPCR and Western blot in the similar sample from figure 5D.
(D and E) Representative rectal cancers showing low (left panel) and high (right panel) TRAF6 immunoreactivity in pre-treatment (Pre-Tx) biopsy specimens were linked to low p21 (left panel) and high p21 (right panel) expression, respectively. In the pre-treatment (Pre-Tx) biopsy specimens, TRAF6 immunoexpression is significantly and positively associated with p21 expression.
Meta-analysis of microarray data sets further revealed that E1A binding protein p300 (EP300 or p300), a critical transcriptional co-activator for p53, may be responsible for the TRAF6-mediated gene expression pattern (Figure 6A). Since p300 is known to critically regulate p53 activity by acetylating p53 protein on the C-terminal lysine (K) residues(Grossman, 2001), we investigated whether p53 acetylation is regulated by TRAF6. Remarkably, Traf6 deficiency in thymus, MEFs and human cancer cell lines impaired p53 acetylation upon IR treatment (Figure 6B, S5D and S6A–C). Mechanistically, we found that the endogenous interaction between p53 and p300 upon IR stimulation was markedly reduced in Traf6−/− MEFs (Figure 6C), while p300 protein level remains unaffected by TRAF6. Hence, TRAF6 serves as an essential regulator for p53 transactivation by recruiting p300 to p53 and subsequent acetylation of p53. As p53 is ubiquitinated by TRAF6 in the cytoplasm, we found that p53 in nucleus is also ubiquitinated through K63 linkage (Figure 6D). This ubiquitination modification is induced by genotoxic stress and is TRAF6 dependent (Figure 6D and S1G–J), which is correlated with the genotoxic stress induced phosphorylation of TRAF6 at S13/T330 sites and subsequent S13/T330 phosphorylation dependent localization of TRAF6 in nucleus (Figure 1G–J and S1G–P). p53 K24R mutant that is deficient in K63-linked ubiquitination also did not interact with p300 in the co-IP assay and was insufficient to induce p21 and Gadd45 expression in the p53-deficient H1299 cells (Figure 6E, 6F and S6D). However, either ubiquitinated p53 and non-ubiquitinated p53 readily binds to p300 in vitro (Figure 6E), suggesting that the mechanism of how ubiquitinated p53 gains advantage in recruiting p300 in the cellular context is complicated, which may involve other regulatory factors to be studied in the future. In addition, both p53 WT and K24R mutant binds to p300 similarly (Figure S6F), suggesting that the effect of p53 K24R mutant when expressing in the cells is probably due to the loss of K63-linked ubiquitination rather than change in its protein structure caused by K24 mutation. Together, these data suggest that genotoxic stress induced K63-linked p53 ubiquitination at K24 by TRAF6 in nucleus is needed for p300 recruitment, p53 acetylation and the expression of p53 target genes such as p21 and Gadd45.
Figure 6. TRAF6 is required for p300 recruitment and p53 acetylation.

(A) Predicted regulators for TRAF6-dependent transcription regulation.
(B) Thymus prepared as described in Figure 5D was analyzed by Western blot.
(C) Immunoprecipitation assay was performed for WT and Traf6−/− MEFs treated with or without IR (10 Gy) for 4 hours.
(D) Immunoprecipitation assay was performed for the nuclear fraction of WT and Traf6−/− primary MEFs to access p53 ubiquitination.
(E) Immunoprecipitation assay was performed for the H1299 cells transfected with p53 WT or K24R to access its binding with p300.
(F) Western blot was performed for H1299 cells transfected with p53 WT or K24R to detect p21 and GADD45.
