

Abstract
Eukaryotic genomes contain many repetitive DNA sequences that exhibit size instability. Some repeat elements have the added complication of being able to form secondary structures, such as hairpin loops, slipped DNA, triplex DNA or G-quadruplexes. Especially when repeat sequences are long, these DNA structures can form a significant impediment to DNA replication and repair, leading to DNA nicks, gaps, and breaks. In turn, repair or replication fork restart attempts within the repeat DNA can lead to addition or removal of repeat elements, which can sometimes lead to disease. One important DNA repair mechanism to maintain genomic integrity is recombination. Though early studies dismissed recombination as a mechanism driving repeat expansion and instability, recent results indicate that mitotic recombination is a key pathway operating within repetitive DNA. The action is two-fold: first, it is an important mechanism to repair nicks, gaps, breaks, or stalled forks to prevent chromosome fragility and protect cell health; second, recombination can cause repeat expansions or contractions, which can be deleterious. In this review, we summarize recent developments that illuminate the role of recombination in maintaining genome stability at DNA repeats.
Keywords: trinucleotide repeat expansion, recombination, replication fork restart, chromosome fragility, DNA structure
INTRODUCTION
Expanded tracts of repetitive DNA sequences are the cause of over 30 genetic diseases and can consist of trinucleotide or larger repetitive units (1–5). The expandable repeats form stable non-B-form DNA structures which impede normal cellular processes like DNA replication and repair. Expanded trinucleotide repeats (TNRs) and other structure-forming repeats break at a greater frequency than non-repetitive DNA; types of DNA breaks that occur include nicks, gaps and double-stranded breaks (DSBs). These lesions must then be repaired in the context of the repetitive DNA. Much of the time the cell will succeed in repairing DNA damage at structure-forming repeats with fidelity, i.e. with no loss or gain of genetic material, thus preserving genome integrity. However, due to both the repetitive nature of the tract as well as the structure-forming potential, mistakes that lead to repeat expansions or contractions are relatively frequent.
There are multiple pathways that repair DNA damage that occurs within TNRs and other repetitive sequences. For example, nicks and gaps can be repaired by base excision repair (BER), or by transcription-coupled repair (TCR) within transcribed regions, both of which can generate TNR expansions (for recent reviews see (2, 5) and the review by Polyzos and McMurray in this issue). Damage that results in DSBs can be repaired by various types of end-joining, by annealing of processed ends, or by recombination-based mechanisms using either a sister chromatid or homolog as the template. In addition, recombination is a primary mechanism used in restarting stalled or collapsed replication forks and in repairing gaps left behind the replication fork. This review will summarize the current knowledge about the role of mitotic recombination in generating genomic changes within repetitive DNA. We will focus on structure-forming triplet repeats, but with comparisons to results found at other biologically relevant repeats and DNA structures.
DNA damage at expanded trinucleotide repeats is repaired by recombination
Deletion of genes required for recombination results in increased breakage of expanded TNRs, suggesting that recombination is normally required for healing these DNA breaks (6, 7). In replicating yeast cells, homologous recombination (HR) and ligase 4-dependent end joining (EJ) both contribute to the repair of breaks at CAG repeats (6). Genome-wide studies to identify novel genes preventing DSBs at GAA and Alu repeats identified several recombinational repair proteins as important, among them the nuclease Mre11, whose absence increased fragility of both repeats (7, 8). Additionally, dividing cells deficient in replication proteins exhibit cell cycle arrest and gross chromosomal rearrangements at Alu repeats because recombination intermediates cannot be resolved, which results in DSBs (8). Failure to heal breaks at expanded TNR repeats can have dire consequences for cells. Yeast cells that lack Rad52 or Ligase 4 and have expanded CAG repeat tracts undergo frequent cell cycle arrest and cell death (9).
Traditionally, DNA repair using recombination has been considered to be an error-free form of repair. However, in actuality, recombination can be highly mutagenic and a source of genomic instability (10–13). Though they are required for repair and cell health, both HR and EJ can be mutagenic when they occur within repetitive DNA, resulting in a loss (contraction) or gain (expansion) of repeat units (14). This is largely due to the challenges of replicating or aligning DNA across a repetitive region, especially one that has formed DNA secondary structures. These DNA structures are varied and include DNA hairpins (common in CAG/CTG and CGG/GCC repeats or inverted repeats), triplexes (formed by purine-rich repeats such as GAA/TTC) and G quadruplexes (for reviews see (5, 15–17)). Though the structures are different, the common theme is that they impede DNA transactions so that replication and repair cannot proceed with fidelity within the repetitive sequence. This inaccurate repair can lead to the incorporation of errors that can range from the aberrant insertion/deletion of DNA bases, as seen in TNR repeat genetic diseases, to genomic rearrangements and loss of heterozygosity, which are commonly seen in cancers. Historically, misalignment of alleles during meiotic crossover was shown to be a mechanism for (GCN)n repeat expansions that code for polyalanine tracts (18), but discounted as a mechanism for length changes of other TNRs, such as (CAG)n repeat tracts encoding polyglutamine. However, these early studies focused on meiotic recombination and did not explore mitotic recombination as a potential mechanism for repairing DNA damage at TNRs and causing repeat instability. The following sections will delve into the various roles of recombination during DNA repair, how each contributes to genomic maintenance of repeat sequences, and the current knowledge of how recombination pathways result in repeat instability.
RECOMBINATION DURING REPLICATION RESULTS IN REPEAT INSTABILITY
Homology-dependent recombinational repair of forks stalled by DNA structures
Addition of repeat units by definition involves DNA synthesis. Incorporation of additional bases might arise as a result of strand slippage either during replication (19) or during fork restart (3). DNA structures formed by repetitive DNA sequences are impediments for DNA synthesis and can cause fork stalling, or gaps behind the replication fork if bypassed. GAA/TCC triplexes and GGC/CCG repeats strongly interfere with replication progression, acting as site-specific barriers (20–22). CAG/CTG repeats are much weaker barriers (23–26) but their replication generates joint molecules that likely represent both reversed fork and sister chromatid recombination intermediates (27, 28). Single stranded gaps occur when leading and lagging strand synthesis becomes uncoupled (reviewed in (29)), and pre-existing DNA nicks or gaps can become DSBs if replicated (5, 30, 31).
After a replication fork stalls at a DNA repeat structure, several types of fork restart can be envisioned (see (32, 33) for reviews on fork restart). First, unwinding of the DNA structure by a helicase may allow replication to continue without replisome dissociation, which would not lead to repeat instability unless slippage occurred (Figure 1C). Second, a fork reversal or template switch mechanism could be used to replicate through the DNA structure (Figure 1A, 1B). The outcome in terms of repeat contraction or expansion will vary depending on where the un-excised hairpin forms (template or nascent strand) and which hairpins are resolved. There are several possibilities for hairpin formation or mis-alignments during the fork restart process, which would likely involve the HR machinery (Figure 1A). Third, a break in the DNA could lead to an HR-dependent stand invasion, either on the same DNA template (broken fork repair (BFR), similar to what is drawn in Figure 1A but initiated from a break) or on a different template (ectopic break-induced replication (BIR; Figure 2). BIR is known to be a mutagenic process (10, 34, 35). Finally, repeat expansions are also known to occur due to hairpin impairment of Okazaki flap processing by the FEN1 endonuclease (Figure 1D; (36)).
Figure 1. Structures formed by DNA repeats cause replication fork stalling and template switch.
