

Abstract
The efficacy of drugs targeting the CNS is influenced by their limited brain access, which can lead to complete pharmacoresistance. We recently reported a tissue-specific and selective upregulation of the multidrug efflux transporter ABCB1 or P-glycoprotein (P-gp) in the spinal cord of both patients and the mutant SOD1-G93A mouse model of amyotrophic lateral sclerosis (ALS), a fatal neurodegenerative disease that prevalently kills motor neurons. Here, we have extended the analysis of P-gp expression in the SOD1-G93A ALS mouse model and found that P-gp upregulation was restricted to endothelial cells of the capillaries, while P-gp expression was not detected in other cells of the spinal cord parenchyma such as astrocytes, oligodendrocytes, and neurons. Using both in vitro human and mouse models of the blood-brain barrier (BBB), we found that mutant SOD1 astrocytes were driving P-gp upregulation in endothelial cells. In addition, we observed a significant increase in reactive oxygen species production, Nrf2 and NFκB activation in endothelial cells exposed to mutant SOD1 astrocytes in both human and murine BBB models. Most interestingly, astrocytes expressing FUS-H517Q, a different familial ALS-linked mutated gene, also drove NFκB-dependent upregulation of P-gp. However, the pathway was not dependent on oxidative stress but rather involved TNFα release. Overall, our findings indicate that nuclear translocation of NFκB is a converging mechanism used by endothelial cells of the BBB to upregulate P-gp expression in mutant SOD1-linked ALS and possibly other forms of familial ALS.
Keywords: amyotrophic lateral sclerosis, endothelial cells, blood-brain barrier, P-glycoprotein, mutant SOD1
INTRODUCTION
Relentless and fatal, amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disorder that selectively kills both upper and lower motor neurons. With a complex pathogenesis, ALS continues to have neither a cure nor treatment that offers more than modest benefits (Grad et al., 2015; Pratt et al., 2012).
A likely culprit underlying the limited effectiveness of riluzole, the only FDA-approved drug for ALS treatment, is the multidrug efflux transporter ABCB1 or P-glycoprotein (P-gp) (Jablonski et al., 2015; Jablonski et al., 2014). P-gp is a membrane-bound protein that works to expel substances out of cells, and is expressed on the apical membrane (blood side) of endothelial cells, which comprise the functional core of the blood-brain barrier (BBB) and blood spinal cord barrier (BSCB) (Abbott et al., 2010; Wong et al., 2013). At these boundaries, P-gp restricts the passage of its substrates, which include a variety of therapeutics, into the central nervous system (CNS), often before they accumulate in meaningful or harmful quantities. In this manner, P-gp can prevent drugs aimed at the CNS from achieving their full therapeutic potential, particularly if P-gp is expressed in higher amounts and/or functioning at a higher capacity (Miller, 2015a). Previously, we identified a tissue-specific and disease progression-driven increase in P-gp expression and activity in the spinal cord of the SOD1-G93A transgenic mouse, a well-characterized and studied mouse model of ALS (Jablonski et al., 2012). These results, together with evidence that riluzole is a P-gp substrate (Milane et al., 2007), suggested that riluzole’s limited efficacy could, at least partially, result from P-gp driven pharmacoresistance that develops over the course of the disease. In a recent proof-of-principle study using SOD1-G93A ALS mice, we showed that co-administration of elacridar, a selective P-gp inhibitor, increased riluzole therapeutic efficacy (Jablonski et al., 2014).
Devising therapeutic strategies that consider P-gp’s role in ALS could thus be a promising means of boosting the efficacy of riluzole and other investigational ALS treatments. Yet, because P-gp serves a protective function at CNS barriers by thwarting the entry of toxic compounds, non-targeted treatments directed at blocking P-gp expression altogether could potentially compromise patient safety (Miller, 2015b; Qosa et al., 2015). To avoid such outcome, it is essential to have an understanding of the specific pathways of P-gp regulation in ALS, so that potential interventions target only the transporter’s pathological responses.
Of the many factors that could be responsible for the ALS-driven increase in P-gp, astrocytes are prime candidates for investigation. While ALS most noticeably leads to the degeneration of motor neurons, it also affects other cell types in the CNS, including astrocytes, which can in turn acquire deleterious properties (Benkler et al., 2013; Ferraiuolo et al., 2011; Foran et al., 2011; Foran et al., 2014; Haidet-Phillips et al., 2011). With respect to the BBB and BSCB, astrocytes expressing mutant SOD1 are particularly suspect to be responsible for P-gp overexpression in ALS, given that these cells are known to secrete factors affecting neighboring cells in the CNS (Abbott et al., 2006; Boillee et al., 2006; Clement et al., 2003; Radford et al., 2015).
Here, we started by further characterizing the P-gp expression profile in the spinal cord of SOD1-G93A ALS mice. We established that P-gp is selectively expressed in higher amounts only in capillary endothelial cells as ALS progresses from asymptomatic to symptomatic stages of disease in these mice. We then examined the potential role of astrocytes harboring ALS-linked SOD1 mutations in regulating the expression and activity of P-gp in endothelial cells. Using both mouse and human in vitro models of BBB, we showed that mutant SOD1-expressing astrocytes promote a transcription-mediated increase in endothelial cells of P-gp expression via nuclear factor kappa beta (NFκB) activation. Finally, we show that astrocytes carrying a different ALS-linked mutation also increase NFκB activation and P-gp expression, but do so through a separate signaling cascade.
MATERIALS AND METHODS
Animals
Transgenic mice expressing the human SOD1-G93A transgene [B6. Cg–Tg (SOD1*G93A)1Gur/J; Stock No: 004435; Jackson Labs] were bred in house. Offspring were assessed for the presence of the human transgene and copy number by polymerase chain reaction (PCR). SOD1-G93A mice were grouped in asymptomatic (Pre; 50 days old), and symptomatic (Symp; 140 days old) stages of ALS. To define asymptomatic and symptomatic SOD1-G93A mice, we used a combination of age and neurological score. Neurological score is a phenotypic scoring system that includes five scores ranging from 0, for symptoms free animals, to 4 for end stage animal where mouse cannot right itself within 30 seconds after being placed on either side. Therefore, for asymptomatic ALS stage, mice below age of 70 days with neurological score of 0 were used, whereas mice over 120 days old with neurological scores of 2–3 have been used as symptomatic ALS mice (Hatzipetros et al., 2015). Our SOD1-G93A mouse colony have 50% survival rate at 157.1 ± 9.3 days. Non-transgenic littermates were used as reference. All animals were housed in accordance with Thomas Jefferson University Institutional Animal Care and Use Committee (IACUC) and the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals.
Immunofluorescence analyses of mouse tissue
Spinal cord sections were embedded in OCT®, sectioned at 10 μm thickness and then probed by dual immunofluorescence staining. Sections were rinsed once with 1.5X Tris-Buffered Saline (TBS), fixed with 4% paraformaldehyde for 10 minutes at room temperature, and subsequently treated with antigen unmasking solution for 2 minutes at −20°C (33% acetic acid and 66% ethanol). Sections were washed and incubated with 2% Bovine Serum Albumin (BSA) blocking solution containing 0.3% Triton-X100, 5% horse serum prepared in 1.5X TBS buffer, and incubated overnight at 4°C with primary antibodies directed against P-gp (C219, Covance, MA) and either CD-31 as marker for brain capillaries (BD biosciences, CA), GFAP for astrocytes (Waco, CA), Olig-2 for oligodendrocytes (Millipore, CA) or NeuN for neurons (Abcam, MA) at dilutions of 1:50, 1:100, 1:300, 1:100, and 1:1000, respectively. At the end of incubation, sections were washed with 1.5X TBS and incubated with fluorescent secondary antibodies for 1 hour at room temperature. Sections were mounted with Prolong Gold DAPI antifade solution (Life technologies, CA) and images were captured using Olympus fluoview laser scanning confocal microscope (Olympus, PA) at total magnification of 600X.
Mouse cell cultures
Primary mouse brain astrocytes were co-cultured with either endothelial cells derived from the immortalized mouse brain cell line, brain endothelial cells line 3 (bEnd3), or with primary mouse brain endothelial cells (pMBEC) in transwell plates (Corning Biocoat, PA). Brain End3 cells (ATCC, Cat# CRL-2299) were used at passage 28–32, and maintained in DMEM supplemented with 10% fetal bovine serum (FBS), 1% w/v nonessential amino acids, glutamine 2 mM and the antibiotics penicillin G (100 IU/ml) and streptomycin (100 μg/ml). pMBEC were isolated from 6–10 weeks old mice as described (Wuest, Wing, & Lee, 2013). Briefly, wild-type mice (C57BL/6, n = 10) were euthanized, the harvested brains rolled over Whatman filter paper to remove meninges, and midbrain and white matter removed. Cortices were digested with type II collagenase (0.69 mg/ml) and DNase I (37.6 U/ml) in DMEM in a 37°C shaker (225 rpm) for one hour. The solution was then diluted in DMEM and centrifuged at 1000x g for 8 minutes. The resulting pellet was re-suspended in 20% BSA in DMEM and centrifuged at 1000x g for 20 minutes to remove myelin. The pellet was re-suspended in DMEM with collagenase-dispase (0.69 mg/ml) and DNase I (28.3 U/ml) and digested in a 37°C shaker (225 rpm) for one hour. The solution was diluted in DMEM and centrifuged at 700x g for 6 minutes. To isolate the microvessels, the pellet was resuspended and separated using a 33% continuous Percoll gradient, which was centrifuged at 1000x g for 10 minutes. The microvessel layer was collected, centrifuged, resuspended in 6 ml of culture media, and then plated in two 60 mm cell culture dishes (coated with 10% fibronectin and 10% collagen type IV). Endothelial cells were maintained in DMEM supplemented with 20% bovine platelet-poor plasma derived serum, human fibroblast growth factor (1 ng/ml), heparin (1 μg/ml), L-glutamine (2 mM), and 1% antibiotic solution (100 U/ml penicillin, 100 μg/ml streptomycin, and 0.25 μg/ml amphotericin). For the first 3 days in culture, media was supplemented with puromycin (4μg/ml) to prevent contamination from other cell types.
Primary mouse brain astrocytes were isolated as previously described (Foran et al., 2011). Astrocyte purity was determined with GFAP immunostaining. Cells in monolayers were > 95% GFAP+. Cells were maintained in a humidified atmosphere (5%CO2/95% air) at 37°C and medium was changed every other day.
