

Abstract
Background
Hypertensive ventricular remodeling is a common cause of heart failure. However, the molecular mechanisms regulating ventricular remodeling remain poorly understood.
Methods
We used a discovery-driven/nonbiased approach to indentify increased ATF3 expression in hypertensive heart. We employed loss/gain of function approaches to understand the role of ATF3 in heart failure. We also examine the mechanisms through transcriptome, CHIP-seq analysis and in vivo and vitro experiments.
Results
ATF3 expression increased in murine hypertensive heart and human hypertrophic heart. Cardiac fibroblast cells are the primary cell type expressing high ATF3 levels in response to hypertensive stimuli. ATF3 knockout (ATF3KO) markedly exaggerated hypertensive ventricular remodeling, a state rescued by lentivirus-mediated/miRNA-aided cardiac fibroblast-selective ATF3 overexpression. Conversely, conditional cardiac fibroblast cell-specific ATF3 transgenic overexpression significantly ameliorated ventricular remodeling and heart failure. We identified Map2K3 as a novel ATF3 target. ATF3 binds with the Map2K3 promoter, recruiting HDAC1, resulting in Map2K3 gene-associated histone deacetylation, thereby inhibiting Map2K3 expression. Genetic Map2K3 knockdown rescued the pro-fibrotic/hypertrophic phenotype in ATF3KO cells. Finally, we demonstrated that p38 is the downstream molecule of Map2K3 mediating the pro-fibrotic/hypertrophic effects in ATF3KO animals. Inhibition of p38 signaling reduced TGF-β signaling-related pro-fibrotic and hypertrophic gene expression, and blocked exaggerated cardiac remodeling in ATF3KO cells.
Conclusion
Our study provides the first evidence that ATF3 upregulation in cardiac fibroblasts in response to hypertensive stimuli protects heart by suppressing Map2K3 expression and subsequent p38-TGF-β signaling. These results suggest that positive modulation of cardiac fibroblast ATF3 may represent a novel therapeutic approach against hypertensive cardiac remodeling.
Keywords: activating transcription factor 3, cardiac fibroblast, p38 MAPK, heart failure
Introduction
Despite technological and pharmaceutical advances, heart failure (HF) remains a leading cause of mortality and morbidity. Hypertensive ventricular remodeling is a common cause of heart failure. Two key aspects of hypertensive ventricular remodeling are cardiac hypertrophy and fibrosis, caused by mechanical stress from blood pressure elevation, neurohormonal alteration, growth factors, and cytokines1. Short-term hypertrophy diminishes wall stress, and is adaptive, but persistent hypertrophy is detrimental. Fibrosis (surplus extracellular matrix) promotes both contractile dysfunction and rhythm disturbances2. Therefore, suppressing hypertensive ventricular remodeling may impede heart failure progression.
The clinical management of pathologic ventricular remodeling includes select pharmacologic agents. Targeting the renin-angiotensin-aldosterone system or β-adrenergic receptor in both animal models and humans attenuates hypertrophy and fibrosis, and extends the life span of heart failure patients3. Ca2+ channel blockers reverse cardiac hypertrophy in some animal models4. Atop managing hypertension, these agents reduce pathological ventricular remodeling by limiting neuroendocrine circuitry and intracellular transduction pathways - the molecular basis of both cardiac hypertrophy and fibrosis. Disappointingly, the overall efficacy of these agents remains limited. While some patients are simply refractory to therapy, cardiovascular disease may progress even in agent-responsive patients5, 6. Clearly, a deeper understanding of the responsible signaling pathways is necessary to derive improved treatment options.
Regulation of gene expression is central for heart development and disease. Cardiac hypertrophy can be viewed as a pro- and anti-hypertrophic gene regulatory disorder. Context specific regulation of gene expression is achieved largely by transcription factor (TF). Cardiac TF have been reported to precisely control a cardiac gene program involved in the generation of cardiac hypertrophy and fibrosis, such as GATA4, nuclear factor-activated T-cell (NFAT), myocyte enhancer factor 2 (MEF2)7-10. As TF orchestrate the numerous gene expressions in the adult heart in response to hypertrophic stimulation, cardiac TF may be promising therapeutic targets. The development of drug molecules or decoy oligodeoxynucleotides that will selectively suppress the activity of pro-hypertrophic TF may be of therapeutic interest. Augmenting the activity of anti-hypertrophic TF is also a promising approach. Identifying the endogenous protective TF counterbalancing the pro-hypertrophic TF may be crucial in designing novel therapeutics against hypertensive remodeling and heart failure.
Utilizing a discovery-driven/unbiased approach, we have identified ATF3 (a member of the cyclic adenosine monophosphate response element binding family of basic leucine zipper TF11) to be a cardiac fibroblast-enriched transcription factor that is cardioprotective against hypertensive remodeling, via suppression of Map2K3 expression and p38 MAPK signaling. Our results suggest positive modulation of cardiac fibroblast ATF3, a “hub” of the cellular adaptive-response network, may represent a novel therapeutic approach against hypertensive cardiac remodeling.
Methods
Relevant citations have been provided for previously published protocols employed in the current study.
Whole-body ATF3-knockout mice were kindly provided by Dr. Hongliang Li (generation/characterization of this mouse strain described previously12). We established a conditional cardiac fibroblast-specific ATF3-transgenetic mouse (ATF3fl/flTG) and miRNA-aided/lentivirus-mediated non-cardiac myocyte ATF3 overexpression model in our laboratory. Detailed methods are provided in Supplemental Data. All animals were maintained under pathogen-free conditions. All experiments were performed using 10-12 week-old male mice. In the angiotensin II (Ang II) infusion model, mice were infused with Ang II subcutaneously (1000 ng/kg/min) for 7 or 28 days with ALZET miniosmotic pumps (DURECT, Palo Alto, CA)13,14. Blood pressure was measured daily via tail-cuff method. In the pressure-overload model, transverse aortic constriction (TAC) or sham operation was performed15. Cardiac function was evaluated by the Vevo 770 high-resolution micro-imaging system (Visualsonic, Toronto, Canada). All animal handling complied with animal welfare regulations of Capital Medical University. The Animal Subjects Committee of Capital Medical University approved the animal study protocol.
Detailed methodology for all protocols utilized in this study, including immunofluoresence, in situ hybridization, quantitative RT-PCR, RNA-sequencing, microarray, chromatin immunoprecipitation (CHIP) assay, CHIP-sequencing, immunoprecipitation, immunoblotting, cardiac fibroblasts and cardiomyocytes culturing, cell sorting, Map2K3 knockdown, ATF3 overexpression, and cytokines measurement, is provided in Supplemental Methods.
Statistical analysis
Data are presented as means±SEM of n independent experiment. Normality analysis was performed before any testing, via the Shapiro-Wilk test. Differences were evaluated using unpaired Student's t test between two groups. One-way ANOVA was conducted followed by Bonferroni post hoc test for comparisons between >2 groups. The non-normal distributed data were analyzed using non-parametric testing (Mann-Whitney U test for two groups and Krushal-Wallis H test for >2 groups). Differences between groups over time were evaluated using repeated measures analysis of variance. The transcriptome data was analyzed by multiple testing. P values less than 0.05 were considered statistically significant. Statistical analysis was performed using GraphPad Prism 4.0 (GraphPad Software Inc., San Diego, CA) and SPSS 13.0 (SPSS Inc., Chicago, IL) software.