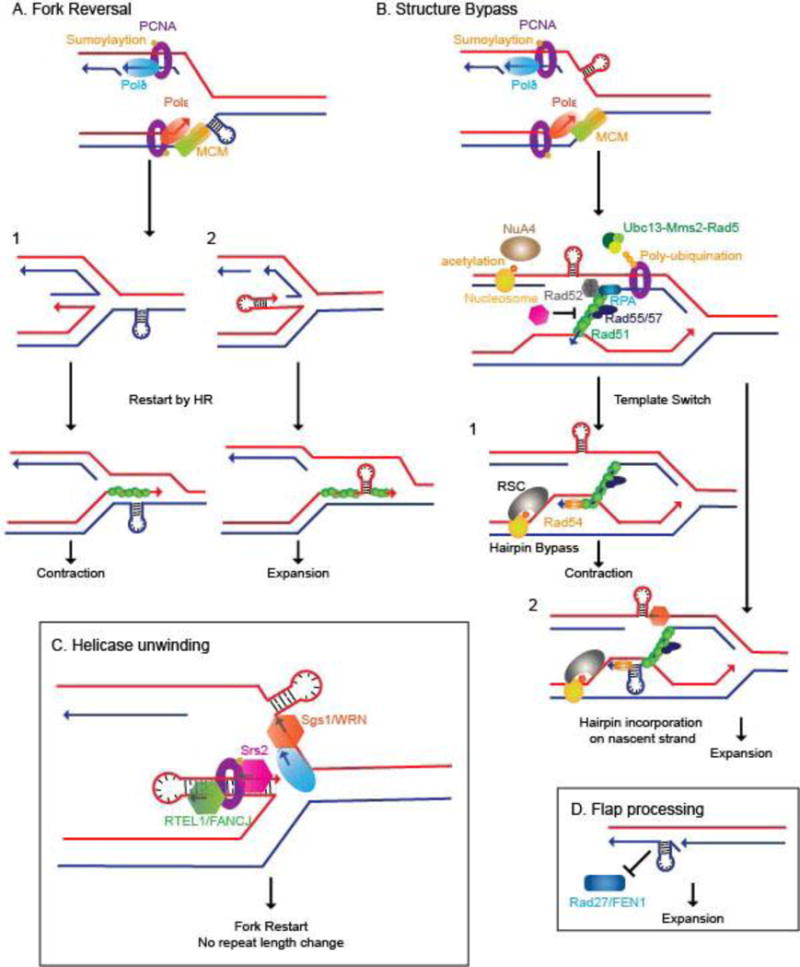
During replication, leading or lagging strand hairpins may cause fork stalling. To recover from these fork stalls, there are two recombinational mechanisms that can be employed: first, by restarting the replication fork, which may involve fork reversal (A), and second, by structure bypass (B). For simplicity, fork reversal is only shown for a leading strand hairpin and structure bypass for a lagging strand hairpin, though either mechanism could be triggered by each barrier. Alternatively, helicase unwinding of the DNA structures could allow fork restart without recombination (C). A. 1) Fork reversal and template switch to the lagging strand may occur without hairpin resolution; the hairpin could then be bypassed during fork restart resulting in a repeat contraction. 2) If fork reversal results in the fold-back of the leading strand (or excess synthesis from the lagging strand without hairpin formation), DNA synthesis would result in an expansion upon replication fork restart. Both mechanisms of fork restart are predicted to utilize recombination proteins such as Rad51 for strand invasion. B. If DNA structures are bypassed, ssDNA gaps must be filled in after replication by using the sister chromatid as a template. Repair of post-replication gaps is dependent on poly-ubiquitination of PCNA by Ubc13-Mms21-Rad5 to initiate the template switch, which is mediated by recombination proteins Rad51, Rad52, Rad55/57 and Rad54 for strand invasion. In addition, the histone H4 acetyltransferase NuA4 and the RSC chromatin remodeler are required for efficient sister chromatid recombination and accurate repair within repetitive DNA (77). Upon strand invasion into the sister chromatid, repeat instability can occur by two mechanisms. 1) strand invasion can result in bypass of the DNA structure which will result in a contraction. 2) Alternatively, D-loop extension during template switching may be prone to replication slippage leading to a repeat expansion. Repeat expansions and contractions could also occur by misalignment of repeat units during either (A) or (B) (not shown). C. Helicases can prevent repeat instability by unwinding DNA structures, which will allow replication to continue without replisome dissociation, or prevent hairpin formation during template switch (e.g. as in B.2). The helicase Srs2 is a 3′-5′ helicase that can unwind hairpins to facilitate fork progression through hairpin structures. This function is dependent on PCNA interaction but independent of its role in inhibiting recombination. Sgs1/WRN is another 3′-5′ helicase that can unwind hairpins as well as G4 DNA. Sgs1/WRN interacts with Polδ and is thought to unwind DNA structures on the lagging strand template during replication. RTEL1 and FANCJ are 5′-3′ helicases that can also unwind DNA structures like hairpins and G4 DNA sequences. D. During replication, structure formation on the lagging strand can impede the 5′ flap endonuclease activity of FEN1/Rad27 which can lead to inefficient or “alternative” flap cleavage and repeat expansions.
Recently it was shown that expanded CAG repeats, which are natural replication fork barriers, result in the transient localization of chromosomes to the nuclear pore during S-phase (37). This relocation was dependent on replication, occurred in late S phase and was resolved by G2, and prevented repeat fragility. Yeast chromosomes exposed to both the alkylating agent MMS and the fork stalling drug hydroxyurea similarly relocate to the nuclear periphery (38). Interestingly, failure to relocate to the pore led to increased Rad52-dependent CAG repeat expansions and contractions. Taken together, these results suggest that relocation to the nuclear pore facilitates fork restart, and this may protect against DSBs and mutagenic Rad52-dependent repair (37, 39). Posttranslational modification of key repair proteins by sumoylation may be important in the re-localization and fork restart process, as deletion of the Slx5/8 SUMO-dependent ubiquitin ligase resulted in an increase in repeat instability and a decrease in nuclear localization of the expanded CAG repeat.
Recombination-mediated repeat instability at the replication fork is not unique to TNR repeats. In S. pombe, Swi1 promotes replication fork progression through telomeric repeats and prevents telomeric instability and aberrant recombination at telomeres (40). Additionally, in human cells, impaired replication of telomeric repeats results in fragile telomeres (41) and efficient replication requires the telomeric binding protein TRF1 and the helicases BLM and WRN to unwind G4 structures that can impede replication machineries (41, 42). Interestingly, replication in the context of HR repair or HR-dependent fork restart proceeds with less fidelity and more mutations than normal replication, even without the complication of copying DNA repeats (43–45). Recently, GAA repeats have been shown to induce mutagenesis up to 8 kb away from the repeat site in yeast, presumably through an HR-mediated repair event (22, 46–48). The authors hypothesize that a barrier to replication caused by a GAA secondary structure recruits the low-fidelity Polζ polymerase. DSB formation or fork stalling at the repeat leads to strand invasion of the homolog, where synthesis with Polζ leads to mutagenesis (46, 48). This repeat-induced mutagenesis (RIM) has also been observed for H-DNA and Z-DNA forming sequences introduced into mammalian cells (49–51). Taken together, expanded TNRs and other structure-forming repeat sequences are sites of replication fork collapses that are repaired by HR, and this repair may result in an increase in the mutation rate.
The role of helicases in replication of structure-forming repeats
Helicases have been shown to be important in preventing replication-associated repeat instability. One important helicase that helps to resolve repeat-induced replication fork stalls in yeast is the helicase Srs2 (Figure 1C). Using direct visualization of fork stalling in vivo by 2D gel electrophoresis, Srs2 has been shown to facilitate replication past a (CGG)45 repeat that causes a barrier to replication via hairpin formation (52). Srs2 had no activity on replication barriers due to G-quadruplex structures or protein binding, thus it is specific to DNA hairpins. Srs2 function at stalled forks was unique among the helicases tested (Sgs1, Pif1, Rrm3), and was dependent on its helicase activity and its ability to interact with PCNA, but not on its Rad51 displacement motif. Srs2 can also unwind CAG hairpins in vitro and prevent expansions that occur during template switch (53, 54) and during sister-chromatid recombination (27). Recently, separation of function alleles were used to determine that Srs2 requires its helicase and PCNA interaction domains to protect against chromosome fragility, for example by hairpin unwinding at the replication fork (Figure 1C), whereas its anti-recombinase function prevents repeat instability (Figure 1B) (28). These results further underscore the ability of replication-associated recombination events to generate repeat expansions.
In humans, unwinding of hairpins can be performed by the RTEL1 helicase as knockdown resulted in an increase in CAG expansion frequency to a similar level as knockdown of Rad18 and HLTF, homologs of yeast Rad18 and Rad5 (55). Strikingly, RTEL1 could substitute for Srs2 in yeast cells to prevent both CAG repeat fragility and instability (55). Though RTEL1 and Srs2 lack protein sequence homology and have opposite DNA unwinding polarities, these results indicate a strong functional conservation between the two enzymes with respect to CAG repeat replication. Both helicases are able to unwind CAG and CTG hairpin structures in vitro, though RTEL1 additionally acts at G4 DNA and is important in telomere maintenance (55–57). In S. cerevisiae, replication through G4 DNA is facilitated by the Pif1 helicase (Pfh1 in S. pombe) (58, 59). In a system used to detect gross chromosomal rearrangements due to G4 DNA Pif1 was shown to be important in preventing genomic instability, suggesting that Pif1-mediated unwinding of G4 DNA prevents error-prone replication or repair (60). Another helicase involved in TNR instability in mammalian cells is the 5′-3′ helicase FANCJ, a member of the Fanconia Anemia pathway, which can unwind G4 DNA and CAG/CTG hairpins during replication to prevent repeat instability (61, 62). Unwinding non-B form DNA structures to prevent instability is a unique role for FANCJ as other members of the Fanconi Anemia (FA) pathway did not exhibit similar roles (62). Similarly, yeast Sgs1 can unwind hairpins and G4 DNA in vitro, as can its human homologs WRN and BLM (52, 63–65). Sgs1 and WRN interact with Polδ, and thus are well positioned to unwind structures on the lagging strand template (Figure 1C). Indeed, deletion of Sgs1 led to a large increase in repeat contractions, consistent with such a role (27), and WRN was identified in a screen in HeLa nuclear extracts for proteins that were able to stimulate repair of a CTG hairpin on the template strand (66). The WRN helicase efficiently unwound CTG hairpins in this system to promote Polδ-catalyzed DNA synthesis across the gap and prevent deletions (66). Thus Srs2/RTEL1 and Sgs1/WRN may work together to resolve hairpins on different strands (Figure 1C). This mechanism could also be relevant during gap repair in non-dividing cells.
Template switch is a mechanism for trinucleotide repeat instability
Template switch repair is used to fill-in ssDNA gaps that result when the replication fork bypasses a fork-blocking lesion (Figure 1B) (reviewed by (67)). Given the ability of TNRs to form secondary structures that impair fork progression, its not surprising that proteins required for template switch have been shown to be involved in the stability of repetitive sequences. Repair of post-replication gaps is dependent on ubiquitination of PCNA, and can be subdivided into two categories: translesion synthesis (TLS) and error-free post-replication repair (PRR) or template switching (reviewed by (68, 69)). The TLS branch is dependent on the PCNA ubiquitin ligases, Rad6 and Rad18, which together monoubiquitinate Lys164 of PCNA. Monoubiquitinated PCNA recruits translesion polymerases (e.g. Polζ or Polη) that synthesize across the lesion. Interestingly, mutations in the TLS polymerases had no effect on CAG repeat instability (70, 71) or GAA repeat stability (22) in wild-type budding yeast, indicating that the TLS pathway is not a significant source of expansions. However recent data suggests that when replicative polymerases are compromised, some GAA repeat expansions do occur by a Polζ-dependent mechanism (22), as do short duplications initiated by small hairpins (72).
