

Summary
The multidomain CCCTC-binding factor (CTCF), containing a tandem array of 11-zinc fingers (ZF), modulates the three-dimensional organization of chromatin. We crystallized the human CTCF DNA binding domain in complex with a known CTCF binding site. While ZF2 does not make sequence-specific contacts, each finger of ZF3-7 contacts three bases of the 15-bp consensus sequence. Each conserved nucleotide makes base-specific hydrogen bonds with a particular residue. Most of the variable base pairs within the core sequence also engage in interactions with the protein. These interactions compensate for deviations from the consensus sequence, allowing CTCF to adapt to sequence variations. CTCF is sensitive to cytosine methylation at position 2, but insensitive at position 12 of the 15-bp core sequence. These differences can be rationalized structurally. Although included in crystallizations, ZF10-11 are not visible, while ZF8-9 span the backbone of the DNA duplex, conferring no sequence specificity but adding to overall binding stability
Keywords: Epigenetics, CTCF, DNA methylation, protein-DNA recognition, C2H2 zinc finger arrays, DNA binding adaptability to sequence variations
Graphical abstract
Hiashimoto et al. described several protein-DNA complex structures of human CTCF tandem zinc-finger array, explaining the adaptability of CTCF to sequence variations, the position-dependent effect of differential DNA methylation at two cytosine residues, and revealing a potential function of C-terminal ZF8-9 spanning across DNA phosphate backbone.
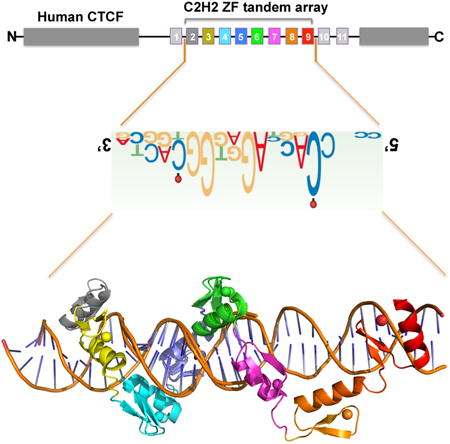
Introduction
The CCCTC-binding factor (CTCF) plays a critical role in organizing genome structure and establishing gene expression patterns in higher eukaryotes. Together with cohesin, CTCF facilitates interactions between enhancers and their cognate promoters by forming loops, while buffering interactions between sequences located inside and outside the loops (Ong and Corces, 2014). The multidomain CTCF protein, conserved in most bilaterian phyla (Heger et al., 2012), influences global chromatin architecture by sequence specific DNA binding, via a central tandem array of eleven Cys2-His2 (C2H2) zinc fingers (ZFs), and protein-protein or protein-RNA interactions (Hadjur et al., 2009; Kung et al., 2015; Parelho et al., 2008; Saldana-Meyer et al., 2014; Wendt et al., 2008; Xiao et al., 2011) (Figure S1A). CTCF is present at ∼80,000 sites on mammalian chromosomes (Chen et al., 2012; Maurano et al., 2015). Experiments using ChIP-exo uncovered a broad CTCF-binding motif that contains a 12-to-15 bp consensus sequence 5′-NCA-NNA-G(G/A)N-GGC-(G/A)(C/G)(T/C)-3′ (Nakahashi et al., 2013; Rhee and Pugh, 2011) (Figure 1A). This consensus is common to most CTCF-binding sites including, for example, the one derived from ChIP-seq data (Jothi et al., 2008) (Figure 1B).
Figure 1. CTCF ZF3-7 binds the 15-bp CORE sequence.
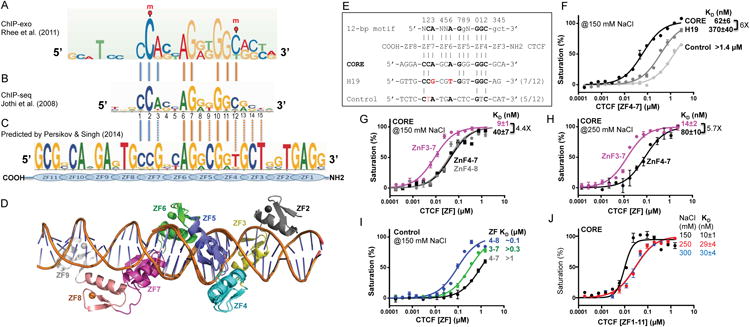
(A) CTCF-binding consensus sequence as determined by ChIP-exo (Rhee and Pugh, 2011). DNA cytosine methylation (indicated by red circles and letters m) occurs at positions 2 and 12 of the consensus sequence in a subset of CTCF-binding sites (Wang et al., 2012).
(B) CTCF consensus binding motifs as determined by ChIP-seq (Jothi et al., 2008).
(C) Predicted CTCF DNA binding specificity (Persikov and Singh, 2014). The notable differences from the consensus sequence involves a Gua (instead of Ade) at position 3 and a Thy (instead of Cyt) at position 12. We note that both Thy (5-methyluracil) and methylated Cyt (5-methylcytosine) contain a methyl group at ring carbon C5.
(D) A model of CTCT ZF2-9 binding DNA, generated by superimposing the common four fingers (4-7) from structures of ZF2-7 and ZF4-9.
(E) Three DNA sequences used for binding assays (CORE, H19 and control).
(F) Binding affinities of CTCF ZF4-7 against the three oligos defined in panel E.
(G-H) ZF3 increases binding affinity against the CORE sequence under two different NaCl concentrations. Because ZF3-7 binds too tightly against CORE (KD being close to probe concentration of 5 nM), we increased NaCl concentration in the binding assays from 150 mM (G) to 250 mM (H).
(I) ZF8 increases non-specific binding affinity.
(J) The 11-ZF DNA binding domain binds the CORE sequence depending on the ionic strength. DNA binding data represent the mean ± SEM of two independent determinations performed in duplicate.
It was more than 25 years ago that the first structure was reported for a C2H2 ZF protein in complex with DNA (Pavletich and Pabo, 1991). In conventional C2H2 ZF proteins, each finger interacts mainly with three adjacent DNA base pairs (Choo and Klug, 1997), which we term the ‘triplet’ element. When bound to DNA, side chains from specific amino acids within the N-terminal portion of each helix and the preceding loop make major groove contacts. These amino acids are the principal determinants of DNA sequence recognition (Persikov and Singh, 2014) (Figures 1C, S1B). ZFs can be linked linearly in tandem, for occupying DNA of varying lengths, usually recognizing one strand of double-stranded DNA in a linear polarity from 3′ to 5′, with their protein sequence proceeding from N to C termini. The predicted DNA-binding specificity of CTCF ZF3-7 is a five-triplet sequence, which matches partially to the 15 bp consensus sequence (Figure 1C) (Persikov and Singh, 2014). To date, it is not known how CTCF recognizes such a large number of degenerate DNA sequences. Using the recombinant DNA binding domain of human CTCF, we here report the structure of ZF2-9, out of 11-zinc fingers of human CTCF, bound to DNA (Figure 1D). We also investigated the binding of CTCF to the H19/Igf2 sequence, which differs from the consensus at positions 3 and 6, and the effect of cytosine methylation at positions 2 and 12 on binding affinity.
