

SUMMARY
Drug-resistant bacterial pathogens pose an urgent public-health crisis. Here, we report the discovery, from microbial-extract screening, of a nucleoside-analog inhibitor that inhibits bacterial RNA polymerase (RNAP) and exhibits antibacterial activity against drug-resistant bacterial pathogens: pseudouridimycin (PUM). PUM is a natural product comprising a formamidinylated, N-hydroxylated Gly-Gln dipeptide conjugated to 6′-amino-pseudouridine. PUM potently and selectively inhibits bacterial RNAP in vitro, inhibits bacterial growth in culture, and clears infection in a mouse model of Streptococcus pyogenes peritonitis. PUM inhibits RNAP through a binding site on RNAP (the NTP addition site) and mechanism (competition with UTP for occupancy of the NTP addition site) that differ from those of the RNAP inhibitor and current antibacterial drug rifampin (Rif). PUM exhibits additive antibacterial activity when co-administered with Rif, exhibits no cross-resistance with Rif, and exhibits a spontaneous resistance rate an order-of-magnitude lower than that of Rif. PUM is a highly promising lead for antibacterial therapy.
Graphical Abstract
INTRODUCTION
There is an urgent need for new antibacterial drugs effective against bacterial pathogens resistant to current drugs (reviewed in Marston et al., 2016; Brown and Wright, 2016).
Bacterial RNAP is a proven target for broad-spectrum antibacterial therapy (reviewed in Mariani and Maffioli, 2009; Ho et al., 2009; Aristoff et al., 2010; Srivastava et al. 2011). The suitability of bacterial RNAP as a target for broad-spectrum antibacterial therapy follows from the fact that bacterial RNAP is an essential enzyme (permitting efficacy), the fact that bacterial RNAP subunit sequences are highly conserved (providing a basis for broad-spectrum activity), and the fact that bacterial RNAP-subunit sequences are not highly conserved in eukaryotic RNAP I, RNAP II, and RNAP III (providing a basis for therapeutic selectivity).
Bacterial RNA polymerase (RNAP) is the target of two classes of antibacterial drugs currently in clinical use: (1) rifamycins (rifampin, rifapentine, rifabutin, and rifamixin), which function by binding to a site adjacent to the RNAP active center and sterically inhibiting extension of short RNA products (Campbell et al., 2001 Feklistov et al., 2008; Lin et al., 2017); and (2) lipiarmycins (fidaxomicin), which function by binding to a site distant from the RNAP active-center and allosterically inhibiting initial RNAP-DNA interaction (Ebright, 2005; Srivastava et al., 2011). Bacterial RNAP also is the target of a class of antibacterial agents currently in preclinical development: myxopyronins, which function by binding to a site distant from the RNAP active-center and allosterically inhibiting opening of, and loading of DNA into, the RNAP active-ceneter cleft (Mukhopadhyay et al., 2008; Srivastava et al., 2011). Rifamycins, lipiarmycins, and myxopyronins are subject to spontaneous resistance emergence (Mariani and Maffioli, 2009; Ho et al., 2009; Aristoff et al., 2010; Srivastava et al. 2011, 2012). Resistance to rifamycins, lipiarmycins, and myxopyronins arises from mutations that result in substitution of the respective binding sites on RNAP for the compounds, preventing binding of the compounds.
Nucleoside-analog inhibitors (NAIs) that selectively inhibit viral nucleotide polymerases have had transformative impact on treatment of HIV (e.g., AZT, DDI, DDC, 3TC, d4T, and tenofovir; reviewed in Cihlar and Ray, 2010) and HCV (e.g., sofosbuvir; reviewed in Summers et al., 2014). NAIs that selectively inhibit bacterial RNAP potentially could have an analogous impact on the treatment of bacterial infections, particularly because functional constraints on substitution of RNAP nucleoside-triphosphate (NTP) binding sites could limit substitutions that confer resistance (Summers et al., 2014; Zhang et al., 2014).
Here, we report the discovery, from microbial-extract screening, of the first NAI that selectively inhibits bacterial RNAP.
RESULTS AND DISCUSSION
Identification of PUM
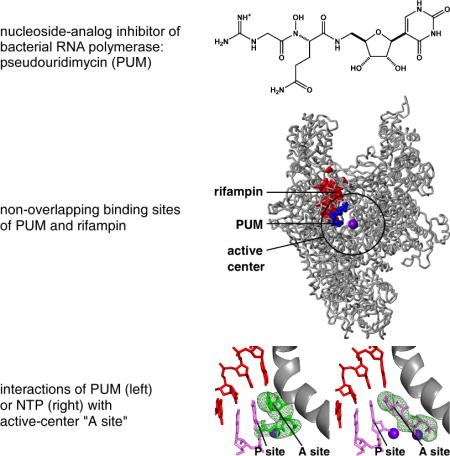
We screened a library of 3,000 Actinobacterial (Landwehr et al., 2016) and fungal culture extracts for the ability to inhibit RNAP, and we identified two extracts that inhibited bacterial RNAP (E. coli RNAP) but did not inhibit a structurally unrelated bacteriophage RNAP (SP6 RNAP) and did not contain a previously characterized inhibitor of bacterial RNAP (see Methods). Fractionation of the two extracts by reversed-phase chromatography and structure elucidation of active components by mass spectrometry and multidimensional NMR spectrometry revealed that the extracts contained the same active component: pseudouridimycin (PUM; Figs. 1A, S1).
Figure 1. Structure, RNAP-inhibitory activity, and antibacterial activity of PUM.
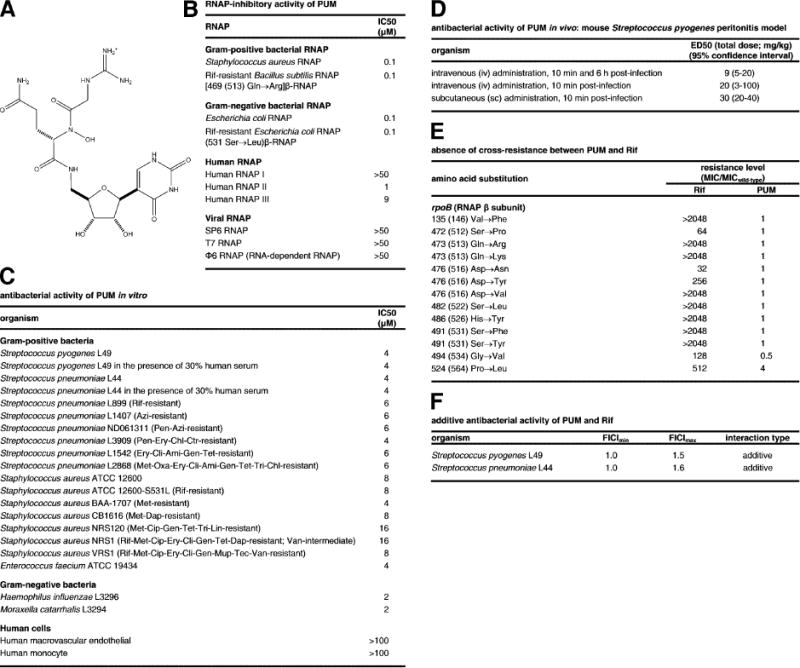
(A) Structure of PUM. (B) RNAP-inhibitory activity of PUM. (C) Antibacterial activity of PUM in vitro. (D), Antibacterial activity of PUM in vivo. Drug resistances are as follows: Ami. amikacin; Azi, azithromycin; Cip, ciprofloxacin; Ctr, ceftriaxone; Dap, daptomycin; Ery, erythromycin; Chl, chloramphenicol; Cli, clindamycin; Gen, gentamycin; Lin, linezolid; Met, methicillin; Mup, mupirocin; Pen, penicillin; Ox, oxacillin; Rif, rifampin; Tec, teicoplanin; Tet, tetracycline; Tri, trimethoprim; Van, vancomycin. (E) Absence of cross-resistance between PUM and Rif (data for S. pyogenes Rif-resistant mutants; residues numbered as in S. pyogenes and, in parentheses, E. coli). (F) Additive antibacterial activity of PUM and Rif.
RNAP-inhibitory and antibacterial activity of PUM
PUM selectively inhibits bacterial RNAP (IC50 = 0.1 μM; selectivity >4- to >500-fold; Figs. 1B, S2; Table S1), selectively inhibits bacterial growth (IC50 = 2 to 16 μM; selectivity >6- to >60-fold; Fig. 1C), and clears infection in vivo in a mouse Streptococcus pyogenes peritonitis model (ED50 = 9 mg/kg; Fig. 1C; Table S2). PUM exhibits antibacterial activity against both Gram-positive and Gram-negative bacteria and against both drug-sensitive and drug-resistant bacterial strains, including rifamycin-, β-lactam-, fluoroquinolone- macrolide-, tetracycline-, aminoglycoside-, lincosamide-, chloramphenicol-, oxazolidinone-, trimethoprim-, glycopeptide-, lipopeptide-, mupirocin-, and multi-drug-resistant strains (Fig. 1C).
PUM exhibits no cross-resistance with the classic RNAP inhibitor Rif (Figs. 1B–C,E), exhibits additive antibacterial activity when co-administered with Rif (Fig. 1F), and exhibits spontaneous resistance rates an order-of-magnitude lower than those of Rif (Fig. 2A), suggesting that PUM inhibits RNAP through a binding site and mechanism different from those of Rif.
Figure 2. Target of PUM: RNAP NTP addition site.
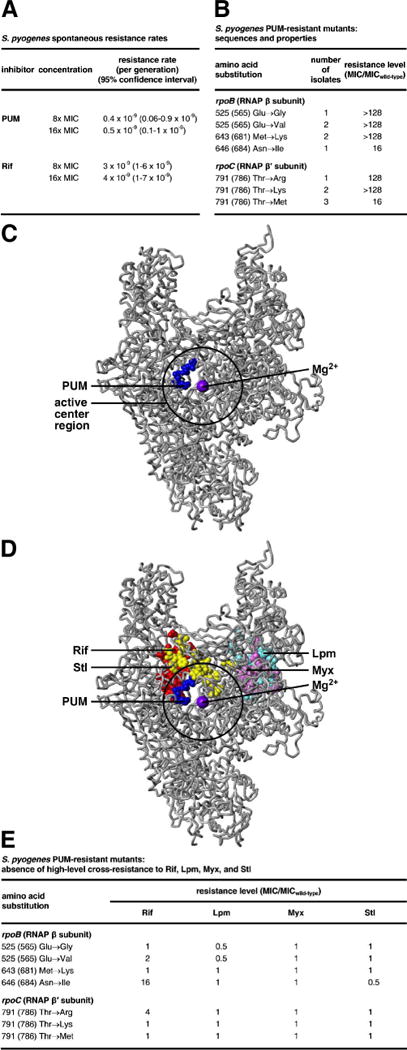
(A) Spontaneous resistance rates for PUM and Rif. (B) S. pyogenes spontaneous PUM-resistant mutants. (C) Location of PUM target (blue) in three-dimensional structure of bacterial RNAP (Mukhopadhyay et al., 2008; gray; black circle for active-center region; violet sphere for active-center Mg2+(I); β′ non-conserved region and σ omitted for clarity). (D) Absence of overlap between PUM target (blue) and Rif (red), Lpm (cyan), Myx (pink), and Stl (yellow) targets. (E), Absence of high-level cross-resistance for S. pyogenes PUM-resistant mutants to Rif, Lpm, Myx, and Stl.
See Fig. S3.
Target of transcription inihibition by PUM
Gene sequencing indicates that PUM-resistant mutants contain mutations in the rpoB gene (encodes RNAP β subunit) or the rpoC gene (encodes RNAP β′ subunit), confirming that RNAP is the functional cellular target of PUM (Figs. 2B, S3A–B). In the Gram-positive bacterium S. pyogenes, substitutions conferring ≥4x PUM-resistance are obtained at four sites: β residues 565, 681, and 684, and β′ residue 786 (numbered as in E. coli RNAP; Fig. 2B). In the Gram-negative bacterium E. coli, substitutions conferring PUM-resistance are obtained at two sites: β residues 565 and 681 (Fig. S3A–B). The number of sites of substitutions conferring PUM-resistance is an order-of-magnitude lower than the number of sites of substitutions conferring Rif-resistance (2 to 4 vs. 25; Jin and Gross, 1988; Garibyan et al., 2003), consistent with, and accounting for, the observation that spontaneous resistance rates for PUM are an order-of-magnitude lower than those for Rif (Fig. 2A).
Mapping the sites of substitutions conferring PUM resistance onto the three-dimensional structure of bacterial RNAP shows that the sites form a single discrete cluster (“PUM target”; Figs. 2C, S3D). The PUM target is located within the RNAP active-center region and overlaps the RNAP active-center NTP addition site (“A site” also referred to as “i+1 site”; Figs. 2C, S3D), suggesting that PUM inhibits RNAP by interfering with function of the NTP addition site. The PUM target is different from, and does not overlap, the Rif target (Figs. 2D, S3E; Jin and Gross, 1988; Garibyan et al., 2003; Campbell et al., 2001), consistent with, and accounting for, the observation that PUM does not share cross-resistance with Rif (Figs. 1B–C,E, 2E, S3F–G) and the observation that PUM and Rif exhibit additive antibacterial activity (Fig. 1F). The PUM target also is different from, and does not overlap, the targets of the RNAP inhibitors lipiarmycin (Lpm; Ebright, 2005; Srivastava et al., 2011), myxopyronin (Myx; Srivastava et al., 2011; Mukhopadhyay et al., 2008; Belogurov et al., 2009), streptolydigin (Stl; Tuske et al., 2005; Temiakov et al., 2005), CBR703 (CBR; Feng et al., 2015; Bae et al., 2015), and salinamide (Sal; Degen et al., 2014) (Figs. 2D, S3E), and, correspondingly, PUM does not exhibit cross-resistance with Lpm, Myx, Stl, CBR703, and Sal (Figs. 2E, S3F,H–I). The PUM target partly overlaps the target for the RNAP inhibitor GE23077 (GE; Fig. S3M; Zhang et al., 2014), and, correspondingly, PUM exhibits partial cross-resistance with GE (Fig. S3N).
Biochemical basis of transcription inhibition by PUM
The observation that PUM is an NAI that has the same Watson-Crick base-pairing specificity as UTP (Fig. 1A) and the observation that the PUM target overlaps the RNAP NTP addition site (Figs. 2B, S3A) suggest the hypothesis that PUM functions as an NAI that competes with UTP for occupancy of the RNAP NTP addition site. Five biochemical results support this hypothesis. First, PUM inhibits transcription by inhibiting nucleotide addition (Fig. S4). Second, high concentrations of UTP–but not high concentrations of GTP, ATP, or CTP–overcome transcription inhibition by PUM (Fig. 3A). Third, PUM inhibits transcription only on templates that direct incorporation of U (Fig. 3B). Fourth, in single-nucleotide-addition transcription reactions, PUM inhibits incorporation of U, but not G, A, or C (Fig. 3C). Fifth, in multiple-nucleotide-addition transcription reactions, PUM inhibits incorporation of U, but not G, A, or C (Fig. 3D). The results in Fig. 3C–D further establish that transcription inhibition by PUM not only requires a template position that directs incorporation of U, but also strongly prefers a preceding template position that directs incorporation of G, A, or U. We conclude that PUM functions as an NAI that competes with UTP at positions that direct incorporation of U preceded by positions that direct incorporation of G, A, or U.
Figure 3. Mechanism of PUM: competition with UTP for occupancy of RNAP NTP addition site.