Results
ATF3 expression is consistently increased in cardiac tissues from hypertensive animals and heart failure patients
To identify the TFs regulating hypertensive cardiac remodeling in a nonbiased fashion, transcriptome analysis was performed in heart samples from two different hypertensive cardiac remodeling models: Ang II infusion (7 or 28 days, respective time points when significant ventricular remodeling has not and has developed, using RNA-sequencing) and TAC (28 days, using microarrray). Compared to control, 1019 and 647 differentially expressed genes (DEGs, filtering criteria p<0.05, fold change >2.0) were identified 7- or 28-days post-Ang II infusion. Compared to respective controls, 537 DEGs were identified 28 days after Ang II infusion and TAC (filtering criteria: p<0.05; fold change >1.5). To identify DEGs encoding TFs, we compared 1100 candidate TFs from the TRANSFAC database to the DEGs identified in these three groups. 41 (from 7-day post-Ang II infusion group), 29 (from 28-day post-Ang II infusion group), and 42 (from 28-day post-TAC group) DEGs are TF-encoding genes (Figure 1A, B and Supplemental Table 1).
Figure 1. Identification of ATF3 as a key transcription factor in hypertensive cardiac pathogenesis.
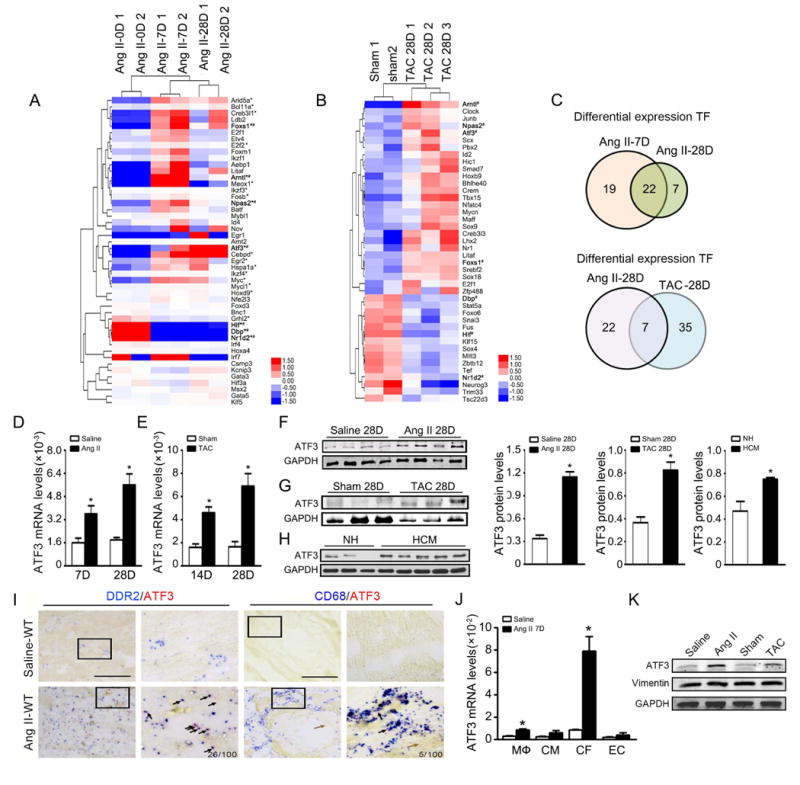
(A, B, C) Global gene expression analysis was performed in hearts from murine models (Ang II infusion for 0, 7, or 28 days; TAC or sham operation for 28 days). Comparative transcriptome profiling was perform in heart tissue, for Ang II infusion model using RNA-seq approach and for TAC model using microarray approach. By comparing DEGs from three groups (Ang II Day 7 vs Day 0, Ang II Day 28 vs Day 0, TAC vs sham operation) with candidate 1100 TFs from TRANSFAC database, 41, 29 and 42 differentially expressed TFs were identified respectively. A clustered heatmap of expression values for differentially expressed TFs is shown. Asterisk (*) indicates 22 TFs continually changing during Ang II infusion for 7 and 28 days. Pound sign (#) indicates 7 TFs with common alteration during Ang II infusion and TAC for 28 days. (D, E) Cardiac ATF3 mRNA expression was evaluated by quantitative RT-PCR (qRT-PCR) during different time points of Ang II infusion or TAC. (n=5 per group) (F,G) ATF3 protein expression was evaluated by Western-blot in hearts after Ang II infusion or TAC. *p<0.05 versus saline or sham operation. (H) Cardiac ATF3 protein expression in a healthy heart donor and hypertrophic cardiomyopathic patient was evaluated by western-blot. *p<0.05 versus normal heart. (I) In situ hybridization analysis demonstrates no ATF3 signal in vascular endothelium or cardiomyocytes (brown arrows). ATF3 signal was markedly detected in cardiac fibroblasts and minimally detected in infiltrated macrophages after Ang II infusion (black arrows). Bars=50 μm. (J) After 7 days of saline or Ang II infusion, hearts were digested and sorted into macrophages (CD45+F4/80+), cardiomyocytes (CD45-PDGFR-α-CD31-), cardiac fibroblasts (CD45-PDGFR-α+), and endothelial cells (CD45-CD31+) via FACS. ATF3 mRNA expression in cells was assessed by qRT-PCR. *p<0.05 versus saline (n=6 per group). (K) ATF3 protein expression in cardiac fibroblasts was assessed by western-blot after Ang II or TAC treatment. *p<0.05 versus saline or sham operation. (n=3 per group). Statistical significance was determined by the unpaired t test (D,E,F) or U test (H, J).
Further analysis revealed that 22 differentially expressed TF genes overlap between 7 and 28 days post-Ang II infusion, and 7 differentially expressed TF genes overlap between Ang II infusion-induced and TAC-induced ventricular remodeling (Figure 1C). Among the differentially expressed and overlapping TF genes in these three groups, we identified ATF3 as the gene most consistently and significant upregulated. Consistent with transcriptome data, both ATF3 mRNA and protein levels were significantly increased in both the Ang II-infused and pressure overloaded heart (Figure 1D-G). Protein-protein interaction (PPI) network predicted that ATF3 interacts with 48 important proteins (Supplemental Figure 1A), including other transcriptional complexes involved in immune response (Rela, Jun, Junb, Jund, Fos), fibrosis (smad2/3), and apoptosis (p53, ATF4, ATF2).
To confirm the translational value of our animal model results, we determined whether ATF3 expression is altered in human cardiac tissue. ATF3 protein expression was significantly increased in hypertrophic cardiomyopathy (HCM) patient heart samples compared to control (Figure 1H). Taken together, these results strongly suggest ATF3 may play a regulatory role in hypertensive ventricular remodeling.