Results
Binding affinities between the CORE and H19 sequences
We generated a series of constructs of human CTCF that included the entire 11-ZF DNA binding domain and fragments with varying number of 3-9 fingers (Figures S1C-S1D). We first compared the binding of CTCF ZF4-7 to three double-stranded oligonucleotides (oligos): a CORE sequence based on an actual CTCF-binding site located in human chromosome 5 (Figures S1E-S1F); a sequence from the human/mouse H19/Igf2 locus known to interact with CTCF (Bell and Felsenfeld, 2000; Hark et al., 2000), particularly ZF4-7 (Renda et al., 2007); and an arbitrary negative control that partially overlaps the consensus (Figure 1E). Fluorescence polarization was used to measure the dissociation constants (KD) towards these oligos (STAR Methods). ZF4-7 displayed approximately 6-fold higher affinity for the CORE sequence than for the H19 sequence (Figure 1F), which shares 7/12 bps with the CORE sequence, and deviates from the 12-bp consensus sequence at two locations: Ade-to-Gua at position 3 and Ade-to-Thy at position 6 (Figure 1E). ZF4-7 bound CORE with >20-fold higher affinity than the negative control (which shares 5/12 bps with the CORE sequence) (Figure 1F). These findings confirm the presumed specificity of ZF4-7.
ZF3, but not ZF8, contributes to the binding of the CORE sequence
Next, we investigated the effects of the immediate neighboring fingers, either the N-terminal ZF3 or the C-terminal ZF8, on the binding affinity of the CORE sequence, which includes additional downstream and upstream triplets with which ZF8 or ZF3 could interact (Figure 1E). The binding affinities of ZF4–8 and ZF4–7 were found to be similar, suggesting that ZF8 does not provide extra binding to the specific sequence (Figure 1G). Addition of ZF3 caused the affinity for CORE to increase by a factor of ∼4 with 150 mM NaCl or ∼6 with 250 mM NaCl (Figures 1G-1H), indicating that ZF3 interacts favorably with the CORE sequence. While ZF8 contributes little to the binding of the specific CORE sequence, it did increase the binding to the control non-specific sequence by a factor of ∼10 (Figure 1I). In addition, we confirmed that the entire 11-ZF domain binds the CORE sequence with varying affinity depending on the ionic strength (Figure 1J).
Structural basis of DNA sequence recognition
To investigate the molecular mechanism by which CTCF recognizes its target DNA sequence, we crystallized six peptide fragments (ZF2-7, 3-7, 4-7, 5-8, 4-10 and 4-11), each bound to oligonucleotides containing the CORE sequence. For larger fragments containing seven or more fingers, ZF1-7 failed to crystallize, whereas for ZF4-10 and ZF4-11, we did not observe the electron densities corresponding to the C-terminal fingers 10 and 11. Thus we obtained structural information for fingers 2 through 9, by superimposing the structures of ZF2-7 and ZF4-9 (Figure 1D). The structures were solved to a resolution of ∼1.7-3.2 Å (Table S1 and Figure S2). These structures were very similar among common fingers shared between structures; for example, with a root mean square deviation (rmsd) of ∼1 Å over 84 pairs of Cα atoms between the common three fingers (5-7) of ZF4-7 and ZF5-8. In addition, we crystallized ZF3-7 in complex with a methylated CpG-containing DNA and ZF6-8 in complex with the H19 sequence (Table S1). We will describe the base-specific interactions using the highest resolution structure of ZF3-7 (1.74 Å; Table 1), and will discuss the differences among them.
Table 1. Structural Statistics, see also Table S1.
| CTCF | ZF3-7 |
|---|---|
| DNA | (5′-3′) GCCAGCAGGGGGCGCTA |
| (3′-5′) CGGTCGTCCCCCGCGAT | |
| PDB | 5KKQ |
| Wavelength (Å) | 1.27046 |
| Space Group | P1 |
| Unit cell (Å) | 41.0, 44.9, 86.7 |
| α, β, γ (°) | 98.4, 92.4, 94.8 |
| * Resolution (Å) | 28.94-1.74 (1.80-1.74) |
| a Rmerge | 0.075 (0.549) |
| b <I/σI> | 12.4 (1.6) |
| Completeness (%) | 92.6 (64.1) |
| Redundancy | 3.6 (1.8) |
| CC 1/2, CC | (0.637 / 0.882) |
| Reflections (observed) | 204,410 |
| Reflections (unique) | 57,383 (4,006) |
| Phasing | Zn-SAD |
| Bijvoet pairs | 53,575 |
| FOM | 0.9 |
| Refinement | |
| Resolution (Å) | 1.74 |
| No. Reflections | 57,333 |
| c Rwork/ d Rfree | 0.170 / 0.199 |
| No. Atoms | |
| Protein | 2395 |
| DNA | 1453 |
| Zn | 10 |
| Solvent | 328 |
| B-factors (Å2) | |
| Protein | 41.7 |
| DNA | 38.3 |
| Zn | 37.5 |
| Solvent | 41.5 |
| R.m.s. deviations | |
| Bond length (Å) | 0.02 |
| Bond angles (°) | 1.5 |
| All atom clashscore | 3.7 |
| Ramachandran (%) | |
| Favored | 98.6 |
| Allowed | 1.4 |
| Cβ deviation | 0 |
Values in parenthesis correspond to highest resolution shell
Rmerge = Σ | I - <I>| /ΣI, where I is the observed intensity and <I> is the averaged intensity from multiple observations
<I/σI> = averaged ratio of the intensity (I) to the error of the intensity (σI)
Rwork = Σ | Fobs - Fcal | /Σ | Fobs |, where Fobs and Fcal are the observed and calculated structure factors, respectively
Rfree was calculated using a randomly chosen subset (5%) of the reflections not used in refinement.
ZF3-7 makes base-specific contacts
Combining structural information from ZF2-7, 3-7 and 4-7, the six-fingers (2-7) interact with DNA exclusively in the major groove (Figure 2A). The convention that we used for numbering nucleotides and amino acids is that the 15 base pairs of the CORE sequence are numbered 1–15 from 5′ (left) to 3′ (right), with the recognition sequence as the “top” strand (colored orange in Figure 2B), whereas the protein sequence runs in the opposite direction from C to N. ZF7 interacts with the 5′ sequence (CCA), ZF6 with the second triplet (GCA), ZF5 with the third triplet (GGG), ZF4 with the fourth triplet (GGC) and ZF3 with the 3′ sequence (GCT). In the structure of ZF2-7 in complex with DNA, ZF2 continues to follow in the major groove (Figure 2A), but the side chains within the DNA-interacting helix were too far away (> 4 Å) to make base-specific hydrogen bonds.
Figure 2. CTCF ZF3-7 forms base-specific contacts.
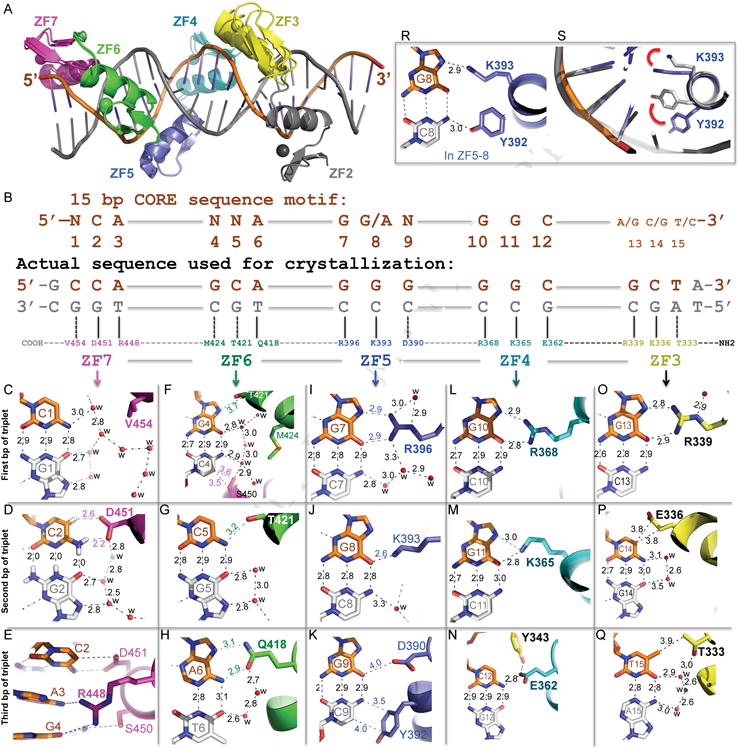
(A) Structural superimposition of ZF2-7 and ZF3-7.