Discussion
p53 displays potent tumor suppressive effect by eliciting apoptosis, senescence, cell cycle arrest and metabolism regulation(Bieging et al., 2014; Levine and Oren, 2009). Among these, p53 executes apoptosis through its transcriptional activation and mitochondria translocation. Notably, p53 readily translocates from cytosol to mitochondria to trigger cytochrome C release and subsequent apoptosis in response to genotoxic stresses. However, how genotoxic stress induces mitochondrial translocation of p53 remains largely unclear. Although one earlier study proposed that Mdm2 may be involved in genotoxic stress-induced p53 mitochondria translocation and apoptosis(Marchenko et al., 2007), such concept may conflict with well-established genetic evidence that loss of Mdm2 in mice causes mouse embryonic lethality through eliciting p53-dependent apoptosis(Jones et al., 1995; Montes de Oca Luna et al., 1995). Thus, a fundamental mechanism accounting for such phenomenon is still lacking and yet to be discovered. Since cytosolic p53 is still detectable in the unstressed cells (Figure S1A and S1B) and unmodified recombinant p53 readily binds to and triggers apoptosis pathway in mitochondria in vitro(Chipuk et al., 2004; Mihara et al., 2003), we hypothesized that there may be a cytosolic gatekeeper keeping p53 away from mitochondria under the unstressed condition. Our study offers the great insight into how such regulatory mechanism is operated in the cells. We identify TRAF6 E3 ligase as a critical factor to elicit such inhibitory mechanism for cytosolic p53. Mechanistically, we show that TRAF6 interacts with cytosolic p53 and triggers K63-linked ubiquitination of p53 at K24, which prevents p53 from binding to BAK/MCL-1 in the mitochondria, thereby keeping p53 away from the mitochondria and protect cells from spontaneous apoptosis. Importantly, we uncover that such inhibitory mechanism by TRAF6 is shut off upon genotoxic stress, empowering p53 to move to the mitochondria to initiate apoptosis, by genotoxic stress induced TRAF6 phosphorylation and subsequent translocation. Interestingly, our result is consistent with a study showing that the amount of ubiquitinated p53 in mitochondria is too low to be detected by Western blot, while ubiquitinated p53 in the cytoplasm is readily detectable (Marchenko et al., 2007). Remarkably, loss of TRAF6 in various mouse tissues and human cancer cells is sufficient to drive p53 mitochondria localization and spontaneous apoptosis even without any genotoxic stress. Thus, our study defines the critical mechanism by which genotoxic stress drives cytosolic p53 mitochondria translocation and subsequent apoptosis by inhibiting TRAF6-mediated K63-linked ubiquitination of p53 at K24 in cytosol. Also, this inhibitory mechanism may be potentially heightened in colorectal cancer patients with WT p53, since TRAF6 overexpression in such setting indicates less apoptosis and could predict poor patient survival outcome. In addition, p53 K24R mutant displays heightened cell-killing effect under TRAF6 overexpression condition as compared to p53 WT, while p53 K24R displays deficiency in p53 target gene expression. Thus, mitochondrial action may be the major tumor suppressive pathway of p53 when it is overexpressed in the in vitro cell growth assay and in vivo xenograft tumor growth assay using H1299 cell line without the treatment of genotoxic agents.
Besides our finding showing a direct role of K63-linked ubiquitination in inhibiting the binding between p53 and BAK, mitochondrial BAK is also suppressed by MCL-1 (Leu et al., 2004). Our data show that K63-linked ubiquitination also inhibited the binding between p53 and MCL-1, resulting in the BAK suppression by MCL-1. In vitro binding assay further indicated that p53 ubiquitination could directly inhibit the binding of p53 to both BAK and MCL-1, suggesting that p53 ubiquitination inhibits p53-BAK binding both directly and indirectly through MCL-1. As BCL-2/BCL-xL is also reported to bind and sequester cytosolic p53 and/or BAK, our study does not rule out the possibility that TRAF6-mediated cytosolic p53 ubiquitination may also regulate the crosstalk between BCL-2/BCL-xL and p53 and/or BAK.