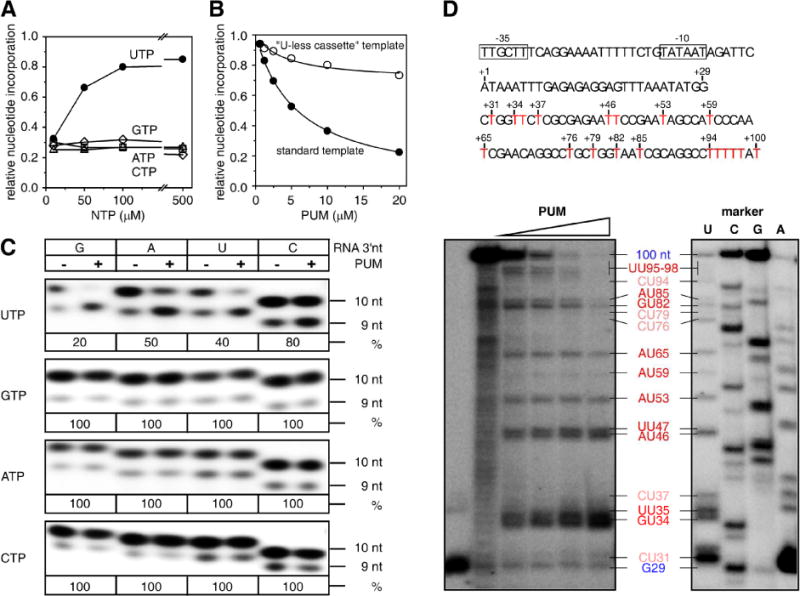
(A) Suppression of inhibition by PUM by high [UTP], but not high [GTP], [ATP], or [CTP] (E. coli RNAP). (B) Inhibition by PUM of transcription directing incorporation of U+G+A+C, but not “U-less” transcription specifying incorporation of G+A+C (E. coli RNAP). (C) Single-nucleotide-addition reactions showing that inhibition by PUM requires template positions directing incorporation of U (row 1) and prefers preceding template positions directing incorporation of G, A, or U (columns 1–3) (E. coli RNAP; 2.5 μM NTPs). UTP, GTP, ATP, or CTP (left), incoming NTP; G, A, U, or C (top), nucleotide at RNA 3′ end; 9 nt RNA (right), precursor for single-nucleotide addition; 10 nt RNA (right), product of single-nucleotide addition; % (right), percent yield of 10 nt RNA in presence of PUM vs. in absence of PUM. (D) Multiple-nucleotide-addition reactions showing that inhibition by PUM requires template positions directing incorporation of U (red GU, AU, or UU, and pink CU) and prefers preceding template positions directing incorporation of G, A, or U (red GU, AU, or UU) (E. coli RNAP; 10 μM NTPs). See Fig. S4.
Structural basis of transcription inhibition by PUM
To define the structural basis of transcription inhibition by PUM, we determined a crystal structure of a transcription initiation complex containing PUM (RPo-GpA-PUM; Fig. 4A) and, for comparison, a crystal structure of a corresponding transcription initiation complex containing CMPcPP, a non-hydrolysable NTP analog shown previously to be able to stably occupy the RNAP NTP addition site (Zhang et al., 2014) (RPo-GpA-CMPcPP; Fig. 4B). The results establish that PUM is an NAI that competes for occupancy of the RNAP NTP addition site (Fig. 4). PUM binds to the NTP addition site (Fig. 4A). The PUM base makes Watson-Crick H-bonds with a DNA template-strand A in a manner equivalent to an NTP base; the PUM sugar moiety makes interactions with the NTP addition site in a manner nearly equivalent to an NTP sugar; the PUM glutamine moiety makes interactions that mimic interactions made by an NTP triphosphate; and the PUM N-hydroxy and guanidinyl moieties interact with the RNA nucleotide base-paired to the preceding template position (RNA 3′-nucleotide), with the N-hydroxy donating an H-bond to the 3′-OH of the RNA 3′-nucleotide, and the guanidinyl moiety donating one H-bond to the 5′-phosphate of the RNA 3′-nucleotide and another to the base of the RNA 3′-nucleotide (Fig. 4A).
Figure 4. Structural basis of transcription inhibition by PUM.
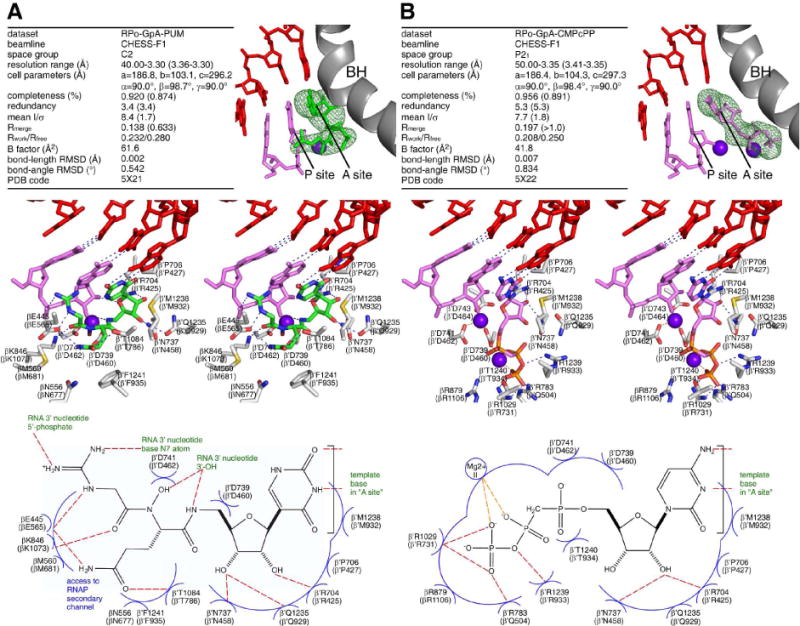
Structures of T. thermophilus transcription initiation complexes containing PUM (A) and CMPcPP (B). Top, crystallization and refinement statistics (left) and experimental electron density and fit (right). Green, PUM; pink, RNA and CMPcPP; red, DNA template strand; violet sphere between RNAP product (P) and addition (A) sites, Mg2+(I); violet sphere in RNAP addition (A) site, Mg2+(II); gray; RNAP bridge helix; green mesh, mFo-DFc omit map (contoured at 2.5σ). Middle, stereodiagram of interactions. Green, PUM carbon atoms; pink, RNA and CMPcPP carbon atoms; gray, RNAP carbon atoms; red, blue, yellow, and orange, oxygen, nitrogen, sulfur, and phosphorous atoms; dashed lines, H-bonds; other colors, as above. Bottom, summary of interactions. Red dashed lines, H-bonds; blue arcs, van der Waals interactions. Residues numbered as in T. thermophilus RNAP and, in parentheses, E. coli RNAP.
The structure of the PUM-inhibited complex accounts for the observed specificity of inhibition for template positions that direct incorporation of U preceded by template positions that direct incorporation of G, A, or U. The Watson-Crick base pair by the PUM base moiety with the DNA template strand provides absolute specificity for a position directing incorporation of U (Fig. 4A). The H-bond donated by the PUM guanidinyl moiety with the base of the RNA 3′-nucleotide confers specificity for a preceding position directing incorporation of G, A, or U (each of which contains an H-bond acceptor at the appropriate position; Fig. 4A).
The structure also explains the selectivity of transcription inhibition by PUM. All RNAP residues contacted by PUM are highly conserved across Gram-positive and Gram-negative bacterial RNAP (Fig. S5), accounting for the inhibition of both Gram-positive and Gram-negative bacterial RNAP. In contrast, four RNAP residues important for PUM are not conserved in human RNAP I, II, and III (β residues 677, 681, and 684, and β′residue 932; Fig. S5), accounting for selectivity for bacterial RNAP over human RNAP I, II, and III.
The structure also explains the small size of the PUM resistance spectrum (four residues in S. pyogenes RNAP; two residues in E. coli RNAP; Figs. 2B, S3A–B). PUM makes direct contacts with RNAP residues at which PUM-resistant substitutions are obtained (Fig. S5). However, PUM also makes direct contacts with ten other RNAP residues that comprise functionally critical residues of the RNAP active center that cannot be substituted without compromising RNAP activity (Zhang et al., 2014; Sagitov et al., 1993; Svetlov et al., 2004; Sosunov et al., 2005; Jovanovic et al., 2011; Yuzenkova et al., 2012), and thus which cannot be substituted to yield viable, fully fit, resistant mutants (Fig. 4A; Zhang et al., 2014). We infer that PUM interacts with a “privileged target” for which most residues (10–12 of 14 residues) have functional constraints that limit substitution to yield viable resistant mutants. Similar results have been reported for the RNAP inhibitor GE, a non-nucleoside-analog inhibitor that binds to the RNAP active center (Zhang et al., 2014) and that exhibits a small target-based resistance spectrum (Zhang et al., 2014) (but that, unlike PUM, exhibits high non-target-based resistance, presumably at the level of uptake or efflux, precluding development as an antibacterial drug).
The structure enables structure-based design of PUM analogs with increased potency and increased selectivity. Initial lead-optimization efforts corroborate the importance of the PUM N-hydroxy, glutamine, and guanidinyl moieties and demonstrate that the PUM glutamine C(O)NH2 can be replaced by C(O)NHR while retaining RNAP inhibitory and antibacterial activity (Fig. 5).
Figure 5. Semi-synthesis, synthesis, and analysis of PUM derivatives.
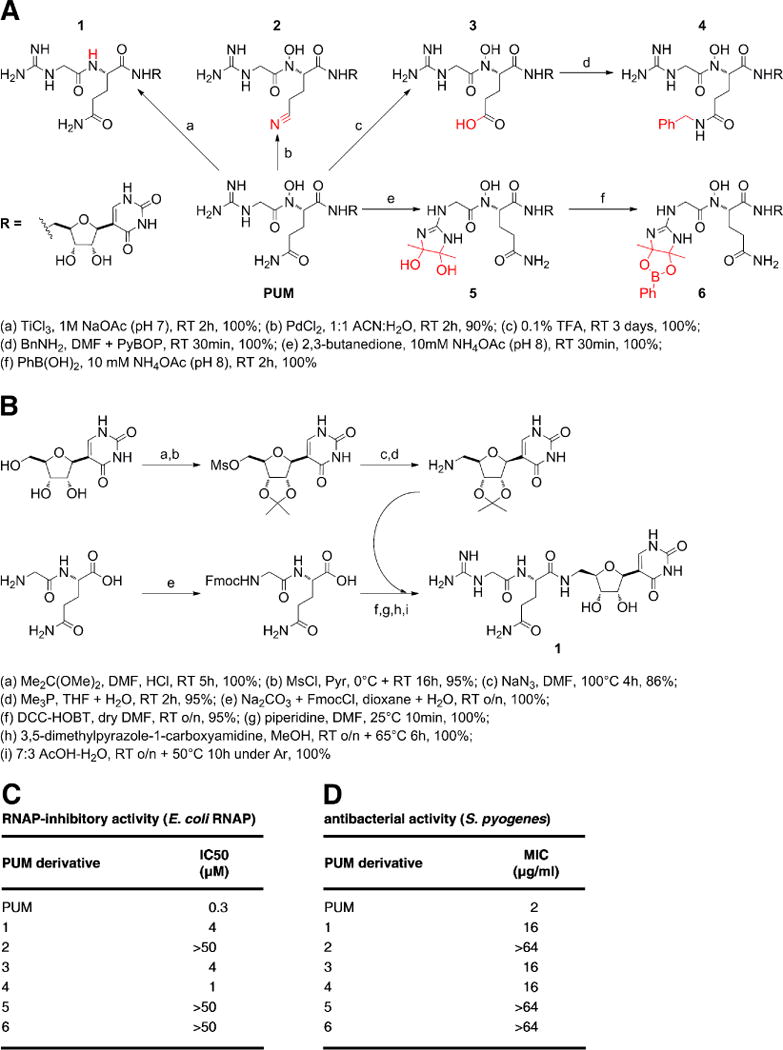
(A) Semi-synthesis of PUM derivatives lacking PUM N-hydroxy group (1), having alterations of PUM glutaminyl sidechain (2–4), or having alterations of PUM guanidinyl sidechain (5–6). (B) Synthesis of PUM derivative lacking PUM N-hydroxy group (1). (C–D) RNAP inhibitory activities and antibacterial activities of PUM derivatives.
Prospect
Our results provide a new class of antibiotic with activity against Gram-positive and Gram-negative bacteria in vitro and in vivo, no cross-resistance with current antibacterial drugs, and low rates of resistance emergence. Our discovery of this class of antibiotic from conventional microbial extract screening indicates that, contrary to widespread belief (Marston et al., 2016), conventional microbial extract screening has not been exhausted as a source of antibacterial lead compounds.
Our results provide a selective NAI of bacterial RNAP. NAIs of viral nucleotide polymerases have been of immense importance for development of anti-HIV (Cihlar and Ray, 2010) and anti-HCV (Summers et al., 2014) drugs. NAIs of bacterial RNAP may show comparable promise for development of antibacterial drugs.
STAR*METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Requests for further information or reagents should be directed to Richard H. Ebright (ebright@waksman.rutgers.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Female ICR mice were obtained from Harlan Laboratories, Italy. All mice weighed 23–25 g when tested. Mice were adapted to standardized environmental conditions (temperature = 23±2°C; humidity = 55%±10%) for one week prior to infection. Procedures were performed in accordance with the institution’s guidelines for the humane handling, care and treatment of research animals.
Cell line and cell culture
HeLa cells were grown to 70–80% confluence in DMEM, high glucose, 2 mM L-glutamine medium containing 10% fetal bovine serum and 1% penicillin-streptomycin. Cells were maintained at 37°C in a 5% CO2 incubator.
METHOD DETAILS
E. coli RNAP core enzyme
For experiments in Fig. 3C, E. coli RNAP core enzyme was prepared from E. coli strain BL21(DE3) (Invitrogen/ThermoFisher) transformed with plasmids pEcABC-H6 (Hudson et al., 2009) and pCDFω (Vrentas et al., 2005), using culture and induction procedures as in Hudson et al., 2009, and using polyethylenimine precipitation, ammonium sulfate precipitation, immobilized-metal-ion affinity chromatography on Ni-NTA agarose (Qiagen), and anion-exchange chromatography on Mono Q (GE Healthcare), as in Mukhopadhyay et al. 2003.
For experiments assessing promoter-independent transcription in Table S1, E. coli RNAP core enzyme was prepared from E. coli strain XE54 (Tang et al., 1994) transformed with plasmid pRL706 (Severinov et al., 1997), using culture and induction procedures, polyethylenimine precipitation, ammonium sulfate precipitation, immobilized-metal-ion affinity chromatography on Ni-NTA agarose (Qiagen), and anion-exchange chromatography on Mono Q (GE Health Sciences), as in Niu et al., 1996.
E. coli RNAP σ70 holoenzyme
For experiments in Fig. 1B, [531Ser→Leu]β-RNAP σ70 holoenzyme was prepared from E. coli strain XE54 (Tang et al., 1994) transformed with plasmid pRL706-531L [constructed from plasmid pRL706 (Severinov et al., 1997) by use of site-directed mutagenesis (QuikChange Site-Directed Mutagenesis Kit; Agilent)], using culture and induction procedures, polyethylenimine precipitation, ammonium sulfate precipitation, immobilized-metal-ion affinity chromatography on Ni-NTA agarose (Qiagen), and anion-exchange chromatography on Mono Q (GE Healthcare), as in Niu et al., 1996.
For experiments in Fig. S3C, E. coli RNAP σ70 holoenzyme and [565Glu→Asp]β-RNAP σ70 holoenzyme were prepared from E. coli strain XE54 (Tang et al., 1994) transformed with plasmid pRL706 (Severinov et al., 1997) or pRL706-565D (Zhang et al., 2014), using the same procedures.
For experiments in Fig. 3D, E. coli RNAP σ70 holoenzyme was prepared from E. coli strain XE54 (Tang et al., 1994) transformed with plasmid pREII-NHα (Niu et al., 1996), using culture and induction procedures, polyethylenimine precipitation, ammonium sulfate precipitation, immobilized-metal-ion affinity chromatography on Ni-NTA agarose (Qiagen), and anion-exchange chromatography on Mono Q (GE Healthcare), as in Degen et al., 2014.