Cardiac fibroblasts are the primary cellular type expressing ATF3 in response to stimuli
It is increasingly recognized that molecules expressed by different heart cell types may differentially regulate global cardiac function116. We next determined the cellular origin of ATF3 expression in the hypertensive heart. In situ hybridization (ISH) performed upon sections of saline or Ang II infused hearts revealed ATF3 staining was very weak in saline-treated animals, intensifying after Ang II infusion (Figure 1I). Importantly, abundant ATF3 staining was noted in the cardiac interstitium, but not within cardiomyocytes or vascular cells. Focusing upon the cardiac interstitium, we co-stained tissue sections with antibodies against ATF3 and DDR2 or Col1a1 (cardiac fibroblasts) or CD68 (macrophages). Double staining revealed an intense ATF3 signal in fibroblasts (DDR2+,Col1a1+), but not in macrophages (CD68+) (Figure 1I and Supplemental Figure 1B). ATF3 signal in a whole heart section was presented (Supplemental Figure 1C). Finally, to obtain more evidence supporting cardiac fibroblasts as the major ATF3-expressing cells post Ang II infusion, we separated heart cells into populations of CD45-PDGFR-α-CD31- cardiac myocytes, CD45-PDGFR-α+ cardiac fibroblasts17, CD45-CD31+ endothelial cells, and CD45+F4/80+ macrophages by our fluorescence-activated cell sorting protocol13. Subsequent qRT-PCR demonstrated low ATF3 expression in all cardiac cell populations in saline-treated animals. Ang II infusion had no significant effect upon ATF3 expression in cardiomyocytes and endothelium, modestly increased ATF3 expression in macrophages, and markedly increased ATF3 expression in cardiac fibroblasts (Figure 1J). ATF3 protein was tested in cardiac fibroblasts. Compared to cardiac fibroblasts of control mice, ATF3 protein expression significantly increased in cardiac fibroblasts isolated from mice after Ang II infusion or TAC (Figure 1K).
Upregulation of ATF3 in cardiac fibroblast cells is a compensatory self-protective mechanism against hypertensive ventricular remodeling and heart failure
Having demonstrated that ATF3 is significantly upregulated in cardiac fibroblast during heart failure development, we next determined whether ATF3 upregulation contributes to or protects against ventricular remodeling. We employed four separate approaches to obtain the most conclusive evidence.
First, we utilized a loss-of-function approach. Whole body ATF3 knockout (ATF3KO) and wild-type (WT) mice were subjected to Ang II infusion. Ang II infusion induced a similar increase in blood pressure in WT and ATF3KO mice (Figure 2A), with significantly increased collagen deposition in ATF3KO mice compared to WT (Figure 2B). ATF3KO mice also exhibited increased α-smooth muscle actin (α-SMA, a marker of myofibroblast-positive cells) α-SMA protein expression, collagen I, and TGF-β mRNA (Figures 2C-E). There was no difference in MYH11-positive vascular smooth muscle cells from WT versus ATF3KO heart sections, which suggested increased α-SMA expression due to the presence of myofibroblast not new vessel formation (Supplemental Figure 2A). ATF3KO mouse hearts gained significantly more weight after Ang II infusion, with increased cardiomyocyte hypertrophy (Figures 2F-G). The mRNA levels of Myh7 and ANP (hypertrophy-associated genes) were increased in ATF3KO mice after Ang II infusion (Figure 2H). These data suggest ATF3 upregulation during heart failure development is a self-protective mechanism against hypertensive ventricular remodeling.
Figure 2. ATF3 deficiency increases Ang II-induced cardiac fibrosis and hypertrophy.
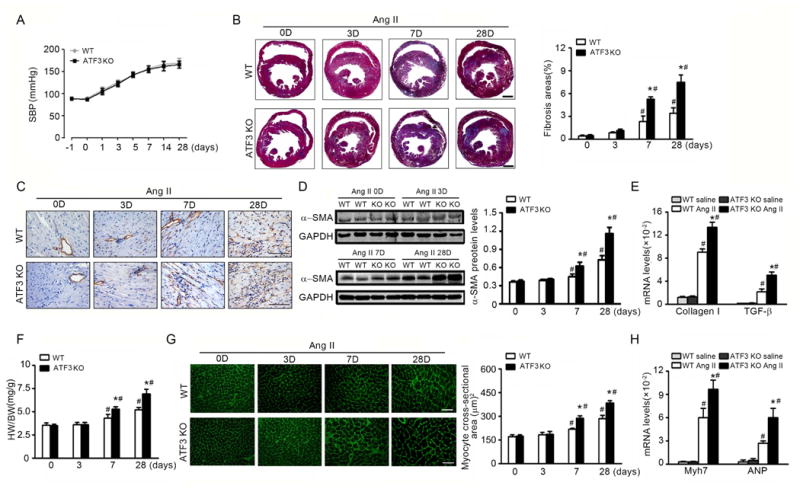
WT and ATF3-KO mice were infused with Ang II (1000 ng/min/kg) for 3, 7, or 28 days. (A) Noninvasive systolic blood pressure (SBP) was measured. (B) Representative cardiac Masson trichrome staining and quantification. Bars=500 μm. (C, D) α-smooth muscle actin (SMA) expression was assessed by immunohistochemistry and western blot. Bars=50 μm. (E) Cardiac expression of fibrosis markers collagen I and TGF-β, by qRT-PCR analysis. (F) Heart to body weight ratios. (G) Wheat germ agglutinin (WGA) stained section of left ventricles and quantification of myocyte cross-sectional area. Bars=50 μm. (H) Cardiac expression of the fetal hypertrophy markers Myh7 and ANP by qRT-PCR analysis. *P<0.05 compared to WT+AngII; #P<0.05 compared to saline. (n=6 per group). Statistical significance was determined by repeated measures analysis (A) or by 1-way ANOVA test (B-H).