The error-free branch of PRR, template switch, requires additional ubiquitination action by Ubc13-Mms2-Rad5 E2–E3 ubiquitin ligases (mammalian HLTF/SHPRH), leading to a poly-ubiquitinated PCNA molecule (Figure 1B) (68, 69). Template switch further requires the action of the HR proteins Rad51, Rad52, Rad57 and Rad54 (73–75) (Figure 1B). The requirement for HR proteins is consistent with the use of the undamaged sister chromatid as a template for synthesis, though the precise mechanism is poorly understood (68, 69, 76, 77). In S. cerevisiae, spontaneous sister chromatid recombination (SCR) is proposed to occur because of gaps formed behind the replication fork (78), and is induced by both CAG repeats and inverted repeats (79). It is the propensity of these sequences to form secondary structures that is thought to impede replication and induce sister chromatid exchange, which was dependent on the presence of Rad52 (79).
Rad5, Rad18, HTLF, and PCNA ubiquitination have all been shown to inhibit (CTG)13 or (CAG)25 expansions in yeast and human cells (53, 55). At a longer (CAG)85 repeat, deletion of RAD5 also increased expansions 3-fold over wild-type (77), though at this repeat size, template switch can also cause repeat expansions (see chromatin section below). The role of Rad5 at short CAG repeats (e.g. less than 35 repeats) was epistatic to a deletion of SRS2, implicating a role for the Srs2 helicase, which was hypothesized to unwind 3′ hairpins occurring during the template switch (53, 54). Altogether, these data indicate that CAG tracts induce both SCR and template switch events, and that in yeast, the Srs2 helicase is important to prevent instability during this process. This template switch event must play an important role in repairing TNR-related gaps, because in its absence (e.g. in rad5Δ or rad18Δ strains) expansions occur, by an alternative unknown pathway.
In contrast to short (CAG)13–25 repeats, ATTCT and GAA repeat expansions are promoted by the presence of Rad5 in yeast (80, 81). ATTCT repeats, which expand to cause SCA10, do not form structured DNA, but instead are DNA unwinding elements (82). In addition, a rad5Δ mutant displays decreased ATTCT fragility (80), suggesting that template switching events can lead to chromosomal fragility at these repeats. Rad5-dependent expansions of the GAA repeat were proposed to occur by a template switching mechanism in which the GAA repeat expansions arise from dissociation of the leading strand from its normal template and aberrant copying from the newly synthesized Okazaki fragment (81). This model predicts that copying would not be dependent on DNA structure per se, but would be facilitated by pausing of the replication fork (80, 81).
How can one explain the different dependencies on Rad5 observed for different types and sizes of repeats? We previously proposed a model to account for the somewhat contradictory roles of proteins in the template switch pathway on repeat instability (5). For longer or more “slippery” repeats (GAA, ATTCT, longer CAGs), the fork stall could be strong enough to mediate a template switching event directly at the stalled fork, hypothesized to be facilitated by Rad5 (Figure 1A). There is experimental evidence for fork reversal at both CAG and GAA repeats by direct visualization of replication intermediates by 2D gel electrophoresis and electron microscopy (27, 28, 83, 84). For CAG repeats, the size needed to produce a fork stall stable enough to be visualized on a 2D gel is approximately 90–100 CAGs (26, 28). After the stall there are two models for generating expansions: for hairpin-forming sequences, fold-back of the leading strand would allow DNA synthesis from the leading strand, resulting in a repeat expansion upon fork restart (Figure 1A, right pathway) (first proposed by (3)). For non-hairpin forming sequences, copying off of the lagging nascent strand provides the extra DNA synthesis, as proposed in (81) for large-scale GAA expansions (see (85) for review). On the other hand, a single hairpin is more likely to be bypassed, leading to a post-replicative template switch that initiates from a gap, and looks more like SCR (Figure 1B) (27, 77). This latter event may be more common for mid-length CAGs, above the expansion threshold of 35 repeats but still less than the size needed to produce a stable fork stall (e.g. ~45–85 repeats). For very short (CAG)13–25 tracts, post-replicative hairpin unwinding by the Srs2 protein could be sufficient most of the time, with less engagement of the full recombination pathway (86). This idea of length-dependent differences is supported by the fact that Tof1, a subunit of the replication-pausing complex, protects against instability for both GAA and ATTCT repeats (80, 81). Thus the replication pausing complex may act to limit the template switching events at stalled forks that can allow for repeat expansions.
Chromatin modifications influence repair fidelity during template switch
The chromatin environment at gaps also contributes to efficient repair by error-free template switch (reviewed in (87, 88)). The absence of Anc1, a subunit of the chromatin modifying complexes INO80, SWI/SNF, and NuA3, leads to an increase in (CAG)25 expansion frequency that is equivalent to the increase in rad5Δ and mms2Δ mutants (53, 89). Histone H4-K16 acetylation by the chromatin remodeling complex NuA4 is specifically enriched at expanded CAG repeats in yeast, and is required for high-fidelity template switch and (CAG)85 repeat maintenance: in the absence of proper histone modification, Rad5 and Rad52-dependent expansions occur more frequently (Figure 1B) (77). Further, Rsc2, an acetyl-lysine binding subunit of the RSC chromatin remodeler, was recruited to the CAG repeat coincident with the peak in H4-K16 acetylation, suggesting a recruitment mechanism for this remodeler and a role in promoting template switch without expansions (Figure 1B) (77). Thus, although template switch is a protective pathway, it can be a source of repeat length changes if it occurs without accessory factors such as chromatin remodelers or modifiers (RSC, H4-K16ac, Anc1) that allow it to occur with fidelity.
RECOMBINATION DURING DSB REPAIR DRIVES REPEAT INSTABILITY
HR-dependent instability can cause large repeat expansions
Structure-forming DNA, including expanded CAG, CGG, GAA, and ATTCT repeats as well as palindrome-forming sequences, are natural fragile sites that cause chromosomal DSBs (reviewed in (5, 15, 90–92)). Consistently, expanded GAA/TTC repeats, Alu repeats, and internal telomeric repeats stimulate mitotic crossovers in yeast (93–95) and recombination in E. coli (96–98). In yeast, the effect of DSB repair by HR on CAG repeat stability was assessed directly by induction of a DSB and selection for repair events that used an ectopically provided (CAG)98 tract for repair (99). This experiment showed that repeat instability occurred during HR-mediated DSB repair, resulting in a much higher percentage of expansions (13%) and contractions (30%), than a control that did not undergo break induction (0% expansions, 10% contractions). Mre11, an endonuclease important in generating a single-stranded 3′ end and initiating HR, was required for efficient repair, and the proportion of expansions increased in its absence (99, 100). In a second study without an induced break, (CAG)70 expansions were also increased 11-fold in the absence of Mre11 with a bias to large expansions, pointing to a key role for this protein in preventing repeat instability (6). A nuclease-dead allele of Mre11 had a lesser effect, arguing that the structural function of the MRX complex was the most important factor in preventing expansions. These expansions were suppressed in the absence of Rad52, and therefore occurred through aberrant recombination (6). Mre11 has yet a different role at short (CTG)20 repeats, as the presence (not absence) of the protein drives expansions during template switch, independent of both its nucleolytic function and its role in HR (101). At DSBs, one of the functions of Mre11 is to associate with Rad50 and Xrs2/Nbs1 to initiate DSB end processing (Figure 2A) and to activate the DNA damage checkpoint kinases Tel1/ATM and Mec1/ATR which are required for efficient DSB repair (102). At the expanded CGG repeat fragile site in a Fragile X mouse model, loss of one Atm allele increased the expansion frequency in both males and females (103), and similarly loss of one copy of the Atr gene was associated with an increased risk of expansion on maternal transmission (104). Additionally cutting by the CRISPR-Cas9 nickase at expanded CAG/CTG repeats in an ATR-deficient cell line resulted in increases in expansions and contractions (105). Although in these mammalian cells the mechanism creating expansions is not known, it is consistent with the data from yeast and bacteria that synthesis-dependent HR from a DSB that is lacking proper MRX/MRN scaffolding is a mechanism that can produce both repeat expansions and contractions (Figure 2E).
Figure 2. Double stranded break repair can drive repeat instability.
