

Abstract
Importance
Sarcomere mutations and left ventricular hypertrophy (LVH) are cardinal features of hypertrophic cardiomyopathy (HCM). However, little is known about the full spectrum of phenotypic manifestations, or how LVH impacts disease expression.
Objective
Genotyped individuals with or at risk for HCM were studied to: (1) characterize sarcomere mutation carriers (G+), including assessing collective phenotypic burden; and (2) investigate the relationship between left ventricular wall thickness (LVWT) and other disease features.
Design
Cross-sectional multicenter observational study
Setting
Network of hypertrophic cardiomyopathy specialty clinical centers (HCMNet).
Participants
Mutation carriers with LVH (G+/LVH+), without LVH (G+/LVH-), and healthy related controls (G-/LVH-) were enrolled through HCMNet sites. A total of 193 participants were enrolled and underwent study procedures. Analyses were performed on 178 participants (G+/LVH+ n=81, G+/LVH- n=55, G-/LVH- n=42) with mean age 26.9, 20.4 and 17.5 years, respectively.
Exposure
The primary stratifying variables were the presence of a sarcomere mutation and measures of LV wall thickness.
Main Outcomes and Measures
Parameters from standardized exercise testing, echocardiography, cardiac magnetic resonance, serum biomarker measurement, and electrocardiography (ECG) were compared across study cohorts.
Results
G+/LVH- subjects had smaller LV cavity size and higher LV wall thickness (LVWT):diastolic diameter ratio and LV ejection fraction than controls. Phenotypic burden was determined as the cumulative number of 7 traits (ECG changes; decreased LV systolic, diastolic diameter, or septal E′; higher LVWT:diameter ratio; serum troponin, natriuretic peptide levels) in each individual. Mean burden was 4.9 in G+/LVH+, 2.4 in G+/LVH-, and 1.3 in controls (p<0.001). Classification and regression tree analysis identified an LVEDD z-score < -1.85 or the combination of LVEDD z-score ≥ -1.85 and Septal E′ z-score < -0.52 as having 74% accuracy in discriminating G+/LVH- subjects from controls. In mutation carriers, these traits and other parameters demonstrated a continuous relationship with LVWT; generally without clear cutoff signifying pathologic transition.
Conclusions and Relevance
G+/LVH- individuals demonstrate altered cardiac dimensions and function and higher burden of early phenotypes. Two methods discriminated phenotypic subgroups – a sum across seven criteria and a tree-based rule that identifies constellations of distinguishing factors. Greater LVWT is associated with more prominent cardiac abnormalities in a continuous, although not always linear manner. A single value of LVWT could not dichotomize the presence or absence of disease.
Introduction
Hypertrophic cardiomyopathy (HCM) is caused by mutations in sarcomere genes. Clinically, left ventricular hypertrophy (LVH) has been the focal point for diagnosing and describing disease. In addition to being the most obvious phenotype, LVH carries prognostic importance. Symptoms and adverse clinical sequelae are exceedingly rare in sarcomere mutation carriers with normal left ventricular wall thickness (LVWT). However, LVH is not specific for HCM and does not fully encompass the entire range of disease. Wall thickness is usually normal in early childhood, and may remain so for decades despite carrying a pathogenic sarcomere mutation1,2. Moreover, other conditions such as infiltrative and metabolic cardiomyopathies, hypertension and athletic remodeling, can mimic the crude phenotype of HCM. These distinct clinical entities have fundamentally different biology and therefore divergent management strategies, prognosis, and implications to family members.
Mutation carriers (genotype-positive, G+) without LVH (LVH-) are referred to as having “preclinical” or “subclinical” HCM. Studies on G+/LVH- cohorts have demonstrated that myocardial function and biochemistry are perturbed in the absence of LVH. Impaired LV relaxation3-5, increased left ventricular ejection fraction (LVEF)3,4, altered myocardial energetics6, electrocardiographic (ECG) abnormalities7, increased mitral valve leaflet length8,9, myocardial crypts by cardiac magnetic resonance (CMR) imaging 9-12, and evidence of a pro-fibrotic state 13,14 may be present in mutation carriers when LVWT is normal. However, very little is currently known about the full spectrum of phenotypic manifestations in mutation carriers.
A collaborative network of HCM specialty centers (HCMNet) was established to advance understanding of disease pathogenesis and foster development of novel disease-modifying therapies. To better characterize the phenotypic spectrum of sarcomere mutations, beyond LVH, rigorous and standardized evaluations were performed on a diverse genotyped population. Rather than focusing on individual traits, cumulative phenotypic burden was also assessed as a potentially more accurate reflection of disease expression. Similarly, recognizing the limitations of diagnosing HCM based on a single relatively arbitrary threshold for pathological LVH, the impact of greater LVWT on sarcomere mutation carriers was analyzed treating LVWT as a continuous rather than a binary variable.
Methods
Study Design and Participants
A cross-sectional, multicenter observational study was performed from 2009 to 2011 to evaluate sarcomere mutation carriers with clinically overt HCM (G+/LVH+), mutation carriers with normal maximal LVWT (G+/LVH-), and healthy relatives who do not carry the family's mutation (G-/LVH- controls). LVH was based on echocardiographic core laboratory measurements and defined as a maximal LVWT ≥12 mm in adults or a z-score ≥ 3 in participants <18 years of age. These criteria were chosen rather than those standardly used to diagnose HCM clinically to avoid including mutation carriers with borderline LVH and potentially emerging or mild HCM in the G+/LVH- group.