(B) Schematic representation of the ZF3–7 interactions with DNA. The top line indicates the 15-bp consensus sequence. The second line indicates the base pair positions (1–15). The third and the fourth lines are the sequence of the double-stranded oligo used for crystallization, shown with the top strand (orange) matching the consensus sequence. Amino acids of each finger interact specifically with the DNA bases shown below.
(C-Q) DNA base specific interactions involve a particular residue of each ZF, colored according to panel A. Atoms are colored dark blue for nitrogen, red for oxygen, and carbon atoms are decorated with finger specific colors. The numerical numbers indicate the inter-atomic distance in angstroms, ‘w’ is water molecules (small red spheres). Note in panel D, hydrogen atoms on the G2:C2 base pair were shown to illustrate the C-H…O type of hydrogen bonds between C2 ring carbon atom C5-H and D451.
(R) In the structure of ZF5-8, side chains of Y392 and K393 interact respectively with the cytosine and guanine of the C8:G8 base pair.
(S) Side chains of Y392 and K393 in the structure of ZF5-8 (grey) adopted different conformations from that of ZF3-7 (blue; as shown in panel J). The red arrows indicate a concerted movement of the two side chains.
Analysis of the CTCF consensus sequences obtained from ChIP data (Figures 1A-1B) suggests that the most conserved base pairs occur at positions 2 and 3 of the first triplet (NCA), position 6 of the second triple (NNA), positions 7 and 8 of the third triple (GRN, where R=A or G), and positions 10-12 of the fourth triplet (GGC). The other base pairs are denoted as variable (N) at position 1 of the 5′ sequence, positions 4 and 5 of the second triplet and position 9 of the third triplet. Interactions with the variable base pairs of the CORE sequence involve water-mediated H-bonds (G1:C1 of triplet 1, Figure 2C), weak H-bonds with T421 and S450 (G4:C4 and C5:G5 of triplet 2, Figures 2F, 2G; Ser and Thr can each act as an H-bond donor or acceptor, explaining how they might accommodate alternative base pairs), hydrophobic interaction with Y392 (G9:C9 of triplet 3, Figure 2K), or a gap in the protein-DNA interface with hydrophobic residue M424 positioned well away from the base (Figure 2F).
The eight conserved base pairs in the consensus sequence are recognized primarily by H-bonds between the bases of the top strand and residues of ZF4-7. The terminal Nη1 and Nη2 groups of R396 and R368 donate H-bonds to the O6 and N7 atoms of guanines at positions 7 and 10, respectively (Figures 2I, 2L). Many sequence-specific proteins recognize Gua in this same manner. For example, the SfiI endonuclease (recognition sequence: GGCCN5GGCC), has three of the four guanines in each half-site form identical H-bonds with Arg; while the fourth Gua H-bonds with a lysine residue in almost the same manner (Vanamee et al., 2005) as we observed for the K365 of ZF4 with Gua at position 11 (Figure 2M). A cancer-associated mutation of Lys365-to-Thr (K365T), found in endometrial cancer cells (Cancer Genome Atlas Research et al., 2013), results in a 20-fold loss of DNA binding (Figure 3A).
Figure 3. Differential cytosine methylation influences CTCF binding.
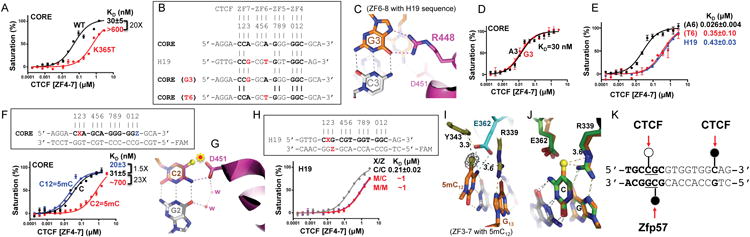
(A) A cancer-associated mutation (K365T) shows diminished DNA binding.
(B) The H19 sequence deviates from the CORE consensus sequence at two locations, a Gua instead of Ade at position 3 and a Thy instead of Ade at position 6.
(C) R448 of ZF7 makes bidentate contacts with the Gua at position 3 in the structure of ZF6-8 in complex with the H19 sequence.
(D) The Ade-to-Gua change at position 3 of the CORE sequence does not affect DNA binding affinity by ZF4-7.
(E) The Ade-to-Thy change at position 6 of the CORE sequence shows much reduced DNA binding by ZF4-7.
(F) Methylation at C2 of the CORE sequence abolishes DNA binding, whereas methylation of C12 enhances DNA binding by ZF4-7.
(G) Modeling a methyl group onto unmodified C2 potentially results in repulsion (indicated by a star) with D451 of ZF7.
(H) Methylation (hemi- or fully) of the CpG dinucleotide at position 2 of the H19 sequence shows reduced DNA binding affinity by ZF4-7.
(I) In the structure of ZF3-7 in complex with the methylated DNA, the omit electron density (grey mesh), contoured at 5σ above the mean, is shown for the 5mC methyl group (in yellow sphere).
(J) Structural comparison of ZF3-7 in complex with methylated DNA (in orange) and unmethylated DNA (in green).
(K) A model of strand-specific interaction associated with differential methylation at C2 by CTCF (open circle: un/de-methylated) and Zfp57 (filled circle: methylated). DNA binding data represent the mean ± SEM of two independent determinations performed in duplicate. See also Figure S3.
The G:C base pair at position 8 forms a single H-bond with K393, via either the guanine O6 atom in the structures of ZF2/3/4-7 (Figure 2J) or the guanine N7 atom in the structure of ZF5-8 (Figure 2R). In the structure of ZF5-8 in complex with DNA, an additional H-bond was formed with the opposite paired cytosine N4 atom and Y392 (Figure 2R); the side chain of which, together with K393, displayed conformational changes between structures of ZF5-8 and ZF2/3/4-7 (Figure 2S). Nevertheless these interactions could also occur to an A:T base pair, a feature that likely contributes to purine specificity (G or A) at that position (Figures 1A-1B).
R448 of ZF7 bridges between two neighboring bases, A3 of the first triplet and G4 of the second triplet (Figure 2E). As mentioned above, apposition of Arg with Gua is the most common mechanism for G:C base pair recognition (Luscombe et al., 2001). Consistent with the prediction (Figure 1C), the H19 sequence has a Gua at the corresponding position 3 (Figure 3B) and in the structure of ZF6-8 in complex with the H19 sequence, R448 forms the orthodox bidentate interaction with the Gua (Figure 3C). We also replaced the A:T base pair at position 3 with a G:C base pair in the CORE sequence, in essence to mimic the H19 sequence. ZF4-7 binds the two oligos indistinguishably (Figure 3D), probably because in both cases R448 involves two H-bonds. Our results suggest that the hydrogen bonds involving the variable bases are adaptable in the sense that the participating amino acids can alter conformation to suit the substrate and, in this way, intimately fit the ZF array to a variety of different sequences. The R448 of ZF7 is an example of such adaptability. Other examples of adaptability are also evident in our structure, such as K393 of ZF5 (for Gua or Ade), T421 of ZF6 (for variable base), and hydrophobic residues Y392 of ZF5 and V454 of ZF7 (for variable bases) as discussed.