S. aureus RNAP σA holoenzyme
S. aureus RNAP core enzyme was prepared from E. coli strain BL21(DE3) (Invitrogen/ThermoFisher) transformed with plasmids pCOLADuet-Sau-BC, pACYCDuet-Sau-H10-A, and pCDFDuet-Sau-Z, using polyethylenimine precipitation, ammonium sulfate precipitation, immobilized-metal-ion affinity chromatography on Ni-NTA agarose (Qiagen), and cation-exchange chromatography on HiPrep Heparin (GE Healthcare); S. aureus σA was prepared from E. coli strain BL21(DE3) transformed with pET21a-Sau-H6-sigA, using immobilized-metal-ion affinity chromatography on Ni-NTA agarose (Qiagen) and gel-filtration chromatography on Superdex 200 (GE Healthcare); and S. aureus RNAP core enzyme and S. aureus σA were combined to yield S. aureus RNAP σA holoenzyme, as in Srivastava et al., 2011.
B. subtilis RNAP σA holoenzyme
Rif-resistant B. subtilis [469(513)Gln→Arg]β-RNAP σA holoenzyme was prepared from B. subtilis strain MH5636-Q469R [spontaneous Rif-resistant mutant of B. subtilis strain MH5636 (Qi et al., 1998); selected on LB agar containing 2 μg/ml Rif; confirmed by PCR amplification and sequencing of rpoB], using immobilized-metal-ion affinity chromatography on Ni-NTA agarose (Qiagen), as in Qi et al., 1998.
T. thermophilus RNAP σA holoenzyme
T. thermophilus RNAP core enzyme was prepared was prepared from T. thermophilus strain H8 (DSM 579; DSMZ), using polyethylenimine precipitation, ammonium sulfate precipitation, cation-exchange chromatography on SP Sepharose FF (GE Healthcare), anion-excahnge chromatography on Mono Q (GE Healthcare), and cation-exchange chromatography on Mono S (GE Healthcare); T. thermophilus σA was prepared from E. coli strain BL21(DE3) transformed with pET28a-Tt-σA, using immobilized-metal-ion affinity chromatography on Ni-NTA agarose (Qiagen) and anion-exchange chromatography on Mono Q (GE Healthcare); T. thermophilus RNAP core enzyme and T. thermophilus σA were combined to yield T. thermophilus RNAP σA holoenzyme; and T. thermophilus RNAP σA holoenzyme was further purified using size-exclusion chromatography on Superdex 200 (GE Healthcare), as in Zhang et al., 2014.
Microbial extract screening
A sub-library of ~3,000 microbial extracts (prepared as in Donadio et al., 2009) with growth-inhibitory activity against Staphylococcus aureus ATCC 6538 was screened for the ability to inhibit E. coli RNAP and bacteriophage SP6 RNAP. Screening was performed using 96-well microplates. Reactions contained (50 μl): 5 μl extract (dissolved in 10% DMSO), 0.2 U E. coli RNAP σ70 holoenzyme (Sigma-Aldrich) or 0.2 U SP6 RNAP (Promega), 0.2 nM plasmid pUC18 (Clontech/Takara; for assays with E. coli RNAP) or 0.2 nM plasmid pGEM-3Z (Promega; for assays with SP6 RNAP), 500 μM ATP, 500 μM GTP, 500 μM CTP, and 2 μM [α32P]UTP (0.2 Bq/fmol; PerkinElmer), in 20 mM Tris-acetate (pH 7.9), 50 mM KCl, 4 mM magnesium acetate, 0.1 mM EDTA, 5 mM dithiothreitol, and 100 μg/ml bovine serum albumin. Reaction components except DNA were pre-equilibrated 10 min at 22°C. Reactions were initiated by addition of DNA, were allowed to proceed 1 h at 22°C, and were terminated by addition of 150 μl ice-cold 10% (w/v) trichloroacetic acid (TCA). After 1 h at 4°C, resulting TCA precipitates were collected on glass-fiber filters (UniFilter GF/B; PerkinElmer) using a 96-well harvester (Packard/PerkinElmer) and were washed once with water. Radioactivity was quantified using a TopCount scintillation counter (Packard/PerkinElmer), and % inhibition was calculated as:
where Rsample, Rpos and Rneg are observed radioactivity levels in a reaction, in a control reaction without extract, and in a control reaction without plasmid, respectively.
Two extracts that inhibited the reaction with E. coli RNAP by ≥80%, that did not inhibit the reaction with SP6 RNAP, and that did not contain mass-spectrometry signals indicative of a previously characterized RNAP inhibitor, were designated as “hit extracts.”
Characterization of producer strains
The producer strains of the hit extracts were strains ID38640 and ID38673. ID38640 and ID38673 are Actinobacterial isolates from soil samples collected in Italy and France, respectively. ID38640 and ID38673 exhibit cell morphologies consistent with the genus Streptomyces and exhibit 16S rRNA gene sequences (determined as in Mazza et al., 2003; GenBank accession numbers GI: JQ929050 and GI: JQ929051) that were 99.9% identical over 1.4 kB to each other and were highly similar to those of a cluster of closely-related Streptomyces species (S. nigrescens, S. tubercidicus, S. rimosus subsp. rimosus, S. hygroscopicus subsp. angustmyceticus, and S. libani subsp. libani).
Isolation and purification of pseudoridimycin (PUM)
For each producer strain of a hit extract, the strain was cultured on a 55 mm BTT agar (Donadio et al., 2009) plate for 4–7 days at 30°C, the mycelium was scraped from the plate and used to inoculate a 50 ml Erlenmeyer flask containing 15 ml of seed medium (20 g/L dextrose monohydrate, 2 g/L yeast extract, 8 g/L soybean meal, 1 g/L NaCl, and 4 g/L CaCO3, pH 7.3), and the resulting culture was incubated 48 h at 30°C on a rotary shaker (200 rpm agitation) for 48 h. Following initial incubation, 5 ml of the culture was used to inoculate 100 ml of fresh seed medium in a 500-ml flask, and the resulting culture was incubated 72 h under the same conditions. A 5% (v/v) inoculum was transferred into 2 L of production medium (10 g/L dextrose monohydrate, 24 g/L maize dextrin, 8 g/L soy peptone, 5 g/L yeast extract, and 1 g/L NaCl, pH 7.2) in a 3-L vessel, and the resulting culture was grown in a BioFlo 115 Fermentor (Eppendorf) 96 h at 30°C, with aeration at 0.5 volume air per volume medium per min stirring at 600 rpm. The culture was filtered through 10.25 in disc filter paper (Scienceware/Bel-Art), and the resulting cleared broth was concentrated to ~1 L in vacuo and loaded onto a column of 500 mg of Dowex 50W × 400 mesh (previously activated with two bed volumes of 5% HCl and washed with H2O until neutralization). After washing with 5 bed volumes each of 20 mM sodium acetate at pH 6 and sodium acetate at pH 7, PUM was eluted using six bed volumes of 100 mM NH4OAc at pH 9. PUM-containing fractions were desalted by reversed-phase medium-pressure liquid chromatography on Combiflash Rf (Teledyne Isco) using a 30 g C18 RediSep Rf column (Teledyne Isco) with linear gradient from 0 to 20% phase B in 20 min (phase A, 0.02% trifluoroacetic acid in H2O; phase B, acetonitrile) and flow rate of 35 ml/min. PUM-containing fractions were pooled, concentrated, and lyophilized twice to yield 196 mg of a white solid highly soluble in water, DMSO, and methanol.
Structure elucidation of PUM
Ion-trap ESI-MS (Bruker Esquire 3000 Plus showed a protonated molecular ion at m/z 487 [M+H]+ and a bimolecular ion at 973 [2M+H]+ (Fig. S1A). Ion-trap ESI-MS/MS (Bruker Esquire 3000 Plus) showed major peaks at m/z 334, 353, 371, 389, 452, and 479 [M+H]+. HR-MS (Thermo Fisher Exactive) showed an exact mass of 487.18865, consistent with the molecular formula C17H26N8O9.
Reversed-phase HPLC (Shimadzu Series 10 with SPD-M10Avp diode array detector; Waters Symmetry Shield RP8 5μm, 250 × 4.6 mm, column; phase A = 2 mM heptafluorobutyric acid in water; phase B = 2 mM heptafluorobutyric acid in acetonitrile; gradient = 0% B at 0 min, 10% B at 20 min, 95% B at 30 min; flow rate = 1 ml/min) showed a single peak with a retention time of 12 minutes. The UV-absorbance spectrum showed maxima at 200 nm and 262 nm, consistent with the presence of a pyrimidine moiety (Ploeser and Loring, 1949; Fig. S1A).
The 1H NMR spectrum (600 MHz Bruker spectrometer in DMSO-d6 at 25°C) revealed one olefinic (H6), five amide (H6′, H3, H5, Hα, and Hε-Gln), four methylene (Hβ-Gln, Hγ-Gln, Hα-Gly, and H5′), and five methine (H1′, H2′, H3′, H4′, and Hα-Gln) signals (Fig. S1B). The 2D 1H–13C-HSQC and -HMBC NMR spectra (600 MHz Bruker spectrometer in DMSO-d6 at 25°C) identified five carboxyl-amide groups (C2, C4, C=O Gln, C=O Gly, and Cδ-Gln), two olefinic carbons (C1 and C6), four methine carbons belonging to a sugar ring (C1′, C2′, C3′, and C4′), one other methine carbon (Cα-Gln), and four methylene groups (Cα-Gly, Cβ-Gln, Cγ-Gln, and C5′) (Fig. S1C–D).
The COSY NMR spectrum (600 MHz Bruker spectrometer in DMSO-d6 at 25°C) identified correlations between the five sugar protons: H1′ (δH 4.35), H2′ (δH 3.92), H3′ (δH 3.65), H4′ (δH 3.65) and H5′ (δH-A 3.24; H-B 3.17). The chemical shift of C5′ and ribose 15N-HSQC correlations between H6′ and H5′ indicated the presence of 6′-amino-ribose. Nitrogen signals N2 and N3 in the 15N HSQC spectrum, HMBC correlations of H1′ to C1, C2 and C6, and HMBC correlations of H6 to C1′, C1, C2, and C6, indicated the presence of uracil C-linked to C1′ of 6′-amino-ribose. 13C-NMR spectra and COSY indicated the presence of a glutaminyl moiety, and HMBC correlations of H6′ and the methine at δH 4.72 (Hα-Gln) to the carbonyl at δc 169.5 indicated linkage of the glutaminyl moiety to N6′ of 6′-amino-ribose. Nε at δ 108.9 was assigned by 15N HSQC. The absence of a glutaminyl Nα signal in the 15N HSQC spectrum suggested the possible presence of an hydroxamic acid, and this was confirmed by reduction of PUM with aqueous TiCl3 (Mattingly and Miller, 1980) to yield desoxy-PUM (1 in Fig. 5A), exhibiting ion-trap ESI-MS mass of 471 [M+H]+, ion-trap ESI-MS/MS major peaks at m/z 355 and 372 [M+H]+, and a new nitrogen peak at δ117.7 in the 15N HSQC spectrum assignable as glutaminyl Nα, with corresponding NH at δH 8.34 coupling with δH 4.24 (Hα-Gln). The presence of two protons (Hα1 and Hβ2) coupling to a HN (δN 75) in COSY, the HMBC correlations of Hα1 and Hβ2 to a carbonyl at δc 157, and a NOESY NMR spectrum (600 MHz Bruker spectrometer in DMSO-d6 at 25°C) of desoxy-PUM indicated the presence of glycine C-linked to glutamine Nα. The chemical shift HN (δN 75) with corresponding δH 7.43 indicated the presence of formamidine C-linked to glycine Nα.
The stereochemistry of the glutamine residue was established to be (L) by total hydrolysis followed by chiral GC-MS (Hewlett-Packard HP5985B GC-MS; procedures as in Kettenring et al., 1991). The stereochemistry of the ribose sugar was inferred to be D by analogy to the natural product pseudouridine and was confirmed to be D by comparison of desoxy-PUM prepared by reduction of PUM with TiCl3 to desoxy-PUM prepared by total synthesis using a D-ribose precursor (Fig. 5A–B).
Effects of PUM on macromolecular synthesis
Cultures of Staphylococcus simulans strain M22 in 0.5x Mueller Hinton II broth (BD Biosciences) were incubated at 37°C with shaking until OD600 = 0.5; diluted in the same medium to OD600 = 0.1–0.2; supplemented with 6 kBq/ml [2-14C]-thymidine (Hartmann Analytic), 40 kBq/ml [5-3H]-uridine (Hartmann Analytic), or 6 kBq/ml L-[14C(U)]-isoleucine (Hartmann Analytic); and further incubated at 37°C with shaking. After 15 min, cultures were divided into two equal aliquots, PUM was added to one aliquot to a final concentration of 100–200 μM, and cultures were further incubated at 37°C with shaking. At time points 0, 10, 20, and 40 min following addition of PUM, 200μl aliquots were removed, mixed with 2 ml ice-cold 10% TCA containing 1 M NaCl, and incubated 30–60 min on ice. The resulting TCA precipitates were collected by filtration on glass-microfiber filters (GF/C; Whatman/GE Healthcare), and filters were washed with 5 ml 2.5% TCA containing 1 M NaCl, transferred to scintillation vials, and dried. Filtersafe scintillation fluid was added (2 ml; Zinnser Analytic), and radioactivity was quantified by scintillation counting (Tri-Carb 3110TR Liquid Scintillation Analyzer; PerkinElmer).
RNAP-inhibitory activity in vitro
For experiments in Fig. 1B and Table S1 assessing promoter-dependent transcription by E. coli RNAP and S. aureus RNAP, reaction mixtures contained (25 μl): 0–20 μM PUM, RNAP [1 U E. coli RNAP σ70 holoenzyme (Epicentre), 20 nM [531Ser→Leu]β-RNAP σ70 holoenzyme, or 20 nM S. aureus σA RNAP holoenzyme], 10 nM DNA fragment carrying positions −112 to −1 of E. coli recA promoter (Sancar et al., 1980) followed by transcribed-region positions +1 to +363 of HeLaScribe Positive Control DNA (Promega; sequence at https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4021065/bin/1471-2199-15-7-S1.docx; yields 363 nt transcript), 20 μM [α32P]GTP (0.3 Bq/fmol; PerkinElmer), 400 μM ATP, 400 μM CTP, and 6.25 μM UTP (6.25 μM, 50 μM, or 250 μM UTP for Table S1), in 10 mM Tris-HCl (pH. 7.8), 2 mM HEPES-NaOH, 40 mM KCl, 3 mM MgCl2, 0.2 mM dithiothreitol, 0.09 mM EDTA, 0.2 mM phenylmethylsulfonyl fluoride, and 10% glycerol. Reaction components except RNAP were pre-equilibrated 10 min at 30°C. Reactions were initiated by addition of RNAP, were allowed to proceed 15 min at 30°C, and were terminated by addition of 175 μl HeLa Extract Stop Solution (Promega). Samples were phenol extracted and ethanol precipitated (procedures as in Sambrook and Russell, 2001), and pellets were resuspended in 10μl 47.5% formamide, 10 mM EDTA, 0.025% bromophenol blue, and 0.01% xylene cyanol and heated 5 min at 95°C. Products were applied to 6% polyacrylamide (19:1 acrylamide:bisacrylamide; 7 M urea) slab gels (Sambrook and Russell, 2001), and gels were electrophoresed in TBE (Sambrook and Russell, 2001) at 10 V/cm for 1.5 h, dried using a gel dryer (BioRad), and analyzed by storage-phosphor imaging (Typhoon; GE Healthcare).