Inclusion Criteria
Likely pathogenic or pathogenic sarcomere mutation, OR healthy genotype-negative relative
Age > 5 years old
Exclusion Criteria
Hypertension (systolic >140 and/or diastolic pressure>90 mmHg, or on treatment)
Coronary artery disease
>Mild intrinsic valvular heart disease
Congenital heart disease
Prior invasive septal reduction
Medical conditions associated with increased collagen turnover
Pregnant/lactating
Age >40 years for G+/LVH- and controls
Participants had full sequencing of at least the 8 sarcomere genes definitively associated with HCM (MYH7, MYBPC3, TNNT2, TNNI3, MYL2, MYL3, TPM1, ACTC). Variants were classified using standard criteria accounting for segregation, conservation, literature review, review of publicly available databases (including ClinVar15) and very low frequency in appropriate control populations16,17 (Exome Aggregation Consortium (ExAC), Cambridge, MA (URL: http://exac.broadinstitute.org) [Accessed September 2015]). Mutations were required to be classified as likely pathogenic or pathogenic at enrollment. Prior to data analysis, genotypes were reviewed and only participants with variants fulfilling current criteria as pathogenic or likely pathogenic were included.
Participating sites included 11 HCM specialty centers in the United States (Supplemental Table 1). The institutional review boards at all participating sites approved the study protocol. All participants provided written informed consent, or parental consent/youth assent.
Study Procedures
Participants underwent history and physical examination, ECG, standardized echocardiography, and exercise testing using a symptom-limited standard Bruce treadmill exercise protocol. Standardized CMR imaging was performed when possible based on the presence of intracardiac devices, local IRB regulations, and participant/parental assent/consent. Blood was drawn for serum biomarker assessment.
Echocardiographic Analysis
Standard two-dimensional images, spectral and color Doppler, and tissue Doppler interrogation were obtained following a standardized protocol. Measurements were determined from the mean of 3 cardiac cycles in accordance with guidelines of the American Society of Echocardiography18,19. Further details are available in the Supplemental Methods. All echocardiographic studies were analyzed offline by the echocardiographic core laboratory (The Johns Hopkins Echocardiography Research Laboratory), blinded to genotype status.
CMR Analysis
Images were acquired utilizing ECG-gating and breath-holding following a standardized protocol consisting of cine steady-state free precession (SSFP) imaging for LV function and mass20. Late gadolinium enhancement (LGE) imaging was used to detect focal myocardial fibrosis20. LGE was quantified using a semi-automated gray-scale threshold technique with manual correction21,22. Further details are available in the Supplemental Methods. The CMR core laboratory (Radiology and Imaging Sciences Department of the National Institutes of Health Clinical Center) performed offline analyses blinded to genotype.
Electrocardiography and Exercise Testing
Standard 12-lead ECGs were obtained at rest in the supine position. Symptom-limited treadmill exercise tolerance testing was performed utilizing a standard Bruce protocol. Q waves were considered abnormal if present in at least 2 contiguous leads >1/3 the height of the R wave and >30 ms duration). ST changes consisted of ST segment elevation or ST segment depression present in at least 2 contiguous leads with depth greater than 0.1 mV if up-sloping, or greater than 0.05 mV if horizontal or down-sloping)
Serum Biomarkers Analysis
Serum and K3-EDTA anticoagulated plasma were collected at enrollment. Samples were processed within 60 minutes of phlebotomy and stored at -80°C prior to analysis. Plasma concentrations of the N-terminal pro-B-type natriuretic peptide (NT-pro BNP) and cardiac troponin I (cTnI) were measured in the biomarker laboratory blinded to genotype status using proBNPII (Roche Diagnostics Corporation, Indianapolis, IN) and ultrasensitive single-molecule Erenna cTnI (Singulex, Alameda, CA) assays, respectively. Results of biomarker analyses in this study will be presented in a separate report.
Statistical Analysis
Due to the wide age range of participants, echocardiographic measurements were converted to z-scores (number of standard deviations from the mean) prior to analysis23,24. Normally distributed data were summarized with means and standard deviations, and categorical variables were summarized with frequencies and percentages.
Generalized estimating equations (GEEs) examined differences in echocardiographic and CMR measures, adjusting for age, gender, and correlation within family, assuming an exchangeable correlation structure. Values are expressed as adjusted means ± standard error (SE). Analysis of variance (ANOVA) was used to compare means across the three groups (G+/LVH+, G+/LVH-, G-/LVH- controls), and post-hoc Bonferroni-corrected P values < 0.017 (0.05/3 comparison groups) were considered statistically significant. Further details are available in the Supplemental Methods. Complete case analysis (listwise deletion) was used. Subjects with missing data for the variables included in the model were excluded.
The 7 components of the phenotypic burden score were chosen based on: (i) significant differences between G+/LVH+ and G+/LVH- subjects in this cohort (echocardiographic LV end-systolic and end-diastolic dimensions, and LV thickness:diameter ratio; serum troponin and NTproBNP levels); or (ii) previous reports indicating the parameter discriminated G+/LVH- from controls (septal E′ velocity3-5; presence of Q waves or ST changes on ECG7). For septal E′ velocity, LVESD and LVEDD, a z-score <-1.5 was chosen as a clinically relevant cutoff. For LV thickness:diameter ratio and serum biomarkers, the optimal threshold was identified by testing candidate cut-offs and choosing the one with the smallest p-value in a chi-square test comparing the three groups in a covariate-adjusted logistic regression model (LV thickness:diameter ratio >0.23; NTproBNP level >100 pg/ml; cardiac troponin I level >3.0 pg/ml).
Classification and regression tree (CART) analysis was performed to identify subgroups that were most likely to be G+/LVH- versus control subjects to assist in the discrimination of these cohorts. The input candidate variables to the CART were continuous (where applicable) versions of the 7 measures used in the burden score. Further details are available in the Supplemental Methods.