Second, we employed a viral-mediated rescue approach. To avoid ATF3 overexpression in cardiomyocytes, we established a miRNA-aided/lentivirus (LV)-mediated system, as recently reported18, 19. As mir-1a and mir-133a are the most abundant miRNA in cardiac myocytes20, we designed a ATF3 expression cassette driven by a Ubi promoter containing 2 tandem copies of a 22-bp complementary target sequence for the mir-1a-3p and mir-133a-3p strands at the 3′ end of the vector cassette (LV-ATF3-mir1/133TS, Figure 3A). LV-control-mir1/133TS served as a control LV vector. We first transfected primary cardiac myocytes and fibroblast isolated from ATF3KO animals with LV-ATF3-mir1/133TS and LV-control-mir1/133TS (50PFU/cell). ATF3 expression was markedly increased in LV-ATF3-mir1/133TS transfected cardiac fibroblast, but only modestly increased in cardiomyocytes (Figure 3B). The LV vectors were administered via tail vein injection to the ATF3KO mice (dose 3×108 PFU per mouse). The efficiency of viral transduction in heart was evaluated in mice injected with saline, LV-Con-mir1/133TS, LV-ATF3-mir1/133TS by flow cytometry. GFP+ cells accounted for about 5-10% of total cells in heart with viral transduction (Supplemental Figure 3A-B). To elevate the specificity of the approach, we isolated the GFP+ cardiac fibroblasts and GFP+ cardiomyocytes from ATF3KO mice injected with LV-Con-mir1/133TS or LV-ATF3-mir1/133TS (Supplemental Figure 3A and C). ATF3 proteins were significantly increased in cardiac fibroblast, but only slightly increased in cardiomyocytes in mice with LV-ATF3-mir1/133TS (Supplemental Figure 3D). ATF3KO mice were administrated with LV-control, LV-Con-mir1/133TS or LV-ATF3-mir1/133TS followed by Ang II infusion for 7 days. After 7 days, the heart tissues were harvested for histopathology and isolation of cardiac fibroblasts. ATF3 expression was significantly increased in the hearts of LV-ATF3-mir1/133TS transduced ATF3KO mice (Figure 3C). Immunostaining revealed ATF3 expression in col1a2-positive fibroblasts in ATF3KO mice transduced with LV-ATF3-mir1/133TS, but not in α-actinin-positive cardiomyocytes. There is no ATF3 expression in mice transduced with LV-Con-mir1/133TS. (Figure 3D). Using LV-ATF3-mir1/133TS transduction, ATF3 expression in ATF3KO mice reached 60% of WT mouse expression treated with LV-Con-mir1/133TS followed by Ang II infusion for 7 days (Supplemental Figure 3E). Importantly, compared to LV-control or LV-Con-mir1/133TS group, transduction with LV-ATF3-mir1/133TS inhibited cardiac fibrosis and hypertrophy in ATF3KO mice after Ang II infusion (Figure 3 E-G). Taken together, these data demonstrated the ATF3KO mouse phenotype was successfully rescued by miRNA-aided/lentivirus-mediated cardiac fibroblast ATF3 overexpression.
Figure 3. LV-ATF3-mir1/133TS inhibit Ang II-induced cardiac remodeling.
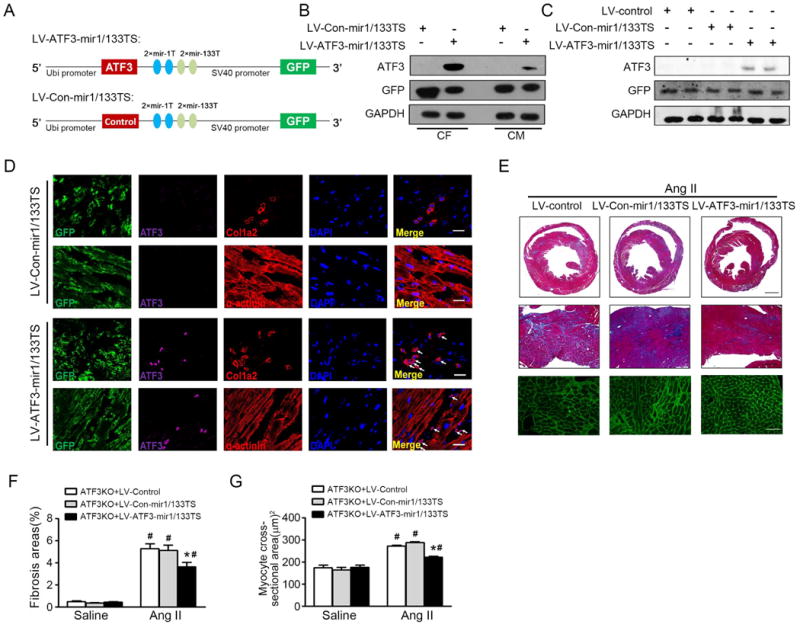
(A) Schematic representation of ATF3 expression cassette in the LV vectors engineered to inhibit overexpression of exogenous ATF3 or negative control in cardiomyocytes expression mir-1a-3p and mir-133a-3p. (B) CM and CF were transfected with indicated LV vector. ATF3 expression was assessed in CM and CF by western-blot. (C) ATF3 KO mice were injected with LV-control, LV-Con-mir1/133TS, or LV-ATF3-mir1/133TS then followed by Ang II infusion for 7 days. ATF3 expression was assessed in heart by western-blot. (D) Representative sections of heart immunostained for GFP, ATF3, and Col1a2 or α-actinin in ATF3KO mice transduced with LV-Con-mir1/133TS or LV-ATF3-mir1/133TS. (E) Representative Masson trichrome and WGA-stained sections. Bars=500 μm (top); 100 μm (middle); 50 μm (bottom). (F) The fibrosis area was measured and expressed as a percentage of total area of left ventricular myocardium. (G) WGA stained section of left ventricles and quantification of myocyte cross-sectional area. *p<0.05 compared with ATF3 KO + Ang II+LV-control (or LV-Con-mir1/133TS). #p<0.05 compared with ATF3 KO+saline. (n=5-8 mice per group). Statistical significance was determined by 1-way ANOVA test (F, G).
Third, we utilized a gain-of-function approach. To obtain the most conclusive date supporting a notion that increased ATF3 expression in cardiac fibroblast is cardioprotective, a conditional cardiac fibroblast-specific ATF3-transgenetic mouse model was generated (ATF3fl/flTG) (Supplemental Figure 4A-B). ATF3fl/fl TG mice were crossed with inducible fibroblast-specific Cre mice (col1a2-Cre/ERT). Tamoxifen was administered intraperitoneally to activate gene expression. To assess the transgenic leakiness, we determined Col1a2 and ATF3 expression in heart, liver, lung, kidney, spleen, and vessels. High level Col1a2 expression was only observed in heart and vessel, low level Col1a2 expression was observed in kidney, and no significant Col1a2 expression was observed in other organs. No significant ATF3 expression was observed in any organ observed in ATF3fl/flTGCre- or WT mice. In ATF3fl/flTGCre+ mice, ATF3 proteins were significantly elevated only in the heart and vessel, slightly elevated in the kidney, and not observed in other organs. These results demonstrated that there was no significant transgenic leakiness in our model (Supplemental Figure 4C). Compared to ATF3fl/flTGCre- mice, ATF3 mRNA expression was significantly increased in cardiac fibroblasts (but not cardiomyocytes) from ATF3fl/fl TG Cre+ mice (Supplemental Figure 4D). To identify which cell population is actually expressing the transgene, we tested the ATF3 proteins expression in isolated cardiac fibroblasts, cardiomyocytes and endothelial cells. The cardiac fibroblast was the only cell types with significantly increased ATF3 expression (Supplemental Figure 4E). Using Lineage tracing technique, a previous study demonstrated that over 96% Col1a2+ cells were cardiac fibroblasts and not other cardiovascular cell types21. We determined the co-location of Col1a2 expression with Col1a1(cardiac fibroblasts), α-actinin (cardiomyocytes), CD31(endothelial), SM22 (smooth muscle cells) and Col2a1(valve cells). Col1a2+ cells clearly co-localized with Col1a1 positive cells, but not with CD31, SM22, α-actinin, or Col2a1 positive cells (Supplemental Figure 4F). We further assessed the cell types of ATF3 expression in ATF3fl/flTGCol1a2-Cre+. Double immunofluorescent staining revealed that ATF3 was dominantly expressed in Col1a1+ or Col1a2+ cardiac fibroblasts in ATF3fl/fl TG Cre+ mice (Supplemental Figure 4G). Taken together, these data strongly suggest that the Col1a2+ cells are specific for cardiac fibroblasts and ATF3 is specifically expressed in cardiac fibroblasts in ATF3fl/flTGCre+ mice.