Triplet 2 of the CORE sequence includes an invariant Ade base at position 6, which is recognized by Q418 of ZF6. The side chain of Q418 donates one H-bond to adenine N7 and accepts one from adenine N6 (Figure 2H). Juxtaposition of Gln (or Asn) with Ade is a common mechanism for recognition of this base (Luscombe et al., 2001). Interestingly, the corresponding H19 sequence is a thymine or cytosine (Bell and Felsenfeld, 2000; Hark et al., 2000) (Figure 3B). When we substituted the T:A base pair for A:T in triplet 2 of the CORE sequence, this change decreased affinity for ZF4-7 and resulted in a similar affinity to that of the H19 sequence (Figure 3E), explaining the observed difference in affinity between the CORE and H19 sequences (Figure 1F). Interestingly, CTCF-binding sites are frequently mutated in cancer, and the mutations are clustered predominately at the A:T base pair, which is changed to any of the three alternative base pairs (Katainen et al., 2015).
Finally, the N-terminal ZF3 recognizes the 3′ sequence - (G/A)(C/G)(T/C) - at base pair positions 13-15, which are more variable based on the ChIP data (Figures 1A-1B). We used sequence GCT in the co-crystallization. The protein-DNA contacts involve R339 interaction with Gua, E336 interaction with a cytosine, and T333 interaction with a thymine (Figures 2O-2Q). Like R448 of ZF7, R339 might accommodate an adenine as well. In addition, the aliphatic side chain Cγ and Cδ atoms of E336 form van der Waals contacts with the ring carbon-5 atom of cytosine at position 14, while the side chain methyl group of T333 makes a van der Waals contact with the methyl group of thymine at position 15. We note that a negatively charged residue is observed in the interactions with C2 (D451 of ZF7; Figure 2D), C12 (E362 of ZF4; Figure 2N), and in the vicinity of a variable base pair at position 9 (D390 of ZF5; Figure 2K).
Methylation-sensitive and methylation-insensitive DNA binding
The two invariant Cyt at positions 2 and 12 are recognized primarily by D451 of ZF7 and E362 of ZF4, respectively (Figures 2D, 2N). For both cytosine residues, the following 3′ nucleotide is either a Gua or an Ade, forming a CpG or CpA dinucleotide, the canonical sites for cytosine methylation in mammalian DNA (Lister et al., 2009). Parallel comparison with bisulfite sequencing data of various human cell types indicated that approximately 40% of variable CTCF binding is linked to differential DNA methylation, concentrated at the two conserved Cyt positions within the 15-bp recognition sequence (Wang et al., 2012). In a binding assay with oligos containing 5-methylcytosine (5mC) in place of cytosine at position 2 or position 12 in the top strand of the CORE recognition sequence, the affinity for the oligo methylated at C2 was drastically reduced, by a factor of 23, while affinity was slightly increased (by a factor 1.5) when methylated at C12 (Figure 3F). This ∼23-fold difference in binding affinity associated with C2 methylation is approximately the same as the difference observed between the CORE and control sequences (Figure 1F), which harbors a thymine (5-methyluracil) at the corresponding position (Figure 1E). The presence of a methyl group at the C5 atom of C2 would sterically obstruct D451 in the Cyt-specific conformation (Figure 3G), perhaps explaining the diminished binding to the C2 methylated oligo. CTCF binding to the H19 sequence was inhibited by DNA methylation at a single CpG site corresponding to the C2 position (Bell and Felsenfeld, 2000; Hark et al., 2000; Renda et al., 2007). As expected, the binding affinity to the H19 oligo was reduced equally regardless of whether the CpG is hemi-methylated or fully-methylated (Figure 3H) – in agreement with the major effect of cytosine methylation involving the top strand. Methylation of the bottom strand has little effect on the interaction (Renda et al., 2007) because ZF7 contacts only the top strand guanine of the G3:C3 base pair (Figure 3C).
In contrast, the methylation at C12 (or at C13 of the opposite strand) does not interfere with the conformation of E362. In the structure of ZF3-7 in complex with DNA containing a 5mC at position 12, the methyl group is in a van der Waals cage surrounded by the side chain carbon Cδ atom of E362, the aromatic side chain of Y343, and the guanidine group of R339, which recognizes the 3′ Gua at position 13 (Figure 3I). The distinct effects of methylation at C2 and C12 on binding affinity are due to the difference in the amino acid (D452 or E362) used in the interaction, with Asp preferring to bind unmodified cytosine and Glu preferring methylated cytosine (Choo and Klug, 1997; Hashimoto et al., 2016; Liu et al., 2013) (Figure S3). This Glu preference for 5mC could also apply to methylated C14 and E336 (Figure 2P). Our analyses show that binding of CTCF is affected differently by the methylation at C2 and C12 – two cytosine residues separated by one helical turn. Superimposition of the structures of ZF3-7 in complex with methylated and unmethylated DNA reveals that the local DNA structure is largely unchanged, but suggests that the gained cation-pi interactions play an important role in the binding, with Arg providing the cationic moiety and 5mC the pi electrons in the 5mC-Arg-G triad (Figure 3J) (Liu et al., 2013; Zou et al., 2012).
Although the experiments were performed in vitro, the results imply that CTCF can respond in modulated ways to alternative modifications at different Cyt positions. CTCF binding could be disrupted by increased C2 methylation or enhanced by increased C12 methylation. Furthermore, the methylation level could also be influenced by the adjacent 3′ sequence, i.e. whether it is a CpG or a non-CpG site, suggesting additional mechanisms to modulate the interaction of CTCF with different sequences in the genome. Approximately a third (29%) of CTCF recognition sequences genome-wide contain a CpG dinucleotide at positions 2 and/or 12 (Wang et al., 2012).
The strand-specific asymmetric recognition by the C2H2 ZF proteins adds yet another layer of regulatory control. For example, CTCF recognizes the top strand of the H19 sequence and is sensitive to the top strand modification. In contrast, Zfp57, an allele-specific binding protein of imprinted loci, recognizes a 6-bp sequence overlapping with the C2 within the H19 sequence (Quenneville et al., 2011) (Figure 3K). The binding of Zfp57 is enhanced by methylation of the bottom strand (Liu et al., 2012). If the two DNA strands can be modified independently (i.e., strand-biased DNA modification or transiently generated during semiconservative DNA replication), then the different modification states could affect binding affinities of at least two different DNA binding proteins.
ZF8-9 span the DNA duplex beyond the 15-bp CORE sequence
Among the CTCF-binding motifs, there is no specific sequence observed immediately upstream to the highly conserved 15-bp consensus sequence (Figures 1A-1B), where ZF8 is predicted to bind (Figure 1C). We show that ZF8 has no effect on the DNA binding to the specific CORE sequence (Figure 1G), but the presence of ZF8 does increase nonspecific binding of a control sequence (Figure 1I). As mentioned earlier, we crystallized the seven-finger fragment ZF4-10 and eight-finger fragment ZF4-11 in complex with DNA (Table S1). In both cases, the C-terminal fingers 10 and 11 were not observed in the electron density and ZF9 has higher averaged crystallographic thermal B-factor (yellow color in Figure 4A), an indication that this part of the structure is more flexible than rest. While ZF4-7 occupies a length of 12-bp CORE sequence, ZF8-9 spans approximately the same length along the DNA phosphate backbone (Figure 4A).
Figure 4. (A) CTCF ZF8-9 spans across the DNA phosphate backbone (A).