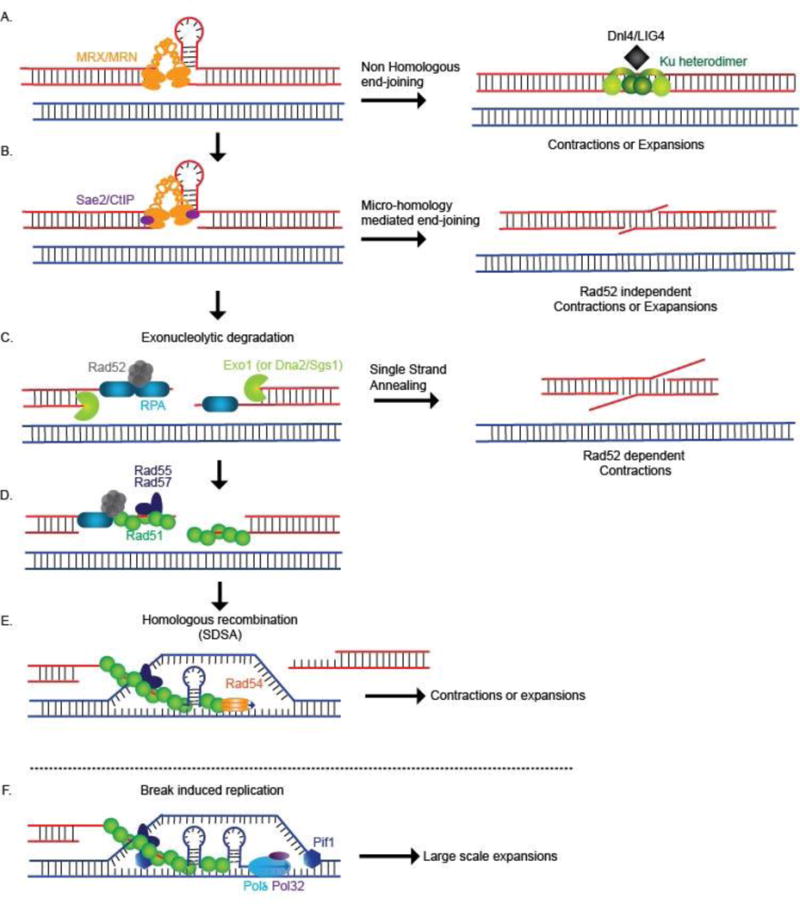
DSBs occurring within repetitive DNA may be repaired by multiple pathways. A. DSB ends are initially bound by the MRX/MRN complex which can mediate either end-joning or HR pathways. Post initial processing by MRX/N, repair via NHEJ may occur. NHEJ requires the Ku heterodimer and Ligase 4 to re-ligate two ends of a DSB that has undergone minimal end processing. NHEJ can result in TNR contractions or expansions B. Sae2 interacts with MRX to initiate end processing and resection; structure-forming repeats may have hairpin capped ends that require additional processing. After initial processing by the MRX/N complex and Sae2/CtIP, one mechanism for repair of breaks at repeats is MMEJ which can result in TNR contractions, and perhaps expansions. C. After initial processing, long range resection occurs by the 5′-3′ exonuclease Exo1 or the RecQ helicase Sgs1 (WRN/BLM) in concert with the nuclease Dna2. After extensive resection by exonucleases, repair can occur via SSA, which requires Rad52 and favors repeat contractions. D. To prevent secondary structure formation, the RPA complex binds to the resected ssDNA. Rad52 mediates the removal of RPA and the nucleation of Rad51 bound filaments; accessory proteins Rad55/57 form co-filaments with Rad51 which increases the stability of the filament (136). E. The Rad51 filaments initiate the homology search and invade homologous DNA which initiates D-loop formation; hairpin formation during D-loop extension provides an opportunity for slippage and repeat expansions. Upon completion of DNA synthesis, repair via SDSA results in re-ligation with the original chromosome end which can result in contractions or expansions depending on the location of hairpin formation and alignment of repeat units. F. For break induced replication, polymerase δ along with Pol32 and Pif1 binds to the D-loop and proceeds with long range conservative DNA replication, which can result in extensive polymerase slippage and large scale TNR expansions
One subpathway of HR is BIR, a highly mutagenic form of HR where a DSB end invades a region of homology and replicates for several kilobases, potentially to the telomere end (10, 34, 35). Pol32, a non-essential subunit of Polδ, as well as the helicase Pif1 are essential for BIR mediated repair in yeast (106, 107). Replication during BIR is highly mutagenic as it proceeds via a migrating bubble of conservative DNA synthesis (108, 109). Recent work has shown that large-scale CAG/CTG expansions (defined as addition of more than 20 repeats) of a (CAG)140 repeat tract in a yeast model utilizes traditional HR machinery as well as Pol32 and Pif1, implicating BIR as a mechanism for generation of large-scale repeat expansions (Figure 2F) (110). These large-scale expansions depended on replication, therefore they could also be generated though a mechanism similar to broken fork repair (111) or HR-dependent fork restart that occurs at protein barriers to replication (33) (Figure 1A). Intriguingly, such large-scale CAG/CTG expansions occur in replicating cells of the pre-meiotic male germline in humans with pre-mutation Huntington’s disease alleles and in a DM1 mouse model (112, 113). Expansions that occur in non-dividing cells could also arise by BIR, as BIR readily occurs in non-dividing cells; in this case DSBs could be generated during mismatch repair or transcription rather than by replication (7, 44). At critically short telomeres, BIR may serve a similar function in extending difficult to replicate sequences, as BIR has recently been shown to be a mechanism for alternative lengthening of telomeres (114, 115). It was hypothesized that PCNA can load at alternative structures with recessed 3′ ends to recruit the Polδ polymerase for telomere synthesis (114).
Despite the strong evidence for HR-induced repeat instability in yeast, in the above cited DM1 mouse model, loss of the Rad54 gene product did not significantly suppress CTG instability, and the absence of the mouse Rad52 gene decreased the size of expansions, but did not eliminate them (116). These results have been interpreted to mean that HR has no effect on CAG/CTG instability in mammalian cells. However, because elimination of HR in mammalian cells is lethal, the Rad52−/− and Rad54−/− lines used in the DM1 mouse studies do not eliminate HR: Rad52 knockouts exhibit only a slightly reduced HR frequency and are not hypersensitive to DSB-inducing agents; Rad54 is not an essential HR protein, but serves to facilitate chromatin remodeling during HR (117, 118). The fact that the mean size of expansions was significantly decreased in the Rad52−/− DM1 mouse model supports the idea that rare large expansions could actually be arising during HR in these mice. Due to the technical difficulties of eliminating HR in mouse models, the role of HR in repeat instability in mammals remains to be fully assessed. Study of HR in mammalian cell culture models where repeat expansions can be detected would be one way to bridge this gap.
Repair of DSBs by single-strand annealing (SSA) and end-joining mechanisms (NHEJ, MMEJ) can cause repeat instability, predominantly contractions
In addition to HR, DSBs occurring at fragile repeat sequences can be repaired by various types of end joining pathways. At an expanded CAG repeat in yeast, deletion of both Rad52 and Dnl4 (Ligase 4) led to an additive level of fragility (6). Also, breaks at Z- and H-DNA forming sequences are repaired by NHEJ in mammalian cells, causing deletions (49, 50, 90, 119)(Figure 2A). In the absence of Dnl4, CAG repeat contraction frequency is significantly increased in yeast and remains so in dnl4Δrad52Δ cells, indicating that these contractions are not occurring through HR (6). The absence of HR proteins Rad52, Rad51 and Rad54 also increased repeat contractions ~2.5-fold (6). Because broken DNA ends undergo greater resection in both rad52 and dnl4 backgrounds, end processing followed by SSA between repeats is an attractive mechanism to explain these contractions; another possibility is microhomology-mediated end joining (MMEJ), which uses short stretches of homology (5–25 bp) to align the broken DNA ends and promote ligation to resolve the lesion (120)(Figure 2B, C). Indeed, in a yeast system where breaks are induced within a CAG repeat, contraction via SSA is a prominent outcome (121), and induction of a DSB by a CAG/CTG-specific TALEN induced 100% contractions in a highly specific manner (122). The same mechanism appears to be operating in human cells, as a CAG-targeted DSB by a zinc finger nuclease also induced frequent contractions (123). Additionally, a nickase version of CRISPR-Cas9 directed toward CAG or CTG repeats resulted in increased large-scale contractions that depended on activation of the ATM branch of the DNA damage response to promote repair via SSA (105). This makes nuclease-directed cleavage of TNRs an attractive method for inducing contractions, which could potentially be used therapeutically (124).
Although more rare, repeat expansions also appear to occur during some end joining events (Figure 2A, 2B). In the yeast study by Sundararajan et al. (6), more than half of the expansions in a rad52Δ strain were eliminated in the rad52Δdnl4Δ background, indicating that an end-joining pathway contributes to expansions in this system. In addition, CRISPR-Cas9 induced DSBs resulted in an increase in both CAG repeat expansions and contractions, further supporting the possibility that end-joining can result in repeat expansions (105). The observation of large expansions created during repair of DSBs by HR or end-joining repair is intriguing, as large expansions that occur during maternal transmission of the DM1 CTG and Fragile X syndrome CGG repeats appears to happen during oogenesis. A prominent stage of oogenesis is meiosis, where many breaks occur and are repaired. In yeast, breaks occur frequently at CAG repeats during meiosis and are repaired to give both expansions and contractions (125, 126). Similarly, while MMEJ favors contraction events, it can also result in templated insertions during Polϴ-dependent fill-in in metazoans (120, 127). Using human proteins in a reconstituted in vitro system, it was revealed that Polβ- and Polλ-dependent microhomology-mediated strand annealing promotes CAG expansions, and the frequency of these events is limited by the 9-1-1 (Rad9/Hus1/Rad1) DNA damage checkpoint complex (128).
Homologous recombination is a mechanism for genome rearrangement
Repetitive DNA sequences can present challenges to maintaining genomic stability. Genomic changes that result from breakage at DNA structures can range from small-scale insertions/deletions to loss of heterozygosity or other large-scale genomic rearrangement, which is a hallmark of cancer genomes (11, 129). Given that structure-forming repeats break at a higher frequency than non-repetitive DNA, they are ideal DNA substrates for the machineries that drive genomic rearrangement.
DNA hairpins formed by some TNRs and long inverted repeats are thought to be cleaved at the base to form DSBs that have hairpin-capped ends (130). These ends must then be processed by the MRX/MRN complex in concert with Sae2/CtIP in order to promote recombination and repair fidelity. In the absence of Sae2 or the MRX complex, recombination is inhibited at inverted Alu repeats, which leads to inverted duplications (94). Another factor that is key to preventing genomic rearrangements is replication protein A (RPA), which coats the 3′ ssDNA that is revealed during end resection of DSBs (Figure 2C) and thus prevents DNA secondary structure formation by intramolecular base pairing at the 3′ overhang. In the absence of RPA, the 3′ DNA overhang can form hairpin-capped ends that recruit Pol32, suggesting that DNA synthesis is required to synthesize a foldback structure (131). The coordination between the MRX complex, Sae2 and RPA is important as it has been demonstrated that loss of this coordination results in palindromic gene amplification and gross chromosomal rearrangements of the type that are commonly seen in cancer cells (131, 132).