To determine the effect of ATF3 overexpression upon cardiac remodeling, Cre+-alone, ATF3fl/flTGCre-, and ATF3fl/flTGCre+ mice were injected with tamoxifen, and then subjected to saline or Ang II infusion for 12 weeks. Compared to Cre+-alone and ATF3fl/flTGCre- mice, ATF3 protein expression was significantly increased in cardiac fibroblasts from ATF3fl/flTGCre+ mice subjected to Ang II (Figure 4A). Ang II-induced gross and microscopic changes were significantly attenuated in ATF3fl/fl TGCre+ mice; specifically, the heart was grossly smaller, the left ventricle was less dilated, cardiac fibrosis was reduced, and heart weight/body weight ratios were decreased (Figures 4B-D). Additionally, the cardiomyocyte cross-sectional area was significantly smaller (Figures 4E/F). The mRNA levels of ANP and Myh7 decreased in ATF3fl/flTGCre+ mice compared to Cre+-alone and ATF3fl/flTGCre- mice (Figures 4G/H). Finally, cardiac function was significantly improved and ventricular chamber dilation was significantly decreased in ATF3fl/flTGCre+ mice subjected to Ang II infusion (Figures 4I-K).
Figure 4. Cardiac fibroblast-specific ATF3 attenuates cardiac fibrosis and hypertrophy after Ang II infusion.
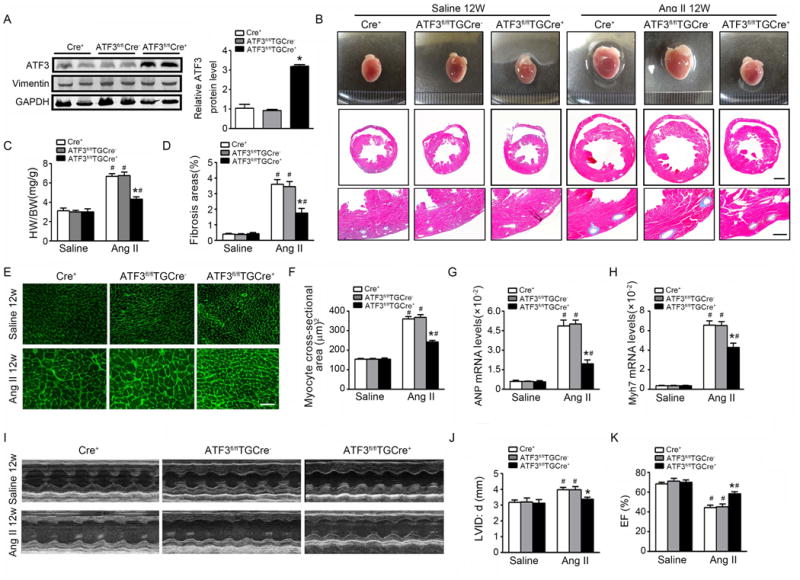
Col1a2-Cre+, ATF3fl/flTGCre-, and ATF3fl/flTGCre+ mice were administered tamoxifen and then subjected to saline or Ang II infusion for 12 weeks. (A) ATF3 expression was measured in cardiac fibroblasts from Col1a2-Cre+, ATF3fl/flTGCre-, and ATF3fl/flTGCre+ mice after Ang II infusion by western-blot. (B) Whole-mount cardiac representation of Col1a2-Cre+, ATF3fl/flTGCre-, and ATF3fl/flTGCre+ mice after saline or Ang II infusion. Representative image of heart sections stained with Masson trichrome. Bars=500 μm (middle); 100 μm (bottom). (C) Heart weight/body weight ratios. (D) The fibrosis area was measured and expressed as a percentage of total area of left ventricular myocardium. (E-F) WGA stained section of left ventricles and quantification of myocyte cross-sectional area. Bars=50 μm. (G-H) Cardiac expression of the fetal hypertrophy markers ANP and Myh7 by qRT-PCR analysis. (I-K) M-mode echocardiographic imaging of heart after saline or Ang II infusion for 12 weeks. Analysis of LVID;d (left ventricular internal dimension in end-diastole) and EF (ejection fraction) % of hearts subjected to Ang II.*p<0.05 compared with Cre+ or ATF3fl/flTG Cre- mice after Ang II infusion. #P<0.05 compared to saline infusion (n=5-6 per group). All statistical significance was determined by 1-way ANOVA test.
Fourth and finally, a bone marrow (BM) transplantation approach was utilized. As Ang II infusion modestly increased macrophage ATF3 expression compared to saline control (Figure 1J), we determined whether macrophage ATF3 expression after Ang II infusion may contribute to cardiac protection. We performed BM transplantation (BMWT→ATF3KO mice or BMATF3KO→WT mice), and subjected animals to Ang II infusion for 28 days. Compared to BMWT→WT mice, ATF3 mRNA was significantly reduced in BMWT→ATF3KO mice, but not in BMATF3KO→WT mice after Ang II infusion (Figure 5A). More importantly, ATF3KO recipient mice manifest increased ventricular remodeling (increased cardiac fibrosis and hypertrophy) regardless of BM origin (either BMWT or BMATF3KO). In contrast, WT mice receiving either WT or ATF3KO donor BM did not exhibit accelerated ventricular remodeling, as seen in BMATF3KO→ATF3KO mice (Figures 5B-G). Finally, we subjected BMWT→WT, BMWT→ATF3KO, and BMATF3KO →ATF3KO chimeric mice to prolonged Ang II infusion (12 week duration), and determined cardiac function. BMWT→ATF3KO and BMATF3KO →ATF3KO mice manifested increased left ventricle internal dimension in end-diastole (LVID;d) and decreased ejection fraction compared to BMWT→WT mice after Ang II infusion (Figure 5H-J). Taken together, these results demonstrated that ATF3 expression in cardiac resident cells, not BM-derived cells, determined susceptibility to Ang II-induced cardiac remodeling.
Figure 5. BM-derived cells ATF3 deficiency is not critical for Ang II-induced ventricular remodeling and heart failure.