Human Induced Pluripotent Stem Cells (iPSC) Culture
Astrocytes were differentiated from human induced pluripotent stem cells (iPSC) derived from one control, non-diseased subject and two ALS patients carrying either the SOD1-A4V or FUS-H517Q mutation as reported (Boulting et al., 2011; Haidet-Phillips et al., 2014). Differentiated astrocytes were used between DIV 90–100 in culture in 50/50 % DMED/F12 medium supplemented with 1% (FBS), 1% w/v nonessential amino acids, L-glutamine 2 mM, Heparin (2 μg/ml), 2% B27 supplement, and the antibiotics penicillin G (100 IU/ml) and streptomycin (100 μg/ml). Human iPS endothelial cells derived from one control, non-diseased subject were purchased from Cellular Dynamics International (Cellular Dynamics, WI, Cat# ECM-100-030-001). Human iPS endothelial cells were maintained and used between passages 2–5 in VascuLife basal medium (Lifeline cell technology, MD) supplemented with 5 ng/ml rh FGF basic, 50 μg/ml ascorbic acid, 1 μg/ml hydrocortisone sulfate, 10 mM L-glutamine, 15 ng/ml rh IGF-1, 5 ng/ml rh EGF, 5 ng/ml rh VEGF, 0.75 IU/ml heparin sulfate, the antibiotics penicillin G (100 IU/ml) and streptomycin (100 μg/ml), and 10% iCell endothelial cells medium supplement (Cellular Dynamics International, WI).
Co-cultures
Human iPSC astrocytes derived from control subject or ALS patients were cultured in 24-well plates. Primary mouse astrocytes were plated in a 24-well plate until confluent, after which they were transduced with adenovirus (Adv) constructs encoding either human SOD1-WT or human SOD1-G93A (Multiplicity of Infection [MOI] = 3). To prepare endothelial cell monolayers, transwell polyester membrane inserts (6.5 mm diameter with 0.4 μm pores; Corning, NY) were coated with fibronectin (60 μg/ml) for 90 min at 37°C. Endothelial cells were plated at a seeding density of 50,000 cells/cm2 on coated inserts, which were then placed atop of the astrocyte layer; medium in the resulting transwell system was changed every other day. hiPS endothelial cells were used between passages 2–5 (Lippmann et al., 2012) and co-cultured with hiPSC astrocytes of different genotypes or control, while bEnd3 and pMBEC were co-cultured with primary mouse astrocytes transduced with GFP-tagged human SOD1-WT or human SOD1-G93A viral constructs. On day 6 of co-culture with astrocytes, endothelial cell monolayers were used for P-gp transport assay, western blot analysis and immunofluorescence staining as indicated.
Permeability Studies
Astrocyte/endothelial cell co-cultures at DIV6 were used to measure apical to basolateral (A→B) transport of NaF and LD800. 200 μl of fresh pre-warmed transport buffer (141 mM NaCl, 4 mM KCl, 2.8 mM CaCl2, 1 mM MgSO4, 10 mM HEPES, and 10 mM D-glucose, pH 7.4) containing 250 μM LD800 and 250 μM NaF were added to the apical chamber and 800 μl of fresh transport buffer were added to the basolateral chambers. Cells were incubated in a humidified atmosphere (5%CO2/95% air) at 37°C for 1 h. At the end of incubation period, the buffer from both chambers was separately collected. LD800 and NaF concentrations in apical and basolateral compartments were determined by measuring their fluorescence intensities at excitation/emission wavelengths of 680/720 nm and 485/529 nm, respectively (Fluostar Optima plate reader; BMG Labtech, NC). Apparent permeation coefficient (Pc) was calculated from the following equation:
| Equation 1 |
where Vb is the volume of basolateral side (800 μl), Cb is the concentration of LD800 or NaF (μM) in the basolateral side, Ca is the concentration of LD800 or NaF (μM) in the apical side, A is the membrane area (0.33 cm2), and T is the time of transport (3600 sec). In addition to measuring the permeability of each compound, we calculated the apical to basolateral transport quotient (TQA→B) for LD800 to normalize for its paracellular permeation, using equation 2.
| Equation 2 |
NFκB activity measurement in cultures
Endothelial cells, either primary mouse brain endothelial cells (pMBEC) or iPS-derived, were seeded on transwell filter membranes and co-cultured for 6 days with astrocytes as described, which were previously transduced with Adv-hSOD1-WT or Adv-hSOD1-G93A constructs (MOI 3). For NFκB inhibition study, transwells were removed from co-culture and incubated with either the active or the inactive SN50 peptide (1μM, Calbiochem, CA, Cat# 481480) for 30 minutes to allow permeation inside the cells. The SN50 peptide was then washed out and endothelial cells were again placed in co-culture. At the end of co-culture period, endothelial cells were immune-stained for NFκB as described in the section bellow. The ratio of nuclear NFκB to cytoplasmic signal was calculated to determine the degree of NFκB activation via nuclear translocation. Endothelial cells were then collected for western blot analysis as described.
P-gp activity in biologically active capillaries
Capillaries were isolated from brain and spinal cord of wild type mice as described previously (Hartz et al., 2004). Briefly, white matter, meninges, midbrain, and olfactory lobes were removed before brain tissues were homogenized in HBSS buffer supplemented with CaCl2 and MgCl2. Spinal cords were flushed out the vertebral column and homogenized using HBSS buffer. One volume of Ficoll solution (30%) was added to the homogenates to a final concentration of 15%. Homogenates were vortexed and then centrifuged (40,000 rpm for 15 min, 4°C). Pellets were suspended in HBSS buffer and passed over glass bead columns. Capillaries adhering to the beads were collected by gentle agitation in HBSS containing 1% bovine serum albumin (BSA). Isolated capillaries were suspended in endothelial cell medium contains 2% Matrigel, plated in 24-well plates over a glass coverslip pre-coated with Matrigel and incubated in a humidified atmosphere (5%CO2/95% air) at 37°C. After 4 hours, the medium was removed and medium composed of 30% endothelial cells medium and 70% astrocytes conditioned medium (ACM) collected from SOD1-WT, SOD1-A4V, FUS-WT or FUS-H517Q astrocytes was added to capillaries for 20 hours. For collection of astrocytes medium, astrocytes were grown in 75-cm2 flask and allowed to reach 80% confluency. At this confluency, medium were changed with fresh one and cells incubated in a humidified atmosphere (5%CO2/95% air) at 37°C. After three days, medium was collected and either used directly to treat isolated capillaries or aliquoted and stored at −80 °C. At the end of incubation periods, 2 μM NBD-Cyclosporin (NBD-CSA) prepared in HBSS buffer supplemented with CaCl2 and MgCl2 was added to capillaries and removed after 1 hour. Capillaries were washed twice with ice-cold HBSS buffer and imaged using Leica TCS SP8 laser scanning confocal microscope at a total magnification of 400X. Luminal fluorescence was quantitated by NIH ImageJ software as described previously (Hartz et al., 2004). For Western blotting analysis, capillaries were collected, spun down at 5,000 rpm, and lysed using RIPA buffer containing complete protease inhibitors (Sigma-Aldrich, MO).
Western Blot analysis
Capillaries and cell homogenates were collected in RIPA buffer with 1% SDS. Samples containing equal total protein amounts were separated by SDS-PAGE using 12% or 4–15% gels (Bio-Rad, CA) and transferred to polyvinylidine fluoride (PVDF) membranes (GE Healthcare, NJ). Membranes were blocked by 5% milk or 3% BSA prepared in TBS with Tween (TBST) and exposed to primary antibody for 1 h at room temperature or overnight at 4 °C. Primary antibodies were anti-P-gp (mouse monoclonal C219, Covance; 1:50), anti-GAPDH (monoclonal mouse, Fitzgerald 1:20,000), anti-GFP (rabbit polyclonal, Living Colors 1:3,000), anti-phosphorylated protein kinase-CβII, anti-PKCβII (Rabbit polyclonal 1:2000, Cell Signaling, MA), anti-tumor necrosis factor-1, anti-TNFR1 (Rabbit polyclonal 1:2000, Abcam, MA), or anti-actin (Mouse monoclonal 1:4000, Abcam, MA). Following three washes with TBST, 15 min each, membranes were incubated with secondary antibody (1:1000 for anti-mouse and anti-rabbit IgG) for 1 h at room temperature, washed again and developed using ECL Prime reagents for chemiluminescent detection (Bio-Rad Quantity-One Detection System). Protein bands were quantified using Quantity-One software (Bio-Rad, CA).
Immunocytochemistry
At the end of co-cultured period, transwells with human iPSC endothelial cells were rinsed twice with ice-cold PBS, fixed with 4% paraformaldehyde for 20 minutes at room temperature, and washed again twice with PBS. Cells were permeabilized in 0.2% Triton-X100 prepared in DPBS for 10 minutes, washed twice with PBS, and blocked in 5% donkey serum prepared in PBS contains 0.1% tween-20 for one hour. Mouse monoclonal P-gp antibody (C219 1:50, Covance; MA), rabbit monoclonal NFκB antibody (1:100, Abcam, MA), rabbit polyclonal peroxisome proliferator-activated receptor (PPARα) antibody (1:100, Abcam, MA), rabbit polyclonal pregnane X receptor (PXR) antibody (1:100, Abcam, MA), rabbit polyclonal constitutive androsterone receptor (CAR) antibody (1:50, Abcam, MA), mouse monoclonal Nrf2 antibody (1:100, R and D systems, MN) were prepared in 5% donkey serum. Cells were incubated with primary antibodies overnight at 4°C followed by 3 washes with PBS and incubation with secondary antibodies (Alexa Fluor 568-donkey anti-mouse IgG or anti-rabbit at 1:200 dilution) for one hour at room temperature on shaker and cells were washed three times with PBS. Transwell membranes were collected and mounted with Prolong Gold DAPI antifade solution (Life technologies, CA) and images were captured using Leica TCS SP8 laser scanning confocal microscope at a total magnification of 400X. Total signal of P-gp and Nuclear and cytoplasmic signals of NFκB, PXR, PPAR, CAR and Nrf2 were quantified using NIH ImageJ software as described previously (Noursadeghi et al., 2008).