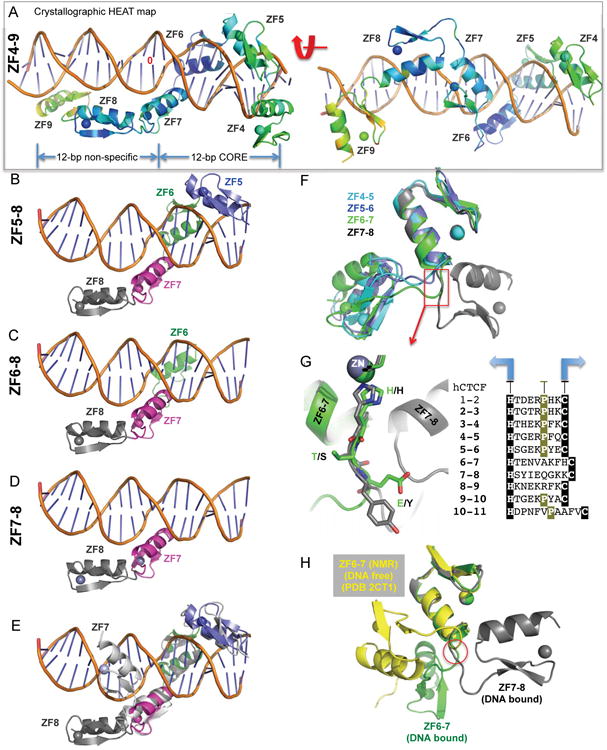
Two views of ZF4-9, displayed by the crystallographic heat map, from low-to-high thermal B-factor (blue, cyan, green, and yellow).
(B-D) Aligned structures of ZF5-8 (B), ZF6-8 (C), and ZF7-8 (D) against a reference DNA molecule.
(E) Superimposition of 4-finger structures of ZF4-7 and ZF5-8 indicates that the C-terminal ZF7 lies in the major groove whereas ZF8 spans across the minor groove of DNA. See also Figure S4.
(F) Superimposition of four fragments of two-finger structures, ZF4-5, ZF5-6, ZF6-7 and ZF7-8, reveals that the C-terminal ZF8 swings to the right whereas the rest swing to the left.
(G) Enlarged linker regions between ZF6-7 and ZF7-8, with the alignment of linker sequences.
(H) Superimposition of DNA-bound ZF6-7 and DNA-free ZF6-7 (PDB 2CT1).
We also used smaller ZF8-containing fragments (ZF5-8 and ZF6-8), and crystallized ZF5-8 with the CORE sequence plus the predicted triplet sequence for ZF8 (GTG or TTG) in two different space groups, P65 and P41212. In both space groups, crystallized under different conditions and having different lattice contacts, ZF5-7 are located in the major groove of DNA while ZF8 does not make any base specific contact (Figure 4B). The two structures are highly similar with an rmsd of ∼1 Å throughout 112 pairs of Cα atoms shared between the two.
Additionally, we solved a structure of ZF6-8 in complex with the H19 sequence. The crystallographic asymmetric unit of this structure is comprised of two protein-DNA complexes: one containing ZF6-8 (Figure 4C) and the second containing ZF7-8 with no electron density observable for the N-terminal ZF6 (Figure 4D). Alignment of the four ZF8-containing complexes indicates that the inter-domain orientation between ZF7 and ZF8 is the same (Figures 4A-4D). Superimposing the first three fingers of the 4-finger structures of ZF4-7 and ZF5-8 revealed that the C-terminal ZF7 is located in the DNA major groove whereas the C-terminal ZF8 is across the minor groove of DNA (Figure 4E).
We do not know whether the ZF8 conformation in relation to ZF7 is induced by DNA binding or the intrinsic feature of inter-finger interactions. We generated four fragments of two-finger structures, ZF4-5, ZF5-6, ZF6-7 and ZF7-8, and superimposed the N-terminal fingers pairwise (rmsd=0.5-1 Å for 30 pairs of Cα atoms). The resulting C-terminal fingers following ZF4, 5 and 6 are located to the left, while ZF8 is located to the right side of the N-terminal finger (Figure 4F). The two conformations of the C-terminal finger are approximately 180° rotation apart. This could be achieved via a series of rotations of main-chain torsion angles along the two residues immediately after the last Zn-ligand histidine of the N-terminal finger (Figure 4G), and regardless of the size of the linker between the two fingers or whether the linker contains a proline downstream (Figure 4G). Comparison of the DNA bound ZF6-7 to a solution NMR structure available for ZF6-7 in the absence of DNA, revealed yet another conformation with a switch point at the same linker residues (Figure 4H). Thus, the flexibility of the linker between the two fingers may allow the multidomain CTCF ZF array to span a greater length of the DNA duplex beyond the 15-bp CORE sequence, without additional sequence specific binding (occurring in ∼85% of CTCF-binding sites (Nakahashi et al., 2013)), as exemplified by the transcription factor TFIIIA (Nolte et al., 1998) (Figure S4).
Discussion
As the first step towards a mechanistic view of the process of forming CTCF-associated DNA loops (Fudenberg et al., 2016; Nichols and Corces, 2015; Sanborn et al., 2015), we obtained a high-resolution structure of the tandem ZF array of CTCF in complex with DNA. The results give important insights into the biology of CTCF and the mechanisms by which mutations in the binding site or the protein may lead to disease states. While the CTCF-bound DNA conformation is largely B-form, we do not know whether the linear, naked DNA structure used in this study represents the chromatin bound form where the local DNA structure could have distortions. Additional experiments using the full-length CTCF and nucleosomal DNA are needed.
Results from in vitro studies of interactions between human CTCF and DNA provide a structural explanation for the sequence adaptability of this protein, which can bind to DNA with high affinity while recognizing sequences with high variability in the 15-bp core sequence motif. This property of CTCF can be traced to the ability of specific residues in the ZFs to adopt alternative conformations to establish versatile H-bonds with some bases but not with others. The structure of the ZF domain of CTCF suggests that ZF8 can span the entire minor groove resulting in no additional sequence specific binding immediately beyond the 15-bp CORE sequence. Importantly, the structural information also seems to indicate a lack of a specific function in DNA recognition/binding for the terminal zinc fingers, i.e., ZF1 and ZF10-11. It is possible that these ZFs together with the C-terminal domain are involved in interactions with other proteins or, alternatively, binding to RNA (Kung et al., 2015; Saldana-Meyer et al., 2014).
Insights gained from the structure of the ZFs also help explain the position-dependent effect of differential DNA methylation at two cytosine residues on the binding affinity of the protein. Interestingly, the two cytosines in the consensus sequence that can be methylated have opposite effects on CTCF-DNA interactions. These results suggest that CTCF binding sites may exist in the genome in four different methylation states with varied affinities for the protein. One study suggested that the C12 methylation in a CTCF-binding site (5′-TCCACCAGGGGCMG-3′, where M=5mC) is associated with tissue-specific PRR15 (proline-rich 15) gene expression (Yu et al., 2013).
Our study suggests that gene expression could plausibly be controlled by a combination of DNA sequence variations in the recognition sequence, patterns of DNA methylation, and variable structural architectures of DNA-binding proteins, such as C2H2 ZF proteins, basic leucine-zipper and basic-helix–loop–helix transcription factors. These observations imply exciting new levels of subtlety and versatility in epigenetic regulatory processes.
Contact for Reagent and Resource Sharing
Please contact X.C. (xcheng5@mdanderson.org) for reagent and resources generated in this study.
Experimental Model and Subject Details
GST-tagged human CTCF (NP_006556.1) fragments were cloned into pGEX6P-1, generating expression plasmids covering the entire 11-ZnFs (Figure S1). Fragments were expressed in the Escherichia coli strain BL21-CodonPlus(DE3)-RIL (Stratagene).
Method Details
Protein expression and purification
Typically, 2–3 L of cultures were grown at 37 °C to log phase (OD600=0.5–0.8) and then shifted to 16 °C, ZnCl2 was added to a final concentration of 25 μM, expression was induced by the addition of isopropyl β-D-1-thiogalactopyranoside to 0.2 mM, and the cultures were incubated overnight at 16°C. Cells were harvested by centrifugation, resuspended in lysis buffer containing 20 mM Tris-HCl (pH 7.5), 500 mM NaCl, 5% (v/v) glycerol, 0.5 mM Tris(2-carboxyethyl)phosphine hydrochloride (TCEP), and 25 μM ZnCl2, and lysed by sonication. Lysates were mixed with polyethylenimine (Sigma) at pH 7 to a final concentration of 0.3-0.4% (w/v) before centrifugation at 16,500 rpm.