While our study reveals that TRAF6 is a negative regulator for p53-mediated apoptosis in the mitochondrial pathway, we also find that TRAF6 is required for p53 acetylation, transcriptional activation and the expression of its target genes, including p21 and Gadd45 upon genotoxic stress. It should be noted that the basal expression of p53 target genes before genotoxic stress and the expression of non-IR inducible p53 target genes are not affected by TRAF6 (Figure S5A), consistent with the fact that K63-linked ubiquitination of p53 by TRAF6 in nucleus is only induced upon genotoxic stress. These data also demonstrate that the spontaneous apoptosis in TRAF6 deficient cells before genotoxic stress is not related to the transcriptional function of p53, since TRAF6 does not regulate p53 target expression at this time. The impairment in the induction of p21 and Gadd45 expression upon genotoxic stress in TRAF6 deficient cells sensitize the cells to genotoxic stress, which is also consistent with the clinical data showing that TRAF6 is up regulated, positively correlated with the expression of p21 and predicts poor response of colorectal cancer patients to chemotherapy and IR treatment (Figure 4D–H). We have also noticed that the TRAF6 knockout affected many other p53 target genes involved in multiple biological processes, including apoptosis pathway (Puma and Bax) in the microarray datasets upon genotoxic stress (Figure S5A). Firstly, this observation should not be confused with the role of TRAF6 in protecting cells from apoptosis before genotoxic stress, since the expression of those apoptosis genes are not affected by TRAF6 before genotoxic stress. More importantly, TRAF6 deficiency does not completely wipe out their expression, but rather reduces their expression to the level comparable to the WT cells without genotoxic stress, which is still sufficient to mediate cellular apoptosis process. Thus, the biological effect of TRAF6 knockout should not be simply considered as equal to the knockout of those apoptotic genes, which are demonstrated as essential in p53 mediated apoptosis (Jeffers et al., 2003). For example, the protein level of Bax is not much induced by genotoxic stress or affected by TRAF6 deficiency upon genotoxic stress (Data not shown), and such basal level of Bax in TRAF6 deficiency may be sufficient to mediate apoptosis. In this regard, it is not surprising to see the increased cell death upon genotoxic stress in TRAF6 deficient cells, since it mainly reflects the loss of protective effect from p21 and Gadd45 against apoptosis. Thus, it is possible that the net outcome of TRAF6 regulated p53 transactivation upon genotoxic stress favors cell survival and could also contribute to the poor response to the CCRT in the colorectal cancer patients.
TRAF6 is shown to be involved in the innate immune response and tumorigenesis by regulating TLR4/IL-1R and growth factor induced AKT signaling (Chen, 2005; Yang et al., 2009; Yang et al., 2010a; Yang et al., 2010b). In this study, we further extend its role in regulating both transcription dependent and independent function of p53 in apoptosis. Interestingly, TRAF6 is responsible for K63-linked ubiquitination of p53 at K24 residue in the nucleus upon genotoxic stresses. This event facilitates the interaction between p53 and p300 and is required for cell survival upon genotoxic stress through p21 and Gadd45 expression. This is in sharp contrast to Mdm2, which triggers K48-linked ubiquitination and degradation of p53. Consistent with the important role of TRAF6 in NF-κB signaling pathway(Dickson et al., 2004), NF-κB is predicted as one of the top potential transcription factors regulated by TRAF6 in our microarray data sets (Figure 6A). However, NF-κB and p53 are shown to antagonize each other in their target gene expression (Webster and Perkins, 1999). Thus it is unlikely that TRAF6 activates p53 target genes indirectly through its well-established role in NF-κB pathway. It is also unlikely that AKT signaling pathway promotes the p53 transactivation, since AKT is shown to enhance MDM2 mediated degradation of p53 (Ogawara et al., 2002). To exclude any potential effect of AKT and NF-κB pathway on the mitochondrial translocation of p53, we overexpressed constitutive active AKT (myristoylated AKT) or p65 (S276D) in TRAF6 knockdown cells and found that they cannot rescue the accumulation of p53 in the mitochondria in TRAF6 deficient cells (Figure S7A), further supporting that TRAF6 may directly regulate p53 mitochondria translocation and such regulation is independent of NF-κB and AKT pathway. However, we are not excluding the possibility that TRAF6 protect cells from genotoxic stress likely through AKT and NF-κB pathway, since NF-κB or AKT activation upon genotoxic stress may also contribute to cell survival. We also demonstrated that TRAF6 is phosphorylated upon genotoxic stress at the ATM and ATR substrate consensus sites as shown in the immunoprecipitation assay (Figure 1H, 1I and S1O). However, without further experimental evidence, we cannot pinpoint which kinases are directly involved in genotoxic stress induced TRAF6 phosphorylation and translocation. Thus, it would be of interest and significance to further explore this aspect in the future to discover novel pathways in the DNA damage response signaling.