TNF-α ELISA
TNF-α levels in the basolateral chamber of the co-culture system were measured using a sandwich enzyme linked immunosorbent assay (ELISA) kit as described by manufacturer (TNFα ELISA Kit, KHC3011, Life technologies, CA). Briefly, 100 μL of basolateral media were added to each well of a 96-well ELISA plate. The plate was incubated at room temperature for 2 hrs followed by overnight incubation at 4°C. At the end of the incubation periods, wells were washed 5 times with washing buffer and 100 μL of biotinylated anti-TNF-α was added into each well. After 2 hrs incubation at room temperature, the solution in each well was aspirated and the wells were washed 5 times. Then, 100 μL Streptavidin-HRP was added to each well and incubated at room temperature for 1 hr. After washing the plate from Streptavidin-HRP solution, 100 μL of chromogen solution was added to each well and incubated for 30 minutes at room temperature and in the dark. Absorbance of each well was measured after addition of stop solution at 450 nm using a plate reader (Fluostar Optima plate reader; BMG Labtech, NC).
Oxidative stress measurement
Oxidative stress in endothelial cells was measured by using CellROX© reagent (Life Technologies, CA). Briefly, medium bathing endothelial cells in transwell was removed and fresh pre-warmed medium containing 2 μM CellROX© was added back to the cells. After 30 min. incubation in a humidified atmosphere (5%CO2/95% air) at 37°C, the medium was removed and the cells washed twice with ice-cold PBS, fixed with 4% paraformaldehyde for 20 minutes at room temperature, and washed again twice with PBS. Transwell membranes were collected and mounted with Prolong Gold DAPI antifade solution (Life technologies, CA) and images were captured using Leica TCS SP8 laser scanning confocal microscope at a total magnification of 400X. Total green signal of CellROX© was quantified using NIH ImageJ software.
Statistical analysis
Data are expressed as mean ± standard error of the mean (s.e.m.), unless otherwise indicated. The experimental results were statistically analyzed for significant difference using a two-tailed Student’s t-test. Values of P < 0.05 were considered statistically significant.
RESULTS
Expression profile of P-gp in the spinal cord of SOD1-G93A mice
Using western blot and transport assay, we recently reported that expression and activity levels of the drug efflux transporter ABCB1 (P-glycoprotein or P-gp) was upregulated in the spinal cord of the SOD1-G93A mouse model of ALS and in ALS patients (Jablonski et al., 2012). Here, we performed a more detailed immunofluorescence analysis of P-gp expression focusing on the ventral horn region of the spinal cord of SOD1-G93A mice to examine the distribution profile of P-gp in the different cell types of this ALS-affected part of the CNS parenchyma. In particular, we considered P-gp expression in endothelial cells, astrocytes, oligodendrocytes and neurons in asymptomatic and symptomatic SOD1-G93A mice, comparing results with age-matched wild type littermate mice. P-gp immunofluorescence signal in the ventral spinal cord of asymptomatic SOD1-G93A mice was exclusively detected in the capillary endothelium immunostained with an antibody against the endothelial marker, CD-31 (PECAM-1), while astrocytes, oligodendrocytes and neurons, although co-stained with their respective markers, did not show detectable expression of P-gp (Figure 1A). Ventral spinal cord of SOD1-G93A symptomatic mice showed similar profile of P-gp distribution (Figure 1B), with endothelial cells in capillaries as the only cells that presented detectable P-gp immunofluorescence signal, at even more intense levels than asymptomatic (Figure 1A) or control littermate mice (Figure 1C).
Figure 1. Expression profile of P-gp in parenchymal cells of the spinal cord of the SOD1-G93A mouse model of ALS at asymptomatic and symptomatic stage of disease.
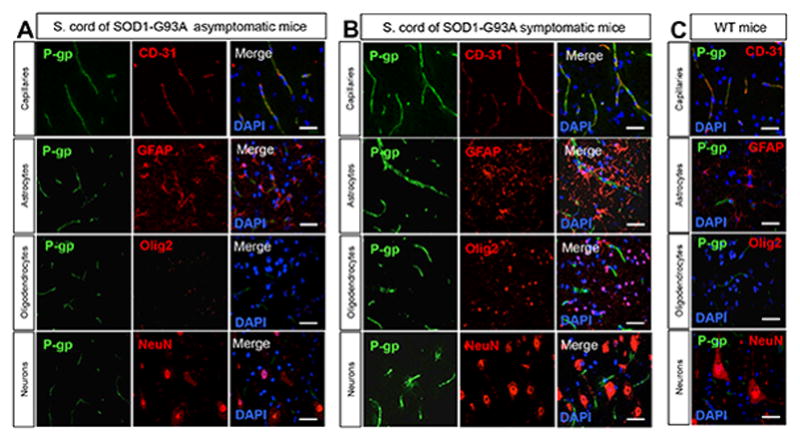
Immunohistochemistry analyses of the mouse spinal cord revealed detectable P-gp expression only in capillaries (elongated tubular structures in the spinal cord parenchyma, identified as CD-31+). P-gp immunofluorescence was absent in bona fide astrocytes (GFAP+ cells), oligodendrocytes (Olig2+ nuclei) and neurons (identified as NeuN+ cells). P-gp immunofluorescence signal was more intense in capillaries of (B) SOD1-G93A symptomatic mice (140 days old) than (A) asymptomatic mice (50 days old) or (C) wild-type control littermate mice, age-matched with symptomatic mice (scale bar = 50 μm).
Upregulation of P-gp in an in vitro model of Blood Brain Barrier
We next sought to unravel the contributing mechanisms and factors in ALS that could lead to the upregulation of P-gp expression and activity in endothelial cells. Astrocytes, through their end feet interaction with endothelial cells of the neurovascular unit, are identified regulators of the BBB properties and integrity (Abbott et al., 2006) and, therefore, prime candidates as potential determinant factors. We used, as the cellular model system of choice for this investigation, primary mouse spinal cord astrocytes overexpressing human SOD1 wild type (SOD1-WT) or the ALS-linked human mutant SOD1-G93A protein. Astrocytes were co-cultured with brain endothelial cells line 3 (bEnd3), an immortalized mouse brain endothelial cell line, or primary mouse brain endothelial cells (pMBECs) in multiwell plates with transwell inserts, as depicted in Figure 2A. Upon co-culture with SOD1-G93A astrocytes, bEnd3 cells expressed higher levels of P-gp than if they were co-cultured with SOD1-WT astrocytes (+41 ± 5.8%; P = 0.0002; Figure 2B), suggesting that mutant SOD1 expression in astrocytes can affect how these cells influence the molecular characteristics of the blood brain barrier. Expression of the human SOD1 transgenes in astrocytes were at comparable levels, indicating that P-gp upregulation mediated by the astrocytes was mutant SOD1 specific and not due to differences in expression levels of the transgenes (Figure 2B). To validate these findings in a more physiological cell model system and to assess whether mutant SOD1 astrocytes were also affecting P-gp activity in endothelial cells, we used pMBECs isolated from adult mice and co-cultured them with the same primary spinal astrocytes in a manner similar to that described for bEnd3 cell experiments. Similar to what was observed for the bEnd3 cell line, pMBECs increased expression levels of P-gp by +85 ± 9% when they were co-cultured with SOD1-G93A astrocytes compared to SOD1-WT astrocyte co-cultures (P = 0.0029; Figure 2B).
Figure 2. Astrocytes expressing mutant SOD1-G93A enhance expression and activity of P-gp in bEnd3 and primary mouse brain endothelial cells (pMBEC).
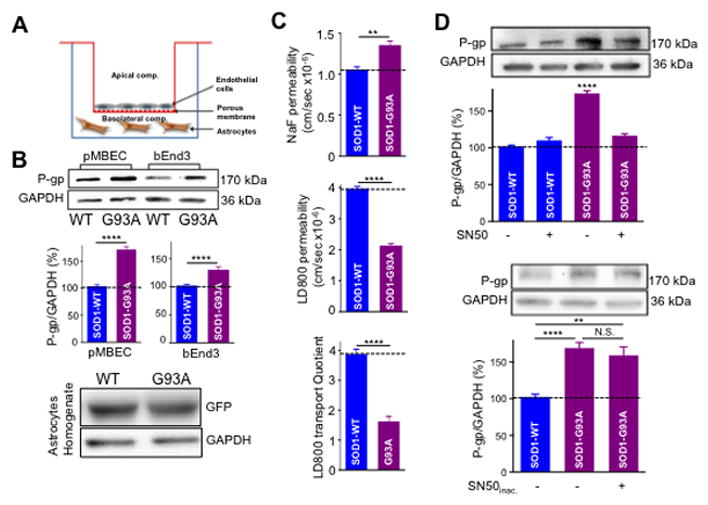
(A) Schematic of the in vitro BBB model. These culture vessels produce an environment that resembles the in vivo state of the brain barriers as closely as possible to enable the growth of specialized cell types such as endothelial cells. The basolateral compartment is represented by the well of the culture plate, which is plated with either mouse or human iPSC-derived astrocytes. The apical compartment is represented by the transwell, which is plated with the endothelial cells. Permeable supports with microporous membranes permit cells to uptake and secrete molecules on both their basal and apical surfaces. Cellular functions such as transport, absorption and secretion can also be studied since cells grown on permeable supports provide convenient, independent access to apical and basolateral plasma membrane domains. (B) Astrocytes plated on wells are transduced for 24 hours with AdV-human SOD1-G93A or AdV-SOD1-WT (both GFP tagged) viral constructs (MOI 3). After virus removal, endothelial cells are plated on transwells and cultured for additional 5 days before analysis. Western blot analysis showed a significant increase in the level of P-gp expression in pMBEC and bEnd3 cells compared to WT (85 ± 9% and 40 + 5.8%, respectively). (C) SOD1-G93A expressing astrocytes significantly increased P-gp function (LD800 permeability) in co-cultured primary endothelial cells compared to SOD1-WT astrocytes. Although SOD1-G93A astrocytes disrupt the integrity of pMBEC monolayer (NaF permeability), the permeability of LD800 was significantly restricted. Reduction in LD800 permeability indicates increased P-gp activity. (D) P-gp upregulation in pMBEC co-cultured with SOD1-G93A astrocytes was blocked by the NFκB inhibitor SN50 (1 μM; in 0.1% DMSO), while addition of inactivated SN50 did not reverse the increase in P-gp expression. Western blot data among the different experimental groups are normalized by expression levels of GAPDH and are plotted in bar graphs as mean ± s.e.m. of three independent experiments (N.S. = not significant; ** p < 0.01; **** p < 0.0001).