The cleared extract was loaded onto a glutathione-Sepharose 4B column (GE Healthcare) pre-equilibrated with the lysis buffer. The GST fusion proteins were eluted with 20 mM glutathione (GSH) in the elution buffer containing 100 mM Tris-HCl (pH 8.0), 250 mM NaCl, 5% (v/v) glycerol, 25 μM ZnCl2, and without TCEP. The GST tag was removed using PreScission protease (purified in-house), leaving five additional N-terminal residues (Gly–Pro– Leu–Gly–Ser) on each protein. The protein solutions were adjusted to 250 mM NaCl and loaded onto tandem HiTrap-Q/HiTrap-SP columns (GE Healthcare) or HiTrap-SP column directly. Most proteins flowed through the Q column onto the SP column from which they were eluted using a linear gradient of NaCl from 0.25-1.0 M. Finally, the pooled protein was concentrated and loaded onto a size exclusion column and eluted as a single peak in the lysis buffer. Final protein concentrations were estimated by absorbance at 280 nm.
Fluorescence-based DNA binding assay
Fluorescence polarization measurements were carried out at 25 °C on a Synergy 4 microplate reader (BioTek). The 6-carboxy-fluorescein (FAM)-labeled double-stranded oligo probe (5 nM) was incubated for 10 min with increasing amounts of protein in 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 5% (v/v) glycerol, and 0.5 mM TCEP. No change in fluorescence intensity was observed with the addition of protein. The oligonucleotide sequences used for DNA binding assays were the CORE sequences of 5′-AGG ACX AGC AGG GGG XGC A-3′ and 3′-TCC TGG TCG TCC CCC GCG T-5′-FAM, the H19 sequences of 5′-GTT GCX GCG TGG TGG CAG-3′ and 3′-CAA CGG XGC ACC ACC GTC-5′-FAM, where X = C or 5-methylcytosine (5mC). The control sequences are 5′-AAT GGA CGA GTC ATA GGA GA-3′ and 3′-TAC CTG CTC AGT ATC CTC TT-5′-FAM.
Crystallography
Purified ZnF fragments were incubated with the double-stranded oligos at equimolar ratio, dialyzed against 20 mM Tris-HCl, pH 8.0, 300 mM NaCl, 1 mM TCEP and concentrated to ∼1 mM protein/DNA complex prior to crystallization. We crystallized CTCF ZnF fragments in the presence of oligos by the sitting-drop vapor diffusion method at 16 °C using equal amounts of protein–DNA mixtures and well solution:
| Crystallization conditions for human CTCF-DNA complexes | |
| ZF2-7 + DNA | 15% (w/v) PEG 4K, 0.2 M ammonium acetate, 0.1 M Na Citrate pH 5.6 |
| ZF3-7 + DNA | 25% (w/v) PEG 3350, 0.2 M ammonium acetate, 0.1 M Tris pH 8.5 |
| ZF3-7 + methylated DNA | 25% (w/v) PEG 3350, 0.2 M NaCl, 0.1 M Hepes pH 7.5 |
| ZF4-7 + DNA | 25% (w/v) PEG 3350, 0.2 M ammonium acetate, 0.1 M Bis-Tris HCl, pH 5.5 |
| ZF5-8 +DNA (P65) | 20% (w/v) PEG 3350, 0.2 M ammonium sulfate, 0.1 M sodium cacodylate pH 6.5 |
| ZF5-8 +DNA (P41212) | 25% (w/v) PEG 3350, 0.2 M ammonium acetate, 0.1 M Bis-Tris HCl, pH 5.5 |
| ZF6-8 + DNA | 20% (w/v) PEG 8000, 0.1 M CHES-NaOH pH 9.5 |
| ZF4-10 + DNA | 28% (w/v) PEG 2K MME, 0.1 M Bis-Tris HCl pH 6.5 |
| ZF4-11 + DNA | 28% (w/v) PEG 2K MME, 0.1 M Bis-Tris HCl pH 6.5 |
Crystals were cryoprotected by soaking in mother liquor supplemented with 20% (v/v) ethylene glycol before plunging into liquid nitrogen. X-ray diffraction data sets were collected at 100 K at the SER-CAT beamlines (22ID-D) at the Advanced Photon Source, Argonne National Laboratory, and processed using HKL2000 (Otwinowski et al., 2003) and/or XDS (Kabsch, 2010). The ZF4-7 data set was severely anisotropic, based on analysis by the UCLA Diffraction Anisotropy Server, and map quality was greatly improved after trimming weak reflections (Strong et al., 2006).
Quantification and Statistical Analysis
DNA binding curves were fit individually using GraphPad Prism 5.0 software (GraphPad Software, Inc.). Binding constants (KD) were calculated as [mP] = [maximum mP] × [C]/(KD + [C]) + [baseline mP], and saturated [mP] was calculated as saturation = ([mP] - [baseline mP])/([maximum mP] - [baseline mP]), where mP is millipolarization and [C] is protein concentration. Saturated [mP] was calculated as saturation = ([mP] – [baseline mP])/([maximum mP] – [baseline mP]). Curves were normalized as percentage of bound oligonucleotides and reported is the mean ± SEM of the interpolated KD from (at least) two independent experiments performed in duplicate. For those binding curves that did not reach saturation, the lower limit of the binding affinity was estimated.
Crystallographic phases were determined by Zn-SAD (Figure S2). Phasing, map production, and model refinement were performed using PHENIX (Adams et al., 2010) and Coot (Emsley et al., 2010). All structures were solved, built, and refined independently. The statistics were calculated for the entire resolution range (Table S1). The Rfree and Rwork values were respectively calculated for 5% (randomly selected) and 95% of the observed reflections (10% and 90%, respectively, for ZF4-7). Molecular graphics were generated using PyMol (DeLano Scientific, LLC).
Data and Software Availablbility
The X-ray structures (coordinates and structure factor files) of CTCF ZFs with bound DNA have been submitted to PDB under accession number 5T0U (ZF2-7), 5KKQ (ZF3-7), 5T00 (ZF3-7 in complex with 5mC DNA), 5K5H (ZF4-7), 5K5I and 5K5J (ZF5-8), 5K5L (ZF6-8 with H19 sequence), and 5UND (ZF4-9).