In summary, our study reveals important insights into how genomic and non-genomic p53 activity is regulated under genotoxic stress. On the basis of our findings, we propose a working model to delineate how genomic and non-genomic p53 activity is regulated in response to genotoxic stress (Figure S7B). Our findings unravel distinct patterns of K63-linked ubiquitination of p53 in various cellular compartments, which serves as an important molecular switch that enables p53 transcriptional activation in the nucleus and p53 transcription independent function in the mitochondrial for apoptosis under stress conditions. This study also identifies TRAF6 as a cytosolic gatekeeper, which restricts p53 mitochondria translocation and subsequent apoptosis. Thus, therapeutically targeting TRAF6 is a promising strategy to elicit p53-mediated apoptosis for cancer therapy and overcoming drug resistance.
Experimental procedures
A complete description of experimental and analysis methods can be found in the supplemental experimental procedures.
Cell culture
Cell culture cells were cultured in DMEM high glucose (Hyclone) containing 10% fetal bovine serum (FBS).
Total thymus cell isolation
Total thymus cells were isolated by mechanical disruption of thymus obtained from mice using two cover slides. Cells were then filtered and used in various assays.
In vivo and in vitro ubiquitination assays
In vivo ubiquitination assay was performed as described (Yang et al., 2009). In brief, cells were transfected with the indicated plasmids for 48 h and harvested by denatured buffer (6 M guanidine-HCl, 0.1 M Na2HPO4/NaH2PO4, 10 mM imidazole). The cell extracts were then incubated with nickel beads (Ni-NTA) for 3 h, washed, and subjected to Western blot analysis. In vitro ubiquitination assay was also performed as described (Yang et al., 2009). Briefly, recombinant TRAF6 (GST tag was removed by thrombin digestion) proteins and GST-p53 protein purified from bacteria were incubated with 0.5ug E1 (Boston Biochem), 1.5ug Ubiquitin (Boston Biochem), 1ug UBC13/UEV1A (Boston Biochem), 1ug UBCH5c (Boston Biochem), 2.5 mM ATP in a final 30 μl reaction buffer (1.5 mM MgCl2, 5 mM KCl, 1 mM DTT, 20 mM HEPES (pH7.4)) at 30°C for 2 h. GST-p53 proteins was then isolated from reaction by glutathione beads and its ubiquitination was detected by Western blot analysis.
Mitochondria fractionation and crosslinking
Cells were cultured in DMEM high glucose and harvested for mitochondria fractionation by mitochondria isolation kit for mammalian cells (Pierce, Thermo scientific) according to the glass homogenizer based fractionation protocol. Purified mitochondria were then re-suspended in MRM-S buffer (250 mM sucrose, 10 mM HEPES, 1 mM ATP, 5 mM succinate, 0.08 mM ADP, 2 mM K2HPO4 at pH 7.4) for further analysis, as previously described (Wei et al., 2000). For crosslinking, mitochondria were incubated in PBS containing 1mM bismaleimidohexane (BMH) (Invitrogen) under room temperature for 30 mins and boiled in 1X SDS sample buffer for subsequent WB analysis.
Gene expression microarray
Total RNA was extracted and purified using RNeasy mini kit (QIAGEN) according to manufacturer’s manual. Microarray analysis was performed for total RNA on Illumina HumanHT12v4 platform or Illumina MouseWG-6v2 following Illumina’s standard procedure.
Cell apoptosis assay
Briefly, cell were labeled by TdT-mediated dUTP Nick End Labeling (TUNEL) assay kit (Roche) according to the manufacturer’s protocol.
Colony forming assay
Cell colonies stained with crystal violet were dissolved in DMSO and quantified by the A600 absorbance.