For experiments in Fig. 1B and Table S1 assessing promoter-dependent transcription by human RNAP I, human RNAP II, and human RNAP III, reaction mixtures contained (25 μl): 0–80 μM PUM, HeLa nuclear extract [3 μl HeLa nuclear extract prepared as in 38, using ~4×107 HeLa cells grown to 70–80% confluence in DMEM, high glucose, 2 mM L-glutamine medium containing 10% fetal bovine serum (Gibco/ThermoFisher) and 1% penicillin-streptomycin (Gibco/ThermoFisher) for assays of human RNAP I; or 6 U HeLaScribe Nuclear Extract (Promega) for assays of human RNAP II and human RNAP III], promoter DNA [4 nM EcoRI-linearized plasmid pHrP2x (Pleiderer et al., 1990) carrying human rDNA promoter for assays of human RNAP I (yields 379 nt transcript; 20 nM HeLaScribe Positive Control DNA (Promega) carrying cytomegalovirus immediate early promoter for assays of human RNAP II (yields 363 nt transcript with same sequence as E.-coli-RNAP-dependent transcript of preceding paragraph); or 2 nM plasmid pVAI (Dean and Berk, 1988) carrying adenovirus VAI promoter for assays of human RNAP III (yields 160 nt transcript)], 20 μM [α32P]GTP (0.3 Bq/fmol: PerkinElmer), 400 μM ATP, 400 μM CTP, and 6.25, 50, or 250 μM UTP, in transcription buffer [12 mM HEPES-NaOH (pH 7.9), 75 mM KCl, 5 mM MgCl2, 10 mM creatine phosphate, 0.5 mM dithiothreitol, 0.1 mM EDTA, and 12% glycerol, for assays with human RNAP I; 10 mM Tris-HCl (pH 7.8), 2 mM HEPES-NaOH, 44 mM KCl, 3 mM MgCl2, 0.2 mM dithiothreitol, 0.09 mM EDTA, 0.2 mM phenylmethylsulfonyl fluoride, and 10% glycerol, for assays with human RNAP II and human RNAP III]. Procedures were as in the preceding paragraph.
For experiments in Fig. 1B assessing transcription by B. subtilis RNAP, SP6 RNAP, and T7 RNAP, reaction mixtures contained (50 μl): 0–200 μM PUM, RNAP [0.2 U nM Rif-resistant B. subtilis [469(513)Gln→Arg]β-RNAP σA holoenzyme (units defined as in Qi et al., 1998), 0.2 U SP6 RNAP (Promega), or 0.2 U T7 RNAP (Promega)], DNA [0.2 nM plasmid pUC18 (Clontech; for assays with B. subtilis RNAP), or 0.2 nM plasmid pGEM-3Z (Promega; for assays with SP6 RNAP and T7 RNAP)], 500 μM ATP, 500 μM GTP, 500 μM CTP, and 6.25 μM [α32P]UTP (0.2 Bq/fmol; PerkinElmer; 6.25, 50, or 250 μM for Table S1), in 40 mM Tris-HCl (pH 7.9), 6 mM MgCl2, 2 mM spermidine, 10 mM NaCl, 10 mM dithiothreitol, and 10μg/ml bovine serum albumin. Reaction components except DNA were pre-equilibrated 15 min at 37°C. Reactions were initiated by addition of DNA, were allowed to proceed 15 min at 37°C, and were terminated by addition of 150 μl ice-cold 10% (w/v) TCA. After 30 min on ice, the resulting TCA precipitates were collected on glass-fiber filters (UniFilter GF/C; PerkinElmer) using a 96-well harvester (Packard/PerkinElmer). filters were washed once with water, and radioactivity was quantified using a TopCount scintillation counter (Packard/PerkinElmer).
For experiments in Fig. 1B assessing transcription by ϕ6 RNA-dependent RNAP, reaction mixtures contained (20 μl): 0–400 μM PUM, 0.5 U ϕ6 RNAP (Thermo Fisher), 2 μg poly(A) ssRNA (GE Healthcare), 1 μM poly(U-15) ssRNA primer (Sigma-Aldrich), 400 μM ATP, 400 μM GTP, 400 μM CTP, and 1.56 μM [α32P]UTP (0.02 Bq/fmol; Perkin-Elmer) in 50 mM Tris-acetate (pH 8.7), 50 mM ammonium acetate, and 1.5 mM MnCl2. Reaction components other than RNA template, RNA primer, and NTPs were pre-incubated 10 min at 30°C. Reactions were initiated by addition of RNA template, RNA primer, and NTPs, reactions were allowed to proceed 1 h min at 30°C, and reactions were terminated by spotting on DE81 filter discs (Whatman; pre-wetted with water) and incubating 1 min at 22°C. Filters were washed with 3×3 ml 0.5 M sodium phosphate dibasic, 2×3 ml water, and 3 ml ethanol using a filter manifold (Hoefer); filters were placed in scintillation vials containing 10 ml Scintiverse BD Cocktail (ThermoFisher); and radioactivity was quantified by scintillation counting (LS6500; Beckman-Coulter).
For experiments in Table S1 assessing promoter-independent transcription by E. coli RNAP and HeLa nuclear extract (human RNAP I/II/II), reaction mixtures contained (20 μl): 0–100 μM PUM, 100 nM E. coli RNAP core enzyme or 8 U HeLaScribe Nuclear Extract (Promega), 1 μg human placental DNA (Sigma-Aldrich; catalog number D7011), 400 μM ATP, 400 μM GTP, and 400 μM CTP, and 1.56, 25, or 400 μM [α32P]UTP (0.1–1 Bq/fmol; PerkinElmer), in 40 mM Tris-HCl (pH 8.0), 7 mM HEPES-NaOH, 70 mM ammonium sulfate, 30 mM KCl, 12 mM MgCl2, 5 mM dithiothreitol, 0.1 mM EDTA, and 12% glycerol. Procedures were as in Degen et al., 2014. Reaction components other than DNA and NTPs were pre-incubated 10 min at 30°C, DNA was added, and reaction components other than NTPs were incubated 15 min at 30°C. Reactions were initiated by addition of NTPs, reactions were allowed to proceed 1 h at 30°C, and reactions were terminated by spotting on DE81 filter discs (Whatman; pre-wetted with water) and incubating 1 min at 22°C. Filters were washed, and radioactivity on filters was quantified, as in the preceding paragraph.
For experiments in Figs. S3C and 5C, fluorescence-detected transcription assays were performed as in Zhang et al., 2014.
Half-maximal inhibitory concentrations (IC50s) were calculated by non-linear regression in SigmaPlot (Systat Software).
Antibacterial activity in vitro
Antibacterial activities in vitro (Fig. 1C, rows 1–20; Fig. 5D) were determined using broth-microdilution growth-curve assays (Holowachuk et al., 2003). [PUM degrades in phosphate-containing media with a half-life of ~12 h. Broth-microdilution endpoint assays (CLSI/NCCLS, 2009), which have a run time of 16–24 h (CLSI/NCCLS, 2009), which corresponds to 1.3 to 2 PUM half-lives, underestimate absolute antibacterial activities of PUM. Broth-microdilution growth-curve assays (Holowachuk et al., 2003), which have shorter run times between assay start and assay signal, more accurately estimate absolute antibacterial activities of PUM.] Colonies of the indicated bacterial strains (5 to 10 per strain) were suspended in 3 ml phosphate-buffered saline (Sambrook and Russell, 2001), suspensions were diluted to 1×105 cfu/ml with growth medium [Todd Hewitt broth (BD Biosciences) for S. pyogenes and S. pneumoniae, aged Mueller Hinton II cation-adjusted broth (BD Biosciences; autoclaved and allowed to stand 2–12 months at room temperature before use) for Staphylococcus aureus and Enterococcus faecium, fresh Mueller Hinton II cation-adjusted broth (autoclaved and used immediately) for Moraxella catarrhalis, or fresh Mueller Hinton II cation-adjusted broth (BD Biosciences; autoclaved and used immediately) containing 0.4% Haemophilus Test Medium (Barry et al., 1993) and 0.5% yeast extract for Haemophilus influenzae], 50 μl aliquots were dispensed into wells of a 96-well microplate containing 50 μl of the same medium or 50 μl of a 2-fold dilution series of PUM in the same medium (final concentrations = 0 and 0.25–256 μM), plates were incubated at 37°C with shaking, and optical densities at 600 nm were recorded at least hourly using a Synergy 2 (BioTek) or GENios Pro (Tecan) microplate reader. For each dilution series, growth curves were plotted, areas under growth curves were calculated, and IC50 was extracted as the lowest tested concentration of PUM that reduced area under the growth curve to 50% that in the absence of PUM (using only time points for rise phase of the growth curve in the absence of PUM).
Identical results were obtained in assays in the absence and presence of 30% human serum (Sigma-Aldrich; Fig. 1C, rows 1–4), indicating that PUM does not bind tightly to human serum proteins (unbound fraction ~100%).
Cytotoxicities for human macrovascular endothelial cells and human monocytes in culture (Fig. 1C, rows 21–22) were determined as in Mazzetti et al., 2012.
Antibacterial activity in vivo
Antibacterial activity in vivo was assessed in a mouse S. pyogenes peritonitis model (Fig. 1D; Table S2). Female ICR mice (weight = 23–25 g; Harlan Laboratories Italy) were adapted to standardized environmental conditions (temperature = 23 ± 2°C; humidity = 55% ± 10%) for one week prior to infection. Infection was induced by intraperitoneal injection of 0.5 ml saline solution (supplemented with 1% peptone) containing 4 × 103 cfu S. pyogenes C203 (an inoculum resulting in ≥95% mortality in untreated controls within 48 to 72 h after infection). Infected mice (eight mice per group; number determined by power calculations; assigned randomly to groups; unblinded) were treated with either: (i) 0.2 ml 5% dextrose or 0.2 ml of a 2.5-fold dilution series of PUM in 5% dextrose, administered intravenously 10 min after infection and again 6 h after infection (total PUM dose = 0 or 3.2–50 mg/kg), (ii) 0.25 ml 5% dextrose or 0.25 ml of a 2.5-fold dilution series of PUM in 5% dextrose, administered intravenously 10 min after infection (total PUM dose = 0 or 1.024–40 mg/kg), or (iii) or 0.25 ml 5% dextrose or 0.25 ml of a 2.5-fold dilution series of PUM in 5% dextrose, administered subcutaneously 10 min after infection (total PUM dose = 0 or 1.024–40 mg/kg). Survival was monitored for 7 days after infection. Experiments were performed in compliance with vertebrate animal ethical regulations and with Institutional Animal Care and Use Committee (IACUC) approval.
ED50s (doses yielding 50% survival at 7 days) and 95% confidence limits were calculated using the trimmed Spearman-Karber method as implemented in the US EPA LC50 Model System (Hamilton et al., 1977; http://sdi.odu.edu/model/lc50.php).
Checkerboard interaction assays
Antibacterial activities of combinations of PUM and Rif were assessed in checkerboard interaction assays (White et al., 1996; Meletiadis et al., 2010; Fig. 1E). Broth-macrodilution assays (procedures as in CLSI/NCCLS, 2009) were performed in checkerboard format, using S. pyogenes strain L49 or S. pneumoniae strain L44, and using Todd Hewitt broth (BD Biosciences) containing pairwise combinations of: (i) PUM at 1x, 0.5x, 0.25x, 0.125x, 0.063x, 0.031x, 0.016x, and 0.0078x MICPUM and (ii) Rif at 0.8x, 0.4x, 0.2x, 0.1x, 0.05x, 0.025x, and 0.0125x MICRif. Fractional inhibitory concentrations (FICs), FIC indices (FICIs), and minimum and maximum FICIs (FICImin and FICImax) were calculated as in Meletiadid et al., 2010. FICImin ≤ 0.5 was deemed indicative of super-additivity (synergism), FICImin > 0.5 and FICImax ≤ 4.0 was deemed indicative of additivity, and FICImax > 4.0 was deemed indicative of sub-additivity (antagonism) (White et al., 1996; Meletiadis et al., 2010).
Spontaneous resistance rate assays
Spontaneous resistance rates were determined in Luria-Delbrück fluctuation assays (Fig. 2A; procedures as in Srivastava et al., 2012). S. pyogenes strain ATCC 12344 (~1 × 109 cfu/plate) was plated on Todd Hewitt agar [Todd Hewitt broth (BD Biosciences) supplemented with 1.5% Bacto agar (BD Biosciences)] containing 64 μg/ml or 128 μg/ml PUM (8x or 16x MIC under these conditions) or 1 μg/ml or 2 μg/ml Rif (8x or 16x MIC under these conditions), and numbers of colonies were counted after 24 h at 37°C (at least six independent determinations each). Resistance rates and 95% confidence intervals were calculated using the Ma-Sandri-Sarkar Maximum Likelihood Estimator (Ma et al., 1992; Sarkar et al., 1992) as implemented on the Fluctuation Analysis Calculator (Hall et al., 2009; http://www.keshavsingh.org/protocols/FALCOR.html).
Spontaneous PUM-resistant mutants, S. pyogenes
To isolate spontaneous PUM-resistant mutants of S. pyogenes (Fig. 2B), a single colony of S. pyogenes ATCC 12344 was inoculated into 5 ml Todd Hewitt broth (BD Biosciences) and incubated 3 h at 37°C with shaking in a 7% CO2/6% O2/4% H2/83% N2 atmosphere (atmosphere controlled using Anoxomat AN2CTS; Advanced Instruments), the culture was centrifuged, and the cell pellet (~1 × 109 cells) was re-suspended in 50 μl Todd Hewitt broth and plated on Todd Hewitt agar (BD Biosciences) containing 16–256 μg/ml PUM (2–32x MIC under these conditions), and plates were incubated 120 h at 37°C in a 7% CO2/6% O2/4% H2/83% N2 atmosphere. PUM-resistant mutants were identified by the ability to form colonies on these media, were confirmed to be PUM-resistant by re-streaking on the same media, and were confirmed to be S. pyogenes, (as opposed to contaminants) using Taxo A differentiation discs (BD Biosciences) and Pyrase strips (Fluka/Sigma-Aldrich).
Genomic DNA was isolated using the Wizard Genomic DNA Purification Kit [Promega; procedures as specified by the manufacturer, but with cells lysed using 1 mg/ml lysozyme (Sigma-Aldrich)] and was quantified by measurement of UV-absorbance (procedures as in Sambrook and Russell, 2001). The rpoC gene and the rpoB gene were PCR-amplified in reactions containing 0.2 μg genomic DNA, 0.4 μM forward and reverse oligodeoxyribonucleotide primers (5′-GGGCAAATGATAACTTAGTTGCGATTTGCTG-3′ and 5′-CCTTTCTGCCTTTGATGACTTTACCAGTTC-3′ for rpoB; 5′-GCTCAAGAAACTCAAGAAGTTTCTGAAACAACTGAC-3′ and 5′-GTCAATGCTTTTTACTGCCAACAAACTCAGAC-3′ for rpoC), 5 U Taq DNA polymerase (Genscript), and 800 μM dNTP mix (200 μM each dNTP; Agilent/Stratagene) (initial denaturation step of 3 min at 94°C; 30 cycles of 30 s at 94°C, 45 s at 53°C, and 4 min at 68°C; final extension step of 10 min at 68°C). PCR products containing the rpoC gene (3.6 kB) or the rpoB gene (3.6 kB) were isolated by electrophoresis on 0.8% agarose gels (procedures as in Sambrook and Russell, 2001), extracted from gel slices using a Gel/PCR DNA Fragments Extraction Kit (IBI Scientific; procedures as specified by the manufacturer), and sequenced (GENEWIZ; Sanger sequencing; seven sequencing primers per gene).
Spontaneous PUM-resistant mutants, E. coli
To isolate spontaneous PUM-resistant mutants of E. coli (Fig. S3A), E. coli uptake-proficient, efflux-deficient strain D21f2tolC (Fralick and Burns-Keliher, 1994) was cultured to saturation in 10 ml LB broth (Sambrook and Russell, 2001) at 37°C, cultures were centrifuged, cell pellets (~1 × 1010 cells) were re-suspended in 50 μl LB broth and plated on LB agar (Sambrook and Russell, 2001) containing 800 μg/ml PUM (~1x MIC under these conditions), and plates were incubated 96–120 h at 37°C. PUM-resistant mutants were identified by the ability to form colonies on this medium.
Genomic DNA was isolated, and rpoB and rpoC genes were PCR-amplified and sequenced as in Degen et al., 2014.