Replication barriers are also a driver of genome rearrangement by promoting template switch between ectopic sequences (133, 134). Repetitive sequences can serve as ideal homologies for template switches to occur. Indeed, simultaneous transcription and replication through a potential G4 DNA-forming sequence resulted in an increase in gross chromosomal rearrangements, where the initiating breakpoint was located within 4 kb of the G4 forming sequence (135). These rearrangements depended on topoisomerase I (Top1) to ease torsional stress associated with transcription through the G4 motifs (135). Taken together, DNA breaks that occur at structure forming sequences must be processed in such a way that DSB repair proteins have access to the broken end, and impairment of end processing can result in large-scale genome rearrangements.
CONCLUSIONS
Many repetitive DNA sequences form secondary structures that serve as constant challenges to DNA replication and repair machineries, resulting in stalled forks, nicks, gaps, and DSBs. Recombination is an important pathway to repair these lesions, and serves as a powerful guardian of the genome. However, recombinational repair at repetitive DNA tracts can be tricky as it can also be a source of mutation, including repeat expansions and contractions. This dichotomy between recombination being a high fidelity form of repair and also a source of instability is important, as it has challenged the canonical idea that HR is error free. Through the study of repeat instability, the scientific community has learned how recombination proteins such as nucleases, helicases, and chromatin remodelers navigate secondary structures in an effort to preserve genomic integrity. A better understanding of how recombination impacts repeat instability could have far-reaching impacts for both inherited genetic disorders, where repeat length correlates to disease severity, and cancers, where structure-forming repeats may initiate genomic instability. Future research will be imperative in dissecting the steps of HR and the exacerbating cellular conditions that drive mutation at repetitive DNAs.
Acknowledgments
Research in CHF’s laboratory is supported by the National Institutes of Health (award P01GM105473 and R01GM122880), National Science Foundation (MCB1330743), and Tufts University.
Abbreviations
- TNR
Trinucleotide repeat
- HR
Homologous recombination
- EJ
End joining
- DSB
Double stranded break
- SCR
Sister chromatid recombination
- NHEJ
Non homologous end-joining
- MMEJ
Microhomology-mediated end-joining
- DM1
Myotonic Dystrophy type I
- HD
Huntington’s Disease
- SCA
Spinocerebellar ataxia
- BER
Base excision repair
- TCR
Transcription-coupled repair
- TLS
Translesion synthesis
- PRR
Post-replication repair
- SSA
Single strand annealing
- G4 DNA
G-quadruplex forming DNA
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors have no conflicts of interest to declare.
For DNA Repair Special Issue DNARR17, Edited by Phillip Hanawalt “Cutting-edge Perspectives in Genomic Maintenance IV”
References
- 1.Gatchel JR, Zoghbi HY. Diseases of unstable repeat expansion: mechanisms and common principles. Nat Rev Genet. 2005;6(10):743–55. doi: 10.1038/nrg1691. doi: nrg1691 [pii]10.1038/nrg1691. [DOI] [PubMed] [Google Scholar]
- 2.McMurray CT. Mechanisms of trinucleotide repeat instability during human development. Nat Rev Genet. 2010;11(11):786–99. doi: 10.1038/nrg2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mirkin SM. DNA structures, repeat expansions and human hereditary disorders. Curr Opin Struct Biol. 2006;16(3):351–8. doi: 10.1016/j.sbi.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 4.Lopez Castel A, Nakamori M, Tome S, Chitayat D, Gourdon G, Thornton CA, Pearson CE. Expanded CTG repeat demarcates a boundary for abnormal CpG methylation in myotonic dystrophy patient tissues. Hum Mol Genet. 2011;20(1):1–15. doi: 10.1093/hmg/ddq427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Usdin K, House NC, Freudenreich CH. Repeat instability during DNA repair: Insights from model systems. Crit Rev Biochem Mol Biol. 2015;50(2):142–67. doi: 10.3109/10409238.2014.999192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sundararajan R, Gellon L, Zunder RM, Freudenreich CH. Double-strand break repair pathways protect against CAG/CTG repeat expansions, contractions and repeat-mediated chromosomal fragility in Saccharomyces cerevisiae. Genetics. 2010;184(1):65–77. doi: 10.1534/genetics.109.111039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang Y, Shishkin AA, Nishida Y, Marcinkowski-Desmond D, Saini N, Volkov KV, Mirkin SM, Lobachev KS. Genome-wide screen identifies pathways that govern GAA/TTC repeat fragility and expansions in dividing and nondividing yeast cells. Mol Cell. 2012;48(2):254–65. doi: 10.1016/j.molcel.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang Y, Saini N, Sheng Z, Lobachev KS. Genome-wide screen reveals replication pathway for quasi-palindrome fragility dependent on homologous recombination. PLoS Genet. 2013;9(12):e1003979. doi: 10.1371/journal.pgen.1003979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sundararajan R, Freudenreich CH. Expanded CAG/CTG repeat DNA induces a checkpoint response that impacts cell proliferation in Saccharomyces cerevisiae. PLoS Genet. 2011;7(3):e1001339. doi: 10.1371/journal.pgen.1001339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Malkova A, Haber JE. Mutations arising during repair of chromosome breaks. Annu Rev Genet. 2012;46:455–73. doi: 10.1146/annurev-genet-110711-155547. [DOI] [PubMed] [Google Scholar]
- 11.Carvalho CM, Lupski JR. Mechanisms underlying structural variant formation in genomic disorders. Nat Rev Genet. 2016;17(4):224–38. doi: 10.1038/nrg.2015.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heyer WD. Regulation of recombination and genomic maintenance. Cold Spring Harb Perspect Biol. 2015;7(8):a016501. doi: 10.1101/cshperspect.a016501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rodgers K, McVey M. Error-Prone Repair of DNA Double-Strand Breaks. J Cell Physiol. 2016;231(1):15–24. doi: 10.1002/jcp.25053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Richard GF, Paques F. Mini- and microsatellite expansions: the recombination connection. EMBO Rep. 2000;1(2):122–6. doi: 10.1038/sj.embor.embor606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vasquez KM, Wang G. The yin and yang of repair mechanisms in DNA structure-induced genetic instability. Mutat Res. 2013;743–744:118–31. doi: 10.1016/j.mrfmmm.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bochman ML, Paeschke K, Zakian VA. DNA secondary structures: stability and function of G-quadruplex structures. Nat Rev Genet. 2012;13(11):770–80. doi: 10.1038/nrg3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mirkin SM. Expandable DNA repeats and human disease. Nature. 2007;447(7147):932–40. doi: 10.1038/nature05977. doi: nature05977 [pii]10.1038/nature05977. [DOI] [PubMed] [Google Scholar]
- 18.Warren ST. Polyalanine expansion in synpolydactyly might result from unequal crossing-over of HOXD13. Science. 1997;275(5298):408–9. doi: 10.1126/science.275.5298.408. [DOI] [PubMed] [Google Scholar]
- 19.Richards RI, Sutherland GR. Simple repeat DNA is not replicated simply. Nat Genet. 1994;6(2):114–6. doi: 10.1038/ng0294-114. [DOI] [PubMed] [Google Scholar]
- 20.Krasilnikova MM, Mirkin SM. Replication stalling at Friedreich’s ataxia (GAA)n repeats in vivo. Mol Cell Biol. 2004;24(6):2286–95. doi: 10.1128/MCB.24.6.2286-2295.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Voineagu I, Surka CF, Shishkin AA, Krasilnikova MM, Mirkin SM. Replisome stalling and stabilization at CGG repeats, which are responsible for chromosomal fragility. Nat Struct Mol Biol. 2009;16(2):226–8. doi: 10.1038/nsmb.1527. doi: nsmb.1527 [pii] 10.1038/nsmb.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shah KA, Shishkin AA, Voineagu I, Pavlov YI, Shcherbakova PV, Mirkin SM. Role of DNA polymerases in repeat-mediated genome instability. Cell Rep. 2012;2(5):1088–95. doi: 10.1016/j.celrep.2012.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pelletier R, Krasilnikova MM, Samadashwily GM, Lahue R, Mirkin SM. Replication and expansion of trinucleotide repeats in yeast. Mol Cell Biol. 2003;23(4):1349–57. doi: 10.1128/MCB.23.4.1349-1357.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cleary JD, Tome S, Lopez Castel A, Panigrahi GB, Foiry L, Hagerman KA, Sroka H, Chitayat D, Gourdon G, Pearson CE. Tissue- and age-specific DNA replication patterns at the CTG/CAG-expanded human myotonic dystrophy type 1 locus. Nat Struct Mol Biol. 2010;17(9):1079–87. doi: 10.1038/nsmb.1876. [DOI] [PubMed] [Google Scholar]
- 25.Liu G, Leffak M. Instability of (CTG)n*(CAG)n trinucleotide repeats and DNA synthesis. Cell Biosci. 2012;2(1):7. doi: 10.1186/2045-3701-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Viterbo D, Michoud G, Mosbach V, Dujon B, Richard GF. Replication stalling and heteroduplex formation within CAG/CTG trinucleotide repeats by mismatch repair. DNA Repair (Amst) 2016;42:94–106. doi: 10.1016/j.dnarep.2016.03.002. [DOI] [PubMed] [Google Scholar]