Cellular fractionation
Cells fractionated by homogenize in hypertonic buffer and followed by centrifuge.
Drug treatment and xenograft mouse model
Pifithrin-μ inhibitor of p53 mitochondria translocation, Cayman Chemical, #10748) or pifithrin-α (inhibitor of p53 dependent transcriptional activation, Cayman Chemical, #13326) dissolved in 1% EtOH/30% PEG/1% Tween-80) was injected intraperitoneally into one-week-old WT and TRAF6 −/− mice (10mg/kg, every other day). After treatment for one week, thymus was then isolated for subsequent analysis. For xenograft model, 0.5 million H1299 cells were injected into each nude mice and tumor size were monitored for 4 weeks (n=5).
Supplementary Material
Figure 7. Schematic model.
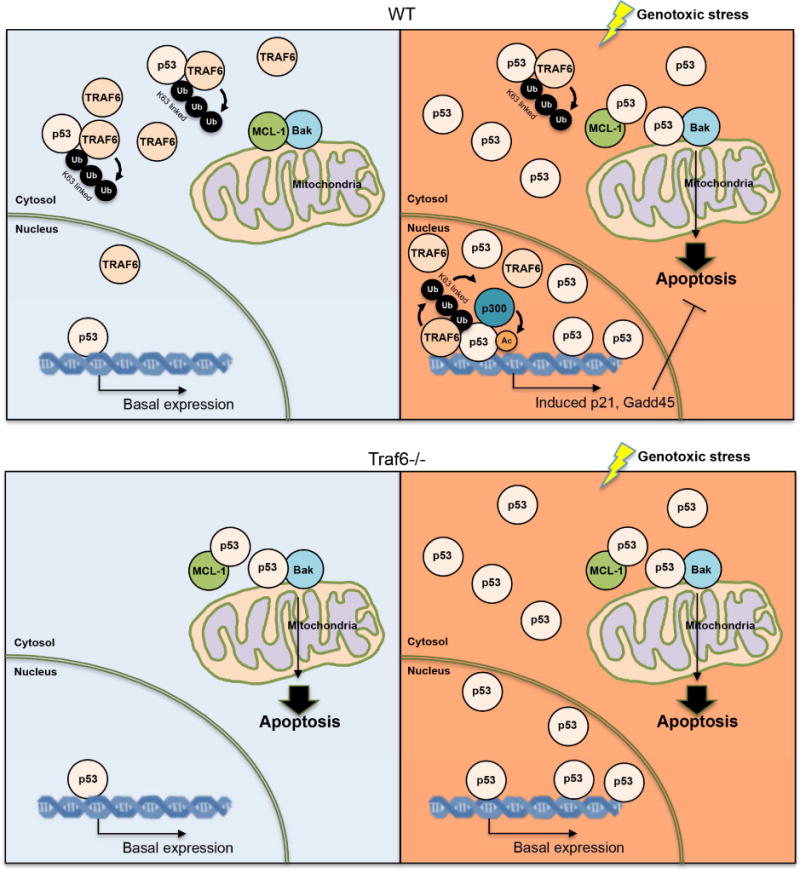
Before genotoxic stress, p53 in the cytosol are bond by TRAF6 and K63-linked ubiquitinated by TRAF6. This ubiquitination inhibited the binding between p53 and BAK/MCL-1, thus keeping p53 away from mitochondria. In the nucleus, the level of TRAF6 and K63-linked ubiqutination of p53 is low, which keeps p53 activity at basal level. Upon genotoxic stress, p53 level is drastically increased, while TRAF6 level is reduced in cytosol, which results in the more non-ubiquitinated p53 and allows p53 translocation to mitochondria for BAK oligomerization and subsequent apoptosis. Meanwhile, genotoxic stress enhances the level of TRAF6 in the nucleus and drives the K63-linked ubiquitination of p53, which facilitates the recruitment of p300 for p53 transactivation. Thus, p53 target genes, including p21 and GADD45 were actively transcribed and exert their functions in cell cycle control and cell survival. In the TRAF6 deficient condition, p53 accumulated in the mitochondria, triggers Bak oligomerization and apoptosis under the unstressed condition. Upon genotoxic stress, TRAF6 deficient cells failed to activate p53 and express p21 and GADD45 and are more sensitive to genotoxic stress.