Resistance and cross-resistance levels
Resistance levels of S. pyogenes and E. coli spontaneous PUM-resistant mutants (Figs. 2B, S3B) were quantified in broth-microdilution assays. A single colony of a mutant strain or the isogenic wild-type parent strain was inoculated into 5 ml Todd Hewitt broth (BD Biosciences; for S. pyogenes) or LB broth (Sambrook and Russell, 2001; for E. coli) and incubated at 37°C with shaking in a 7% CO2/6% O2/4% H2/83% N2 atmosphere (atmosphere controlled using Anoxomat AN2CTS; Advanced Instruments); for S. pyogenes) or in air (for E. coli) until OD600 = 0.4–0.8. Diluted aliquots (~2 × 105 cells in 50 μl same medium) were dispensed into wells of a 96-well microplate containing 50 μl of the same medium or 50 μl of a 2-fold dilution series of PUM in the same medium (final PUM concentration = 0 or 0.098–800 μg/ml), and were incubated 16 h at 37°C with shaking in a 7% CO2/6% O2/4% H2/83% N2 atmosphere (for S. pyogenes) or in air (for E. coli). MIC was defined as the lowest tested concentration of PUM that inhibited bacterial growth by >90%. MIC/MICwild-type was defined as the ratio of MIC for mutant to MIC for isogenic wild-type parent (S. pyogenes MICwild-type = 6.25 μg/ml under these conditions; E. coli MICwild-type = 400 μg/ml under these conditions).
Cross-resistance levels of S. pyogenes and E. coli spontaneous PUM-resistant mutants (Figs. 2E, S3F) were determined as in the preceding paragraph, but using culture aliquots (~1×105 cells) in 97 μl growth medium supplemented with 3 μl methanol or 3 μl of a 2-fold dilution series of Rif (Sigma-Aldrich; S. pyogenes MICwild-type = 0.098 μg/ml; E. coli MICwild-type = 0.20 μg/ml), lipiarmycin A3 (Lpm; BioAustralis; S. pyogenes MICwild-type = 6.25 μg/ml; E. coli MICwild-type = 1.56 μg/ml), myxopyronin B (Myx; prepared as in Ebright and Ebright, 2013; S. pyogenes MICwild-type = 6.25 μg/ml; E. coli MICwild-type = 0.20 μg/ml), streptolydigin (Stl; Sourcon-Padena; S. pyogenes MICwild-type = 3.13 μg/ml; E. coli MICwild-type = 3.13 μg/ml), CBR703 (CBR; Maybridge; E. coli MICwild-type = 6.25 μg/ml), or salinamide A (Sal; W. Fenical, Scripps Institution of Oceanography; E. coli MICwild-type = 0.049 μg/ml) in methanol (final concentrations = 0 and 0.006–50 μg/ml), or using culture aliquots (~2×105) cells in 50 μl growth medium supplemented with 50 μl growth medium or 50 μl of a 2-fold dilution series of GE23077 (GE; prepared as in Ciciliato et al., 2004); E. coli MICwild-type = 500 μg/ml) in growth medium (final concentrations = 0 and 125–8000 μg/ml).
Cross-resistance levels of S. pyogenes Rif-resistant mutants to PUM (Fig. 1E) were determined were determined as described for cross-resistance levels of S. pyogenes spontaneous PUM-resistant mutants, but analyzing a collection of 13 S. pyogenes spontaneous Rif-resistant mutants [isolated and sequenced using the same procedures used for isolation and sequencing of S. pyogenes PUM-resistant mutants (Methods, Spontaneous PUM-resistant mutants, S. pyogenes), but using Todd Hewitt agar containing 1–16x MIC Rif (0.1–2 μg/ml under these conditions)] and the isogenic wild-type parent, and analyzing a 2-fold dilution series of PUM (final concentration = 0 or 1.56–100 μg/ml).
Cross-resistance levels of E. coli Rif-, Lpm-, Myx-, and Sal-resistant mutants to PUM (Fig. S3G–I,L) were determined as described for resistance levels of E. coli spontaneous PUM-resistant mutants, but analyzing a collection of E. coli D21f2tolC derivatives comprising four chromosomal Rif-resistant mutants, five chromosomal Lpm-resistant mutants, five chromosomal Myx-resistant mutants, five chromosomal Sal-resistant mutants, and the isogenic wild-type parent (Degen et al., 2014), and analyzing a 2-fold dilution series of PUM (final concentration = 0 or 25–1600 μg/ml).
Cross-resistance levels of E. coli Stl-, CBR-, and GE-resistant mutants to PUM (Fig. S3J–K,N) were determined analogously, analyzing a collection of E. coli D21f2tolC pRL706 and E. coli D21f2tolC pRL663 derivatives comprising five plasmid-based Stl-resistant mutants, five plasmid-based CBR-resistant mutants, six plasmid-based GE-resistant mutants, and plasmid-based wild-type isogenic parents (Zhang et al., 2014; Tuske et al., 2005; Feng et al., 2015). Single colonies were inoculated into 5 ml LB broth containing 200 μg/ml ampicillin (Sigma-Aldrich), incubated at 37°C with shaking until OD600 = 0.4–0.8, supplemented with IPTG (Gold Bio) to a final concentration of 1 mM, and further incubated 1 h at 37°C with shaking. Diluted aliquots (~2 × 105 cells in 50 μl LB broth containing 200 μg/ml ampicillin and 1 mM IPTG) were dispensed into wells of a 96-well microplate containing 50 μl of the same medium or 50 μl of a 2-fold dilution series of PUM in the same medium (final concentration = 0 or 25–4000 μg/ml), and were incubated 16 h at 37°C with shaking.
Amino-acid substitutions that confer PUM-resistance in the context of S. pyogenes RNAP were re-constructed and re-analyzed in the context of E. coli RNAP using an E. coli plasmid-based resistance assay (Fig. S3B). Site-directed mutagenesis (QuikChange Site-Directed Mutagenesis Kit; Agilent) was used to construct plasmid pRL706 (Severinov et al., 1997) and pRL663 (Wang et al., 1995) derivatives encoding E. coli RNAP β-subunit and β′-subunit derivatives having amino-acid substitutions that confer PUM-resistance in S. pyogenes (sequences from Fig. 2B). The resulting plasmids were introduced by transformation into E. coli strain D21f2tolC (Fralick and Bums-Keliher, 1994), and resistance levels of transformants were determined using the procedures of the preceding paragraph.
Formation of RNAP-promoter open complex
Experiments (Fig. S4A) were performed as in Mukhopadhyay et al., 2008. Reaction mixtures contained (20 μl): test compound (0 or 500 μM PUM, 2 μM Rif, or 100 μM Lpm), 40 nM E. coli RNAP σ70 holoenzyme, 10 nM Cy5-labelled DNA fragment carrying positions −40 to +15 of lacUV5 promoter (lacUV5-[−40;+15]-Cy5; Mukhopadhyay et al., 2008), and 100 μg/ml heparin, in 50 mM Tris-HCl (pH 8.0), 100 mM KCl, 10 mM MgCl2, 1 mM dithiothreitol, 10 μg/ml bovine serum albumin, and 5% glycerol. Reaction components other than DNA and heparin were incubated 10 min at 37°C; DNA was added and reactions were incubated 15 min at 37°C; and heparin was added and reactions were incubated 2 min at 37°C. Products were applied to 5% TBE-polyacrylamide slab gels (Bio-Rad), electrophoresed in TBE (Sambrook and Russell, 2001), and analyzed by fluorescence scanning (Typhoon 9400).
Nucleotide addition in transcription initiation
Experiments were performed as in Zhang et al., 2014, using reaction mixtures that contained no inhibitor, 500 μM PUM, 2 μM Rif, or 100 μM Lpm, and using 5 μM [α32P]UTP (3 Bq/fmol; PerkinElmer) (Fig. S4B).
Nucleotide addition in transcription elongation
Experiments were performed as in Zhang et al., 2014, using reaction mixtures that contained no inhibitor, 500 μM PUM, 2 μM Rif, or 100 μM Lpm (Fig. S4C).
Nucleotide addition at elevated NTP concentrations
Experiments (Fig. 3A) were performed as described above for assays of promoter-dependent transcription by B. subtilis RNAP (Methods, RNAP-inhibitory activity in vitro), using reaction mixtures (50μl) that contained 0 or 6 μM PUM, 0.4 U E. coli RNAP σ70 holoenzyme (Epicentre), 0.4 nM plasmid pUC19 Clontech/Takara), 80 mM HEPES-KOH (pH 7.6), 80 mM KCl, 4 mM MgCl2, 0.1 mM EDTA, 5 mM dithiothreitol, 100 μg/ml bovine serum albumin, and either (i) 100 μM ATP, 100 μM [α32P]CTP (0.2 Bq/fmol; PerkinElmer), 100 μM GTP, and 10–500 μM UTP; (ii) 10–500 μM GTP, 100 μM ATP, 100 μM CTP, and 2 μM [α32P]UTP (0.2 Bq/fmol; PerkinElmer); (iii) 100 μM GTP, 10–500 μM A, 100 μM CTP, and 2 μM [α32P]UTP (0.2 Bq/fmol); or (iv) 100 μM GTP, 100 μM ATP, 10–500 μM CTP, and 2 μM [α32P]UTP (0.2 Bq/fmol). The reaction time was 30 min at 37°C. Relative nucleotide incorporation was defined as the ratio of nucleotide incorporation in the presence of PUM to nucleotide incorporation in the absence of PUM.
Nucleotide addition on standard and “U-less cassette” templates
Experiments (Fig. 3B) were performed as described in the preceding section (Methods, Nucleotide addition at elevated NTP concentrations), using reaction mixtures (50 μl) that contained 0–20 μM PUM, 0.4 U E. coli RNAP σ70 holoenzyme (Epicentre), 2 μM [α32P]CTP (0.2 Bq/fmol; PerkinElmer), 100 μM ATP, 100 μM GTP, and 5 μM UTP, in 40 mM Tris-HCl (pH 7.5), 10 mM MgCl2, 5 mM dithiothreitol, 15 mM KCl, 0.01 % Triton X-100, and 100 μg/ml bovine serum albumin, and either (i) 50 nM DNA fragment carrying positions −112 to +8 of the E. coli recA promoter (Sancar et al., 1980) followed by 5′-CAGGGACAAGTTAGTTCGTTCAGCGACACGCGGCAACAAG-3′ (directs incorporation of U, G, A, and C) or (ii) 50 nM DNA fragment carrying positions −112 to +8 of the E. coli recA promoter followed by 5′-CAGGGACAAGGAGACCAACGCAGCGACACGCGGCAACAAG-3′ (“U-less cassette”; directs incorporation of G, A, and C). The reaction time was 60 min at 37°C. Relative nucleotide incorporation was defined as the ratio of nucleotide incorporation in the presence of PUM to nucleotide incorporation in the absence of PUM.
Template-sequence specificity of inhibition by PUM: single-nucleotide-addition reactions
Template-sequence specificity of inhibition by PUM was assessed in single-nucleotide-addition experiments (Fig. 3C) using E. coli RNAP transcription elongation complexes assembled on the nucleic-acid scaffolds in Table S5. Nucleic-acid scaffolds for single-nucleotide-addition reactions were prepared as follows: 1 μM nontemplate-strand oligodeoxyribonucleotide [5′-ACGCCAGACAGGG-3′ or 5′-TCGCCAGACAGGG-3′; IDT), 1 μM template-strand oligodeoxyribonucleotide [3′-GCCGCGCG-(C or T or A or G)-(A or C or G)-TGCGGTCTGTCCC-5′ or 3′-GCCGCGCG-(C or T or A or G)-(T)-AGCGGTCTGTCCC-5′; IDT], and 1 μM 32P-5′-end-labeled oligoribonucleotide [5′-32P-CGGCGCGC-(U or C or A or G)-3′; 90 Bq/fmol; prepared using T4 polynucleotide kinase (New England Biolabs), [γ32P]ATP (100 Bq/fmol; PerkinElmer), and corresponding unlabelled oligoribonucleotide (IDT); procedures as in Sambrook and Russell, 2001], in 5 mM Tris-HCl (pH 7.7), 200 mM NaCl, and 10 mM MgCl2, were heated 5 min at 95°C, cooled to 4°C in 2°C steps with 1 min/step using a thermal cycler (Applied Biosystems), and stored at −20°C. Reaction mixtures for single-nucleotide-addition reactions contained (10 μl): 0 or 25 μM PUM, 40 nM E. coli RNAP core enzyme, 10 nM nucleic-acid scaffold, and 2.5 μM ATP, GTP, CTP, or UTP, in 50 mM Tris-HCl (pH 8.0), 100 mM KCl, 10 mM MgCl2, 1 mM dithiothreitol, 10 μg/ml BSA, and 5% glycerol. Reaction components except PUM and NTP were pre-incubated 10 min at 37°C, PUM was added and reaction mixtures were incubated 5 min at 37°C, and NTP was added and reaction mixtures were incubated 5 min at 37°C. Reactions were terminated by addition of 5 μl 80% formamide, 10 mM EDTA, 0.04% bromophenol blue, 0.04% xylene cyanol, and 0.08% amaranth red, and heating 2 min at 95°C. Samples were applied to denaturing 15% polyacrylamide (19:1 acrylamide:bisacrylamide, 7 M urea) slab gels (Sambrook and Russell, 2001), electrophoresed in TBE (Sambrook and Russell, 2001), and analyzed by storage-phosphor scanning (Typhoon 9400; GE Healthcare).
Template-sequence specificity of inhibition by PUM: multiple-nucleotide-addition reactions
Template-sequence specificity of inhibition by PUM was assessed in multiple-nucleotide-addition experiments (Fig. 3D), performed by adding PUM to transcription elongation complexes halted at position +29 of a 100 bp transcription unit by omission of CTP, re-starting transcription elongation complexes and allowing transcription of positions +30 to +100 of the transcription unit by addition of CTP, and identifying positions at which PUM inhibits transcription.
Halted transcription elongation complexes were prepared as in Revyakin et al., 2006. Reaction mixtures (20 μl) contained: 40 nM E. coli RNAP σ70 holoenzyme, 10 nM DNA fragment N25-100-tR2 (Revyakin et al., 2006), 100 μg/ml heparin, 5 μM [γ32P]ATP (6 Bq/fmol; PerkinElmer), 5 μM UTP, and 5 μM GTP, in 50 mM Tris-HCl (pH 8.0), 100 mM KCl, 10 mM MgCl2, 1 mM dithiothreitol, 10 μg/ml bovine serum albumin, and 5% glycerol. Reaction components other than heparin and NTPs were pre-incubated 5 min at 30°C; heparin was added and reaction mixtures were incubated 2 min at 30°C; NTPs were added and reaction mixtures were incubated 3 min at 30°C. Halted transcription elongation complexes were provided with PUM (1.25 μl 125 μM PUM, 1.25 μl 250 μM PUM, 1.25 μl 500 μM PUM, or 1.25 μl 1 mM PUM) or, to provide markers, chain-terminating 3′-O-methyl-NTPs (RiboMed; 1.25 μl 400 pM 3′-O-methyl-UTP, 1.25 μl 400 μM 3′-O-methyl-CTP, 1.25 μl 400 μM 3′-O-methyl-GTP, or 1.25 μl 400 μM 3′-O-methyl-ATP), were incubated 3 min at 30°C, were re-started by addition of 0.625 μl 200 μM UTP, 1.25 μl 200 μM CTP, 0.625 μl 200 μM GTP, and 0.625 μl 200 μM ATP, and were further incubated 10 min at 30°C. Reactions were terminated by addition of 12.5 μl 80% formamide, 10 mM EDTA, 0.04% bromophenol blue, 0.04% xylene cyanol, and 0.08% amaranth red, and heating 4 min at 95°C. Samples were applied to 10% polyacrylamide (19:1 acrylamide:bisacrylamide; 7 M urea) slab gels (Sambrook and Russell, 2001), electrophoresed in TBE (Sambrook and Russell, 2001), and analyzed by storage-phosphor scanning (Typhoon 9400; GE Healthcare).
Structure determination: RPo-GpA-PUM
Crystals of T. thermophilus RPo-GpA were prepared as in Zhang et al., 2012. Crystallization drops contained 1 μl 18 μM RPo (prepared from T. thermophilus RNAP σA holoenzyme and synthetic nucleic-acid scaffold as in Zhang et al., 2012) and 1 mM GpA (TriLink) in 20 mM Tris-HCl, pH 7.7, 100 mM NaCl, and 1% glycerol, and 1 μl reservoir buffer (RB; 100 mM Tris-HCl, pH 8.4, 200 mM KCl, 50 mM MgCl2, and 10% PEG4000), and were equilibrated against 400 μl RB in a vapor-diffusion hanging-drop tray (Hampton Research). Rod-like crystals appeared in 1 day, and grew to a final size of 0.1 mm × 0.1 mm × 0.3 mm in 5 days.