- 27.Kerrest A, Anand RP, Sundararajan R, Bermejo R, Liberi G, Dujon B, Freudenreich CH, Richard GF. SRS2 and SGS1 prevent chromosomal breaks and stabilize triplet repeats by restraining recombination. Nat Struct Mol Biol. 2009;16(2):159–67. doi: 10.1038/nsmb.1544. doi: nsmb.1544 [pii] 10.1038/nsmb.1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nguyen JH, Viterbo D, Anand RP, Verra L, Sloan L, Richard GF, Freudenreich CH. Differential requirement of Srs2 helicase and Rad51 displacement activities in replication of hairpin-forming CAG/CTG repeats. Nucleic Acids Res. 2017 doi: 10.1093/nar/gkx088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heller RC, Marians KJ. Replisome assembly and the direct restart of stalled replication forks. Nat Rev Mol Cell Biol. 2006;7(12):932–43. doi: 10.1038/nrm2058. [DOI] [PubMed] [Google Scholar]
- 30.Byun TS, Pacek M, Yee MC, Walter JC, Cimprich KA. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005;19(9):1040–52. doi: 10.1101/gad.1301205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hashimoto Y, Chaudhuri AR, Lopes M, Costanzo V. Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat Struct Mol Biol. 2010;17(11):1305–11. doi: 10.1038/nsmb.1927. doi: nsmb.1927 [pii]10.1038/nsmb.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lambert S, Froget B, Carr AM. Arrested replication fork processing: interplay between checkpoints and recombination. DNA Repair (Amst) 2007;6(7):1042–61. doi: 10.1016/j.dnarep.2007.02.024. [DOI] [PubMed] [Google Scholar]
- 33.Lambert S, Carr AM. Impediments to replication fork movement: stabilisation, reactivation and genome instability. Chromosoma. 2013;122(1–2):33–45. doi: 10.1007/s00412-013-0398-9. [DOI] [PubMed] [Google Scholar]
- 34.Anand RP, Lovett ST, Haber JE. Break-induced DNA replication. Cold Spring Harb Perspect Biol. 2013;5(12):a010397. doi: 10.1101/cshperspect.a010397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Malkova A, Ira G. Break-induced replication: functions and molecular mechanism. Curr Opin Genet Dev. 2013;23(3):271–9. doi: 10.1016/j.gde.2013.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu Y, Kao HI, Bambara RA. Flap endonuclease 1: a central component of DNA metabolism. Annu Rev Biochem. 2004;73:589–615. doi: 10.1146/annurev.biochem.73.012803.092453. [DOI] [PubMed] [Google Scholar]
- 37.Su XA, Dion V, Gasser SM, Freudenreich CH. Regulation of recombination at yeast nuclear pores controls repair and triplet repeat stability. Genes Dev. 2015;29(10):1006–17. doi: 10.1101/gad.256404.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nagai S, Dubrana K, Tsai-Pflugfelder M, Davidson MB, Roberts TM, Brown GW, Varela E, Hediger F, Gasser SM, Krogan NJ. Functional targeting of DNA damage to a nuclear pore-associated SUMO-dependent ubiquitin ligase. Science. 2008;322(5901):597–602. doi: 10.1126/science.1162790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Freudenreich CH, Su XA. Relocalization of DNA Lesions to the Nuclear Pore Complex. FEMS Yeast Res. 2016 doi: 10.1093/femsyr/fow095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gadaleta MC, Das MM, Tanizawa H, Chang YT, Noma K, Nakamura TM, Noguchi E. Swi1Timeless Prevents Repeat Instability at Fission Yeast Telomeres. PLoS Genet. 2016;12(3):e1005943. doi: 10.1371/journal.pgen.1005943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sfeir A, Kosiyatrakul ST, Hockemeyer D, MacRae SL, Karlseder J, Schildkraut CL, de Lange T. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell. 2009;138(1):90–103. doi: 10.1016/j.cell.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Drosopoulos WC, Kosiyatrakul ST, Schildkraut CL. BLM helicase facilitates telomere replication during leading strand synthesis of telomeres. J Cell Biol. 2015;210(2):191–208. doi: 10.1083/jcb.201410061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hicks WM, Kim M, Haber JE. Increased mutagenesis and unique mutation signature associated with mitotic gene conversion. Science. 2010;329(5987):82–5. doi: 10.1126/science.1191125. doi: 329/5987/82 [pii]10.1126/science.1191125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Deem A, Keszthelyi A, Blackgrove T, Vayl A, Coffey B, Mathur R, Chabes A, Malkova A. Break-induced replication is highly inaccurate. PLoS Biol. 2011;9(2):e1000594. doi: 10.1371/journal.pbio.1000594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Iraqui I, Chekkal Y, Jmari N, Pietrobon V, Freon K, Costes A, Lambert SA. Recovery of arrested replication forks by homologous recombination is error-prone. PLoS Genet. 2012;8(10):e1002976. doi: 10.1371/journal.pgen.1002976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Saini N, Zhang Y, Nishida Y, Sheng Z, Choudhury S, Mieczkowski P, Lobachev KS. Fragile DNA motifs trigger mutagenesis at distant chromosomal loci in saccharomyces cerevisiae. PLoS Genet. 2013;9(6):e1003551. doi: 10.1371/journal.pgen.1003551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tang W, Dominska M, Gawel M, Greenwell PW, Petes TD. Genomic deletions and point mutations induced in Saccharomyces cerevisiae by the trinucleotide repeats (GAA.TTC) associated with Friedreich’s ataxia. DNA Repair (Amst) 2013;12(1):10–7. doi: 10.1016/j.dnarep.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shah KA, Mirkin SM. The hidden side of unstable DNA repeats: Mutagenesis at a distance. DNA Repair (Amst) 2015;32:106–12. doi: 10.1016/j.dnarep.2015.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang G, Vasquez KM. Naturally occurring H-DNA-forming sequences are mutagenic in mammalian cells. Proc Natl Acad Sci U S A. 2004;101(37):13448–53. doi: 10.1073/pnas.0405116101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang G, Christensen LA, Vasquez KM. Z-DNA-forming sequences generate large-scale deletions in mammalian cells. Proc Natl Acad Sci U S A. 2006;103(8):2677–82. doi: 10.1073/pnas.0511084103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang G, Vasquez KM. Models for chromosomal replication-independent non-B DNA structure-induced genetic instability. Mol Carcinog. 2009;48(4):286–98. doi: 10.1002/mc.20508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Anand RP, Shah KA, Niu H, Sung P, Mirkin SM, Freudenreich CH. Overcoming natural replication barriers: differential helicase requirements. Nucleic Acids Res. 2012;40(3):1091–105. doi: 10.1093/nar/gkr836. doi: gkr836 [pii]10.1093/nar/gkr836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Daee DL, Mertz T, Lahue RS. Postreplication repair inhibits CAG.CTG repeat expansions in Saccharomyces cerevisiae. Mol Cell Biol. 2007;27(1):102–10. doi: 10.1128/MCB.01167-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dhar A, Lahue RS. Rapid unwinding of triplet repeat hairpins by Srs2 helicase of Saccharomyces cerevisiae. Nucleic Acids Research. 2008;36(10):3366–73. doi: 10.1093/nar/gkn225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Frizzell A, Nguyen JH, Petalcorin MI, Turner KD, Boulton SJ, Freudenreich CH, Lahue RS. RTEL1 inhibits trinucleotide repeat expansions and fragility. Cell Rep. 2014;6(5):827–35. doi: 10.1016/j.celrep.2014.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vannier JB, Pavicic-Kaltenbrunner V, Petalcorin MI, Ding H, Boulton SJ. RTEL1 dismantles T loops and counteracts telomeric G4-DNA to maintain telomere integrity. Cell. 2012;149(4):795–806. doi: 10.1016/j.cell.2012.03.030. [DOI] [PubMed] [Google Scholar]
- 57.Vannier JB, Sandhu S, Petalcorin MI, Wu X, Nabi Z, Ding H, Boulton SJ. RTEL1 is a replisome-associated helicase that promotes telomere and genome-wide replication. Science. 2013;342(6155):239–42. doi: 10.1126/science.1241779. [DOI] [PubMed] [Google Scholar]
- 58.Paeschke K, Capra JA, Zakian VA. DNA replication through G-quadruplex motifs is promoted by the Saccharomyces cerevisiae Pif1 DNA helicase. Cell. 2011;145(5):678–91. doi: 10.1016/j.cell.2011.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sabouri N, Capra JA, Zakian VA. The essential Schizosaccharomyces pombe Pfh1 DNA helicase promotes fork movement past G-quadruplex motifs to prevent DNA damage. BMC Biol. 2014;12:101. doi: 10.1186/s12915-014-0101-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Paeschke K, Bochman ML, Garcia PD, Cejka P, Friedman KL, Kowalczykowski SC, Zakian VA. Pif1 family helicases suppress genome instability at G-quadruplex motifs. Nature. 2013;497(7450):458–62. doi: 10.1038/nature12149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Matsuzaki K, Borel V, Adelman CA, Schindler D, Boulton SJ. FANCJ suppresses microsatellite instability and lymphomagenesis independent of the Fanconi anemia pathway. Genes Dev. 2015;29(24):2532–46. doi: 10.1101/gad.272740.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Barthelemy J, Hanenberg H, Leffak M. FANCJ is essential to maintain microsatellite structure genome-wide during replication stress. Nucleic Acids Res. 2016;44(14):6803–16. doi: 10.1093/nar/gkw433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sun H, Karow JK, Hickson ID, Maizels N. The Bloom’s syndrome helicase unwinds G4 DNA. J Biol Chem. 1998;273(42):27587–92. doi: 10.1074/jbc.273.42.27587. [DOI] [PubMed] [Google Scholar]
- 64.Huber MD, Lee DC, Maizels N. G4 DNA unwinding by BLM and Sgs1p: substrate specificity and substrate-specific inhibition. Nucleic Acids Res. 2002;30(18):3954–61. doi: 10.1093/nar/gkf530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shen J, Loeb LA. Unwinding the molecular basis of the Werner syndrome. Mech Ageing Dev. 2001;122(9):921–44. doi: 10.1016/s0047-6374(01)00248-2. [DOI] [PubMed] [Google Scholar]