In mutation carriers, the relationships between maximal LVWT and clinical parameters were examined using GEE models with these parameters as the outcome (Y) and maximal LVWT z-score as the predictor (X), adjusted for age, gender, and within-family correlation. Linear spline analysis was performed to identify physiologically relevant thresholds of maximal LVWT. Knots for the splines were identified by testing candidates at small increments and choosing the knot that produced the lowest Akaike Information Criterion (AIC).
All analyses were conducted using the Statistical Analysis System version 9.3 (Statistical Analysis System, SAS Institute, Inc., Cary, NC).
Results
Study Population
A total of 193 participants underwent study procedures. At the time of data analysis, the sarcomere variant present in 13 G+ participants was reclassified from likely pathogenic to uncertain significance. Accordingly these participants and 2 participating G-/LVH- relatives were excluded. Analyses were therefore performed on 178 participants with an average age of 23 ± 12 years (range 5-60). Basic demographic and genetic characteristics are summarized in Table 1. Study participants were generally asymptomatic (95% of G+/LVH+ were NYHA I-II; all G+/LVH- and controls were NYHA class I). As typical, the majority (>86%) of mutations were present in MYH7 and MYBPC3. Supplemental Table 2 provides the full listing of genetic variants.
Table 1. Clinical Characteristics and Genetic Background of Participants by Study Group.
| G+/LVH+(n=81) | G+/LVH-(n=55) | Control (n=42) | 3- Group P-Value | G+/LVH+ vs G+/LVH-P-value | G+/LVH+ vs Control P-value | G+/LVH- vs Control P-value | |
|---|---|---|---|---|---|---|---|
| Age at baseline, yrs | 26.9 (14) | 20.4 (10) | 17.5 (8) | < 0.001 | 0.003 | <0.0001 | 0.12 |
| Female, n (%) | 28 (35%) | 30 (55%) | 24 (57%) | 0.019 | 0.02 | 0.02 | 0.80 |
| Race, n (%) | 0.122 | 0.11 | 0.42 | 0.08 | |||
| White | 75 (93%) | 55 (100%) | 39 (93%) | ||||
| Black or African American | 5 (6%) | 0 (0%) | 1 (2%) | ||||
| Asian | 1 (1%) | 0 (0%) | 1 (2%) | ||||
| More than one race | 0 (0%) | 0 (0%) | 1 (2%) | ||||
| Body Surface Area (kg/m2) | 1.83 (0.41) | 1.69 (0.32) | 1.61 (0.40) | 0.006 | 0.02 | 0.005 | 0.33 |
| Height, cm | 168.5 (17.8) | 162.2 (14.4) | 159.6 (18.0) | 0.012 | 0.03 | 0.01 | 0.42 |
| Weight, kg | 72.3 (24.1) | 63.2 (18.3) | 59.5 (22.5) | 0.005 | 0.01 | 0.005 | 0.38 |
| Systolic blood pressure, mmHg | 115 (15) | 113 (12) | 113 (13) | 0.724 | 0.50 | 0.54 | 0.99 |
| Diastolic blood pressure, mmHg | 68 (9) | 68 (9) | 66 (10) | 0.510 | 0.80 | 0.34 | 0.28 |
| NYHA Class, n(%) | |||||||
| Class I | 67 (83%) | 55 (100%) | 42 (100%) | ||||
| Class II | 10 (12%) | 0 (0%) | 0 (0%) | ||||
| Class III | 2 (2%) | 0 (0%) | 0 (0%) | ||||
| Class IV | 2 (2%) | 0 (0%) | 0 (0%) | ||||
| Medication Use, n (%) | |||||||
| Amiodarone | 3 (4%) | 0 (0%) | 0 (0%) | ||||
| β-blockers | 28 (35%) | 1 (2%) | 0 (0%) | ||||
| Calcium channel blockers | 9 (11%) | 1 (2%) | 0 (0%) | ||||
| Disopyramide | 1 (1%) | 0 (0%) | 0 (0%) | ||||
| Coumadin/warfarin | 3 (4%) | 0 (0%) | 0 (0%) | ||||
| No medications | 32 (40%) | 34 (62%) | 32 (76%) | ||||
|
| |||||||
| Genetic Background, n (%) | |||||||
| MYH7 | 26 (32%)a | 22 (40%) | |||||
| MYBPC3 | 44 (54%)b | 29 (53%)e | |||||
| TNNT2 | 7 (9%)c | 1 (2%) | |||||
| TNNI3 | 1 (1%) | 0 | |||||
| MYL2 | 1 (1%) | 0 | |||||
| MYL3 | 2 (3%)d | 2 (3%) | |||||
| ACTC | 0 | 1 (2%) | |||||
Values are reported as mean (SD) or n (%)
P values <0.017 are considered statistically significant, applying post hoc Bonferroni correction.
One patient also had a secondary MYH7 mutation
One patient also had a TNNI3 mutation and one had a secondary MYBPC3 mutation
One patient also had a secondary MYBPC3 mutation
Both patients also had a secondary MYBPC3 mutation
One patient also had a secondary MYBPC3 mutation
Results of Cardiac Imaging Studies
Cardiac imaging findings are summarized in Table 2. Mutation carriers, with and without LVH, had smaller LV cavity size (echocardiographic LV systolic and diastolic diameter; CMR LV volumes), greater LVWT:diameter ratio, and higher echocardiographic LVEF compared with controls (p<0.017 for all comparisons). G+/LVH+ participants additionally had lower mean tissue Doppler early diastolic (E′) velocity z-scores and higher E/ E′ ratios compared with both G+/LVH- and control participants.