Supplementary Material
Key Resources Table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial Strains and expression vectors | ||
| Escherichia coli BL21(DE3) codon plus RIL | Stratagene | 230240 |
| pGEX-6p-1 | GE Healthcare | 28-9546-48 |
| Human CTCF ZF6-8 (residues 405-492) | This paper | pXC1197 |
| Human CTCF ZF5-8 (residues 377-492) | This paper | pXC1199 |
| Human CTCF ZF4-7 (residues 349-465) | This paper | pXC1202 |
| Human CTCF ZF1-3 (residues 263-348) | This paper | pXC1356 |
| Human CTCF ZF4-8 (residues 348-492) | This paper | pXC1357 |
| Human CTCF ZF8-11 (residues 464-581) | This paper | pXC1358 |
| Human CTCF ZF9-11 (residues 493-581) | This paper | pXC1359 |
| Human CTCF ZF1-4 (residues 273-377) | This paper | pXC1417 |
| Human CTCF ZF1-11 (residues 273-581) | This paper | pXC1441 |
| Human CTCF ZF4-7 (residues 349-465) Lys365-to-Thr (K365T) mutant | This paper | pXC1518 |
| Human CTCF ZF3-7 (residues 321-465) | This paper | pXC1551 |
| Human CTCF ZF3-9 (residues 321-518) | This paper | pXC1571 |
| Human CTCF ZF1-7 (residues 263-465) | This paper | pXC1564 |
| Human CTCF ZF2-7 (residues 294-465) | This paper | pXC1565 |
| Human CTCF ZF3-11 (residues 321-581) | This paper | pXC1567 |
| Human CTCF ZF4-11 (residues 348-581) | This paper | pXC1568 |
| Human CTCF ZF4-9 (residues 348-518) | This paper | pXC1573 |
| Human CTCF ZF4-10 (residues 348-547) | This paper | pXC1574 |
| Deposited Data | ||
| ZF2-7 | This paper | PDB: 5T0U |
| ZF3-7 | This paper | PDB: 5KKQ |
| ZF3-7 in complex with 5mC DNA | This paper | PDB: 5T00 |
| ZF4-7 | This paper | PDB: 5K5H |
| ZF5-8 (space group P65) | This paper | PDB: 5K5I |
| ZF5-8 (space group P41212) | This paper | PDB: 5K5J |
| ZF6-8 with H19 sequence | This paper | PDB: 5K5L |
| ZF4-9 | This paper | PDB: 5UND |
| Oligonucleotides | ||
| 5′-GTTGCCGCGTGGTGGCAG-3′ 3′-CAACGGCGCACCACCGTC-5′-FAM |
New England BioLabs | Custom order |
| 5′-GTTGC5mCGCGTGGTGGCAG-3′ 3′-CAACGG5mCGCACCACCGTC-5′-FAM |
New England BioLabs | Custom order |
| 5′-AGGACCAGCAGGGGGCGCA-3′ 3′-TCCTGGTCGTCCCCCGCGT-5′-FAM |
IDT | Custom order |
| 5′-AGGAC5mCAGCAGGGGGCGCA-3′ 3′-TCCTGGTCGTCCCCCGCGT-5′-FAM |
IDT | Custom order |
| 5′-AGGACCAGCAGGGGG5mCGCA-3′ 3′-TCCTGGTCGTCCCCCGCGT-5′-FAM |
IDT | Custom order |
| 5′-AGGACCGGCAGGGGGCGCA-3′ 3′-TCCTGGCCGTCCCCCGCGT-5′-FAM |
IDT | Custom order |
| 5′-AGGACCAGCTGGGGGCGCA-3′ 3′-TCCTGGTCGACCCCCGCGT-5′-FAM |
IDT | Custom order |
| 5′-AATGGACGAGTCATAGGAGA-3′ 3′-TACCTGCTCAGTATCCTCTT-5′-FAM |
IDT | Custom order |
| Software and Algorithms | ||
| HKL2000 | Otwinowski et al. (2003) Acta Crystallogr A 59, 228-234. | www.hkl-xray.com |
| XDS | Kabsch (2010) Acta Crystallogr D Biol Crystallogr 66, 125-132 | xds.mpimf-heidelberg.mpg.de |
| UCLA Diffraction Anisotropy server | Strong et al. (2006) Proc Natl Acad Sci U S A 103, 8060-8065 | services.mbi.ucla.edu/anisoscale |
| Coot | Emsley et al. (2010) Acta Crystallogr D Biol Crystallogr 66, 486-501 | www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot |
| Phenix | Adams et al. (2010) Acta Crystallogr D Biol Crystallogr 66, 213-221 | www.phenix-online.org |
| Pymol | DeLano Scientific LLC | www.pymol.org |
| PRISM 5.0 | Graph pad Software | www.graphpad.com/scientific-software/prism |
Highlights.
ZF3-7 of CTCF recognize the major groove of five triplets of specific nucleotide
CTCF binds to DNA with high affinity while allowing high sequence variability
CTCF forms versatile H-binds that can arise with some bases but not with others
Structure explains the position-dependent effect of DNA methylation on CTCF binding
Acknowledgments
We thank B. Baker of New England Biolabs for synthesizing the oligonucleotides, Yiwei Liu for initial work on CTCF, Yusuf Olatunde Olanrewaju, John Shanks and Alison Setili for help with protein purifications, Robert M. Blumenthal for comments, and Lanlan Shen for discussion. The Department of Biochemistry of Emory University School of Medicine supported the use of SER-CAT beamlines. This work was supported by a grant from the National Institutes of Health GM049245-23 to X.Z. and X.C.
Footnotes
Author Contributions: H.H. performed crystallography, D.W. and H.H. performed protein expression, purification, crystallization, and DNA binding assays, J.R.H. performed data collection and structural determination of ZF4-10 and ZF4-11; V.G.C. initiated this collaborative work and participated in discussion throughout, X.Z. and X.C. organized and designed the scope of the study. All authors were involved in analyzing data and preparing the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bell AC, Felsenfeld G. Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature. 2000;405:482–485. doi: 10.1038/35013100. [DOI] [PubMed] [Google Scholar]
- Buck-Koehntop BA, Stanfield RL, Ekiert DC, Martinez-Yamout MA, Dyson HJ, Wilson IA, Wright PE. Molecular basis for recognition of methylated and specific DNA sequences by the zinc finger protein Kaiso. Proc Natl Acad Sci U S A. 2012;109:15229–15234. doi: 10.1073/pnas.1213726109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research, N. Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, Shen H, Robertson AG, Pashtan I, Shen R, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67–73. doi: 10.1038/nature12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Tian Y, Shu W, Bo X, Wang S. Comprehensive identification and annotation of cell type-specific and ubiquitous CTCF-binding sites in the human genome. PLoS One. 2012;7:e41374. doi: 10.1371/journal.pone.0041374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choo Y, Klug A. Selection of DNA binding sites for zinc fingers using rationally randomized DNA reveals coded interactions. Proc Natl Acad Sci U S A. 1994;91:11168–11172. doi: 10.1073/pnas.91.23.11168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choo Y, Klug A. Physical basis of a protein-DNA recognition code. Curr Opin Struct Biol. 1997;7:117–125. doi: 10.1016/s0959-440x(97)80015-2. [DOI] [PubMed] [Google Scholar]
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fudenberg G, Imakaev M, Lu C, Goloborodko A, Abdennur N, Mirny LA. Formation of Chromosomal Domains by Loop Extrusion. Cell reports. 2016;15:2038–2049. doi: 10.1016/j.celrep.2016.04.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjur S, Williams LM, Ryan NK, Cobb BS, Sexton T, Fraser P, Fisher AG, Merkenschlager M. Cohesins form chromosomal cis-interactions at the developmentally regulated IFNG locus. Nature. 2009;460:410–413. doi: 10.1038/nature08079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hark AT, Schoenherr CJ, Katz DJ, Ingram RS, Levorse JM, Tilghman SM. CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature. 2000;405:486–489. doi: 10.1038/35013106. [DOI] [PubMed] [Google Scholar]
- Hashimoto H, Olanrewaju YO, Zheng Y, Wilson GG, Zhang X, Cheng X. Wilms tumor protein recognizes 5-carboxylcytosine within a specific DNA sequence. Genes Dev. 2014;28:2304–2313. doi: 10.1101/gad.250746.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto H, Wang D, Steves AN, Jin P, Blumenthal RM, Zhang X, Cheng X. Distinctive Klf4 mutants determine preference for DNA methylation status. Nucleic Acids Res. 2016;44:10177–10185. doi: 10.1093/nar/gkw774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heger P, Marin B, Bartkuhn M, Schierenberg E, Wiehe T. The chromatin insulator CTCF and the emergence of metazoan diversity. Proc Natl Acad Sci U S A. 2012;109:17507–17512. doi: 10.1073/pnas.1111941109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jothi R, Cuddapah S, Barski A, Cui K, Zhao K. Genome-wide identification of in vivo protein-DNA binding sites from ChIP-Seq data. Nucleic Acids Res. 2008;36:5221–5231. doi: 10.1093/nar/gkn488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katainen R, Dave K, Pitkanen E, Palin K, Kivioja T, Valimaki N, Gylfe AE, Ristolainen H, Hanninen UA, Cajuso T, et al. CTCF/cohesin-binding sites are frequently mutated in cancer. Nat Genet. 2015;47:818–821. doi: 10.1038/ng.3335. [DOI] [PubMed] [Google Scholar]