PUM was soaked into RPo-GpA crystals by adding 0.2 μl 100 mM PUM in RB to the crystallization drop and incubating 30 min at 22°C. RPo-GpA-PUM crystals were transferred to reservoir solutions containing 2 mM PUM in 17.5% (v/v) (2R,3R)-(−)−2,3-butanediol (Sigma-Aldrich) and were flash-cooled with liquid nitrogen.
Diffraction data for RPo-GpA-PUM were collected from cryo-cooled crystals at Cornell High Energy Synchrotron Source (CHESS) beamline F1. Data were integrated and scaled using HKL2000 (Otwinowski et al., 1997). Structure factors were converted using the French-Wilson algorithm (French and Wilson, 1978) in Phenix (Adams et al., 2010) and were subjected to anisotropy correction using the UCLA MBI Diffraction Anisotropy server (Strong et al., 2006; http://services.mbi.ucla.edu/anisoscale/). The structure of RPo-GpA-PUM was solved by molecular replacement in Molrep (Vagin and Teplyakov, 1997), using one RNAP molecule from the structure of T. thermophilus RPo (PDB 4G7H; Zhang et al., 2012) as the search model. Early-stage refinement included rigid-body refinement of each RNAP molecule, followed by rigid-body refinement of each subunit of each RNAP molecule. Cycles of iterative model building with Coot (Emsley et al., 2010) and refinement with Phenix (Adams et al., 2010) were performed. Atomic models of the DNA nontemplate strand, the DNA template strand, and GpA were built into mFo-DFc omit maps, and further refinement with Phenix was performed. The atomic model of PUM was built into the mFo-DFc omit map and was refined with Phenix. The final crystallographic model of RPo-GpA-PUM at 3.30 Å resolution, refined to Rwork and Rfree of 0.232 and 0.280, has been deposited in the PDB with accession code PDB: 5×21 (Fig. 4A; Table S3).
Structure determination: RPo-GpA-CMPcPP
Crystals of T. thermophilus RPo-GpA-CMPcPP were prepared by co-crystallization. Crystallization drops contained 1 μl 18 μM RPo (prepared from T. thermophilus RNAP σA holoenzyme and synthetic nucleic-acid scaffold as in Zhang et al., 2012), 1 mM GpA (TriLink), and 10 mM CMPcPP (Jena Bioscience) in 20 mM Tris-HCl, pH 7.7, 100 mM NaCl, and 1% glycerol, and 1 μl RB, and were equilibrated against 400 μl RB in a vapor-diffusion hanging-drop tray (Hampton Research). Rod-like crystals appeared in 1 day, and grew to a final size of 0.1 mm × 0.1 mm × 0.3 mm in 5 days. RPo-GpA-CMPcPP crystals were transferred to reservoir solutions containing 2 mM CMPcPP in 17.5% (v/v) (2R,3R)-(−)−2,3-butanediol (Sigma-Aldrich), and were flash-cooled with liquid nitrogen.
Diffraction data for RPo-GpA-CMPcPP were collected from cryo-cooled crystals at CHESS beamline F1. Data were integrated and scaled, structure factors were converted and subjected to anisotropy correction, and the structure was solved and refined using procedures analogous to those in the preceding section. The final crystallographic model of RPo-GpA-CMPcPP at 3.35 Å resolution, refined to Rwork and Rfree of 0.208 and 0.250, has been deposited in the PDB with accession code PDB: 5×22 (Fig. 4B; Table S3).
Semi-synthesis of PUM derivatives
Semi-syntheses of PUM derivatives from PUM corroborate the inferred structure of PUM, provide routes for preparation of novel PUM derivatives, and provide initial structure-activity relationships (Fig. 5A,C–D). Reactions were conducted starting from 1 mg PUM, and products were identified by LC-MS (Agilent 1100 with flow split in 1:1 ratio between UV detector and ion-trap ESI-MS interface of Bruker Esquire 3000 Plus; Waters Atlantis 3 μm, 50 × 4.6 mm, column; phase A = 0.05% trifluoroacetic acid in water; phase B = acetonitrile; gradient = 5–95% B in 6 min; flow rate = 1 ml/min; run temperature = 40°C; PUM retention time = 1.4 min).
Reaction of PUM with TiCl3 in 1 M sodium acetate (pH 7.0) for 2 h at room temperature resulted in reduction of the N-hydroxy moiety, yielding desoxy-PUM (1; m/z = 471 [M+H]+). Reaction of PUM with PdCl2 (Maffioli et al., 2005) in 1:1 acetonitrile:water for 2 h at room temperature resulted in selective dehydration of the PUM glutamine sidechain amide -NH2, yielding nitrile analog 2 (m/z = 469 [M+H]+). Reaction of PUM with 0.1% TFA in water for 3 days at room temperature resulted in hydrolysis of the glutamine sidechain amide, yielding carboxy analog 3 (m/z = 488 [M+H]+). Reaction of 3 with benzylamine in DMF containing benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate (PyBOP) for 30 min at room temperature yielded benzylamide analog 4 (m/z = 577 [M+H]+). Reaction of PUM with 2,3-butanedione (Leitner et al., 2003) in 10 mM ammonium acetate (pH 8.0) for 30 min at room temperature resulted in diolic intermediate 5 (m/z = 573 [M+H]+), which subsequently was trapped by treatment with phenylboronic acid for 2 h at room temperature, yielding phenyl-dioxaborolan analog 6 (m/z = 659 [M+H]+).
Total synthesis of desoxy-PUM
Total synthesis of desoxy-PUM provides a reference compound that corroborates the inferred stereochemistry of PUM [by comparison of desoxy-PUM prepared by total synthesis (1 in Fig. 5B) to desoxy-PUM prepared by semi-synthesis from PUM (1 in Fig. 5A)] and provides an additional route to novel PUM derivatives. Desoxy-PUM was obtained in eight steps by convergent synthesis from commercially available β-D-pseudouridine and glycyl-L-glutamine, as follows (Fig. 5B).
Acetonide protection (reaction a in Fig. 5B)
To a solution of β-D-pseudouridine (Berry & Associates; 400 mg, 1.64 mmol) and 2,2-dimethoxypropane (Sigma-Aldrich; 12 ml) in dimethylformamide (8 ml), concentrated HCl (80 μL) was added, and the reaction mixture was stirred 5 h at room temperature. After neutralization with 2.5 M NaOH, solvent was removed under vacuum. 1H-NMR (400 MHz, D2O, δ-H): 1.35 (s, 3H, CH3), 1.56 (s, 3H, CH3), 3.67 (dd, 1H, J = 12.2, 5.65 Hz, H-5′), 3.75 (dd, 1H, J = 12.2, 3.75 Hz, H-5′), 4.11 (dd, 1H, H-4′), 4.75 (m, 2H), 4.86 (m, 1H), 7.62 (s, 1H, H-6).
Mesylation (reaction b in Fig. 5B)
To a solution of the crude product of the preceding step (419 mg, 1.47 mmol) in pyridine (Sigma-Aldrich; 4.7 ml), methanesulfonyl chloride (Sigma-Aldrich: 95 μL, 1.23 mmol) was added with stirring at 0°C. The reaction mixture was stirred at room temperature until completeness (16 h). Solvent was removed by rotary evaporation, and the raw material was purified by flash chromatography on Combiflash (Teledyne ISCO), yielding 475 mg of a white powder (95% yield). 1H-NMR (400 MHz, acetonitrile-d3, δ-H): 1.32 (s, 3H, CH3), 1.54 (s, 3H, CH3), 4.33 (dd, 1H, J = 11 Hz, H-5′), 4.46 (dd, 1H, J = 11 Hz, H-5′), 4.20 (m, 1H), 4.72 (dd, 1H), 4.80 (m, 2H), 7.55 (s, 1H, H-6), 10.23 (sb, 1H, NH), 10.45 (sb, 1H, NH).
Azidation (reaction c in Fig. 5B)
To a solution of the product of the preceding step (475 mg) in dimethylformamide (24 ml), sodium azide (Sigma-Aldrich: 476 mg) was added, the reaction mixture was stirred 4 h at 100°C, and solvent was removed by rotary evaporation. 1H-NMR (400 MHz, acetonitrile-d3, δ-H): 1.30 (s, 3H, CH3), 1.50 (s, 3H, CH3), 3.52 (d, 2H, J = 5.3 Hz, H-5′), 4.04 (m, 1H, H-3′), 4.69 (dd, 1H, H-4′), 4.75 (d, 1H, J = 3.3 Hz, H-1′), 4.87 (dd, 1H, J = 3.3 Hz, H-2′), 7.58 (s, 1H, H-6).
Azide reduction (reaction d in Fig. 5B)
To a solution of the crude product of the preceding step (193 mg) in tetrahydrofuran (8.8 ml) and water (1.8 ml), 1 M trimethylphosphine in tetrahydrofuran (Sigma-Aldrich; 0.74 ml) was added, the reaction mixture was stirred 2 h at room temperature, and solvent was removed by rotary evaporation. 1H-NMR (400 MHz, D2O, δ-H): 1.47 (s, 3H, CH3), 168 (s, 3H, CH3), 3.40 (dd, 1H, H-5′), 3.49 (dd, 1H, H-5′),4.38 (m, 1H, H-4′), 4.90 (dd, 1H, H-1′), 4.94 (d, 1H, H-3′), 5.05 (dd, 1H, H-2′), 7.76 (s, 1H, H-6).
Fmoc protection (reaction e in Fig. 5B)
To a solution of the crude product of the preceding step (22 mg, 0.11 mmol) in dioxane (150 μL) and water (250 μL) sodium carbonate (26.5 mg) was added, followed by Fmoc chloride (Sigma-Aldrich; 31 mg, 1.3 eq), and the reaction mixture stirred overnight at room temperature. After addition of water (5 ml), the reaction was extracted with ethyl acetate (3 × 5 ml), the combined organic extracts were extracted with saturated sodium bicarbonate (3 × 5 ml), the combined aqueous extracts were acidified to pH 1 with 1 M HCl and extracted with ethyl acetate (3 × 5 ml), and the combined organic extracts were treated with sodium sulfate and evaporated to dryness, providing Fmoc-glycl-L-glutamine in quantitative yield. 1H-NMR (400 MHz, D2O, δ-H): 1.98 (m, 1H, Asn-β), 2.18 (m, 1H, Asn-β), 2.33 (m, 2H, Asn-γ), 3.90 (m, 2H, Gly-α), 4.23 (m, 1H), 4.31 (m, 1H), 4.47 (dd, 1H, Asn-α), 7.31 (m, 2H, Ar), 7.38 (m, 2H, Ar), 7.69 (m, 2H, Ar), 7.81 (m, 2H, Ar).
Coupling, Fmoc deprotection, and formamidinylation (reactions f–h in Fig. 5B)
To a solution of the product of the preceding step (20 mg) and the product of the azide-reduction reaction (30 mg, 1.1 eq) in dry dimethylformamide (1.5 ml), N,N′-dicyclohexylcarbodiimide (Sigma-Aldrich; 18 mg, 1.2 eq) and 1-hydroxybenzotriazole (Sigma-Aldrich; 19.5 mg, 2 eq) were added, and the reaction mixture was stirred overnight at room temperature, and the solvent was evaporated under reduced pressure. To a solution of the crude coupled product (12 mg) in dimethylformamide (800 μl), piperidine (200 μl) was added, and the reaction mixture was stirred 10 min at 25°C, the solvent was evaporated under reduced pressure, and the residue was washed with methylene chloride (2 × 5 ml). To a solution of the crude Fmoc-deprotected product (22 mg) in methanol (300 μl), 3,5-dimethylpyrazole-1-carboxamidine (Sigma-Aldrich; 45 mg, 10 eq) was added, and the reaction mixture was stirred overnight at room temperature, followed by 6 h under reflux at 65°C to complete the reaction. The solvent was evaporated under reduced pressure, and the solid residue was washed with methylene chloride (2 × 10 ml). 1H-NMR (400 MHz, D2O/CD3OD, δ-H): 1.33 (s, 3H, CH3), 1.54 (s, 3H, CH3), 2.01 (m, 1H, Asn-β), 2.17 (m, 1H, Asn-β), 2.37 (m, 2H, Asn-γ), 3.37 (m, 1H, H-5′), 3.65 (m, 1H, H-5′), 4.04 (s, 2H, Gly-α), 4.03 (m, 1H), 4.11 (m, 1H), 4.42 (m, 1H), 4.63 (m, 1H), 7.53 (s, 1H, H-6).
Acetonide deprotection (reaction i in Fig. 5B)
A solution of the crude product of the preceding step (17 mg) in acetic acid:water (7:3; 2 ml) was stirred overnight at room temperature and then heated to 50°C for 10 h under argon. The solvent was evaporated under reduced pressure, and the solid residue was washed with methylene chloride (2 × 5 ml) and methanol (2 ml), yielding a white solid that, when analyzed by LC-MS [performed as described for LC-MS of PUM (Methods, Structure Elucidation of PUM); retention time = 14 min], 1D- and 2D-NMR, was indistinguishable from desoxy-PUM obtained by reduction of PUM with TiCl3 (1 in Fig. 5A). 1H-NMR (600 MHz, DMSO-d6/D2O, δ-H): 1.75 (m, 1H, Asn-β), 1.90 (m, 1H, Asn-β), 2.10 (m, 2H, Asn-γ), 3.29 (m, 2H, H-5′), 3.72 (m, 2H), 3.87 (s broad, 2H, Gly-α), 3.96 (m, 1H, H-2′), 4.24 (m, 1H, Asn-α), 4.40 (d, 1H, J=5.3 Hz, H-1′), 6.73 (s broad, CONH2), 7.32 (s broad, CONH2), 7.40 (s, 1H), 8.11 (t broad, 1H, NH), 8.34 (d broad, 1H, NH-Asn).13C-NMR (DMSO-d6 δ-H): 28.4, 31.9, 41.4, 44.0, 53.0, 72.3, 73.7, 79.9, 81.6, 110.4, 141.5, 152.2, 158.2, 164.2, 168.0, 171.3, 173.7.
QUANTITATION AND STATISTICAL ANALYSIS
Data for RNAP-inhibitory activities, growth-inhibitory activities, resistance, and cross-resistance are means of at least two technical replicates. Data for mouse infection models, resistance-rate assays, and checkerboard interaction assays are means and 95% confidence intervals for eight biological replicates, at least six biological replicates, and at least five technical replicates, respectively.
DATA AVAILABILITY
Atomic coordinates and structure factors for crystal structures of RPo-GpA-PUM and RPo-GpA-CMPcPP have been deposited in the Protein Data Bank with accession numbers PDB: 5×21 and PDB: 5×22. 16S rRNA gene sequences of PUM producer strains ID38640 and ID38673 have been deposited in GenBank with accession numbers GI: JQ929050 and GI: JQ929051. PUM producer strain ID38640 has been deposited in the Deutsche Sammlung von Mikroorganismen und Zellkulturen patent depository collection with accession number DSMZ: DSM-26212. Both PUM producer strains, ID38640 and ID38673, can be obtained from NAICONS under a Material Transfer Agreement.
Supplementary Material
(A) Chromatographic profile of Streptomyces sp. ID38640 culture extract, showing peaks for PUM and lidicamycin, and UV-absorbance and mass spectra for PUM. (B) Structure and 1H-NMR spectrum of PUM in DMSO-d6 at 25°C at 400 MHz. (C) 2D-HSQC spectrum of PUM in DMSO-d6 at 25°C at 400 MHz. (D) 2D-HMBC spectrum of PUM in DMSO-d6 at 25°C at 400 MHz. (D) Summary of 1H-,13C-, and 15N-NMR data for PUM in DMSO-d6.