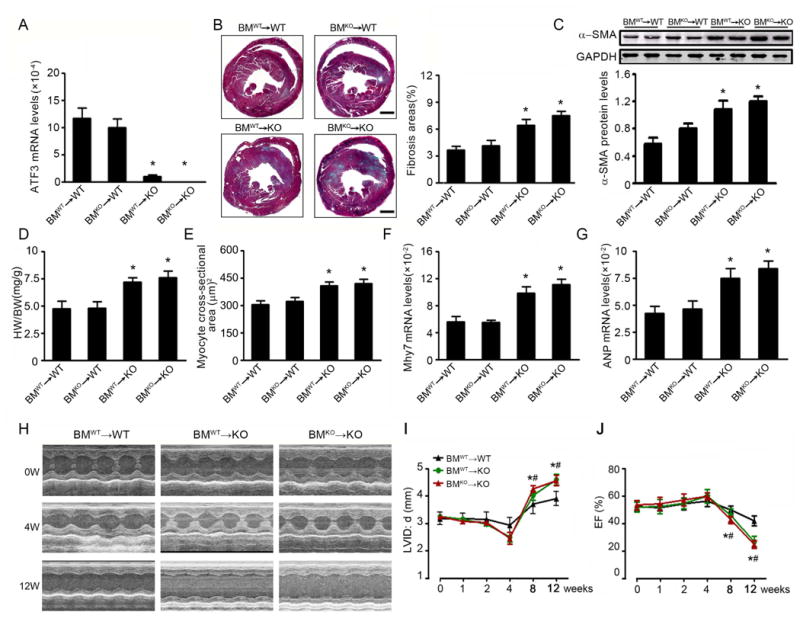
After 10 weeks of bone marrow reconstitution, BM chimeric mice were infused Ang II for 28 days. (A) ATF3 mRNA expression in the hearts of Ang II-infused chimeric mice. (B) Representative Masson trichrome staining and quantification in chimera hearts. Bars=500 μm. (C) The α-SMA protein expression was determined by western blot. (D) Heart to body weight ratios. (E) Quantification of myocyte cross-sectional area. (F, G) Cardiac expression of the fetal hypertrophy markers Myh7 and ANP by qRT-PCR analysis. (H) BMWT→WT, BMWT→KO and BMKO→WT mice were infused with Ang II for 12 weeks. M-mode echocardiographic imaging of heart before (0) and after (4 and 12 weeks) Ang II infusion. (I, J) Analysis of LVID;d and EF % of hearts subjected to long-term Ang II infusion. *p<0.05 BMWT→KO vs BMWT→WT; #p<0.05 BMKO→KO vs BMWT→WT (n=5 per group). Statistical significance was determined by 1-way ANOVA test (A-G) or by by repeated measures analysis (I, J).
Map2K3 is a novel ATF3 target gene whose inhibition by ATF3 is responsible for ATF3-mediated cardioprotection
To explore how ATF3 regulates ventricular remodeling, two strategies were utilized to identify the Map2K3 regulated genes responsible for ATF3-Map2K3 downstream signaling. First, cardiac fibroblasts were isolated from Ang II-infused WT or ATF3KO mice, and subjected to transcriptome analysis. RNA-Seq analysis identified 701 upregulated and 468 downregulated DEGs (FDR<0.05; fold change>1.2) in ATF3KO cardiac fibroblasts compared to WT (Supplemental Table 2). To confirm the accuracy of RNA-Seq identification of DEGs, ten DEGs were randomly selected. qRT-PCR was performed. Nine out ten DEGs demonstrated completely concordant expression patterns between RNA-Seq and qRT-PCR results (Supplemental Figure 5). ATF3KO cardiac fibroblast exhibited increased mRNA expression of various pro-fibrotic, hypertrophic, and inflammatory genes (Supplemental Table 3). Pathway analysis of DEGs revealed that upregulated signaling pathways were closely related to hypertensive cardiac remodeling, specifically ECM deposition (focal adhesion and ECM-receptor interaction)22; hypertrophy (insulin, MAPK, and mTOR signaling)23, and inflammation (cytokine-cytokine receptor interaction and leukocyte transendothlial migration) (Supplemental Figures 6A/B). Finally, Path-Net analysis was performed on 47 significant pathways and their interactions to determine the signaling pathways most likely responsible for ATF3-mediated protection. In this analysis, the MAPK signaling pathway genes exhibited greatest enrichment among the upregulated pathways in ATF3KO cells (Supplemental Figure 6C).
Second, cardiac fibroblasts were isolated from ATF3KO mice, transfected with either ATF3-overexpressing (Ad-ATF3) or empty (Ad-con) adenovirus, and subjected to Ang II stimulation in vitro. After upregulation of ATF3 expression was confirmed in the transfected cells (Figure 6A), ATF3 CHIP-seq was performed. We identified 727 ATF3 target genes selectively enriched in ATF3-overexpressed cardiac fibroblasts (Figure 6B). To narrow the number of ATF3-mediated genes, we performed a two-tiered bioinformatics analysis: 1) transcriptome differential analysis between WT and ATF3-depleted fibroblasts for genes related to ATF3; and 2) differential ATF3 ChiP sequencing in ATF3-depleted fibroblasts transfected with Ad-con and Ad-ATF3 for ATF3-bound genes. 31 genes exhibiting significant mRNA alteration between WT and ATF3-depleted cardiac fibroblasts were identified to directly bind with ATF3 (Figure 6C and Supplemental Table 4).
Figure 6. CHIP analysis identifies Map2K3 as an ATF3 target gene.
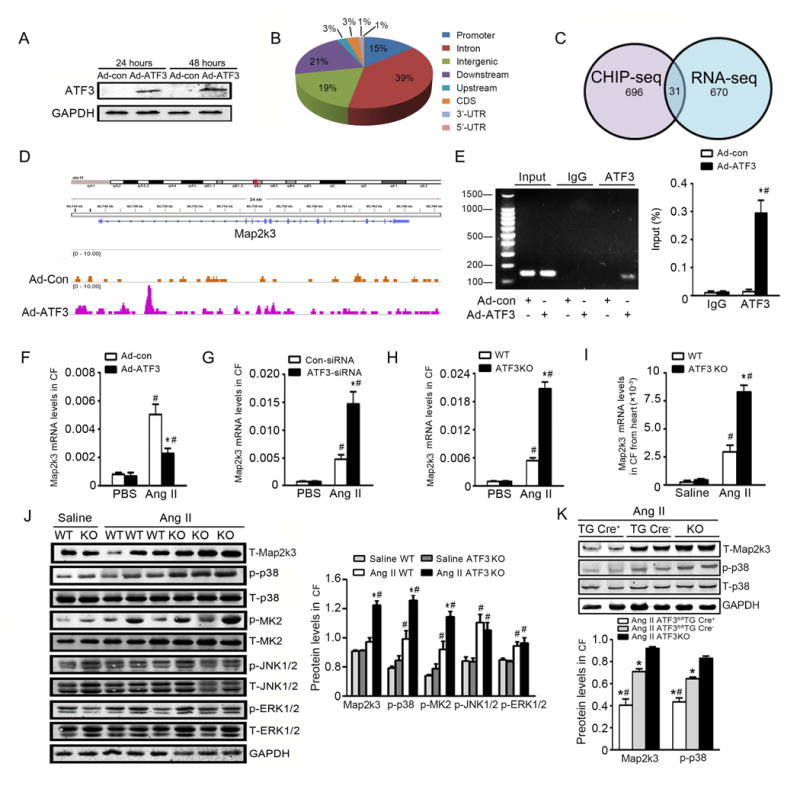
(A) Cardiac fibroblasts from ATF3KO mice were transfected with ATF3-overexpressing (Ad-ATF3) or empty (Ad-con) adenovirus for 48 hours, and plus with Ang II for another 6 hours. ATF3 protein expression was determined by western blot. (B) CHIP-seq analysis was performed in cardiac fibroblasts infected with Ad-con or Ad-ATF3 with an ATF3 antibody. Chart demonstrates percent of peaks within each category. (C) ATF3-binded genes identified in the CHIP dataset (left circle) were overlaid upon genes exhibiting ATF3-dependent expression in RNA-seq analysis (right circle) using WT or ATF3KO fibroblasts. 