- 66.Chan NL, Hou C, Zhang T, Yuan F, Machwe A, Huang J, Orren DK, Gu L, Li GM. The Werner syndrome protein promotes CAG/CTG repeat stability by resolving large (CAG)(n)/(CTG)(n) hairpins. J Biol Chem. 2012;287(36):30151–6. doi: 10.1074/jbc.M112.389791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Heller RC, Marians KJ. Replication fork reactivation downstream of a blocked nascent leading strand. Nature. 2006;439(7076):557–62. doi: 10.1038/nature04329. doi: nature04329 [pii]10.1038/nature04329. [DOI] [PubMed] [Google Scholar]
- 68.Boiteux S, Jinks-Robertson S. DNA repair mechanisms and the bypass of DNA damage in Saccharomyces cerevisiae. Genetics. 2013;193(4):1025–64. doi: 10.1534/genetics.112.145219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Saugar I, Ortiz-Bazan MA, Tercero JA. Tolerating DNA damage during eukaryotic chromosome replication. Exp Cell Res. 2014 doi: 10.1016/j.yexcr.2014.07.009. [DOI] [PubMed] [Google Scholar]
- 70.Freudenreich CH, Kantrow SM, Zakian VA. Expansion and length-dependent fragility of CTG repeats in yeast. Science. 1998;279(5352):853–6. doi: 10.1126/science.279.5352.853. [DOI] [PubMed] [Google Scholar]
- 71.Dixon MJ, Lahue R. Examining the potential role of DNA polymerases η and ζ in triplet repeat instability in yeast. DNA Repair. 2002 doi: 10.1016/s1568-7864(02)00095-2. [DOI] [PubMed] [Google Scholar]
- 72.Northam MR, Moore EA, Mertz TM, Binz SK, Stith CM, Stepchenkova EI, Wendt KL, Burgers PM, Shcherbakova PV. DNA polymerases zeta and Rev1 mediate error-prone bypass of non-B DNA structures. Nucleic Acids Res. 2014;42(1):290–306. doi: 10.1093/nar/gkt830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gangavarapu V, Prakash S, Prakash L. Requirement of RAD52 group genes for postreplication repair of UV-damaged DNA in Saccharomyces cerevisiae. Mol Cell Biol. 2007;27(21):7758–64. doi: 10.1128/MCB.01331-07. doi: MCB.01331-07 [pii] 10.1128/MCB.01331-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vanoli F, Fumasoni M, Szakal B, Maloisel L, Branzei D. Replication and recombination factors contributing to recombination-dependent bypass of DNA lesions by template switch. PLoS Genet. 2010;6(11):e1001205. doi: 10.1371/journal.pgen.1001205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Minca EC, Kowalski D. Multiple Rad5 activities mediate sister chromatid recombination to bypass DNA damage at stalled replication forks. Mol Cell. 2010;38(5):649–61. doi: 10.1016/j.molcel.2010.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhang H, Lawrence CW. The error-free component of the RAD6/RAD18 DNA damage tolerance pathway of budding yeast employs sister-strand recombination. Proc Natl Acad Sci U S A. 2005;102(44):15954–9. doi: 10.1073/pnas.0504586102. doi: 0504586102 [pii]10.1073/pnas.0504586102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.House NC, Yang JH, Walsh SC, Moy JM, Freudenreich CH. NuA4 Initiates Dynamic Histone H4 Acetylation to Promote High-Fidelity Sister Chromatid Recombination at Postreplication Gaps. Mol Cell. 2014;55(6):818–28. doi: 10.1016/j.molcel.2014.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mozlin AM, Fung CW, Symington LS. Role of the Saccharomyces cerevisiae Rad51 paralogs in sister chromatid recombination. Genetics. 2008;178(1):113–26. doi: 10.1534/genetics.107.082677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nag DK, Suri M, Stenson EK. Both CAG repeats and inverted DNA repeats stimulate spontaneous unequal sister-chromatid exchange in Saccharomyces cerevisiae. Nucleic Acids Res. 2004;32(18):5677–84. doi: 10.1093/nar/gkh901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cherng N, Shishkin AA, Schlager LI, Tuck RH, Sloan L, Matera R, Sarkar PS, Ashizawa T, Freudenreich CH, Mirkin SM. Expansions, contractions, and fragility of the spinocerebellar ataxia type 10 pentanucleotide repeat in yeast. Proc Natl Acad Sci U S A. 2011;108(7):2843–8. doi: 10.1073/pnas.1009409108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shishkin AA, Voineagu I, Matera R, Cherng N, Chernet BT, Krasilnikova MM, Narayanan V, Lobachev KS, Mirkin SM. Large-scale expansions of Friedreich’s ataxia GAA repeats in yeast. Mol Cell. 2009;35(1):82–92. doi: 10.1016/j.molcel.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Potaman VN, Bissler JJ, Hashem VI, Oussatcheva EA, Lu L, Shlyakhtenko LS, Lyubchenko YL, Matsuura T, Ashizawa T, Leffak M, Benham CJ, Sinden RR. Unpaired structures in SCA10 (ATTCT)n.(AGAAT)n repeats. J Mol Biol. 2003;326(4):1095–111. doi: 10.1016/s0022-2836(03)00037-8. [DOI] [PubMed] [Google Scholar]
- 83.Fouche N, Ozgur S, Roy D, Griffith JD. Replication fork regression in repetitive DNAs. Nucleic Acids Res. 2006;34(20):6044–50. doi: 10.1093/nar/gkl757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Follonier C, Oehler J, Herrador R, Lopes M. Friedreich’s ataxia-associated GAA repeats induce replication-fork reversal and unusual molecular junctions. Nat Struct Mol Biol. 2013;20(4):486–94. doi: 10.1038/nsmb.2520. [DOI] [PubMed] [Google Scholar]
- 85.Kim JC, Mirkin SM. The balancing act of DNA repeat expansions. Curr Opin Genet Dev. 2013;23(3):280–8. doi: 10.1016/j.gde.2013.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bhattacharyya S, Lahue RS. Saccharomyces cerevisiae Srs2 DNA helicase selectively blocks expansions of trinucleotide repeats. Mol Cell Biol. 2004;24(17):7324–30. doi: 10.1128/MCB.24.17.7324-7330.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.House NC, Koch MR, Freudenreich CH. Chromatin modifications and DNA repair: beyond double-strand breaks. Front Genet. 2014;5:296. doi: 10.3389/fgene.2014.00296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lahue RS, Frizzell A. Histone deacetylase complexes as caretakers of genome stability. Epigenetics. 2012;7(8):806–10. doi: 10.4161/epi.20922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Erlich RL, Fry RC, Begley TJ, Daee DL, Lahue RS, Samson LD. Anc1, a protein associated with multiple transcription complexes, is involved in postreplication repair pathway in S. cerevisiae. PLoS One. 2008;3(11):e3717. doi: 10.1371/journal.pone.0003717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kha DT, Wang G, Natrajan N, Harrison L, Vasquez KM. Pathways for double-strand break repair in genetically unstable Z-DNA-forming sequences. J Mol Biol. 2010;398(4):471–80. doi: 10.1016/j.jmb.2010.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Thys RG, Lehman CE, Pierce LC, Wang YH. DNA secondary structure at chromosomal fragile sites in human disease. Curr Genomics. 2015;16(1):60–70. doi: 10.2174/1389202916666150114223205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lobachev KS, Rattray A, Narayanan V. Hairpin- and cruciform-mediated chromosome breakage: causes and consequences in eukaryotic cells. Front Biosci. 2007;12:4208–20. doi: 10.2741/2381. [DOI] [PubMed] [Google Scholar]
- 93.Tang W, Dominska M, Greenwell PW, Harvanek Z, Lobachev KS, Kim HM, Narayanan V, Mirkin SM, Petes TD. Friedreich’s ataxia (GAA)n*(TTC)n repeats strongly stimulate mitotic crossovers in Saccharomyces cerevisae. PLoS Genet. 2011;7(1):e1001270. doi: 10.1371/journal.pgen.1001270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lobachev KS, Gordenin DA, Resnick MA. The Mre11 complex is required for repair of hairpin-capped double-strand breaks and prevention of chromosome rearrangements. Cell. 2002;108(2):183–93. doi: 10.1016/s0092-8674(02)00614-1. [DOI] [PubMed] [Google Scholar]
- 95.Aksenova AY, Greenwell PW, Dominska M, Shishkin AA, Kim JC, Petes TD, Mirkin SM. Genome rearrangements caused by interstitial telomeric sequences in yeast. Proc Natl Acad Sci U S A. 2013;110(49):19866–71. doi: 10.1073/pnas.1319313110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pluciennik A, Iyer RR, Napierala M, Larson JE, Filutowicz M, Wells RD. Long CTG.CAG repeats from myotonic dystrophy are preferred sites for intermolecular recombination. J Biol Chem. 2002;277(37):34074–86. doi: 10.1074/jbc.M202127200. [DOI] [PubMed] [Google Scholar]
- 97.Napierala M, Parniewski P, Pluciennik A, Wells RD. Long CTG.CAG repeat sequences markedly stimulate intramolecular recombination. J Biol Chem. 2002;277(37):34087–100. doi: 10.1074/jbc.M202128200. [DOI] [PubMed] [Google Scholar]
- 98.Hebert ML, Wells RD. Roles of double-strand breaks, nicks, and gaps in stimulating deletions of CTG.CAG repeats by intramolecular DNA repair. J Mol Biol. 2005;353(5):961–79. doi: 10.1016/j.jmb.2005.09.023. [DOI] [PubMed] [Google Scholar]
- 99.Richard GF, Goellner GM, McMurray CT, Haber JE. Recombination-induced CAG trinucleotide repeat expansions in yeast involve the MRE11-RAD50-XRS2 complex. EMBO J. 2000;19(10):2381–90. doi: 10.1093/emboj/19.10.2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mimitou EP, Symington LS. DNA end resection–unraveling the tail. DNA Repair (Amst) 2011;10(3):344–8. doi: 10.1016/j.dnarep.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ye Y, Kirkham-McCarthy L, Lahue RS. The Saccharomyces cerevisiae Mre11-Rad50-Xrs2 complex promotes trinucleotide repeat expansions independently of homologous recombination. DNA Repair (Amst) 2016;43:1–8. doi: 10.1016/j.dnarep.2016.04.012. [DOI] [PubMed] [Google Scholar]