Effects of PUM on DNA synthesis (A; [14C]-thymidine incorporation), RNA synthesis (B; [3H]-uridine incorporation), and protein synthesis (C; [14C]-isoleucine incorporation) in Staphylococcus simulans in culture. Results match characteristic pattern for inhibition of RNAP-dependent RNA-synthesis (Degen et al., 2014; Lancini et al., 1968,1969; Sergio et al., 1975; Irschik et al., 1983, 1985, 1995): i.e., rapid and strong inhibition of RNA synthesis, slower and weaker inhibition of protein synthesis, and little or no inhibition of DNA synthesis.
(A) E. coli spontaneous PUM-resistant mutants. (B) Effects of S. pyogenes PUM-resistant mutants (sequences from Fig. 2B) when analyzed in E. coli plasmid-based resistance assay. Two substitutions confer moderate or higher (≥4x) resistance in E. coli plasmid-based resistance assay: β565 Glu→Gly and β681 Met→Lys. (C) PUM-resistant phenotype of purified E. coli RNAP containing β565 Glu→Asp. (D) Location of E. coli PUM target (sequences from A–B) in three-dimensional structure of bacterial RNAP (colors as in Fig. 2C). (E) Absence of overlap between PUM target (blue) and Rif (red), Lpm (cyan), Myx (pink), Stl (yellow), CBR (light blue), and Sal (green) targets. (F) Absence of cross-resistance of E. coli PUM-resistant mutants (sequences from A–B) to Rif, Lpm, Myx, Stl, CBR, and Sal. (G)–(L), Absence of cross-resistance of E. coli Rif-, Lpm-, Myx-, Stl-, CBR-, and Sal-resistant mutants to PUM. (M) Location of GE target (blue) in structure of bacterial RNAP. PUM target (D) shows partial overlap with GE target (M). (N) Partial cross-resistance of E. coli GE-resistant mutants to PUM.
(A) Absence of inhibition by PUM of formation of catalytically-competent RNAP-promoter open complex, RPo (E. coli RNAP). (B) Inhibition by PUM of nucleotide addition in transcription initiation (E. coli RNAP). (C) Inhibition by PUM of nucleotide addition in transcription elongation (E. coli RNAP).
Locations of residues that contact PUM in the sequences of RNAP β subunit (A) and RNAP β′ subunit (B). Sequence alignments for β and β′ subunits of bacterial RNAP (top 24 sequences in each panel) and corresponding subunits of human RNAP I, RNAP II, and RNAP III (bottom three sequences in each panel), showing locations of RNAP residues that contact PUM (black rectangles; numbered as in S. pyogenes and, in parentheses, as in E. coli; identities from Fig. 4A), locations of residues at which substitutions conferring PUM-resistance are obtained in both S. pyogenes and E. coli (red asterisks; identities from Figs. 2B, S3A–B), locations of residues at which substitutions conferring PUM-resistance are obtained in S. pyogenes but not E. coli (black asterisks; identities from Figs. 2B, S3A–B), locations of RNAP structural elements (Weinzierl, 201; Hein and Landick, 2010; top row of black bars), and RNAP conserved regions (Sweetser et al., 1987; Lane and Darst, 2010; Jokerst et al., 1989; next two rows of black bars). Species are as follows: E. coli (ECOLI), Salmonella typhimurium (SALTY), Klebsiella pneumoniae (KLEP7), Enterococcus cloacae (ENTCC), Vibrio cholerae (VIBCH), Haemophilus influenzae (HAEIN), Campylobacter jejuni (CAMJE), Neisseria gonorrhoeae (NEIG1), Stenotrophomonas maltophilia (STPMP), Moraxella catarrhalis (MORCA), Acinetobacter baumannii (ACIBC), Pseudomonas aeruginosa (PSEAE), Staphylococcus aureus (STAAU), Staphylococcus epidermidis (STAEQ), Enterococcus faecalis (ENTFA), Streptococcus pyogenes (STRP1), Streptococcus pneumoniae (STRP2), Clostridium difficile (CDIFF), Mycobacterium tuberculosis (MYCTU), Mycobacterium avium (MYCA1), Mycobacterium abscessus (MYCA9), Thermus aquaticus (THEAQ), Thermus thermophilus (THETH), Deinococcus radiodurans (DEIRA), and Homo sapiens (HUMAN).
HIGHLIGHTS.
new antibiotic from microbial extract screening
selective nucleoside-analog inhibitor of bacterial RNA polymerase
competition with UTP for occupancy of RNAP active-center NTP addition site
low resistance rate due to functional constraints on substitution of active center
In Brief.
Pseudouridimycin competes with incoming nucleotides to inhibit bacterial RNA polymerase, effectively blocking growth of a broad range of pathogens.
Acknowledgments
Work was supported by NIH grants GM041376 and AI104660 to RHE and Italian Ministry of Research and Regione Lombardia grants to NAICONS. We thank former colleagues at Vicuron Pharmaceuticals for early characterization of PUM, S. Parapini for cytotoxicity assays, W. Fenical for Sal, A. Berk and I. Grummt for plasmids, J. Silverman, NARSA, and BEI Resources for strains, and the Cornell High Energy Synchrotron Source for beamline access.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
S.M. performed structure elucidation of PUM and semi-syntheses and syntheses of PUM derivatives. Y.Z. purified RNAP, assessed template-specificity of inhibition and determined crystal structures. D.D. purified RNAP, and assessed RNAP-inhibitory activities, antibacterial activities, resistance, and cross-resistance. T.C., S.S., and G.D. assessed RNAP-inhibitory activities. G.D.G. and C.M. purified PUM and participated in semi-syntheses and syntheses of PUM derivatives. P.M. performed characterization and fermentation of the PUM producer strain. P.G. and G.C. performed mouse infection studies. A.I.C. and H.-G.S. performed macromolecular synthesis assays. G.F. and P.K. performed microbial extract screening and de-replication. S.D. and R.H.E. designed the study, analyzed data, and wrote the paper.
References
- Adams P, Afonine P, Bunkóczi G, Chen V, Davis I, Echols N, Head J, Hung L, Kapral G, Grosse-Kunstleve R, McCoy A, Moriarty N, Oeffner R, Read R, Richardson D, Richardson J, Terwilliger T, Zwart P. PHENIX: a comprehensive Python-based system for macromolecular structure. Acta Cryst D. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aristoff P, Garcia GA, Kirchoff P, Showalter H. Rifamycins–obstacles and opportunities. Tuberculosis. 2010;90:94–118. doi: 10.1016/j.tube.2010.02.001. [DOI] [PubMed] [Google Scholar]
- Bae B, Nayak D, Ray A, Mustaev A, Landick R, Darst S. CBR antimicrobials inhibit RNA polymerase via at least two bridge-helix cap-mediated effects on nucleotide addition. Proc Natl Acad Sci USA. 2015;112:E4178–E4187. doi: 10.1073/pnas.1502368112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry A, Pfaller M, Fuchs P. Haemophilus test medium versus Mueller-Hinton broth with lysed horse blood for antimicrobial susceptibility testing of four bacterial species. Eur J Clin Microbiol Infect Dis. 1993;12:548–553. doi: 10.1007/BF01970963. [DOI] [PubMed] [Google Scholar]
- Belogurov G, Vassylyeva M, Sevostyanova A, Appleman J, Xiang A, Lira R, Webber S, Klyuyev S, Nudler E, Artsimovitch I, Vassylyev D. Transcription inactivation through local refolding of the RNA polymerase structure. Nature. 2009;45:332–335. doi: 10.1038/nature07510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown E, Wright G. Antibacterial drug discovery in the resistance era. Nature. 2016;529:336–343. doi: 10.1038/nature17042. [DOI] [PubMed] [Google Scholar]
- Campbell E, Korzheva N, Mustaev A, Murakami K, Nair S, Goldfarb A, Darst S. Structural mechanism for rifampicin inhibition of bacterial RNA polymerase. Cell. 2001;104:901–912. doi: 10.1016/s0092-8674(01)00286-0. [DOI] [PubMed] [Google Scholar]
- Ciciliato I, Corti E, Sarubbi E, Stefanelli S, Gastaldo L, Montanini N, Kurz M, Losi D, Marinelli F, Selva E. Antibiotics GE23077, novel inhibitors of bacterial RNA polymerase I. taxonomy, isolation and characterization. J Antibiot. 2004;57:210–217. doi: 10.7164/antibiotics.57.210. [DOI] [PubMed] [Google Scholar]
- Cihlar T, Ray A. Nucleoside and nucleotide HIV reverse transcriptase inhibitors: 25 years after zidovudine. Antiviral Res. 2010;85:39–58. doi: 10.1016/j.antiviral.2009.09.014. [DOI] [PubMed] [Google Scholar]
- Clinical and Laboratory Standards Institute (CLSI/NCCLS) Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically; Approved Standard. (Eighth) 2009 CLIS Document M07-A8 Wayne, PA. [Google Scholar]
- Dean N, Berk A. Ordering promoter binding of class III transcription factors TFIIIC1 and TFIIIC2. Mol Cell Biol. 1988;8:3017–3025. doi: 10.1128/mcb.8.8.3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degen D, Feng Y, Zhang Y, Ebright K, Ebright Y, Gigliotti M, Vahedian-Movahed H, Mandal S, Talaue M, Connell N, Arnold E, Fenical W, Ebright RH. Transcription inhibition by the depsipeptide antibiotic salinamide A. eLife. 2014;3:e02451. doi: 10.7554/eLife.02451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donadio S, Monciardini P, Sosio M. Approaches to discovering novel antibacterial and antifungal agents. Meths Enzymol. 2009;458:3–28. doi: 10.1016/S0076-6879(09)04801-0. [DOI] [PubMed] [Google Scholar]
- Ebright RH. RNA exit channel–target and method for inhibition of bacterial RNA polymerase. 2005 WO/2005/001034. [Google Scholar]
- Ebright RH, Ebright Y. Antibacterial agents: high-potency myxopyronin derivatives. 2013 WO/2012037508. [Google Scholar]
- Emsley P, Lohkamp B, Scott W, Cowtan K. Features and development of Coot. Acta Cryst D. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Degen D, Wang X, Gigliotti M, Liu S, Zhang Y, Das D, Michalchuk T, Ebright Y, Talaue M, Connell N, Ebright RH. Structural basis of transcription inhibition by CBR hydroxamidines and CBR pyrazoles. Structure. 2015;23:1470–1481. doi: 10.1016/j.str.2015.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fralick J, Burns-Keliher L. Additive effect of tolC and rfa mutations on the hydrophobic barrier of the outer membrane of Escherichia coli K-12. J Bacteriol. 1994;176:6404–6406. doi: 10.1128/jb.176.20.6404-6406.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French S, Wilson K. On the treatment of negative intensity observations. Acta Cryst D. 1978;34:517–525. [Google Scholar]
- Friedman L, Alder J, Silverman J. Genetic changes that correlate with reduced susceptibility to daptomycin in Staphylococcus aureus. Antimicrob Agents Chemother. 2006;50:2137–2145. doi: 10.1128/AAC.00039-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garibyan L, Huang T, Kim M, Wolff E, Nguyen A, Nguyen T, Diep A, Hu K, Iverson A, Yang H, Miller J. Use of the rpoB gene to determine the specificity of base substitution mutations on the Escherichia coli chromosome. DNA Repair. 2003;2:593–608. doi: 10.1016/s1568-7864(03)00024-7. [DOI] [PubMed] [Google Scholar]
- Hall B, Ma C, Liang P, Singh K. Fluctuation Analysis CalculatOR: a web tool for the determination of mutation rate using Luria-Delbrück fluctuation analysis. Bioinformatics. 2009;25:1564–1565. doi: 10.1093/bioinformatics/btp253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton M, Russo R, Thurston R. Trimmed Spearman-Karber method for estimating median lethal concentrations in toxicity bioassays. Environ Sci Technol. 1977;11:714–719. [Google Scholar]
- Hein P, Landick R. The bridge helix coordinates movements of modules in RNA polymerase. BMC Biol. 2010;8:141. doi: 10.1186/1741-7007-8-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holowachuk S, Bal’a M, Buddington R. A kinetic microplate method for quantifying the antibacterial properties of biological fluids. J Microbiol Meth. 2003;55:441–446. doi: 10.1016/s0167-7012(03)00190-8. [DOI] [PubMed] [Google Scholar]
- Hudson B, Quispe J, Lara-González S, Kim Y, Berman H, Arnold E, Ebright RH, Lawson C. Three-dimensional EM structure of an intact activator-dependent transcription initiation complex. Proc Natl Acad Sci USA. 2009;106:19830–19835. doi: 10.1073/pnas.0908782106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irschik H, Gerth K, Hofle G, Kohl W, Reichenbach H. The myxopyronins, new inhibitors of bacterial RNA synthesis from Myxococcus fulvus (Myxobacterales) J Antibiot. 1983;36:1651–1658. doi: 10.7164/antibiotics.36.1651. [DOI] [PubMed] [Google Scholar]
- Irschik H, Jansen R, Hofle G, Gerth K, Reichenbach H. The corallopyronins, new inhibitors of bacterial RNA synthesis from Myxobacteria. J Antibiot. 1985;38:145–152. doi: 10.7164/antibiotics.38.145. [DOI] [PubMed] [Google Scholar]
- Irschik H, Augustiniak H, Gerth K, Hofle G, Reichenbach H. The ripostatins, novel inhibitors of eubacterial RNA polymerase isolated from Myxobacteria. J Antibiot. 1995;48:787–792. doi: 10.7164/antibiotics.48.787. [DOI] [PubMed] [Google Scholar]
- Jin DJ, Gross C. Mapping and sequencing of mutations in the Escherichia coli rpoB gene that lead to rifampicin resistance. J Mol Biol. 1988;202:45–58. doi: 10.1016/0022-2836(88)90517-7. [DOI] [PubMed] [Google Scholar]
- Jokerst R, Weeks J, Zehring W, Greenleaf A. Analysis of the gene encoding the largest subunit of RNA polymerase II in Drosophila. Mol Gen Genet. 1989;215:266–275. doi: 10.1007/BF00339727. [DOI] [PubMed] [Google Scholar]
- Jovanovic M, Burrows P, Bose D, Cámara B, Wiesler S, Weinzierl R, Zhang X, Wigneshweraraj S, Buck M. An activity map of the Escherichia coli RNA polymerase bridge helix. J Biol Chem. 2011;286:14469–14479. doi: 10.1074/jbc.M110.212902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettenring J, Colombo L, Ferrari P, Tavecchia P, Nebuloni M, Vékey K, Gallo G, Selva E. Antibiotic GE2270A: a novel inhibitor of bacterial protein synthesis: I. structure elucidation. J Antibiot. 1991;44:702–715. doi: 10.7164/antibiotics.44.702. [DOI] [PubMed] [Google Scholar]
- Lancini G, Sartori G. Rifamycins LXI: in vivo inhibition of RNA synthesis of rifamycins. Experentia. 1968;24:1106–1106. doi: 10.1007/BF02147783. [DOI] [PubMed] [Google Scholar]
- Lancini G, Pallanza R, Silvestri L. Relationships between bactericidal effect and inhibition of ribonucleic acid nucleotidyltransferase by rifampicin in Escherichia coli K-12. J Bacteriol. 1969;97:761–768. doi: 10.1128/jb.97.2.761-768.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landwehr W, Wolf C, Wink J. Actinobacteria and Myxobacteria: two of the most important bacterial resources for novel antibiotics. Curr Top Microbiol Immunol. 2016;398:273–302. doi: 10.1007/82_2016_503. [DOI] [PubMed] [Google Scholar]
- Lane W, Darst S. Molecular evolution of multisubunit RNA polymerases: sequence analysis. J Mol Biol. 2010;395:671–685. doi: 10.1016/j.jmb.2009.10.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitner A, Lindner W. Probing of arginine residues in peptides and proteins using selective tagging and electrospray ionization mass spectrometry. J Mass Spectrom. 2003;38:891–899. doi: 10.1002/jms.477. [DOI] [PubMed] [Google Scholar]
- Ma W, Sandri G, Sarkar S. Analysis of the Luria-Delbrück distribution using discrete convolution powers. J Appl Probab. 1992;29:255–267. [Google Scholar]