To measure whether the transport activity of P-gp was also affected, we assessed the extent to which two compounds, sodium fluorescein (NaF, 376 Daltons) and LD800 (380 Daltons) permeated through the endothelial cell monolayer. Collectively, the transport of these two molecules represents the means of traversing the BBB/BSCB, and the degree to which each compound crosses the endothelial cell monolayer reflects different aspects of the barrier’s structure and/or function. NaF is a small, water-soluble molecule that travels across the monolayer only through passive paracellular diffusion (i.e. through spaces between cells), and its transport is thus a measure of the strength of the tight junctions forming the barrier’s wall (Tortarolo et al., 2015). By contrast, LD800 is a small, lipophilic molecule that traverses the monolayer exclusively through passive transcellular diffusion (i.e. through cells) and is a specific substrate for P-gp. Because only P-gp extrudes LD800 from cells and limits its passage across the monolayer, the transport of LD800 serves as a proxy for P-gp activity level (On et al., 2011). Passive permeability of NaF significantly increased across pMBEC barriers in co-culture with SOD1-G93A astrocytes, implying a disruption to the integrity of the paracellular barrier. NaF permeability was 1.05 ± 0.23 × 10−6 cm/sec and 1.36 ± 0.18 × 10−6 cm/sec across pMBEC monolayer co-cultured with SOD1-WT and SOD1-G93A astrocytes, respectively (P = 0.0038; Figure 2C). By contrast, the steady-state level of LD800 permeability was lower in the pMBEC monolayer exposed to SOD1-G93A astrocytes (2.14 ± 0.06 × 10−6 cm/sec) compared to SOD1-WT astrocytes (3.97 ± 0.15 × 10−6 cm/sec, p = 0.0024; Figure 2C), indicating that SOD1-G93A astrocytes were capable of enhancing P-gp transporter-mediated efflux activity. Consistent with this, the transport quotient of LD800 was significantly lowered across the pMBEC monolayer exposed to SOD1-G93A astrocytes compared to SOD1-WT astrocytes (1.5 ± 0.2 vs. 3.9 ± 0.6, p = 0.0023; Figure 2C). These results indicate that despite disrupting the pMBEC monolayer, which if anything should increase the barrier’s overall permeability, SOD1-G93A astrocytes decrease the permeability of P-gp substrates, presumably by enhancing P-gp activity coupled to increased P-gp expression. Thus, these findings further confirmed the significant contribution that P-gp could make to the transport of compounds across the endothelial cell monolayer at the blood-brain barrier level.
Regulation of P-gp in endothelial cells has been attributed to the effect of multiple transcription factors (Qosa et al., 2015). The most prominent ones are xenobiotic nuclear transcription factors and NFκB (Miller, 2015a). Confined to the cytosol in their inactive state, these transcription factors can become activated and translocate into the nucleus to influence the transcription of P-gp. As NFκB alterations are also related to ALS pathology, we hypothesized that astrocytes in ALS could initiate P-gp upregulation through NFκB activation. Under this logic, inhibiting NFκB translocation would block the increase in P-gp protein expression in endothelial cells mediated by mutant SOD1-expressing astrocytes. Mouse pMBEC cells co-cultured with SOD1-G93A astrocytes treated with SN50, a selective peptidergic inhibitor of NFκB nuclear translocation, displayed significantly lower levels of P-gp than control, untreated cells (116% ± 14.0% and 174% ± 14.4%, respectively; p = 0.049; Figure 2D). By contrast, treating bEnd3 cells co-cultured with SOD1-WT astrocytes with SN50 did not affect P-gp protein expression levels (110% ± 20.4%) compared to untreated bEnd3 cells in co-culture with SOD1-WT astrocytes (102% ± 9.4%, P = 0.59; Figure 2D). Furthermore, treating bEnd3 cells co-cultured with SOD1-G93A astrocytes with the inactive SN50 peptide that has no measureable effect on NFκB translocation did not block P-gp upregulation (Figure 2D). Taken together, these findings reveal that mutant SOD1 astrocytes regulate expression of P-gp in endothelial cells by impacting the NFκB activation pathway.
Regulation of P-gp in a human iPSC-derived culture model of the Blood Brain Barrier
To determine whether the astrocyte driven upregulation of P-gp is also a relevant mechanism to human ALS, we employed an in vitro model of BBB and tested whether ALS patient-derived iPS astrocytes could alter P-gp expression in human derived iPS endothelial cells. Endothelial cells were differentiated from iPSCs derived from a healthy control individual, whereas astrocytes were differentiated from iPSCs derived from both control (SOD1-WT) and a SOD1-A4V patient. These astrocytes were co-cultured for 5 days with endothelial cells in a transwell culture system (Figure 2A). Formation of human iPSCs endothelial monolayer was monitored and confirmed by TEER measurement (Supplementary Figure 1) Compared to control, SOD1-A4V astrocytes significantly increased P-gp expression in human iPS endothelial cells by about 70%, according to immunocytochemical analyses (P = 0.015, Figure 3A) and about 30% according to western blot analyses (P = 0.0041, Figure 3B). Upregulation of P-gp expression levels was matched by an increase in P-gp efflux activity as measured by LD800 transport assay from apical to the basolateral chamber. Although SOD1-A4V astrocytes significantly increased the passive permeability of NaF across the hiPS endothelial monolayer (Pc = 2.05 ± 0.11 × 10−6 cm/sec) relative to control SOD1-WT astrocytes (Pc =1.74 ± 0.32 × 10−6 cm/sec, P = 0.028; Figure 3C), the endothelial cell monolayer was less permeable to LD800 (Pc = 3.5 ± 0.36 × 10−6 cm/sec) than was the monolayer co-cultured with SOD1-WT astrocytes (Pc = 3.9 ± 0.29 × 10−6 cm/sec, p = 0.001; Figure 3D). Moreover, inhibition of P-gp with 1μM elacridar significantly increased LD800 compound across the endothelial monolayer co-cultured with wild type or SOD-G93A astrocytes (Pc = 4.8 ± 0.34 × 10−6 cm/sec and 4.9 ± 0.22 × 10−6 cm/sec; Figure 3D). Since disturbances to the endothelial cell monolayer, independent of P-gp activity, could potentially increase the passive permeability of LD800, we normalized the LD800 permeability results by calculating the transport quotient of LD800. Consistent with permeability results, LD800 compound TQ was significantly lower across endothelial monolayer co-cultured with SOD1-A4V astrocytes compared to monolayer co-cultured with SOD1-WT (TQ = 1.5 ± 0.25 and 2.1 ± 0.11, respectively, P = 0.042; Figure 3E).
Figure 3. Astrocytes derived from ALS patient iPS cells enhance expression and activity of P-gp in co-cultured iPS brain endothelial cells derived from normal subject.
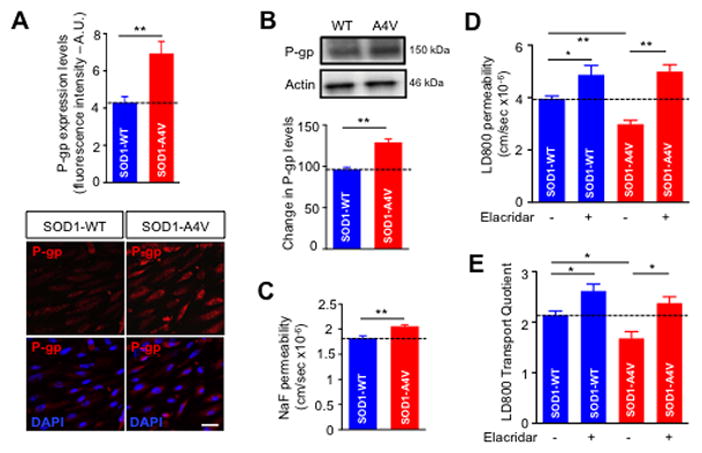
Compared to iPS SOD1-WT astrocytes, iPS SOD1A4V astrocytes increased P-gp expression in iPS brain endothelial cells as measured by (A) immunocytochemical and (B) western blot analyses. While iPS SOD1-A4V astrocytes disrupt the integrity of iPS endothelial cells as shown by (C) increased NaF permeability, they enhance P-gp activity within these same cells as demonstrated by decreased LD800 (D) permeability and (E) transport quotient. Incubation of endothelial cells with the P-gp inhibitor, elacridar (1 μM) abolished upregulation of P-gp activity mediated by SOD1-A4V astrocytes. (Scale bar = 50 μm). Data are mean ± s.e.m. of at least three independent experiments (* p < 0.05 and ** p < 0.01).
Molecular pathways underlying P-gp upregulation in endothelial cells
In addition to NFκB, xenobiotic nuclear receptors such as PPAR, PXP and CAR are known effectors of P-gp regulation (Miller, 2015b; Qosa et al., 2015). We therefore investigated whether the effect of mutant SOD1 astrocytes involved activation of these effectors in endothelial cells. Co-culturing human iPS-derived endothelial cells with iPS-derived SOD1-A4V astrocytes significantly stimulated nuclear translocation of NFκB in endothelial cells, while it did not change the activity of xenobiotic nuclear receptors (Figure 4). Nuclear to cytoplasmic ratio of NFκB increased by 30% when endothelial cells were co-cultured with SOD1-A4V astrocytes (0.44 ± 0.036 in endothelial cell co-cultured with SOD1-A4V astrocytes and 0.32 ± 0.051 in endothelial cells co-cultured with SOD1-WT astrocytes, P = 0.037; Figure 4). To confirm the involvement of NFκB nuclear translocation, endothelial cells were pre-incubated with 1 μM SN50 before being co-cultured with SOD1-A4V astrocytes. We found that this treatment significantly prevented NFκB nuclear translocation (Figure 5A). Consistent with this, SOD1-A4V induced P-gp expression was reversed by SN50, whereas the inactive form of the SN50 peptide was ineffective, confirming the role of activated NFκB in inducing overexpression of P-gp by SOD1-A4V astrocytes (Figure 5B). Moreover, while inhibition of NFκB did not change the effect of SOD1-A4V astrocytes on the integrity of the endothelial cell monolayer as measured by NaF permeability (Figure 6A), it could reverse the effect of SOD1-A4V astrocytes on P-gp activity as assessed by LD800 permeability and transport quotient (Figure 6B, C). Pretreatment with 1 μM SN50 peptide restored the LD800 permeability of the iPS endothelial cell monolayer co-cultured with SOD1-A4V astrocytes to control levels, whereas pretreatment with the inactive SN50 peptide did not prevent P-gp upregulation and LD800 expulsion (Figure 6B). Transport quotient of LD800 was significantly lower across the human iPS endothelial monolayer co-cultured with SOD1-A4V astrocytes (TQ = 0.75 ± 0.2) compared to monolayer co-cultured with hSOD1-WT astrocytes (1.35 ± 0.3, p = 0.009), or SOD1-A4V pre-treated with SN50 peptide (1.5 ± 0.32), but similar to that of human iPS endothelial monolayer co-cultured with SOD1-A4V and treated with inactivated SN50 peptide (0.73 ± 0.26 %) (Figure 6C). These results affirm the important role P-gp plays in the transport of compounds across the endothelial monolayer and support the notion that SOD1-A4V astrocytes upregulate P-gp in endothelial cells by increasing NFκB translocation.