Table 2. Cardiac Imaging Findings.
| Echocardiographic Measure | G+/LVH+N=81 | G+/LVH-N=55 | Control N=42 | G+/LVH+ vs G+/LVH- p-value | G+/LVH+ vs Control p-value | G+/LVH- vs Control p-value |
|---|---|---|---|---|---|---|
| LVEDD z-score | -2.7 (0.2) | -1.8 (0.1) | -1.1 (0.2) | 0.001 | <0.001 | 0.007 |
| LVESD z-score | -2.5 (0.2) | -1.6 (0.2) | -0.6 (0.2) | 0.004 | <0.001 | 0.001 |
| IVS z-score | 6.2 (0.6) | 0.7 (0.2) | 0.4 (0.2) | <0.001 | <0.001 | 0.15 |
| PW z-score | 1.3 (0.3) | -0.5 (0.2) | -0.5 (0.3) | <0.001 | <0.001 | 0.80 |
| Maximal LV wall thickness z-score | 9.6 (0.8) | 1.5 (0.2) | 1.3 (0.3) | <0.001 | <0.001 | 0.18 |
| LV Thickness: Diameter Ratio | 0.41 (0.02) | 0.21 (0.01) | 0.20 (0.01) | <0.001 | <0.001 | 0.001 |
| Left atrial diameter z-score | -0.8 (0.2) | -1.4 (0.1) | -1.6 (0.1) | <0.001 | <0.001 | 0.058 |
| LVEF, % | 66.7 (1.1) | 66.1 (1.1) | 62.1 (1.1) | 0.97 | 0.02 | 0.015 |
| Peak LVOT gradient, mmHg | 10.3 (0.8) | 7.8 (0.4) | 8.0 (0.4) | 0.002 | <0.001 | 0.75 |
| Subjects with peak gradient >30 mmHg (%, n/N) | 5 (4/78) | 0 (0/55) | 0 (0/42) | |||
| Septal E′ z-score | -1.5 (0.2) | -0.3 (0.1) | 0.01 (0.2) | <0.001 | <0.001 | 0.21 |
| Lateral E′ z-score | -1.2 (0.2) | -0.4 (0.1) | -0.5 (0.2) | <0.001 | 0.01 | 0.58 |
| Septal S′ z-score | 0.2 (0.2) | 0.7 (0.2) | 0.4 (0.2) | 0.15 | 0.91 | 0.15 |
| Lateral S′ z-score | -0.3 (0.2) | 0.1 (0.1) | -0.1 (0.2) | 0.07 | 0.76 | 0.16 |
| E/ E′ Septal | 9.7 (0.3) | 7.8 (0.3) | 7.4 (0.3) | <0.001 | <0.001 | 0.45 |
| Lateral E/ E′ | 6.8 (0.3) | 5.8 (0.2) | 6.0 (0.3) | 0.001 | 0.013 | 0.51 |
| CMR Measurea | N=42 | N=34 | N=23 | |||
| Age, years (SD) | 29.9 (13) | 21.4 (8) | 21.6 (7) | |||
| Female, n (%) | 13 (31%) | 16 (47%) | 15 (65%) | |||
| LVEF, % | 67.8 (1.1) | 60.5 (2.2) | 58.3 (1.2) | 0.006 | <0.001 | 0.38 |
| LV Mass, g | 158 (5.8) | 115 (4.0) | 121 (3.7) | <0.001 | <0.001 | 0.24 |
| LVEDV, ml | 138 (5.1) | 130 (3.5) | 146 (4.1) | 0.17 | 0.25 | <0.001 |
| LVESV, ml | 45.2 (2.4) | 48.9 (2.0) | 60.8 (2.4) | 0.23 | <0.001 | <0.001 |
| Maximal LV Wall Thickness, mm | 15.4 (.77) | 10.1 (.35) | 9.9 (.31) | <0.001 | <0.001 | 0.64 |
| LGE present (%Yes)b | 43% (18/41) | 0% (0/31) | 0% (0/23) | <0.001 | 0.01 | 0.99 |
| Mass of LGE, g | 12.0 (14) | NA | NA | |||
| LGE, % LV mass | 5.9 (8) | NA | NA | |||
LV, left ventricular; LVEDD, left ventricular end-diastolic dimension; LVESD, left ventricular end-systolic dimension; IVS, interventricular septum thickness; PW, posterior wall thickness; LVOT, left ventricular outflow tract; LVEF, left ventricular ejection fraction; E′, early diastolic myocardial relaxation velocity; S′, systolic myocardial velocity; E, mitral inflow E wave velocity; CMR, cardiac magnetic resonance; SD, standard deviation; LVEDV, left ventricular end-diastolic volume; LVESV, left ventricular end-systolic volume; LGE, late gadolinium enhancement
Results are shown as means and standard error (unless otherwise specified), adjusted for family correlation, gender, and age. Echo parameters additionally adjusted for the manufacturer of the echocardiographic equipment used.
CMR dimensions additionally adjusted for age-squared and BSA;
Adjusted by exact logistic regression for age and gender, but not family.
P values <0.017 are considered statistically significant, applying post hoc Bonferroni correction.
Ninety-nine participants (56% of the cohort) underwent CMR (42 G+/LVH+, 34 G+/LVH-, 23 Controls). Based on pediatric IRB guidelines, CMR studies were not performed on G-/LVH- and most G+/LVH- children. LGE was present in 44% (18/41) of G+/LVH+ participants but absent from all G+/LVH- and control participants. Older age was significantly related to the presence but not extent of LGE (mean age 35 (range 13-56) years for the 18 overt HCM patients with LGE versus 26 (range 10-54) years for the 23 patients without LGE; p=0.02).