Highlights.
TRAF6 restricts p53 mitochondria localization and spontaneous apoptosis.
TRAF6 promotes K63-linked ubiquitination of p53 at K24.
K63-linked ubiquitination of p53 in cytosol attenuates tumor suppressive function.
Genotoxic stress promotes TRAF6 phosphorylation and TRAF6 translocation.
Acknowledgments
We thank Drs. Wei Gu, Susan Taylor, Tom Hamilton, Ed Harhaj and Yi-Chieh Nancy for reagents. We also thank the members from Dr. Hui-Kuan Lin’s laboratory for their support and critical comments on this study. This work was supported by Start-up funds from Wake Forest School of Medicine, the Endowed Professorship fund from the Anderson Family, and NIH grants to Hui-Kuan Lin. This study is also partly supported by Ministry of Health and Welfare (DOH102-TD-M-111-102001 and MOHW103-TD-B-111-05 to Chien-Feng Li. The authors are grateful to the BioBank of Chi Mei Medical Center to provide the tumor samples.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
X.Z. and H.K.L. designed all the experiments and wrote the manuscript. X.Z., C.F.L., L.Z., C.Y.W., L.H., G. J., A.H.R., F.H., C.L., C.X. and X.X. performed the experiments. C.Y.H., F.J.T, C.H.T. and K.W. provided scientific inputs and suggestions. All authors have read and discussed the manuscript.
References
- Arnoult D. Mitochondrial fragmentation in apoptosis. Trends in cell biology. 2007;17:6–12. doi: 10.1016/j.tcb.2006.11.001. [DOI] [PubMed] [Google Scholar]
- Bieging KT, Mello SS, Attardi LD. Unravelling mechanisms of p53-mediated tumour suppression. Nature reviews Cancer. 2014;14:359–370. doi: 10.1038/nrc3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady CA, Jiang D, Mello SS, Johnson TM, Jarvis LA, Kozak MM, Kenzelmann Broz D, Basak S, Park EJ, McLaughlin ME, et al. Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell. 2011;145:571–583. doi: 10.1016/j.cell.2011.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks C, Wei Q, Feng L, Dong G, Tao Y, Mei L, Xie ZJ, Dong Z. Bak regulates mitochondrial morphology and pathology during apoptosis by interacting with mitofusins. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:11649–11654. doi: 10.1073/pnas.0703976104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks CL, Gu W. p53 ubiquitination: Mdm2 and beyond. Molecular cell. 2006;21:307–315. doi: 10.1016/j.molcel.2006.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Ko LJ, Jayaraman L, Prives C. p53 levels, functional domains, and DNA damage determine the extent of the apoptotic response of tumor cells. Genes & development. 1996;10:2438–2451. doi: 10.1101/gad.10.19.2438. [DOI] [PubMed] [Google Scholar]
- Chen ZJ. Ubiquitin signalling in the NF-kappaB pathway. Nature cell biology. 2005;7:758–765. doi: 10.1038/ncb0805-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chipuk JE, Green DR. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends in cell biology. 2008;18:157–164. doi: 10.1016/j.tcb.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M, Green DR. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science. 2004;303:1010–1014. doi: 10.1126/science.1092734. [DOI] [PubMed] [Google Scholar]
- David R. Apoptosis: A lipid trigger of MOMP. Nature reviews Molecular cell biology. 2012;13:208–209. doi: 10.1038/nrm3316. [DOI] [PubMed] [Google Scholar]
- Dickson KM, Bhakar AL, Barker PA. TRAF6-dependent NF-kB transcriptional activity during mouse development. Developmental dynamics: an official publication of the American Association of Anatomists. 2004;231:122–127. doi: 10.1002/dvdy.20110. [DOI] [PubMed] [Google Scholar]