Figure 4. Nuclear translocation of NFκB in human iPS brain endothelial cells underlies the increase in P-gp expression.

Immunocytochemical analysis was used to estimate nuclear to cytoplasmic ratios of xenobiotic nuclear receptors (PPARα, PXR and CAR) and NFκB. Images were analyzed with NIH ImageJ software. 6–9 image fields were analyzed/group. (Scale bar = 50 μm). Data are mean ± s.e.m. of three independent experiments (*** p < 0.001).
Figure 5. Inhibition of NFκB nuclear translocation blocks upregulation of P-gp in human iPS brain endothelial cells co-cultured with iPS-SOD1-A4V astrocytes.
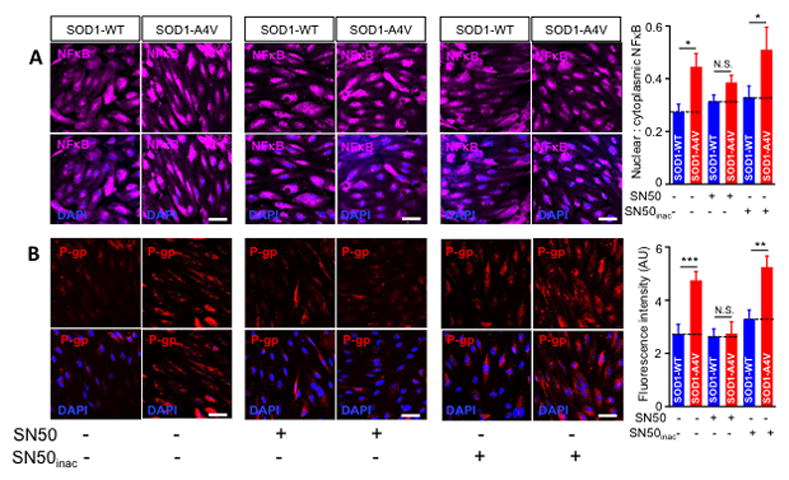
(A) Preincubation with SN50 1 μM effectively decreased the nuclear to cytoplasmic ratio of NFκB in human iPS brain endothelial cells co-cultured with iPS-SOD1-A4V astrocytes, whereas the inactive SN50 peptide was ineffective. Data are mean ± s.e.m. of three independent experiments (N.S. = not significant; * p < 0.05). 6–9 image fields were analyzed/group. (B) P-gp upregulation in endothelial cells co-cultured with SOD1-A4V astrocytes correlates with NFκB activity. Preincubation of endothelial cells with SN50 (1 μM) abolished the increase in P-gp, bringing it down to control levels. As expected, inactivated SN50 failed to reverse P-gp upregulation in endothelial cells co-cultured with hSOD1-A4V astrocytes. (Scale bar = 50 μm). 6–9 image fields were analyzed/group. Data are mean ± s.e.m. of three independent experiments (* p < 0.05, ** p < 0.01, (*** p < 0.001).
Figure 6. Upregulation of P-gp activity in human iPS endothelial cells co-cultured with SOD1-A4V astrocytes is connected with NFκB translocation.

(A) NaF permeability of endothelial cells is not affected by preincubation with SN50 (1 μM), indicating that the disruptive effect of SOD1-A4V astrocytes on the integrity of endothelial monolayer is mechanistically disconnected from the mechanism involved in P-gp upregulation. (B) Concomitantly, LD800 permeability increased significantly upon SN50 treatment, indicating reduced P-gp activity. (C) Transport quotient of LD800 compound confirmed that reduction in LD800 compound permeability is related to enhanced P-gp activity. Data are mean ± s.e.m. of three independent experiments (* p < 0.05, ** p < 0.01).
Previous studies suggested that regulation of P-gp expression in endothelial cells mediated by NFκB could be initiated by inflammatory mediators or oxidative stress (Bauer et al., 2007; Hartz et al., 2008; Qosa et al., 2015). We therefore examined the role of both of these pathways in the upregulation of P-gp. First, we determined the levels of reactive oxygen species (ROS) produced by endothelial cells after being co-cultured with iPS-derived astrocytes. Endothelial cells displayed a robust increase in the intensity of the fluorescence signal of CellROX© when these cells were co-cultured with SOD1-A4V vs. SOD1-WT human iPS astrocytes (423 ± 75%; p = 0.0001; Figure 7A). Moreover, we tested the translocation of Nrf2, a transcription factor that was previously shown to stimulate NFκB-mediated P-gp overexpression in endothelial cells exposed to oxidative stress (Wang et al., 2014). Consistent with increased levels of ROS, we measured an increase in nuclear to cytoplasmic ratio of Nrf2 in endothelial cells exposed to SOD1-A4V astrocytes (167 ± 28%; Figure 7B). A previous study showed that under inflammatory stress, tumor necrosis factor-α (TNF-α) levels increased and activated TNF-receptor-1 (TNFR-1), which in turn activated a signaling cascade that converged on NFκB nuclear partition and P-gp overexpression. Our data showed that co-culturing mutant SOD1 astrocytes with endothelial cell did not significantly increase TNF levels in the basolateral chamber (2.9 ± 0.3 pg/ml and 3.1 ± 0.27 pg/ml in SOD1-WT and SOD1-A4V astrocytes, P = 0.85; Figure 7C). Moreover, SOD1-A4V astrocytes did not enhance expression of TNF-R1 or protein kinase-CβII, key protein in TNF-NFκB signaling cascade (Figure 7D). Collectively, our results pointed at oxidative stress as a possible underlying stressor that is induced by SOD1-A4V astrocytes and could activate NFκB and P-gp overexpression in endothelial cells.
Figure 7. Oxidative stress triggers NFκB nuclear translocation in endothelial cells leading to enhanced P-gp expression.
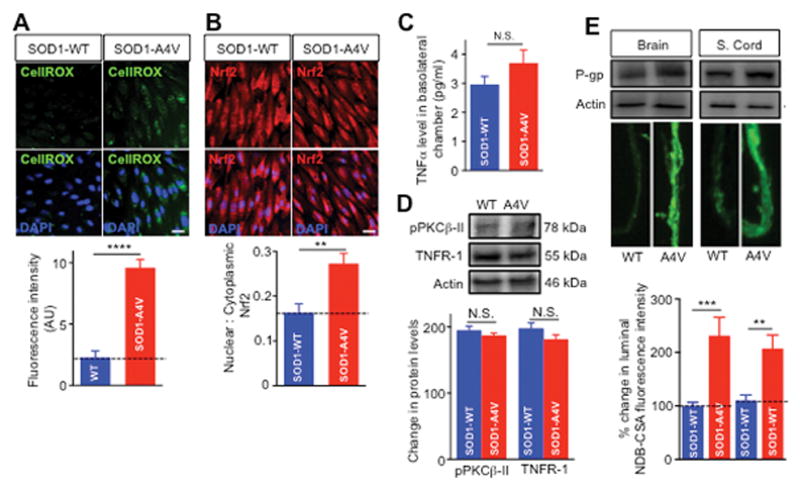
(A) Oxidative stress was assessed using a cell permeable molecule (CellRox©) that fluoresces upon exposure to ROS and oxidation. CellRox© (2 μM) was incubated in a humidified atmosphere (5%CO2/95% air) at 37°C for 30 min. Quantitation of the fluorescence intensity showed significant increase in the level of ROS in endothelial cells co-cultured with SOD1-A4V astrocytes compared to the respective control. (B) Significant increase in Nrf2 nuclear translocation in endothelial cells confirmed the establishment of oxidative stress conditions in endothelial cells induced by hSOD1-A4V astrocytes. (C) ELISA assay for TNF-α showed comparable TNF-α levels in the basolateral media of endothelial cells co-cultured with hSOD1-WT or hSOD1-A4V astrocytes. (D) Western blot of TNFR and PKCβII expression levels were normalized for actin band intensity in each lane. Analysis showed that levels did not significantly differ among homogenates of endothelial cells co-cultured with SOD1-WT or SOD1-A4V astrocytes. (E) NBD-CSA accumulation in active isolated brain and spinal cord capillaries showed higher P-gp expression and activity in capillaries treated with conditioned media (CM) collected from SOD1-A4V astrocytes than CM collected from SOD1-WT astrocytes. Data is presented as mean ± SEM of three independent experiments (** p < 0.01, *** p < 0.001, **** p < 0.0001).
To validate our results using a more physiological and integrated system, we tested the effect of medium collected from SOD1-WT and SOD1-A4V astrocyte on expression and activity of P-gp in biologically active brain and spinal cord capillaries. Results in isolated biologically active capillaries from brain and spinal cords showed significant increase in P-gp expression and activity when these were incubated with human iPS SOD1-A4V astrocytes conditioned medium compared to SOD1-WT astrocytes control medium (Figure 7E). In sum, our results strongly indicated that astrocytes, in mutant SOD1-linked ALS, can enhance P-gp expression and activity in a clinically relevant, human-based in vitro BBB model, and do so at least in part by promoting the translocation and transcriptional activity of NFκB. Additionally, we provide evidence that mutant SOD1 astrocytes induce oxidative stress in endothelial cells, which is what triggers NFκB activation and subsequent P-gp upregulation.