Results from Exercise Testing and Electrocardiography
Results from exercise testing and ECG analysis are summarized in Supplemental Tables 3 and 4. Effort tolerance was preserved, with a mean exercise duration >11 minutes using a standard Bruce protocol. Approximately 65% of G+/LVH- and control participants had normal ECG tracings, compared with 39% of G+/LVH+ participants. No exercise or ECG metrics discriminated G+/LVH- from controls.
Burden of Early Phenotypes of Sarcomere Mutations
A composite measure including 7 parameters (LV thickness:diameter ratio > 0.23,; LVEDD z-score < -1.5; LVESD z-score < -1.5; septal E′ z-score < -1.5 serum high sensitivity troponin I level > 3.0 pg/ml; NTproBNP level > 100 pg/ml; and; Q waves and/or ST changes on ECG) was created to score the number of early manifestations present in each participant. Each parameter was more prevalent in sarcomere mutation carriers compared to mutation-negative controls (Table 3).
Table 3. Prevalence of Early Phenotypic Abnormalities in Mutation Carriers and Controls.
| G+/LVH+(n=81) | G+/LVH-(N=55) | Controls (n=42) | G+/LVH+ vs G+/LVH- P-value | G+/LVH+ vs Control P-value | G+/LVH- vs Control P-value | |
|---|---|---|---|---|---|---|
| LV Thickness:Diameter Ratio > 0.23 | 99% | 35% | 12% | < 0.001 | < 0.001 | 0.04 |
| LVEDD z-score < -1.5 | 88% | 68% | 37% | 0.002 | < 0.001 | 0.009 |
| Singulex Troponin I >3.0 pg/ml | 73% | 40% | 20% | 0.006 | < 0.001 | 0.05 |
| LVESD z-score < -1.5 | 72% | 57% | 30% | 0.09 | < 0.001 | 0.004 |
| NTproBNP>100 pg/ml | 58% | 14% | 8% | < 0.001 | < 0.001 | 0.45 |
| QST Present on ECG | 46% | 14% | 3% | < 0.001 | < 0.001 | 0.09 |
| Septal E′ z-score < -1.5 | 42% | 13% | 5% | < 0.001 | 0.002 | 0.17 |
QST, ECG Q waves or ST changes; LVEDD, left ventricular end-diastolic dimension; LVESD, left ventricular end-systolic dimension; LV, left ventricular
P values and percentages were adjusted for patient age, sex, and familial relationships.
P values <0.017 are considered statistically significant, applying post hoc Bonferroni correction.
Phenotypic burden was scored as the total number of early phenotypes in each participant, ranging from 0 to 7. There was a significant stepwise increase in burden, comparing the 3 cohorts (p<0.001 for all comparisons; Figure 1). Burden was highest in G+/LVH+ participants, with a mean of 4.9 phenotypes/individual. No G+/LVH+ participants had 0 abnormalities and 76% had 4 or more abnormalities, including 21% with all 7 traits. G+/LVH- participants had an intermediate burden score with a mean of 2.4 phenotypes/individual; 12% had 0 abnormalities. Control participants had the lowest burden score with a mean of 1.3 phenotypes/individual; 34% had 0 abnormalities. A burden score of ≥3 had 47% sensitivity and 88% specificity in distinguishing G+/LVH- from controls. Notably, no significant difference in burden scores were identified when comparing MYH7 and MYBPC3 mutation carriers (mean 3.8 vs. 3.9), including separate analyses restricted to G+/LVH+ and G+/LVH- subjects (mean 2.5 vs. 2.3).
Figure 1. Burden of Early Phenotypic Abnormalities in Sarcomere Mutation Carriers Compared to Controls.

Sarcomere mutation carriers had a higher number of early phenotypic abnormalities compared to healthy related controls. Early phenotypic burden was highest in participants with overt HCM, intermediate in preclinical HCM and lowest in controls. The adjusted mean number of early phenotypes was 4.9 ± 0.2 for G+/LVH+, 2.4 ± 0.2 for G+/LVH-, and 1.3 ± 0.2 for controls (p<0.001).
Early Phenotypes: QST on ECG; z-score <-1.5 for Septal E′, LVEDD, LVESD; serum NTproBNP>100 pg/ml; serum cardiac troponin I >3.0 pg/ml; LV thickness:dimension ratio >0.23
Discriminating G+/LVH- and Control Participants
The 7 traits comprising the phenotypic burden score were also evaluated with classification and regression tree (CART) analysis with the goal of discriminating G+/LVH- and control participants without a requirement of prespecified threshold values as input. The model that maximized sensitivity is shown in Supplemental Figure 2 and indicates that using a combination of LVEDD z-score and septal E′ z-score to identify subgroups has a sensitivity of 76% and specificity of 71% in identifying mutation carriers and true non-carriers. Specifically, an LVEDD z-score < -1.85 or the combination of LVEDD z-score ≥ -1.85 and Septal E′ z-score < -0.52 had 74% accuracy in discriminating G+/LVH- subjects from controls.
Increasing Maximal LV wall Thickness and Disease Manifestations in Mutation Carriers
Rather than dichotomizing mutation carriers as LVH+ or LVH, linear regression and spline analysis were used to characterize how phenotypic manifestations and burden change with increasing LVWT, analyzed as a continuous variable. Analyses were restricted to mutation carriers.
Linear regression is shown in Supplemental Table 5. With the exception of the extent of LGE and exercise duration and hemodynamic response, nearly all metrics of LV structure, function, and ECG manifestations become significantly more abnormal, and the overall burden of early phenotypic traits increased with greater maximal LVWT z-score.