- Dornan D, Wertz I, Shimizu H, Arnott D, Frantz GD, Dowd P, O’Rourke K, Koeppen H, Dixit VM. The ubiquitin ligase COP1 is a critical negative regulator of p53. Nature. 2004;429:86–92. doi: 10.1038/nature02514. [DOI] [PubMed] [Google Scholar]
- el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- Gartel AL, Tyner AL. The role of the cyclin-dependent kinase inhibitor p21 in apoptosis. Molecular cancer therapeutics. 2002;1:639–649. [PubMed] [Google Scholar]
- Grossman SR. p300/CBP/p53 interaction and regulation of the p53 response. European journal of biochemistry/FEBS. 2001;268:2773–2778. doi: 10.1046/j.1432-1327.2001.02226.x. [DOI] [PubMed] [Google Scholar]
- Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90:595–606. doi: 10.1016/s0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–299. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- Haupt Y, Rowan S, Shaulian E, Vousden KH, Oren M. Induction of apoptosis in HeLa cells by trans-activation-deficient p53. Genes & development. 1995;9:2170–2183. doi: 10.1101/gad.9.17.2170. [DOI] [PubMed] [Google Scholar]
- Jeffers JR, Parganas E, Lee Y, Yang C, Wang J, Brennan J, MacLean KH, Han J, Chittenden T, Ihle JN, et al. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer cell. 2003;4:321–328. doi: 10.1016/s1535-6108(03)00244-7. [DOI] [PubMed] [Google Scholar]
- Jones SN, Roe AE, Donehower LA, Bradley A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature. 1995;378:206–208. doi: 10.1038/378206a0. [DOI] [PubMed] [Google Scholar]
- Kastan MB, Zhan Q, el-Deiry WS, Carrier F, Jacks T, Walsh WV, Plunkett BS, Vogelstein B, Fornace AJ., Jr A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell. 1992;71:587–597. doi: 10.1016/0092-8674(92)90593-2. [DOI] [PubMed] [Google Scholar]
- Kokontis JM, Wagner AJ, O’Leary M, Liao S, Hay N. A transcriptional activation function of p53 is dispensable for and inhibitory of its apoptotic function. Oncogene. 2001;20:659–668. doi: 10.1038/sj.onc.1204139. [DOI] [PubMed] [Google Scholar]
- Kruse JP, Gu W. Modes of p53 regulation. Cell. 2009;137:609–622. doi: 10.1016/j.cell.2009.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387:299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- Leng RP, Lin Y, Ma W, Wu H, Lemmers B, Chung S, Parant JM, Lozano G, Hakem R, Benchimol S. Pirh2, a p53-induced ubiquitin-protein ligase, promotes p53 degradation. Cell. 2003;112:779–791. doi: 10.1016/s0092-8674(03)00193-4. [DOI] [PubMed] [Google Scholar]
- Leu JI, Dumont P, Hafey M, Murphy ME, George DL. Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex. Nat Cell Biol. 2004;6:443–450. doi: 10.1038/ncb1123. [DOI] [PubMed] [Google Scholar]
- Leu JI, George DL. Hepatic IGFBP1 is a prosurvival factor that binds to BAK, protects the liver from apoptosis, and antagonizes the proapoptotic actions of p53 at mitochondria. Genes Dev. 2007;21:3095–3109. doi: 10.1101/gad.1567107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leu JI, Pimkina J, Frank A, Murphy ME, George DL. A small molecule inhibitor of inducible heat shock protein 70. Molecular cell. 2009;36:15–27. doi: 10.1016/j.molcel.2009.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nature reviews Cancer. 2009;9:749–758. doi: 10.1038/nrc2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Yang C, Hill R, Yin C, Hollander MC, Fornace AJ, Jr, Van Dyke T. Inactivation of gadd45a sensitizes epithelial cancer cells to ionizing radiation in vivo resulting in prolonged survival. Cancer research. 2008;68:3579–3583. doi: 10.1158/0008-5472.CAN-07-5533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ly JD, Grubb DR, Lawen A. The mitochondrial membrane potential (deltapsi(m)) in apoptosis; an update. Apoptosis: an international journal on programmed cell death. 2003;8:115–128. doi: 10.1023/a:1022945107762. [DOI] [PubMed] [Google Scholar]