Astrocytes harboring FUS-H517Q drive P-gp upregulation through inflammatory stress
We next examined whether P-gp upregulation in endothelial cells was the result of a specific effect of mutant SOD1 astrocytes or was a more general effect related to the presence of ALS-linked mutated genes. We tested the effect of astrocytes expressing the fused in sarcoma mutated protein (FUS-H517Q), a RNA/DNA binding protein mutated in familial ALS (Bosco et al., 2010). Figure 8 shows that FUS-H517Q significantly enhanced P-gp expression in endothelial cells. Immunocytochemical and western blot analyses showed ~ 55% and ~27% increase, respectively in P-gp expression in endothelial cells co-cultured with human iPS-derived FUS-H517Q astrocytes compared to FUS-WT (P = 0.0021 and 0.017, respectively, Figure 8A, B). Moreover, isolated brain and spinal cord capillaries incubated with medium collected from FUS-H517Q astrocytes showed significant increase in P-gp expression and activity (Figure 8C). Similar to mutant SOD1 astrocytes, human iPS-derived FUS-H517Q astrocytes increased P-gp expression in endothelial cells through NFκB-dependent pathway (Figure 9A). However, in contrast to mutant SOD1 astrocytes, mutant FUS astrocytes did not induce significant oxidative stress or Nrf2 nuclear translocation in endothelial cells (Figures 9B, C). Rather, mutant FUS astrocytes induced an inflammatory stress response by increasing TNF-α secretion (Figure 9D) and stimulaton of TNFR-1, which in turn activated PKCβII (Figure 9E) that enhanced NFκB nuclear translocation and P-gp over expression.
Figure 8. Astrocytes derived from iPS cells of ALS patient carrying mutant FUS-H517Q enhanced expression and activity of P-gp in endothelial cells and in isolated brain and spinal cord capillaries.

Compared to iPS astrocytes derived from a control subject (iPS FUS-WT astrocytes), iPS FUS-H517Q astrocytes increased P-gp expression in iPS brain endothelial cells as measured by immunocytochemical analysis (A) and western blot analysis (B). NBD-CSA accumulation in active isolated brain and spinal cord capillaries showed higher P-gp expression and activity in capillaries treated with conditioned media (CM) from FUS-H517Q astrocytes than CM from FUS-WT astrocytes. Data are mean ± s.e.m. of three independent experiments (* p < 0.05 and ** p < 0.01). Scale bar = 50 μm.
Figure 9. FUS-H517Q astrocytes induce P-gp expression and activity in the endothelial cells by stimulating NFκB activity via an inflammatory pathway.

(A) Representative images and nuclear to cytoplasmic ratio of NFκB showed significant increase in nuclear to cytoplasmic ratio of NFκB in endothelial cells co-cultured with FUS-H517Q astrocytes. (B) Oxidative stress assessment using CellRox showed comparable level of ROS in endothelial cell co-cultured with FUS-H517Q and FUS-WT astrocytes. (C) Nrf2 nuclear translocation in endothelial cells co-cultured with FUS-H517Q astrocytes is similar to that measured in endothelial cells co-cultured with FUS-WT astrocytes. (D) TNF-α ELSIA showed higher levels of TNF-α in the basolateral media of endothelial cells co-cultured with FUS-H517Q or FUS-WT astrocytes. (E) PKCβII levels were higher in endothelial cells co-cultured with FUS-H517Q compared to endothelial cells co-cultured with FUS-WT astrocytes, while TNFR receptor levels were comparable between two co-culture systems. Data is presented as mean ± SEM of three independent experiments (* p < 0.05, ** p < 0.01, **** p < 0.0001).
DISCUSSION
Numerous studies have established that blood-CNS-barriers are damaged in ALS, a phenotype common to sporadic and familial ALS (sALS and fALS) patients (Winkler et al., 2013; Zlokovic, 2011). Similarly, transgenic mice model of ALS expressing human SOD1 mutations also display injury and alterations of the blood-spinal cord-barrier (BSCB) (Garbuzova-Davis and Sanberg, 2014; Rodrigues et al., 2012; Winkler et al., 2014). Consistent with these findings, we showed that impairment of the BSCB comprised changes to the membrane-bound drug efflux transporter, P-glycoprotein or P-gp (Jablonski et al., 2012). More specifically, we reported that there was a significant increase in activity and expression levels of P-gp in spinal cord capillaries of ALS patients as well as in the human SOD1-G93A mouse, the most studied and a well-established animal model of ALS. As ALS progressed in this mouse model from the onset of symptoms to the end-stage of disease, we found a robust upregulation of P-gp (Jablonski et al., 2012; Jablonski et al., 2014). These findings were the first to suggest the inception of a pharmacoresistance to CNS drug penetration over ALS progression. Most importantly, we showed that counteracting pharmacoresistance by inhibiting the activity of certain multidrug efflux transporters such as BCRP and P-gp could improve the outcome of ALS pharmacotherapy (Jablonski et al., 2014). In the current study we found that the increase in P-gp expression in the SOD1-G93A mouse model was cell-specific and exclusively restricted to the endothelial cells of the capillaries comprising the BSCB, as we did not observe expression of P-gp in other parenchyma cells of the spinal cord such as motor neurons, astrocytes and oligodendrocytes, even when the analysis was extended to disease end-stage. This result is consistent with prior work revealing that P-gp is expressed in brain capillaries and is undetectable in neurons and glia cells in normal CNS (Daood et al., 2008; Ronaldson et al., 2004; Volk et al., 2005). These findings are however in contrast to other studies that reported expression of P-gp in astrocytes and in selected neuronal cells (Ashraf et al., 2014). These discrepancies reported in the literature regarding CNS parenchyma localization of P-gp emphasize the many complications, possibly related to issues such as the use of a particular antibody or tissue fixation and immunostaining procedures, when studying localization of P-gp expression in the CNS (Loscher and Potschka, 2005). Several studies reported expression of P-gp to be affected in a variety of CNS disorders (Qosa et al., 2015). However, it is worth mentioning that these studies considered P-gp expression mainly in the brain, while our study is the first to assess P-gp expression in parenchyma cells of the spinal cord. Therefore, regional differences between the various CNS areas could explain the discrepancies between our results and the ones that detected P-gp in parenchymal and neuronal cells of the brain. Nevertheless, unlike endothelial cells, we showed here that expression of P-gp in CNS parenchyma cells of the spinal cord remained undetected over ALS progression.
ALS-driven increase in P-gp did not occur in systemic organs such as the liver and kidney where P-gp is also robustly expressed (Jablonski et al., 2012). Moreover, although expression of mutated SOD1 and FUS proteins is global and endothelial cells express these mutations in ALS patients, our preliminary study showed that expression of ALS mutations by endothelial cells does not affect P-gp expression (Supplementary Figure 2). We therefore reasoned that ALS specific determinants within the CNS—as opposed to more general mechanisms occurring throughout the body—were responsible for the upregulation of P-gp. Astrocytes have an important role in the viability and function of CNS by secreting a number of soluble factors that could influence nearby cells (Weber and Barros, 2015). With regards to ALS, previous studies showed astrocytes as clinically determinant contributors to ALS pathogenesis. Indeed, astrocyte pathology is observed in all cases of familial and sporadic ALS (Haidet-Phillips et al., 2011; Haidet-Phillips et al., 2014; Meyer et al., 2014). Studies showed that primary astrocytes from the SOD1-G93A mouse adversely affected the viability of both wild type and SOD1-G93A expressing motor neurons (Di Giorgio et al., 2007). Consistent with their toxic effects, astrocytes expressing SOD1 mutant protein have several defective functions such as mitochondrial bioenergetics and dynamics and impaired glutamate uptake that could contribute to the death of motor neurons (Cassina et al., 2008; Pardo et al., 2006; Tan et al., 2014). It is surprising, however, how little is known about the effect of ALS astrocytes on non-neuronal cells. For instance, even though perivascular astrocytes are known to maintain the health and regulate normal functions of the endothelial cells of the BBB/BSCB (Abbott et al., 2006), they could release inflammatory mediators under disease conditions that disrupt the very same cells (Hurst & Fritz, 1996). Therefore, it was conceivable to predict that ALS astrocytes could significantly impact the normal physiology of endothelial cells, including expression and activity of P-gp. The current study also points at astrocytes carrying fALS-linked genetic mutations as cellular determinants that could lead to deleterious effect on the BBB, gauged in this study by an increased paracellular NaF permeability. Interestingly, we observed that in spite of this increased permeability of the endothelial monolayer, the transport of LD800 compound (a P-gp substrate) was restricted and significantly decreased when endothelial cells were co-cultured with astrocytes expressing mutated SOD1 protein. Consistent with the reduction in LD800 permeability, we showed that astrocytes expressing two different ALS-linked SOD1 mutations, SOD1-G93A or SOD1-A4V, both drove upregulation of P-gp expression in endothelial cells. This effect suggests that astrocytes may drive in parallel an alternative mechanism to counteract the loss of integrity of the endothelial barrier and protect the CNS from undesirable entry of potentially harmful compounds circulating in the blood (Abbott and Friedman, 2012).