Spline analysis was performed to identify physiologically relevant inflection points of maximal LVWT z-score. Most measures had significant linear associations across the continuum of maximal LVWT, without obvious knots. However, inflection points were identified for septal E′, exercise capacity (peak METs), and phenotypic burden (Figure 2). Each of these metrics initially became more abnormal as LVWT increased, but when maximal LVWT z-scores exceeded 10, 5.5, and 8, respectively (corresponding with an absolute LV wall thickness in the average adult of ∼18, 13.5, and 16 mm, respectively), the relationship leveled off. These metrics no longer worsened with increasing maximal LVWT z-score.
Figure 2. Spline Analysis for the Relationship Between Maximal LV Wall Thickness Z-Score and E′ Velocity, Exercise Capacity and Early Phenotypic Burden in Sarcomere Mutation Carriers.
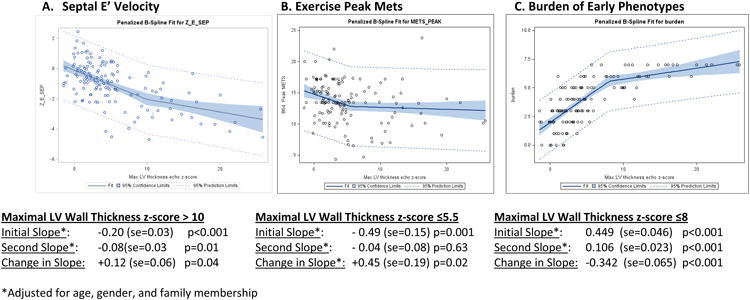
A. Septal E′ velocity tended to worsen with increasing LV wall thickness but the association diminished beyond a maximal LV wall thickness z score of 10 (corresponding to an absolute LV wall thickness of ∼18 mm in the average adult).
B. Peak Mets achieved on exercise testing initially worsened, but beyond a maximal LV wall thickness z-score of 5.5 (absolute adult LV wall thickness of ∼13.5 mm), there is no change in exercise capacity with increasing LV wall thickness.
C. The burden of early phenotypic manifestations increases prominently with increasing LV wall thickness up to a maximal LV wall thickness z-score of 8 (absolute adult LV wall thickness of ∼16 mm) then shows little further incremental change.
Discussion
Determining that sarcomere mutations cause HCM was a seminal discovery in the study of genetic cardiomyopathies. However, much remains unknown regarding the full phenotypic spectrum of sarcomere mutations, the natural history of apparently healthy, at-risk mutation carriers identified from cascade family testing, and how left ventricular hypertrophy relates to disease expression. Key findings from this study include: (1) mutation carriers without LVH have a higher burden of early phenotypes compared with healthy controls; and (2) cardiac abnormalities become more prominent with greater LV wall thickness, however the presence or absence of disease is not accurately reflected by a single measurement.
Early Phenotypes of Sarcomere Mutation: Individual Traits and Collective Burden
Left ventricular hypertrophy is not the initial manifestation of hypertrophic cardiomyopathy. Other facets of LV function and geometry differ significantly between controls and mutation carriers with normal LV wall thickness. For example, echocardiographic LV diameter was almost one standard deviation smaller in G+/LVH- individuals compared to their healthy genotype-negative relatives. Smaller CMR LV volumes and increased LV thickness:diameter ratios of G+/LVH- subjects are further evidence of altered LV geometry in mutation carriers with normal LVWT. These findings expand on earlier, largely single-center studies25-27. Other metrics that differentiated G+/LVH- and G- controls in smaller, more homogeneous studies, including E′ velocity and ECG abnormalities4,7, were not discriminating in this cohort. These results underscore that the G+/LVH- cohort, like the G+/LVH+ cohort, is heterogeneous, with variability in phenotypic expressivity and penetrance throughout all stages of disease development.
We also emphasize that that the presence of a sarcomere mutation is not equivalent to the clinical diagnosis of HCM. Although phenotypic manifestations can be identified in G+/LVH- individuals, these findings are of greater value as markers of underlying disease biology and future clinical course, rather than providing information about current clinical consequences. Careful longitudinal study is critically needed to more fully characterize disease evolution and to accurately identify factors that influence prognosis and disease expression.
Recognizing that individual early phenotypic “abnormalities” can also be found in healthy controls, we tested the hypothesis that mutation carriers manifest a greater composite burden of phenotypes. We constructed a phenotypic burden score that discriminated the three subject groups: G+/LVH+ participants had the highest burden and G- controls the lowest. Similarly, CART analysis demonstrated that preclinical mutation carriers could be differentiated from controls by virtue of smaller LV cavity size and lower E′ velocity. In a smaller single center study, Gandjbakhch, et al 25 described the value of assessing the collective impact of four echocardiographic measures (IVS/PW ratio, relative wall thickness, septal E/ E′ velocity). They found that an echo/TDI score had higher sensitivity and specificity in identifying G+/LVH- individuals from controls compared with looking individually at E′ velocity. Although composite burden approaches will need to be refined and prospectively validated in other genotyped populations, we propose that assessing phenotypic burden will be a more accurate and biologically relevant means of following disease development and/or progression, rather than focusing on any individual trait.
The Impact of LV Wall Thickness on Phenotype in Sarcomere Mutation Carriers
HCM has traditionally been defined by relatively arbitrary and binary thresholds of LV wall thickness based on population norms. Once the threshold is exceeded, pathologic LVH is deemed to be present and, in the appropriate context, HCM is diagnosed. This is an imperfect approach, as disease likely develops along a continuum, and simply measuring LV wall thickness cannot fully capture its presence or absence. Analyzing LVWT as a continuous variable, the relationship with maximal LVWT z-score and other clinical metrics appeared generally continuous and linear in sarcomere mutation carriers. A single threshold of LVWT did not cleanly separate mutation carriers as “normal” or “abnormal”. While practical necessity often drives the use of dichotomous cut points and definitions in clinical practice, our data underscore that HCM does not develop in a binary manner.