- Marchenko ND, Wolff S, Erster S, Becker K, Moll UM. Monoubiquitylation promotes mitochondrial p53 translocation. The EMBO journal. 2007;26:923–934. doi: 10.1038/sj.emboj.7601560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, Moll UM. p53 has a direct apoptogenic role at the mitochondria. Molecular cell. 2003;11:577–590. doi: 10.1016/s1097-2765(03)00050-9. [DOI] [PubMed] [Google Scholar]
- Miyashita T, Krajewski S, Krajewska M, Wang HG, Lin HK, Liebermann DA, Hoffman B, Reed JC. Tumor suppressor p53 is a regulator of bcl-2 and bax gene expression in vitro and in vivo. Oncogene. 1994;9:1799–1805. [PubMed] [Google Scholar]
- Moll UM, Wolff S, Speidel D, Deppert W. Transcription-independent pro-apoptotic functions of p53. Current opinion in cell biology. 2005;17:631–636. doi: 10.1016/j.ceb.2005.09.007. [DOI] [PubMed] [Google Scholar]
- Montes de Oca Luna R, Wagner DS, Lozano G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature. 1995;378:203–206. doi: 10.1038/378203a0. [DOI] [PubMed] [Google Scholar]
- Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Molecular cell. 2001;7:683–694. doi: 10.1016/s1097-2765(01)00214-3. [DOI] [PubMed] [Google Scholar]
- Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, Tokino T, Taniguchi T, Tanaka N. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science. 2000;288:1053–1058. doi: 10.1126/science.288.5468.1053. [DOI] [PubMed] [Google Scholar]
- Ogawara Y, Kishishita S, Obata T, Isazawa Y, Suzuki T, Tanaka K, Masuyama N, Gotoh Y. Akt enhances Mdm2-mediated ubiquitination and degradation of p53. The Journal of biological chemistry. 2002;277:21843–21850. doi: 10.1074/jbc.M109745200. [DOI] [PubMed] [Google Scholar]
- Pietsch EC, Leu JI, Frank A, Dumont P, George DL, Murphy ME. The tetramerization domain of p53 is required for efficient BAK oligomerization. Cancer biology & therapy. 2007;6:1576–1583. doi: 10.4161/cbt.6.10.4719. [DOI] [PubMed] [Google Scholar]
- Tang Y, Zhao W, Chen Y, Zhao Y, Gu W. Acetylation is indispensable for p53 activation. Cell. 2008;133:612–626. doi: 10.1016/j.cell.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaseva AV, Moll UM. The mitochondrial p53 pathway. Biochimica et biophysica acta. 2009;1787:414–420. doi: 10.1016/j.bbabio.2008.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell. 2009;137:413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- Vousden KH, Ryan KM. p53 and metabolism. Nature reviews Cancer. 2009;9:691–700. doi: 10.1038/nrc2715. [DOI] [PubMed] [Google Scholar]
- Webster GA, Perkins ND. Transcriptional cross talk between NF-kappaB and p53. Molecular and cellular biology. 1999;19:3485–3495. doi: 10.1128/mcb.19.5.3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WL, Wang J, Chan CH, Lee SW, Campos AD, Lamothe B, Hur L, Grabiner BC, Lin X, Darnay BG, et al. The E3 ligase TRAF6 regulates Akt ubiquitination and activation. Science. 2009;325:1134–1138. doi: 10.1126/science.1175065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WL, Wu CY, Wu J, Lin HK. Regulation of Akt signaling activation by ubiquitination. Cell cycle. 2010a;9:487–497. doi: 10.4161/cc.9.3.10508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WL, Zhang X, Lin HK. Emerging role of Lys-63 ubiquitination in protein kinase and phosphatase activation and cancer development. Oncogene. 2010b;29:4493–4503. doi: 10.1038/onc.2010.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.