- Kung JT, Kesner B, An JY, Ahn JY, Cifuentes-Rojas C, Colognori D, Jeon Y, Szanto A, del Rosario BC, Pinter SF, et al. Locus-specific targeting to the X chromosome revealed by the RNA interactome of CTCF. Mol Cell. 2015;57:361–375. doi: 10.1016/j.molcel.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Toh H, Sasaki H, Zhang X, Cheng X. An atomic model of Zfp57 recognition of CpG methylation within a specific DNA sequence. Genes Dev. 2012;26:2374–2379. doi: 10.1101/gad.202200.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Zhang X, Blumenthal RM, Cheng X. A common mode of recognition for methylated CpG. Trends Biochem Sci. 2013;38:177–183. doi: 10.1016/j.tibs.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Olanrewaju YO, Zheng Y, Hashimoto H, Blumenthal RM, Zhang X, Cheng X. Structural basis for Klf4 recognition of methylated DNA. Nucleic Acids Res. 2014;42:4859–4867. doi: 10.1093/nar/gku134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscombe NM, Laskowski RA, Thornton JM. Amino acid-base interactions: a three-dimensional analysis of protein-DNA interactions at an atomic level. Nucleic Acids Res. 2001;29:2860–2874. doi: 10.1093/nar/29.13.2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurano MT, Wang H, John S, Shafer A, Canfield T, Lee K, Stamatoyannopoulos JA. Role of DNA Methylation in Modulating Transcription Factor Occupancy. Cell reports. 2015;12:1184–1195. doi: 10.1016/j.celrep.2015.07.024. [DOI] [PubMed] [Google Scholar]
- Nakahashi H, Kwon KR, Resch W, Vian L, Dose M, Stavreva D, Hakim O, Pruett N, Nelson S, Yamane A, et al. A genome-wide map of CTCF multivalency redefines the CTCF code. Cell reports. 2013;3:1678–1689. doi: 10.1016/j.celrep.2013.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols MH, Corces VG. A CTCF Code for 3D Genome Architecture. Cell. 2015;162:703–705. doi: 10.1016/j.cell.2015.07.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolte RT, Conlin RM, Harrison SC, Brown RS. Differing roles for zinc fingers in DNA recognition: structure of a six-finger transcription factor IIIA complex. Proc Natl Acad Sci U S A. 1998;95:2938–2943. doi: 10.1073/pnas.95.6.2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong CT, Corces VG. CTCF: an architectural protein bridging genome topology and function. Nat Rev Genet. 2014;15:234–246. doi: 10.1038/nrg3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parelho V, Hadjur S, Spivakov M, Leleu M, Sauer S, Gregson HC, Jarmuz A, Canzonetta C, Webster Z, Nesterova T, et al. Cohesins functionally associate with CTCF on mammalian chromosome arms. Cell. 2008;132:422–433. doi: 10.1016/j.cell.2008.01.011. [DOI] [PubMed] [Google Scholar]
- Pavletich NP, Pabo CO. Zinc finger-DNA recognition: crystal structure of a Zif268-DNA complex at 2.1 A. Science. 1991;252:809–817. doi: 10.1126/science.2028256. [DOI] [PubMed] [Google Scholar]
- Persikov AV, Singh M. De novo prediction of DNA-binding specificities for Cys2His2 zinc finger proteins. Nucleic Acids Res. 2014;42:97–108. doi: 10.1093/nar/gkt890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quenneville S, Verde G, Corsinotti A, Kapopoulou A, Jakobsson J, Offner S, Baglivo I, Pedone PV, Grimaldi G, Riccio A, et al. In embryonic stem cells, ZFP57/KAP1 recognize a methylated hexanucleotide to affect chromatin and DNA methylation of imprinting control regions. Mol Cell. 2011;44:361–372. doi: 10.1016/j.molcel.2011.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159:1665–1680. doi: 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renda M, Baglivo I, Burgess-Beusse B, Esposito S, Fattorusso R, Felsenfeld G, Pedone PV. Critical DNA binding interactions of the insulator protein CTCF: a small number of zinc fingers mediate strong binding, and a single finger-DNA interaction controls binding at imprinted loci. J Biol Chem. 2007;282:33336–33345. doi: 10.1074/jbc.M706213200. [DOI] [PubMed] [Google Scholar]
- Rhee HS, Pugh BF. Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution. Cell. 2011;147:1408–1419. doi: 10.1016/j.cell.2011.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldana-Meyer R, Gonzalez-Buendia E, Guerrero G, Narendra V, Bonasio R, Recillas-Targa F, Reinberg D. CTCF regulates the human p53 gene through direct interaction with its natural antisense transcript, Wrap53. Genes Dev. 2014;28:723–734. doi: 10.1101/gad.236869.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanborn AL, Rao SS, Huang SC, Durand NC, Huntley MH, Jewett AI, Bochkov ID, Chinnappan D, Cutkosky A, Li J, et al. Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc Natl Acad Sci U S A. 2015;112:E6456–6465. doi: 10.1073/pnas.1518552112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanamee ES, Viadiu H, Kucera R, Dorner L, Picone S, Schildkraut I, Aggarwal AK. A view of consecutive binding events from structures of tetrameric endonuclease SfiI bound to DNA. Embo J. 2005;24:4198–4208. doi: 10.1038/sj.emboj.7600880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Maurano MT, Qu H, Varley KE, Gertz J, Pauli F, Lee K, Canfield T, Weaver M, Sandstrom R, et al. Widespread plasticity in CTCF occupancy linked to DNA methylation. Genome Res. 2012;22:1680–1688. doi: 10.1101/gr.136101.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendt KS, Yoshida K, Itoh T, Bando M, Koch B, Schirghuber E, Tsutsumi S, Nagae G, Ishihara K, Mishiro T, et al. Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature. 2008;451:796–801. doi: 10.1038/nature06634. [DOI] [PubMed] [Google Scholar]
- Xiao T, Wallace J, Felsenfeld G. Specific sites in the C terminus of CTCF interact with the SA2 subunit of the cohesin complex and are required for cohesin-dependent insulation activity. Mol Cell Biol. 2011;31:2174–2183. doi: 10.1128/MCB.05093-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao T, Wongtrakoongate P, Trainor C, Felsenfeld G. CTCF Recruits Centromeric Protein CENP-E to the Pericentromeric/Centromeric Regions of Chromosomes through Unusual CTCF-Binding Sites. Cell reports. 2015;12:1704–1714. doi: 10.1016/j.celrep.2015.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu DH, Ware C, Waterland RA, Zhang J, Chen MH, Gadkari M, Kunde-Ramamoorthy G, Nosavanh LM, Shen L. Developmentally programmed 3′ CpG island methylation confers tissue- and cell-type-specific transcriptional activation. Mol Cell Biol. 2013;33:1845–1858. doi: 10.1128/MCB.01124-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zandarashvili L, White MA, Esadze A, Iwahara J. Structural impact of complete CpG methylation within target DNA on specific complex formation of the inducible transcription factor Egr-1. FEBS Lett. 2015;589:1748–1753. doi: 10.1016/j.febslet.2015.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou X, Ma W, Solov'yov IA, Chipot C, Schulten K. Recognition of methylated DNA through methyl-CpG binding domain proteins. Nucleic Acids Res. 2012;40:2747–2758. doi: 10.1093/nar/gkr1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.