Limitations
Our study has several important limitations. Although our cohort was larger, more comprehensively studied, and more diverse than prior studies, it was relatively small, owing to the rarity of genotyped individuals. We did not specifically select for participants with mild disease, however the G+/LVH+ cohort was young (mean age 27 ± 14 years) and generally healthy, thus may not be fully representative of the adult overt HCM population. Due to the small size of specific genetic subgroups, all mutations were pooled together for analysis. Individual phenotypes could have stronger associations with certain genes or mutations than for others, but these signals could be diluted by pooled analysis. Finally, this was a cross-sectional study. We compared individuals anticipated to be in the early (G+/LVH-) or established (G+/LVH+) stages of disease and used LV wall thickness as a crude reflection of disease severity and progression. Performing true longitudinal studies of larger genotyped populations will be crucial to accurately characterize natural history and the transition to disease.
Conclusions
Abnormalities of LV geometry and a greater burden of phenotypic manifestations can be detected in sarcomere mutation carriers without left ventricular hypertrophy. Two methods that integrated composite features were able to discriminate phenotypic subgroups – a sum across seven criteria and a tree-based rule that identifies constellations of distinguishing factors. Rather than focusing on individual traits, more holistic approaches to studying HCM will likely to be more effective to identify individuals with preclinical HCM who are progressing towards clinically overt HCM and may merit closer follow up, and to monitor the effectiveness of interventions designed to slow or prevent preclinical HCM disease progression. Similarly, a single threshold of LV wall thickness could not adequately define the presence or absence of disease in our population. There is likely a progressive interaction between the underlying sarcomere mutation and the cardiomyocyte that leads to ongoing remodeling and disease development. Shifting phenotypic assessment of HCM from a dichotomous definition based on the presence or absence of LVH, to one that more accurately reflects this continual evolution, will aid future studies that seek to better understand and modify disease, thereby improving clinical management.
Supplementary Material
Acknowledgments
CYH had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis
Funding: This work was supported by the National Heart, Lung, and Blood Institute at the National Institutes of Health [grant number 1P20HL101408 to CYH]. NIH had no role in the conduct of the study or this manuscript.
Footnotes
Disclosures: CES is a founder and owns shares in MyoKardia Inc., a startup company that is developing therapeutics that target the sarcomere
References
- 1.Maron BJ, Seidman JG, Seidman CE. Proposal for contemporary screening strategies in families with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2004 Dec 7;44(11):2125–2132. doi: 10.1016/j.jacc.2004.08.052. [DOI] [PubMed] [Google Scholar]
- 2.Niimura H, Bachinski LL, Sangwatanaroj S, et al. Mutations in the gene for cardiac myosin-binding protein C and late- onset familial hypertrophic cardiomyopathy [see comments] N Engl J Med. 1998;338(18):1248–1257. doi: 10.1056/NEJM199804303381802. [DOI] [PubMed] [Google Scholar]
- 3.Ho CY, Carlsen C, Thune JJ, et al. Echocardiographic Strain Imaging to Assess Early and Late Consequences of Sarcomere Mutations in Hypertrophic Cardiomyopathy. Circulation: Cardiovascular Genetics. 2009;2:314–321. doi: 10.1161/CIRCGENETICS.109.862128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ho CY, Sweitzer NK, McDonough B, et al. Assessment of diastolic function with Doppler tissue imaging to predict genotype in preclinical hypertrophic cardiomyopathy. Circulation. 2002 Jun 25;105(25):2992–2997. doi: 10.1161/01.cir.0000019070.70491.6d. [DOI] [PubMed] [Google Scholar]
- 5.Cardim N, Perrot A, Ferreira T, et al. Usefulness of Doppler myocardial imaging for identification of mutation carriers of familial hypertrophic cardiomyopathy. The American journal of cardiology. 2002 Jul 15;90(2):128–132. doi: 10.1016/s0002-9149(02)02434-7. [DOI] [PubMed] [Google Scholar]
- 6.Crilley JG, Boehm EA, Blair E, et al. Hypertrophic cardiomyopathy due to sarcomeric gene mutations is characterized by impaired energy metabolism irrespective of the degree of hypertrophy. J Am Coll Cardiol. 2003 May 21;41(10):1776–1782. doi: 10.1016/s0735-1097(02)03009-7. [DOI] [PubMed] [Google Scholar]
- 7.Lakdawala NK, Thune JJ, Maron BJ, et al. Electrocardiographic features of sarcomere mutation carriers with and without clinically overt hypertrophic cardiomyopathy. The American journal of cardiology. 2011 Dec 1;108(11):1606–1613. doi: 10.1016/j.amjcard.2011.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maron MS, Olivotto I, Harrigan C, et al. Mitral valve abnormalities identified by cardiovascular magnetic resonance represent a primary phenotypic expression of hypertrophic cardiomyopathy. Circulation. 2011 Jul 5;124(1):40–47. doi: 10.1161/CIRCULATIONAHA.110.985812. [DOI] [PubMed] [Google Scholar]