- 102.Symington LS. Mechanism and regulation of DNA end resection in eukaryotes. Crit Rev Biochem Mol Biol. 2016;51(3):195–212. doi: 10.3109/10409238.2016.1172552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Entezam A, Usdin K. ATM and ATR protect the genome against two different types of tandem repeat instability in Fragile X premutation mice. Nucleic Acids Res. 2009;37(19):6371–7. doi: 10.1093/nar/gkp666. doi: gkp666 [pii]10.1093/nar/gkp666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Entezam A, Usdin K. ATR protects the genome against CGG.CCG-repeat expansion in Fragile X premutation mice. Nucleic Acids Res. 2008;36(3):1050–6. doi: 10.1093/nar/gkm1136. doi: gkm1136 [pii]10.1093/nar/gkm1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cinesi C, Aeschbach L, Yang B, Dion V. Contracting CAG/CTG repeats using the CRISPR-Cas9 nickase. Nat Commun. 2016;7:13272. doi: 10.1038/ncomms13272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lydeard JR, Jain S, Yamaguchi M, Haber JE. Break-induced replication and telomerase-independent telomere maintenance require Pol32. Nature. 2007;448(7155):820–3. doi: 10.1038/nature06047. [DOI] [PubMed] [Google Scholar]
- 107.Wilson MA, Kwon Y, Xu Y, Chung WH, Chi P, Niu H, Mayle R, Chen X, Malkova A, Sung P, Ira G. Pif1 helicase and Pol delta promote recombination-coupled DNA synthesis via bubble migration. Nature. 2013;502(7471):393–6. doi: 10.1038/nature12585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Saini N, Ramakrishnan S, Elango R, Ayyar S, Zhang Y, Deem A, Ira G, Haber JE, Lobachev KS, Malkova A. Migrating bubble during break-induced replication drives conservative DNA synthesis. Nature. 2013;502(7471):389–92. doi: 10.1038/nature12584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Donnianni RA, Symington LS. Break-induced replication occurs by conservative DNA synthesis. Proc Natl Acad Sci U S A. 2013;110(33):13475–80. doi: 10.1073/pnas.1309800110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kim JC, Harris ST, Dinter T, Shah KA, Mirkin SM. The role of break-induced replication in large-scale expansions of (CAG)n/(CTG)n repeats. Nat Struct Mol Biol. 2017;24(1):55–60. doi: 10.1038/nsmb.3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mayle R, Campbell IM, Beck CR, Yu Y, Wilson M, Shaw CA, Bjergbaek L, Lupski JR, Ira G. DNA REPAIR. Mus81 and converging forks limit the mutagenicity of replication fork breakage. Science. 2015;349(6249):742–7. doi: 10.1126/science.aaa8391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yoon SR, Dubeau L, de Young M, Wexler NS, Arnheim N. Huntington disease expansion mutations in humans can occur before meiosis is completed. Proc Natl Acad Sci U S A. 2003;100(15):8834–8. doi: 10.1073/pnas.1331390100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Savouret C, Garcia-Cordier C, Megret J, te Riele H, Junien C, Gourdon G. MSH2-dependent germinal CTG repeat expansions are produced continuously in spermatogonia from DM1 transgenic mice. Mol Cell Biol. 2004;24(2):629–37. doi: 10.1128/MCB.24.2.629-637.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Dilley RL, Verma P, Cho NW, Winters HD, Wondisford AR, Greenberg RA. Break-induced telomere synthesis underlies alternative telomere maintenance. Nature. 2016;539(7627):54–8. doi: 10.1038/nature20099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Roumelioti FM, Sotiriou SK, Katsini V, Chiourea M, Halazonetis TD, Gagos S. Alternative lengthening of human telomeres is a conservative DNA replication process with features of break-induced replication. EMBO Rep. 2016;17(12):1731–7. doi: 10.15252/embr.201643169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Savouret C, Brisson E, Essers J, Kanaar R, Pastink A, te Riele H, Junien C, Gourdon G. CTG repeat instability and size variation timing in DNA repair-deficient mice. EMBO J. 2003;22(9):2264–73. doi: 10.1093/emboj/cdg202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.van Veelen LR, Essers J, van de Rakt MW, Odijk H, Pastink A, Zdzienicka MZ, Paulusma CC, Kanaar R. Ionizing radiation-induced foci formation of mammalian Rad51 and Rad54 depends on the Rad51 paralogs, but not on Rad52. Mutat Res. 2005;574(1–2):34–49. doi: 10.1016/j.mrfmmm.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 118.Suwaki N, Klare K, Tarsounas M. RAD51 paralogs: roles in DNA damage signalling, recombinational repair and tumorigenesis. Semin Cell Dev Biol. 2011;22(8):898–905. doi: 10.1016/j.semcdb.2011.07.019. [DOI] [PubMed] [Google Scholar]
- 119.Lieber MR. NHEJ and its backup pathways in chromosomal translocations. Nat Struct Mol Biol. 2010;17(4):393–5. doi: 10.1038/nsmb0410-393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.McVey M, Lee SE. MMEJ repair of double-strand breaks (director’s cut): deleted sequences and alternative endings. Trends Genet. 2008;24(11):529–38. doi: 10.1016/j.tig.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Richard GF, Dujon B, Haber JE. Double-strand break repair can lead to high frequencies of deletions within short CAG/CTG trinucleotide repeats. Mol Gen Genet. 1999;261(4–5):871–82. doi: 10.1007/s004380050031. [DOI] [PubMed] [Google Scholar]
- 122.Richard GF, Viterbo D, Khanna V, Mosbach V, Castelain L, Dujon B. Highly specific contractions of a single CAG/CTG trinucleotide repeat by TALEN in yeast. PLoS One. 2014;9(4):e95611. doi: 10.1371/journal.pone.0095611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mittelman D, Moye C, Morton J, Sykoudis K, Lin Y, Carroll D, Wilson JH. Zinc-finger directed double-strand breaks within CAG repeat tracts promote repeat instability in human cells. Proc Natl Acad Sci U S A. 2009;106(24):9607–12. doi: 10.1073/pnas.0902420106. doi: 0902420106 [pii] 10.1073/pnas.0902420106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Richard GF. Shortening trinucleotide repeats using highly specific endonucleases: a possible approach to gene therapy? Trends Genet. 2015;31(4):177–86. doi: 10.1016/j.tig.2015.02.003. [DOI] [PubMed] [Google Scholar]
- 125.Jankowski C, Nasar F, Nag DK. Meiotic instability of CAG repeat tracts occurs by double-strand break repair in yeast. Proc Natl Acad Sci U S A. 2000;97(5):2134–9. doi: 10.1073/pnas.040460297. doi: 10.1073/pnas.040460297 040460297 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Schweitzer JK, Reinke SS, Livingston DM. Meiotic alterations in CAG repeat tracts. Genetics. 2001;159(4):1861–5. doi: 10.1093/genetics/159.4.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chan SH, Yu AM, McVey M. Dual roles for DNA polymerase theta in alternative end-joining repair of double-strand breaks in Drosophila. PLoS Genet. 2010;6(7):e1001005. doi: 10.1371/journal.pgen.1001005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Crespan E, Czabany T, Maga G, Hubscher U. Microhomology-mediated DNA strand annealing and elongation by human DNA polymerases lambda and beta on normal and repetitive DNA sequences. Nucleic Acids Res. 2012;40(12):5577–90. doi: 10.1093/nar/gks186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lu S, Wang G, Bacolla A, Zhao J, Spitser S, Vasquez KM. Short Inverted Repeats Are Hotspots for Genetic Instability: Relevance to Cancer Genomes. Cell Rep. 2015 doi: 10.1016/j.celrep.2015.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Darmon E, Eykelenboom JK, Lincker F, Jones LH, White M, Okely E, Blackwood JK, Leach DR. E. coli SbcCD and RecA control chromosomal rearrangement induced by an interrupted palindrome. Mol Cell. 2010;39(1):59–70. doi: 10.1016/j.molcel.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Chen H, Lisby M, Symington LS. RPA coordinates DNA end resection and prevents formation of DNA hairpins. Mol Cell. 2013;50(4):589–600. doi: 10.1016/j.molcel.2013.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Deng SK, Chen H, Symington LS. Replication protein A prevents promiscuous annealing between short sequence homologies: Implications for genome integrity. Bioessays. 2015;37(3):305–13. doi: 10.1002/bies.201400161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hu L, Kim TM, Son MY, Kim SA, Holland CL, Tateishi S, Kim DH, Yew PR, Montagna C, Dumitrache LC, Hasty P. Two replication fork maintenance pathways fuse inverted repeats to rearrange chromosomes. Nature. 2013;501(7468):569–72. doi: 10.1038/nature12500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Mizuno K, Miyabe I, Schalbetter SA, Carr AM, Murray JM. Recombination-restarted replication makes inverted chromosome fusions at inverted repeats. Nature. 2013;493(7431):246–9. doi: 10.1038/nature11676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Yadav P, Owiti N, Kim N. The role of topoisomerase I in suppressing genome instability associated with a highly transcribed guanine-rich sequence is not restricted to preventing RNA:DNA hybrid accumulation. Nucleic Acids Res. 2016;44(2):718–29. doi: 10.1093/nar/gkv1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Liu J, Renault L, Veaute X, Fabre F, Stahlberg H, Heyer WD. Rad51 paralogues Rad55-Rad57 balance the antirecombinase Srs2 in Rad51 filament formation. Nature. 2011;479(7372):245–8. doi: 10.1038/nature10522. [DOI] [PMC free article] [PubMed] [Google Scholar]