31 genes were present in both datasets. (D) Map of the Map2K3 locus revealing ATF3 binding in CF infected with Ad-con or Ad-ATF3; aligned reads were visualized by Integrated Genomics Viewer 2.024. The signal of the Ad-con or Ad-ATF3 cardiac fibroblasts is represented with yellow or pink peaks, respectively. (E) The independent CHIP assay was performed upon Ad-con- or Ad-ATF3-infected cardiac fibroblasts to confirm ATF3 binding to the promoter regions of the Map2K3 gene. *P<0.05 vs Ad-con in ATF3 Ab. #P<0.05 vs IgG Ab. Data are from 3 independent experiments. (F, G) WT cardiac fibroblast was transfected with Ad-con or Ad-ATF3 (F), control-siRNA or ATF3-siRNA (G), and treated with PBS or Ang II for 24 hours. Map2K3 mRNA levels were evaluated by qRT-PCR. *P<0.05 vs Ad-con+Ang II in (F); *P<0.05 vs control-siRNA+Ang II in (G). #P<0.05 vs PBS. Data are from 3 independent experiments. (H) Map2K3 mRNA expression was determined in WT or ATF3KO cardiac fibroblasts treated with or without Ang II. *P<0.05 vs WT +Ang II. #P<0.05 vs PBS. Data are from 3 independent experiments. (I) Cardiac fibroblasts were isolated from WT or ATF3 KO mice infused with Ang II for 7 days. Map2K3 mRNA was evaluated by qRT-PCR. (J) The key MAPK pathway kinases were evaluated by Western-blot in cardiac fibroblasts isolated from WT or ATF3 KO mice infused with Ang II. *P<0.05 vs WT+Ang II. #P<0.05 vs Saline. (n=6-10 mice per group). (K) Mapk2K3 and p38 signals were assessed in cardiac fibroblasts from ATF3fl/flTGCre+, ATF3fl/flTGCre-, and ATF3KO mice infused with Ang II. *P<0.05 vs ATF3KO. #P<0.05 vs ATF3fl/flTG Cre-. Statistical significance was determined by the 1-way ANOVA test (E-K).
Combining the findings from these two strategies, we focused upon MAPK pathway genes. ChiP-seq analysis revealed a significant ATF3-binding promoter region in the Map2K3 gene (Figure 6D). Independent CHIP assays confirmed marked enrichment of Map2K3 over normal IgG following ATF3 overexpression (Figure 6E). Interestingly, Map2K3 mRNA expression is increased in ATF3KO cardiac fibroblasts, and decreased in Ad-ATF3 transfected cardiac fibroblasts (Figure 6F-H), suggesting this transcription factor inhibits, rather than stimulates Map2K3 expression. Consistent with these in vitro findings, Map2K3 mRNA (Figure 6I) and protein (Figure 6J, lane 1) expression was significantly increased in cardiac fibroblasts isolated from Ang II-infused ATF3KO mice. Map2K3 expression decreased in cardiac fibroblasts of ATF3fl/flTGCre+ mice compared to TGCre- mice after Ang II infusion (Figure 6K, lane 1).
To establish a causative relationship between Map2K3 upregulation and exacerbated hypertensive cardiac remodeling in ATF3KO animals, we performed several experiments. First, cardiac fibroblasts were isolated and treated with Ang II in vitro. Compared to control, Ang II-induced α-SMA and collagen I expression in cardiac fibroblast to greater extent in ATF3KO mice (Figure 7A). Inhibition of Map2K3 (by a lentivirus expressing a Map2K3 shRNA, LV-Map2K3-shRNA, Supplemental Figure 7A-B) significantly attenuated Ang II-induced α-SMA and collagen I expression in ATF3KO cardiac fibroblasts (Figure 7B). Cardiac fibroblasts regulate cardiac myocytes via paracrine communication25. We next co-cultured cardiac myocytes with cardiac fibroblasts isolated from ATF3KO animals, and determined the effect of Map2K3 inhibition upon Ang II-induced cardiac myocyte hypertrophy. Genetic inhibition of Map2K3 with LV-Map2K3-shRNA significantly inhibited Ang II-induced ANP expression and decreased cardiac myocyte size (Figure 7C/D).
Figure 7. ATF3 deficiency-induced ventricular remodeling is dependent upon Map2K3.
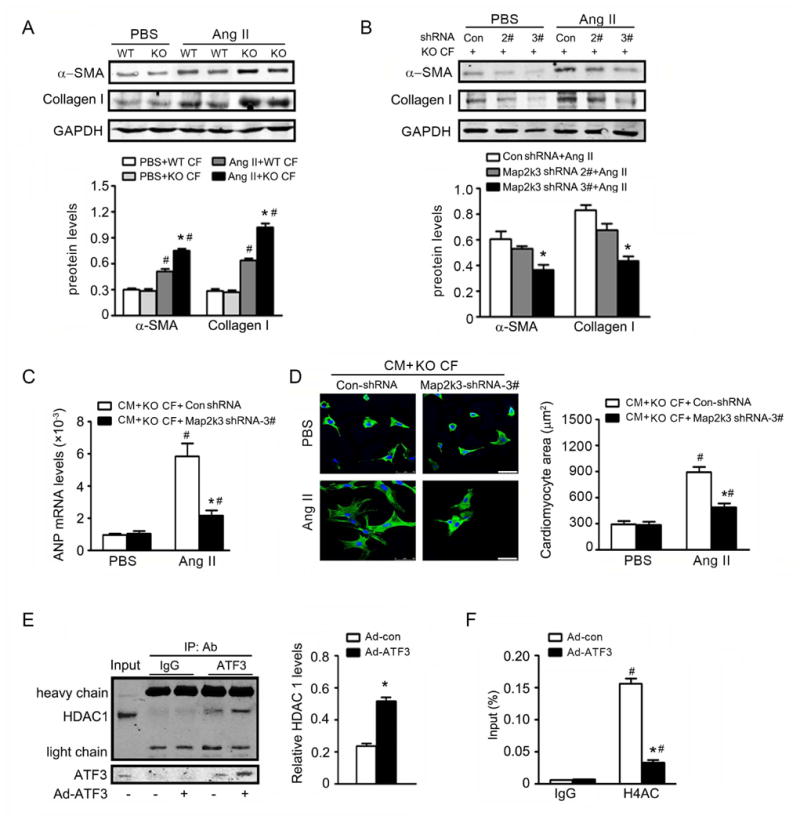
(A) Cardiac fibroblasts from WT and ATF3 KO mice were treated Ang II for 48 hours. The expression of α-SMA and collagen I was detected by western-blot. *p<0.05 vs WT CF+Ang II. #P<0.05 vs PBS. (B) Cardiac fibroblasts from ATF3KO mice were transfected with lentivirus-medicated Map2K3 shRNA (LV-Map2K3-shRNA) or lentivirus-Control-shRNA (LV-con-shRNA) for 48 hours, and subjected to Ang II for 48 additional hours. The expression of α-SMA and collagen I was detected. *p<0.05 compared with LV-con-shRNA+Ang II. (C-D) CM was treated with supernatant from ATF3 KO cardiac fibroblasts infected with LV-Map2K3-shRNA or LV-con-shRNA, with or without Ang II. ANP mRNA expression and cardiomyocyte area were evaluated. *p<0.05 vs CM+KO CF+LV-con-shRNA+Ang II. #P<0.05 vs CM+KO CF+PBS. (E) Cardiac fibroblasts were transfected with Ad-ATF3 or empty Ad-con adenovirus for 48 hours, and pulsed with Ang II for another 6 hours. Nuclear extracts were isolated and immunoprecipitated with anti-IgG or anti-ATF3 antibody, and immunoblotted with anti-HDAC1 or anti-ATF3. *P<0.05 vs Ad-con transfection. (F) Cardiac fibroblast was transfected with Ad-ATF3 or Ad-con for 48 hours and plus with Ang II for 6 hours. CHIP assays were performed with anti-acetyl H4 antibody, then Map2K3 binding was assessed. *P<0.05 vs Ad-conin H4AC Ab. #P<0.05 vs Ig Ab. All data are from 3 independent experiments. Statistical significance was determined by 1-way ANOVA test (A-D, F) or by the unpaired t test (E).