- 9.Captur G, Lopes LR, Mohun TJ, et al. Prediction of sarcomere mutations in subclinical hypertrophic cardiomyopathy. Circulation. Cardiovascular imaging. 2014 Nov;7(6):863–871. doi: 10.1161/CIRCIMAGING.114.002411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Germans T, Wilde AA, Dijkmans PA, et al. Structural abnormalities of the inferoseptal left ventricular wall detected by cardiac magnetic resonance imaging in carriers of hypertrophic cardiomyopathy mutations. J Am Coll Cardiol. 2006 Dec 19;48(12):2518–2523. doi: 10.1016/j.jacc.2006.08.036. [DOI] [PubMed] [Google Scholar]
- 11.Maron MS, Rowin EJ, Lin D, et al. Prevalence and clinical profile of myocardial crypts in hypertrophic cardiomyopathy. Circulation Cardiovascular imaging. 2012 Jul;5(4):441–447. doi: 10.1161/CIRCIMAGING.112.972760. [DOI] [PubMed] [Google Scholar]
- 12.Brouwer WP, Germans T, Head MC, et al. Multiple myocardial crypts on modified long-axis view are a specific finding in pre-hypertrophic HCM mutation carriers. European heart journal cardiovascular Imaging. 2012 Apr;13(4):292–297. doi: 10.1093/ehjci/jes005. [DOI] [PubMed] [Google Scholar]
- 13.Ho CY, Lopez B, Coelho-Filho OR, et al. Myocardial fibrosis as an early manifestation of hypertrophic cardiomyopathy. N Engl J Med. 2010 Aug 5;363(6):552–563. doi: 10.1056/NEJMoa1002659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ho CY, Abbasi SA, Neilan TG, et al. T1 measurements identify extracellular volume expansion in hypertrophic cardiomyopathy sarcomere mutation carriers with and without left ventricular hypertrophy. Circulation Cardiovascular imaging. 2013 May 1;6(3):415–422. doi: 10.1161/CIRCIMAGING.112.000333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Landrum MJ, Lee JM, Benson M, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016 Jan 4;44(D1):D862–868. doi: 10.1093/nar/gkv1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alfares AA, Kelly MA, McDermott G, et al. Results of clinical genetic testing of 2,912 probands with hypertrophic cardiomyopathy: expanded panels offer limited additional sensitivity. Genet Med. 2015 Nov;17(11):880–888. doi: 10.1038/gim.2014.205. [DOI] [PubMed] [Google Scholar]
- 17.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015 May;17(5):405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schiller NB, Shah PM, Crawford M, et al. Recommendations for quantitation of the left ventricle by two-dimensional echocardiography. American Society of Echocardiography Committee on Standards, Subcommittee on Quantitation of Two-Dimensional Echocardiograms. J Am Soc Echocardiogr. 1989 Sep-Oct;2(5):358–367. doi: 10.1016/s0894-7317(89)80014-8. [DOI] [PubMed] [Google Scholar]
- 19.Park SH, Shub C, Nobrega TP, Bailey KR, Seward JB. Two-dimensional echocardiographic calculation of left ventricular mass as recommended by the American Society of Echocardiography: correlation with autopsy and M-mode echocardiography. J Am Soc Echocardiogr. 1996;9(2):119–128. doi: 10.1016/s0894-7317(96)90019-x. [DOI] [PubMed] [Google Scholar]
- 20.Kramer CM, Barkhausen J, Flamm SD, Kim RJ, Nagel E. Society for Cardiovascular Magnetic Resonance Board of Trustees Task Force on Standardized P. Standardized cardiovascular magnetic resonance (CMR) protocols 2013 update. J Cardiovasc Magn Reson. 2013;15:91. doi: 10.1186/1532-429X-10-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Young AA, Cowan BR, Thrupp SF, Hedley WJ, Dell'Italia LJ. Left ventricular mass and volume: fast calculation with guide-point modeling on MR images. Radiology. 2000 Aug;216(2):597–602. doi: 10.1148/radiology.216.2.r00au14597. [DOI] [PubMed] [Google Scholar]
- 22.Schulz-Menger J, Bluemke DA, Bremerich J, et al. Standardized image interpretation and post processing in cardiovascular magnetic resonance: Society for Cardiovascular Magnetic Resonance (SCMR) board of trustees task force on standardized post processing. J Cardiovasc Magn Reson. 2013;15:35. doi: 10.1186/1532-429X-15-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sluysmans T, Colan SD. Theoretical and empirical derivation of cardiovascular allometric relationships in children. J Appl Physiol. 2005 Aug;99(2):445–457. doi: 10.1152/japplphysiol.01144.2004. [DOI] [PubMed] [Google Scholar]
- 24.Colan SD. Normal echocardiographic values for cardiovascular structures. In: Lai WWCM, Geva T, Mertens L, editors. Echocardiography in Pediatric and Congenital Heart Disease. 2. West Sussex, UK: Wiley-Blackwell; 2015. [Google Scholar]
- 25.Gandjbakhch E, Gackowski A, Tezenas du Montcel S, et al. Early identification of mutation carriers in familial hypertrophic cardiomyopathy by combined echocardiography and tissue Doppler imaging. Eur Heart J. 2010 Jul;31(13):1599–1607. doi: 10.1093/eurheartj/ehq101. [DOI] [PubMed] [Google Scholar]
- 26.Hagege AA, Dubourg O, Desnos M, et al. Familial hypertrophic cardiomyopathy. Cardiac ultrasonic abnormalities in genetically affected subjects without echocardiographic evidence of left ventricular hypertrophy. Eur Heart J. 1998 Mar;19(3):490–499. doi: 10.1053/euhj.1997.0735. [DOI] [PubMed] [Google Scholar]
- 27.Jensen MK, Havndrup O, Christiansen M, et al. Echocardiographic evaluation of pre-diagnostic development in young relatives genetically predisposed to hypertrophic cardiomyopathy. Int J Cardiovasc Imaging. 2015 Dec;31(8):1511–1518. doi: 10.1007/s10554-015-0723-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.