Mutant SOD1 astrocytes could impact nearby endothelial cells by secreting soluble factors. Studies done in astrocytes derived from iPSCs from ALS patients and cultured from ALS animal models have revealed a wide variety of biologically active and potentially toxic factors that could be released in disease, including inflammatory mediators (TNFα), chemokines (IL-1β), reactive oxygen species (ROS) and other yet undetermined molecules that could also adversely affect motor neurons located in proximity (Boillee et al., 2006; Clement et al., 2003; Haidet-Phillips et al., 2011; Pirooznia et al., 2014). Moreover, several in vitro, in vivo and clinical studies confirmed the extensive cross talk between astrocytes and endothelial cells of the neurovascular unit at the BBB and BSCB carried out by several astrocyte-released factors, which mediate an intricate network of signaling pathways (Abbott et al., 2006). Some of these factors directly regulate P-gp expression and activity. For instance, recent studies pointed at NFκB as a transcriptional factor that is activated by oxidative and inflammatory stressors and enhances P-gp expression (Bauer et al., 2007; Hartz et al., 2008). With respect to ALS, NFκB protein expression is increased in the spinal cords of sporadic ALS patients compared to controls, and it was reported that inhibiting NFκB in the TDP43-G348C mouse model reduced disease symptoms and prevented denervation at the neuromuscular junctions (Frakes et al., 2014; Swarup and Julien, 2011). Moreover, mutant SOD1 expressing astrocytes initiate oxidative and inflammatory stress responses that are known to ultimately activate NFκB (Cassina et al., 2008; Tortarolo et al., 2015). In line with these observations, our findings showed that upregulation in endothelial cells of P-gp expression and activity concurred with an increased cellular production of reactive oxygen species (ROS) and increased NFκB activity. Expression of mutant SOD1 proteins in iPSCs astrocytes derived from ALS patient weakens antioxidant activity of astrocytes and enhance production and release of ROS (Vehvilainen et al., 2014). These ROS affect the normal biological functions of endothelial cells directly or by increasing endothelial cells ROS cytoplasmic levels (Miller et al., 2006; Parfenova and Leffler, 2008). Moreover, astrocytes could release a wide range of toxic factors that could stimulate endothelial ROS level by increasing ROS production or interfering with ROS scavenging systems (Abbott et al., 2006; Freeman and Keller, 2012). Therefore, it is difficult to identify the exact source of ROS in endothelial cells. Overall, ROS produced in endothelial cells or released from astrocytes could be implicated in endothelial cell injury and vascular damage as well as in regulation of endothelial cell physiological function such as expression of P-gp. Functional BBB integrity can be disrupted by ROS (Freeman and Keller, 2012; Krizbai et al., 2005; Pun et al., 2009). Production of ROS and subsequent oxidative stress has been previously shown to alter BBB expression of tight junction proteins leading to increased paracellular permeability of endothelial monolayer (Kim et al., 2008; Krizbai et al., 2005). Previous study demonstrated that ROS-induced tyrosine phosphorylation of the BBB tight junctional complexes results in intracellular gap formation and increased vascular permeability (Haorah et al., 2007). In addition, alterations of the tight junctional complexes responsible for disruption of the BBB are also elicited by ROS-dependent phospholipase C activation and changes in the intracellular levels of calcium (Van der Goes et al., 2001)(Van der Goes et al. 2001). Overall, mounting evidence prove that oxidative stress contributes to endothelial dysfunction and BBB disruption by several mechanisms. In addition to ROS-mediated activation, NFκB can also be activated in response to inflammatory stress (Bauer et al., 2007). In this study we reported that NFκB activation in endothelial cells was associated with high TNFα level in the basolateral medium of endothelial cells co-cultured with astrocytes expressing mutated FUS protein. This association suggested that mutant FUS astrocytes initiated a TNFα-mediated inflammatory response in endothelial cells that resulted in NFκB activation to stimulate P-gp upregulation. It is intriguing that astrocytes in ALS could mediate P-gp upregulation by different mechanisms, through induction of specific stressors in endothelial cells. Our findings are consistent with the fact that P-gp expression and activity is regulated by several molecular signaling pathways that respond to different stimuli by different stressors.
Collectively, our results support the hypothesis of a fundamental role of astrocytes in controlling P-gp upregulation and the development of pharmacoresistance in ALS, further underscoring the critical role of this cell type in the multi-systemic nature of ALS. Additionally, our findings suggest that different ALS-causative mutations trigger distinct mechanisms that ultimately converge to increase P-gp expression.
Supplementary Material
Main Points.
ALS-linked mutant SOD1 astrocytes increase endothelial P-glycoprotein expression and activity via oxidative stress
Mutant FUS astrocytes upregulate endothelial P-glycoprotein via TNFα release
Pathways of upregulation converge on NFκB activation in endothelial cells
Acknowledgments
This work was supported by the National Institute of Health grant RO1-NS074886 (to DT), by a program grant from Target ALS at Columbia University (to DT and PP), by The Maryland Stem Cell Research Fund 2014-MSCRFE-0636 (to NJM). The Jefferson Weinberg ALS Center is also supported by the Farber Family Foundation. Yuqing Huo and Ni-Meng provided technical support for iPSC-derived astrocyte cultures and transgenic mice.
Footnotes
Conflict of interest: The authors declare no conflict of interest.
References
- Abbott NJ, Friedman A. Overview and introduction: the blood-brain barrier in health and disease. Epilepsia. 2012;53(Suppl 6):1–6. doi: 10.1111/j.1528-1167.2012.03696.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiol Dis. 2010;37:13–25. doi: 10.1016/j.nbd.2009.07.030. [DOI] [PubMed] [Google Scholar]
- Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7:41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- Ashraf T, Kao A, Bendayan R. Functional expression of drug transporters in glial cells: potential role on drug delivery to the CNS. Adv Pharmacol. 2014;71:45–111. doi: 10.1016/bs.apha.2014.06.010. [DOI] [PubMed] [Google Scholar]
- Bauer B, Hartz AM, Miller DS. Tumor necrosis factor alpha and endothelin-1 increase P-glycoprotein expression and transport activity at the blood-brain barrier. Mol Pharmacol. 2007;71:667–75. doi: 10.1124/mol.106.029512. [DOI] [PubMed] [Google Scholar]
- Benkler C, Ben-Zur T, Barhum Y, Offen D. Altered astrocytic response to activation in SOD1(G93A) mice and its implications on amyotrophic lateral sclerosis pathogenesis. Glia. 2013;61:312–26. doi: 10.1002/glia.22428. [DOI] [PubMed] [Google Scholar]
- Boillee S, Vande Velde C, Cleveland DW. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006;52:39–59. doi: 10.1016/j.neuron.2006.09.018. [DOI] [PubMed] [Google Scholar]
- Bosco DA, Lemay N, Ko HK, Zhou H, Burke C, Kwiatkowski TJ, Jr, Sapp P, McKenna-Yasek D, Brown RH, Jr, Hayward LJ. Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum Mol Genet. 2010;19:4160–75. doi: 10.1093/hmg/ddq335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulting GL, Kiskinis E, Croft GF, Amoroso MW, Oakley DH, Wainger BJ, Williams DJ, Kahler DJ, Yamaki M, Davidow L, et al. A functionally characterized test set of human induced pluripotent stem cells. Nat Biotechnol. 2011;29:279–86. doi: 10.1038/nbt.1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassina P, Cassina A, Pehar M, Castellanos R, Gandelman M, de Leon A, Robinson KM, Mason RP, Beckman JS, Barbeito L, et al. Mitochondrial dysfunction in SOD1G93A-bearing astrocytes promotes motor neuron degeneration: prevention by mitochondrial-targeted antioxidants. J Neurosci. 2008;28:4115–22. doi: 10.1523/JNEUROSCI.5308-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement AM, Nguyen MD, Roberts EA, Garcia ML, Boillee S, Rule M, McMahon AP, Doucette W, Siwek D, Ferrante RJ, et al. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science. 2003;302:113–7. doi: 10.1126/science.1086071. [DOI] [PubMed] [Google Scholar]
- Daood M, Tsai C, Ahdab-Barmada M, Watchko JF. ABC transporter (P-gp/ABCB1, MRP1/ABCC1, BCRP/ABCG2) expression in the developing human CNS. Neuropediatrics. 2008;39:211–8. doi: 10.1055/s-0028-1103272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giorgio FP, Carrasco MA, Siao MC, Maniatis T, Eggan K. Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model. Nat Neurosci. 2007;10:608–14. doi: 10.1038/nn1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraiuolo L, Higginbottom A, Heath PR, Barber S, Greenald D, Kirby J, Shaw PJ. Dysregulation of astrocyte-motoneuron cross-talk in mutant superoxide dismutase 1-related amyotrophic lateral sclerosis. Brain. 2011;134:2627–41. doi: 10.1093/brain/awr193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foran E, Bogush A, Goffredo M, Roncaglia P, Gustincich S, Pasinelli P, Trotti D. Motor neuron impairment mediated by a sumoylated fragment of the glial glutamate transporter EAAT2. Glia. 2011;59:1719–31. doi: 10.1002/glia.21218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foran E, Rosenblum L, Bogush A, Pasinelli P, Trotti D. Sumoylation of the astroglial glutamate transporter EAAT2 governs its intracellular compartmentalization. Glia. 2014;62:1241–53. doi: 10.1002/glia.22677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frakes AE, Ferraiuolo L, Haidet-Phillips AM, Schmelzer L, Braun L, Miranda CJ, Ladner KJ, Bevan AK, Foust KD, Godbout JP, et al. Microglia induce motor neuron death via the classical NF-kappaB pathway in amyotrophic lateral sclerosis. Neuron. 2014;81:1009–23. doi: 10.1016/j.neuron.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman LR, Keller JN. Oxidative stress and cerebral endothelial cells: regulation of the blood-brain-barrier and antioxidant based interventions. Biochim Biophys Acta. 2012;1822:822–9. doi: 10.1016/j.bbadis.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbuzova-Davis S, Sanberg PR. Blood-CNS Barrier Impairment in ALS patients versus an animal model. Front Cell Neurosci. 2014;8:21. doi: 10.3389/fncel.2014.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grad LI, Fernando SM, Cashman NR. From molecule to molecule and cell to cell: prion-like mechanisms in amyotrophic lateral sclerosis. Neurobiol Dis. 2015;77:257–65. doi: 10.1016/j.nbd.2015.02.009. [DOI] [PubMed] [Google Scholar]