- Maffioli S, Marzorati E, Marazzi A. Mild and reversible dehydration of primary amides with PdCl2 in aqueous acetonitrile. Org Lett. 2005;7:5237–5239. doi: 10.1021/ol052100l. [DOI] [PubMed] [Google Scholar]
- Mariani R, Maffioli S. Bacterial RNA polymerase inhibitors: an organized overview of their structure, derivatives, biological activity and current clinical development status. Curr Med Chem. 2009;16:430–454. doi: 10.2174/092986709787315559. [DOI] [PubMed] [Google Scholar]
- Marston H, Dixon D, Knisely J, Palmore T, Fauci A. Antimicrobial resistance. JAMA. 2016;316:1193–1204. doi: 10.1001/jama.2016.11764. [DOI] [PubMed] [Google Scholar]
- Mattingly P, Miller M. Titanium trichloride reduction of substituted N-hydroxy-2-azetidinones and other hydroxamic acids. J Org Chem. 1980;45:410–411. [Google Scholar]
- Mazza P, Monciardini P, Cavaletti L, Sosio M, Donadio S. Diversity of Actinoplanes and related genera isolated from an Italian soil. Microb Ecol. 2003;45:362–372. doi: 10.1007/s00248-002-2038-4. [DOI] [PubMed] [Google Scholar]
- Mazzetti C, Ornaghi M, Gaspari E, Parapini S, Maffioli S, Sosio M, Donadio S. Halogenated spirotetronates from Actinoallomurus. J Nat Prod. 2012;75:1044–1050. doi: 10.1021/np300003n. [DOI] [PubMed] [Google Scholar]
- Meletiadis J, Pournaras S, Roilides E, Walsh T. Defining fractional inhibitory concentration index cutoffs for additive interactions based on self-drug additive combinations, Monte Carlo simulation analysis, and in vitro-in vivo correlation data for antifungal drug combinations against Aspergillus fumigatus. Antimicrob Agents Chemother. 2010;54:602–609. doi: 10.1128/AAC.00999-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay J, Mekler V, Kortkhonjia E, Kapanidis A, Ebright Y, Ebright R. Fluorescence resonance energy transfer (FRET) in analysis of transcription-complex structure and function. Meths Enzymol. 2003;371:144–159. doi: 10.1016/S0076-6879(03)71010-6. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay J, Das K, Ismail S, Koppstein D, Jang M, Hudson B, Sarafianos S, Tuske S, Patel J, Jansen R, Irschik H, Arnold E, Ebright RH. The RNA polymerase “switch region” is a target for inhibitors. Cell. 2008;135:295–307. doi: 10.1016/j.cell.2008.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu W, Kim Y, Tau G, Heyduk T, Ebright RH. Transcription activation at Class II CAP-dependent promoters: two interactions between CAP and RNA polymerase. Cell. 1996;87:1123–1134. doi: 10.1016/s0092-8674(00)81806-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Meths Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Pfleiderer C, Smid A, Bartsch I, Grummt I. An undecamer DNA sequence directs termination of human ribosomal gene transcription. Nucl Acids Res. 1990;18:4727–4736. doi: 10.1093/nar/18.16.4727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ploeser J, Loring H. The ultraviolet absorption spectra of the pyrimidine ribonucleosides and ribonucleotides. J Biol Chem. 1949;178:431–437. [PubMed] [Google Scholar]
- Qi Y, Hulett FM. PhoP~P and RNA polymerase σA holoenzyme are sufficient for transcription of Pho regulon promoters in Bacillus subtilis: PhoP~P activator sites within the coding region stimulate transcription in vitro. Mol Microbiol. 1998;28:1187–1197. doi: 10.1046/j.1365-2958.1998.00882.x. [DOI] [PubMed] [Google Scholar]
- Revyakin A, Liu C, Ebright RH, Strick T. Abortive initiation and productive initiation by RNA polymerase involve DNA scrunching. Science. 2006;314:1139–1143. doi: 10.1126/science.1131398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagitov V, Nikiforov V, Goldfarb A. Dominant lethal mutations near the 5′ substrate binding site affect RNA polymerase propagation. J Biol Chem. 1993;268:2195–2202. [PubMed] [Google Scholar]
- Sambrook J, Russell D. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 2001. [Google Scholar]
- Sancar A, Stachelejk C, Konigsberg W, Rupp W. Sequences of the recA gene and protein. Proc Natl Acad Sci USA. 1980;77:2611–2615. doi: 10.1073/pnas.77.5.2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S, Ma W, Sandri G. On fluctuation analysis: a new, simple and efficient method for computing the expected number of mutants. Genetica. 1992;85:173–179. doi: 10.1007/BF00120324. [DOI] [PubMed] [Google Scholar]
- Schreiber E, Matthias P, Müller M, Schaffner W. Rapid detection of octamer binding proteins with ‘mini-extracts’, prepared from a small number of cells. Nucl Acids Res. 1989;17:6419. doi: 10.1093/nar/17.15.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergio S, Pirali G, White R, Parenti F. Lipiarmycin, a new antibiotic from Actinoplanes III. Mechanism of action. J Antibiot. 1975;1975:543–549. doi: 10.7164/antibiotics.28.543. [DOI] [PubMed] [Google Scholar]
- Severinov K, Mooney R, Darst SA, Landick R. Tethering of the large subunits of Escherichia coli RNA polymerase. J Biol Chem. 1997;272:24137–24140. doi: 10.1074/jbc.272.39.24137. [DOI] [PubMed] [Google Scholar]
- Sosunov V, Zorov S, Sosunova E, Nikolaev A, Zakeyeva I, Bass I, Goldfarb A, Nikiforov V, Severinov K, Mustaev A. The involvement of the aspartate triad of the active center in all catalytic activities of multisubunit RNA polymerase. Nucl Acids Res. 2005;33:4202–4211. doi: 10.1093/nar/gki688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava A, Talaue M, Liu S, Degen D, Ebright RY, Sineva E, Chakraborty A, Druzhinin S, Chatterjee S, Mukhopadhyay J, Ebright Y, Zozula A, Shen J, Sengupta S, Niedfeldt R, Xin C, Kaneko T, Irschik H, Jansen R, Donadio S, Connell N, Ebright RH. New target for inhibition of bacterial RNA polymerase: “switch region”. Curr Opin Microbiol. 2011;14:532–543. doi: 10.1016/j.mib.2011.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava A, Degen D, Ebright Y, Ebright RH. Frequency, spectrum, and nonzero fitness costs of resistance to myxopyronin in Staphylococcus aureus. Antimicrob Agents Chemother. 2012;56:6250–6255. doi: 10.1128/AAC.01060-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strong M, Sawaya M, Wang S, Phillips M, Cascio D, Eisenberg D. Toward the structural genomics of complexes: crystal structure of a PE/PPE protein complex from Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 2006;103:8060–8065. doi: 10.1073/pnas.0602606103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers B, Beavers J, Klibanov O. Sofosbuvir [Solvadi], a novel nucleotide analogue inhibitor used for the treatment of hepatitis C virus. J Pharm Pharmacol. 2014;66:1653–1666. doi: 10.1111/jphp.12294. [DOI] [PubMed] [Google Scholar]
- Svetlov V, Vassylyev D, Artsimovitch I. Discrimination against deoxyribonucleotide substrates by bacterial RNA polymerase. J Biol Chem. 2004;279:38087–38090. doi: 10.1074/jbc.C400316200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweetser D, Nonet M, Young R. Prokaryotic and eukaryotic RNA polymerases have homologous core subunits. Proc Natl Acad Sci USA. 1987;84:1192–1196. doi: 10.1073/pnas.84.5.1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H, Severinov K, Goldfarb A, Fenyo D, Chait B, Ebright RH. Location, structure, and function of the target of a transcription activator protein. Genes Dev. 1994;8:3058–3067. doi: 10.1101/gad.8.24.3058. [DOI] [PubMed] [Google Scholar]
- Temiakov D, Zenkin N, Vassylyeva M, Perederina A, Tahirov T, Kaihatsu K, Savkina M, Zorov S, Nikiforov V, Igarashi N, Matsugaki N, Wakatsuki S, Severinov K, Vassylyev D. Structural basis of transcription inhibition by antibiotic streptolydigin. Mol Cell. 2005;19:655–666. doi: 10.1016/j.molcel.2005.07.020. [DOI] [PubMed] [Google Scholar]
- Tuske S, Sarafianos S, Wang X, Hudson B, Sineva E, Mukhopadhyay J, Birktoft J, Leroy O, Ismail S, Clark A, Dharia C, Napoli A, Laptenko O, Lee J, Borukhov S, Ebright RH, Arnold E. Inhibition of bacterial RNA polymerase by streptolydigin: stabilization of a straight-bridge-helix active-center conformation. Cell. 2005;122:541–552. doi: 10.1016/j.cell.2005.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. J Appl Cryst. 1997;30:1022–1025. [Google Scholar]
- Vrentas C, Gaal T, Ross W, Ebright RH, Gourse R. Response of RNA polymerase to ppGpp: requirement for the ω subunit and relief of this requirement by DksA. Genes Dev. 2005;19:2378–2387. doi: 10.1101/gad.1340305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Meier T, Chan C, Feng G, Lee D, Landick R. Discontinuous movements of DNA and RNA in RNA polymerase accompany formation of a paused transcription complex. Cell. 1995;81:341–350. doi: 10.1016/0092-8674(95)90387-9. [DOI] [PubMed] [Google Scholar]
- Weinzierl R. The nucleotide addition cycle of RNA polymerase is controlled by two molecular hinges in the Bridge Helix domain. BMC Biol. 2010;8:134. doi: 10.1186/1741-7007-8-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White R, Manduru M, Bosso J. Comparison of three different in vitro methods of detecting synergy: time-kill, checkerboard, and E test. Antimicrob Agents Chemother. 1996;40:1914–1918. doi: 10.1128/aac.40.8.1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuzenkova Y, Roghanian M, Zenkin N. Multiple active centers of multi-subunit RNA polymerases. Transcription. 2012;3:115–118. doi: 10.4161/trns.19887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Feng Y, Chatterjee S, Tuske S, Ho M, Arnold E, Ebright RH. Structural basis of transcription initiation. Science. 2012;338:1076–1080. doi: 10.1126/science.1227786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Degen D, Ho M, Sineva E, Ebright K, Ebright Y, Mekler V, Vahedian-Movahed H, Feng Y, Yin R, Tuske S, Irschik H, Jansen R, Maffioli S, Donadio S, Arnold E, Ebright RH. GE23077 binds to the RNA polymerase ‘i’ and ‘i+1’ sites and prevents the binding of initiating nucleotides. eLife. 2014;3:e02450. doi: 10.7554/eLife.02450. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Chromatographic profile of Streptomyces sp. ID38640 culture extract, showing peaks for PUM and lidicamycin, and UV-absorbance and mass spectra for PUM. (B) Structure and 1H-NMR spectrum of PUM in DMSO-d6 at 25°C at 400 MHz. (C) 2D-HSQC spectrum of PUM in DMSO-d6 at 25°C at 400 MHz. (D) 2D-HMBC spectrum of PUM in DMSO-d6 at 25°C at 400 MHz. (D) Summary of 1H-,13C-, and 15N-NMR data for PUM in DMSO-d6.
Effects of PUM on DNA synthesis (A; [14C]-thymidine incorporation), RNA synthesis (B; [3H]-uridine incorporation), and protein synthesis (C; [14C]-isoleucine incorporation) in Staphylococcus simulans in culture. Results match characteristic pattern for inhibition of RNAP-dependent RNA-synthesis (Degen et al., 2014; Lancini et al., 1968,1969; Sergio et al., 1975; Irschik et al., 1983, 1985, 1995): i.e., rapid and strong inhibition of RNA synthesis, slower and weaker inhibition of protein synthesis, and little or no inhibition of DNA synthesis.
(A) E. coli spontaneous PUM-resistant mutants. (B) Effects of S. pyogenes PUM-resistant mutants (sequences from Fig. 2B) when analyzed in E. coli plasmid-based resistance assay. Two substitutions confer moderate or higher (≥4x) resistance in E. coli plasmid-based resistance assay: β565 Glu→Gly and β681 Met→Lys. (C) PUM-resistant phenotype of purified E. coli RNAP containing β565 Glu→Asp. (D) Location of E. coli PUM target (sequences from A–B) in three-dimensional structure of bacterial RNAP (colors as in Fig. 2C). (E) Absence of overlap between PUM target (blue) and Rif (red), Lpm (cyan), Myx (pink), Stl (yellow), CBR (light blue), and Sal (green) targets. (F) Absence of cross-resistance of E. coli PUM-resistant mutants (sequences from A–B) to Rif, Lpm, Myx, Stl, CBR, and Sal. (G)–(L), Absence of cross-resistance of E. coli Rif-, Lpm-, Myx-, Stl-, CBR-, and Sal-resistant mutants to PUM. (M) Location of GE target (blue) in structure of bacterial RNAP. PUM target (D) shows partial overlap with GE target (M). (N) Partial cross-resistance of E. coli GE-resistant mutants to PUM.
(A) Absence of inhibition by PUM of formation of catalytically-competent RNAP-promoter open complex, RPo (E. coli RNAP). (B) Inhibition by PUM of nucleotide addition in transcription initiation (E. coli RNAP). (C) Inhibition by PUM of nucleotide addition in transcription elongation (E. coli RNAP).
Locations of residues that contact PUM in the sequences of RNAP β subunit (A) and RNAP β′ subunit (B). Sequence alignments for β and β′ subunits of bacterial RNAP (top 24 sequences in each panel) and corresponding subunits of human RNAP I, RNAP II, and RNAP III (bottom three sequences in each panel), showing locations of RNAP residues that contact PUM (black rectangles; numbered as in S. pyogenes and, in parentheses, as in E. coli; identities from Fig. 4A), locations of residues at which substitutions conferring PUM-resistance are obtained in both S. pyogenes and E. coli (red asterisks; identities from Figs. 2B, S3A–B), locations of residues at which substitutions conferring PUM-resistance are obtained in S. pyogenes but not E. coli (black asterisks; identities from Figs. 2B, S3A–B), locations of RNAP structural elements (Weinzierl, 201; Hein and Landick, 2010; top row of black bars), and RNAP conserved regions (Sweetser et al., 1987; Lane and Darst, 2010; Jokerst et al., 1989; next two rows of black bars). Species are as follows: E. coli (ECOLI), Salmonella typhimurium (SALTY), Klebsiella pneumoniae (KLEP7), Enterococcus cloacae (ENTCC), Vibrio cholerae (VIBCH), Haemophilus influenzae (HAEIN), Campylobacter jejuni (CAMJE), Neisseria gonorrhoeae (NEIG1), Stenotrophomonas maltophilia (STPMP), Moraxella catarrhalis (MORCA), Acinetobacter baumannii (ACIBC), Pseudomonas aeruginosa (PSEAE), Staphylococcus aureus (STAAU), Staphylococcus epidermidis (STAEQ), Enterococcus faecalis (ENTFA), Streptococcus pyogenes (STRP1), Streptococcus pneumoniae (STRP2), Clostridium difficile (CDIFF), Mycobacterium tuberculosis (MYCTU), Mycobacterium avium (MYCA1), Mycobacterium abscessus (MYCA9), Thermus aquaticus (THEAQ), Thermus thermophilus (THETH), Deinococcus radiodurans (DEIRA), and Homo sapiens (HUMAN).
Data Availability Statement
Atomic coordinates and structure factors for crystal structures of RPo-GpA-PUM and RPo-GpA-CMPcPP have been deposited in the Protein Data Bank with accession numbers PDB: 5×21 and PDB: 5×22. 16S rRNA gene sequences of PUM producer strains ID38640 and ID38673 have been deposited in GenBank with accession numbers GI: JQ929050 and GI: JQ929051. PUM producer strain ID38640 has been deposited in the Deutsche Sammlung von Mikroorganismen und Zellkulturen patent depository collection with accession number DSMZ: DSM-26212. Both PUM producer strains, ID38640 and ID38673, can be obtained from NAICONS under a Material Transfer Agreement.