- Haidet-Phillips AM, Hester ME, Miranda CJ, Meyer K, Braun L, Frakes A, Song S, Likhite S, Murtha MJ, Foust KD, et al. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat Biotechnol. 2011;29:824–8. doi: 10.1038/nbt.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haidet-Phillips AM, Roybon L, Gross SK, Tuteja A, Donnelly CJ, Richard JP, Ko M, Sherman A, Eggan K, Henderson CE, et al. Gene profiling of human induced pluripotent stem cell-derived astrocyte progenitors following spinal cord engraftment. Stem Cells Transl Med. 2014;3:575–85. doi: 10.5966/sctm.2013-0153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haorah J, Ramirez SH, Schall K, Smith D, Pandya R, Persidsky Y. Oxidative stress activates protein tyrosine kinase and matrix metalloproteinases leading to blood-brain barrier dysfunction. J Neurochem. 2007;101:566–76. doi: 10.1111/j.1471-4159.2006.04393.x. [DOI] [PubMed] [Google Scholar]
- Hartz AM, Bauer B, Block ML, Hong JS, Miller DS. Diesel exhaust particles induce oxidative stress, proinflammatory signaling, and P-glycoprotein up-regulation at the blood-brain barrier. FASEB J. 2008;22:2723–33. doi: 10.1096/fj.08-106997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartz AM, Bauer B, Fricker G, Miller DS. Rapid regulation of P-glycoprotein at the blood-brain barrier by endothelin-1. Mol Pharmacol. 2004;66:387–94. doi: 10.1124/mol.104.001503. [DOI] [PubMed] [Google Scholar]
- Hatzipetros T, Kidd JD, Moreno AJ, Thompson K, Gill A, Vieira FG. A Quick Phenotypic Neurological Scoring System for Evaluating Disease Progression in the SOD1-G93A Mouse Model of ALS. J Vis Exp. 2015 doi: 10.3791/53257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jablonski M, Miller DS, Pasinelli P, Trotti D. ABC transporter-driven pharmacoresistance in Amyotrophic Lateral Sclerosis. Brain Res. 2015;1607:1–14. doi: 10.1016/j.brainres.2014.08.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jablonski MR, Jacob DA, Campos C, Miller DS, Maragakis NJ, Pasinelli P, Trotti D. Selective increase of two ABC drug efflux transporters at the blood-spinal cord barrier suggests induced pharmacoresistance in ALS. Neurobiol Dis. 2012;47:194–200. doi: 10.1016/j.nbd.2012.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jablonski MR, Markandaiah SS, Jacob D, Meng NJ, Li K, Gennaro V, Lepore AC, Trotti D, Pasinelli P. Inhibiting drug efflux transporters improves efficacy of ALS therapeutics. Ann Clin Transl Neurol. 2014;1:996–1005. doi: 10.1002/acn3.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SR, Bae YH, Bae SK, Choi KS, Yoon KH, Koo TH, Jang HO, Yun I, Kim KW, Kwon YG, et al. Visfatin enhances ICAM-1 and VCAM-1 expression through ROS-dependent NF-kappaB activation in endothelial cells. Biochim Biophys Acta. 2008;1783:886–95. doi: 10.1016/j.bbamcr.2008.01.004. [DOI] [PubMed] [Google Scholar]
- Krizbai IA, Bauer H, Bresgen N, Eckl PM, Farkas A, Szatmari E, Traweger A, Wejksza K, Bauer HC. Effect of oxidative stress on the junctional proteins of cultured cerebral endothelial cells. Cell Mol Neurobiol. 2005;25:129–39. doi: 10.1007/s10571-004-1378-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippmann ES, Azarin SM, Kay JE, Nessler RA, Wilson HK, Al-Ahmad A, Palecek SP, Shusta EV. Derivation of blood-brain barrier endothelial cells from human pluripotent stem cells. Nat Biotechnol. 2012;30:783–91. doi: 10.1038/nbt.2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loscher W, Potschka H. Blood-brain barrier active efflux transporters: ATP-binding cassette gene family. NeuroRx. 2005;2:86–98. doi: 10.1602/neurorx.2.1.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer K, Ferraiuolo L, Miranda CJ, Likhite S, McElroy S, Renusch S, Ditsworth D, Lagier-Tourenne C, Smith RA, Ravits J, et al. Direct conversion of patient fibroblasts demonstrates non-cell autonomous toxicity of astrocytes to motor neurons in familial and sporadic ALS. Proc Natl Acad Sci U S A. 2014;111:829–32. doi: 10.1073/pnas.1314085111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milane A, Fernandez C, Vautier S, Bensimon G, Meininger V, Farinotti R. Minocycline and riluzole brain disposition: interactions with p-glycoprotein at the blood-brain barrier. J Neurochem. 2007;103:164–73. doi: 10.1111/j.1471-4159.2007.04772.x. [DOI] [PubMed] [Google Scholar]
- Miller AA, Drummond GR, Sobey CG. Novel isoforms of NADPH-oxidase in cerebral vascular control. Pharmacol Ther. 2006;111:928–48. doi: 10.1016/j.pharmthera.2006.02.005. [DOI] [PubMed] [Google Scholar]
- Miller DS. Regulation of ABC transporters at the blood-brain barrier. Clin Pharmacol Ther. 2015a;97:395–403. doi: 10.1002/cpt.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DS. Regulation of ABC transporters blood-brain barrier: the good, the bad, and the ugly. Adv Cancer Res. 2015b;125:43–70. doi: 10.1016/bs.acr.2014.10.002. [DOI] [PubMed] [Google Scholar]
- Noursadeghi M, Tsang J, Haustein T, Miller RF, Chain BM, Katz DR. Quantitative imaging assay for NF-kappaB nuclear translocation in primary human macrophages. J Immunol Methods. 2008;329:194–200. doi: 10.1016/j.jim.2007.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- On NH, Chen F, Hinton M, Miller DW. Assessment of P-glycoprotein activity in the Blood-Brain Barrier (BBB) using Near Infrared Fluorescence (NIRF) imaging techniques. Pharm Res. 2011;28:2505–15. doi: 10.1007/s11095-011-0478-6. [DOI] [PubMed] [Google Scholar]
- Pardo AC, Wong V, Benson LM, Dykes M, Tanaka K, Rothstein JD, Maragakis NJ. Loss of the astrocyte glutamate transporter GLT1 modifies disease in SOD1(G93A) mice. Exp Neurol. 2006;201:120–30. doi: 10.1016/j.expneurol.2006.03.028. [DOI] [PubMed] [Google Scholar]
- Parfenova H, Leffler CW. Cerebroprotective functions of HO-2. Curr Pharm Des. 2008;14:443–53. doi: 10.2174/138161208783597380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirooznia SK, Dawson VL, Dawson TM. Motor neuron death in ALS: programmed by astrocytes? Neuron. 2014;81:961–3. doi: 10.1016/j.neuron.2014.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt AJ, Getzoff ED, Perry JJ. Amyotrophic lateral sclerosis: update and new developments. Degener Neurol Neuromuscul Dis. 2012;2012:1–14. doi: 10.2147/DNND.S19803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pun PB, Lu J, Moochhala S. Involvement of ROS in BBB dysfunction. Free Radic Res. 2009;43:348–64. doi: 10.1080/10715760902751902. [DOI] [PubMed] [Google Scholar]
- Qosa H, Miller DS, Pasinelli P, Trotti D. Regulation of ABC efflux transporters at blood-brain barrier in health and neurological disorders. Brain Res. 2015 doi: 10.1016/j.brainres.2015.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radford RA, Morsch M, Rayner SL, Cole NJ, Pountney DL, Chung RS. The established and emerging roles of astrocytes and microglia in amyotrophic lateral sclerosis and frontotemporal dementia. Front Cell Neurosci. 2015;9:414. doi: 10.3389/fncel.2015.00414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues MC, Hernandez-Ontiveros DG, Louis MK, Willing AE, Borlongan CV, Sanberg PR, Voltarelli JC, Garbuzova-Davis S. Neurovascular aspects of amyotrophic lateral sclerosis. Int Rev Neurobiol. 2012;102:91–106. doi: 10.1016/B978-0-12-386986-9.00004-1. [DOI] [PubMed] [Google Scholar]
- Ronaldson PT, Bendayan M, Gingras D, Piquette-Miller M, Bendayan R. Cellular localization and functional expression of P-glycoprotein in rat astrocyte cultures. J Neurochem. 2004;89:788–800. doi: 10.1111/j.1471-4159.2004.02417.x. [DOI] [PubMed] [Google Scholar]
- Swarup V, Julien JP. ALS pathogenesis: recent insights from genetics and mouse models. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35:363–9. doi: 10.1016/j.pnpbp.2010.08.006. [DOI] [PubMed] [Google Scholar]
- Tan W, Pasinelli P, Trotti D. Role of mitochondria in mutant SOD1 linked amyotrophic lateral sclerosis. Biochim Biophys Acta. 2014;1842:1295–301. doi: 10.1016/j.bbadis.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tortarolo M, Vallarola A, Lidonnici D, Battaglia E, Gensano F, Spaltro G, Fiordaliso F, Corbelli A, Garetto S, Martini E, et al. Lack of TNF-alpha receptor type 2 protects motor neurons in a cellular model of amyotrophic lateral sclerosis and in mutant SOD1 mice but does not affect disease progression. J Neurochem. 2015;135:109–24. doi: 10.1111/jnc.13154. [DOI] [PubMed] [Google Scholar]
- Van der Goes A, Wouters D, Van Der Pol SM, Huizinga R, Ronken E, Adamson P, Greenwood J, Dijkstra CD, De Vries HE. Reactive oxygen species enhance the migration of monocytes across the blood-brain barrier in vitro. FASEB J. 2001;15:1852–4. doi: 10.1096/fj.00-0881fje. [DOI] [PubMed] [Google Scholar]
- Vehvilainen P, Koistinaho J, Gundars G. Mechanisms of mutant SOD1 induced mitochondrial toxicity in amyotrophic lateral sclerosis. Front Cell Neurosci. 2014;8:126. doi: 10.3389/fncel.2014.00126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volk H, Potschka H, Loscher W. Immunohistochemical localization of P-glycoprotein in rat brain and detection of its increased expression by seizures are sensitive to fixation and staining variables. J Histochem Cytochem. 2005;53:517–31. doi: 10.1369/jhc.4A6451.2005. [DOI] [PubMed] [Google Scholar]
- Wang X, Campos CR, Peart JC, Smith LK, Boni JL, Cannon RE, Miller DS. Nrf2 upregulates ATP binding cassette transporter expression and activity at the blood-brain and blood-spinal cord barriers. J Neurosci. 2014;34:8585–93. doi: 10.1523/JNEUROSCI.2935-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber B, Barros LF. The Astrocyte: Powerhouse and Recycling Center. Cold Spring Harb Perspect Biol. 2015:7. doi: 10.1101/cshperspect.a020396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler EA, Sengillo JD, Sagare AP, Zhao Z, Ma Q, Zuniga E, Wang Y, Zhong Z, Sullivan JS, Griffin JH, et al. Blood-spinal cord barrier disruption contributes to early motor-neuron degeneration in ALS-model mice. Proc Natl Acad Sci U S A. 2014;111:E1035–42. doi: 10.1073/pnas.1401595111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler EA, Sengillo JD, Sullivan JS, Henkel JS, Appel SH, Zlokovic BV. Blood-spinal cord barrier breakdown and pericyte reductions in amyotrophic lateral sclerosis. Acta Neuropathol. 2013;125:111–20. doi: 10.1007/s00401-012-1039-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong AD, Ye M, Levy AF, Rothstein JD, Bergles DE, Searson PC. The blood-brain barrier: an engineering perspective. Front Neuroeng. 2013;6:7. doi: 10.3389/fneng.2013.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat Rev Neurosci. 2011;12:723–38. doi: 10.1038/nrn3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.