As our data suggests ATF3 inhibits Map2K3 gene expression, we investigated the potential mechanism by which ATF3 suppresses Map2K3 transcription. The PPI map demonstrated connectivity of ATF3 with histone deacetylases (HDACs) (Supplemental Figure 1). Interestingly, Co-IP assays demonstrated that HDAC1 is indeed present in the ATF3-immunoprecipitated complex (Figure 7E). Most importantly, levels of Map2K3 promoter-bound histone acetylation (H4ac) in cardiac fibroblasts was significantly increased in response to Ang II, an effect significantly attenuated by ATF3-overexpression (Figure 7F).
p38 MAPK activation mediates ATF3 deficiency-induced fibrosis and hypotrophy
Map2K3 achieves diverse biologic function by activating p38 MAPK. Figure 6J demonstrates the phosphorylation of p38 and its downstream target, MAPK-activated protein kinase 2 (but not JNK or ERK1/2), were significantly increased in cardiac fibroblasts isolated from Ang II-infused ATF3KO mice. p38 phosphorylation significantly decreased in cardiac fibroblasts of ATF3fl/flTGCre+ mice compared to TGCre- mice (Figure 6K). These data suggested that ATF3 may inhibit hypertensive remodeling by suppressing Map2K3 expression and subsequent p38 inhibition. We performed additional experiments to obtain direct evidence supporting this notion. First, cardiac myocytes were co-cultured with cardiac fibroblasts isolated from ATF3KO animals. p38 inhibition significantly attenuated Ang II-induced ANP mRNA expression and reduced cardiac myocyte size (Supplemental Figure 8A/B), similar to results observed during genetic inhibition of Map2K3 (Figure 7C/D). Given the well-recognized role of TGF-β in ventricular remodeling, as well as its identification as one of the foremost 3 downregulated pathways after p38 inhibition (RNA-seq analysis, Supplemental Figure 8C), we determined the effect of p38 inhibition upon Ang II-activated TGF-β signaling in ATF3KO cardiac fibroblasts. qRT-PCR analysis demonstrated the mRNA levels of TGF-β signaling-related genes (including Tgfb2, BMP4, Thbs2, Thbs3, Col1a1, Col3a1, Igf1, Itgb5, and Igfbp5) were significantly upregulated in ATF3-deficient cardiac fibroblasts, and were suppressed after p38 inhibition (Figure 8A/B). Additionally, multiple signaling molecules known to be responsible for TGF-β-mediated fibrosis and cardiac myocyte hypertrophy (including IL-6, endothelin-1, FGF2, and phosphorylated-Smad3) were all significantly increased in ATF3-deficient cardiac fibroblasts after Ang II treatment. These effects were abrogated in the setting of p38 inhibition (Figures 8C-G).
Figure 8. p38 inhibition blocks TGF-β pathway activation.
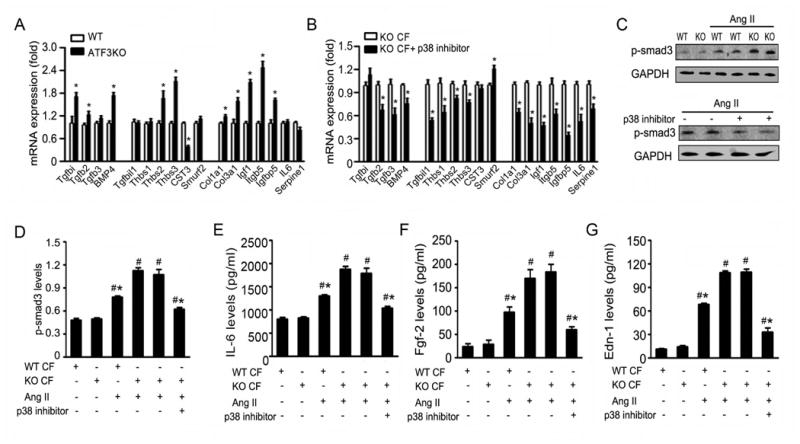
(A, B) qRT-PCR analysis of TGF-β signaling-related genes in Ang II-stimulated cardiac fibroblasts isolated from Ang II-infused WT and ATF3 KO heart (A), or ATF3KO cardiac fibroblasts with or without p38 inhibitor (B). *p<0.05 compared with WT CFs (A).*p<0.05 compared with ATF3 KO CFs+control (B). (C) WT and ATF3KO cardiac fibroblasts were stimulated with Ang II or saline for 4 hours. The phosphorylation of smad3 (p-smad3) expression was detected. (D) ATF3KO cardiac fibroblasts were pretreated with p38 inhibitor or control for 1 hour, and stimulated with Ang II for 4 hours. The p-smad3 expression was detected. (E-G) The levels of IL-6, FGF-2, and Endothlin-1 were assessed by ELISA in WT or ATF3KO cardiac fibroblasts subjected to Ang II stimuli, with or without p38 inhibitor pretreatment. *p<0.05 vs ATF3 KO CF+Ang II. #P<0.05 vs non Ang II treatment. Data from 3 independent experiments. Statistical significance was determined by the unpaired t test (A, B) or by 1-way ANOVA test (C-G).
To determine the translational value of our results thus far, we performed cross-species transcriptome analysis of cardiac tissue from five HCM patients (Supplemental Table 5) and control, and of Ang II-stimulated cardiac fibroblasts pretreated with or without a p38 inhibitor. Hierarchical clustering of the data was performed, yielding 970 upregulated genes and 829 downregulated genes in HCM patient hearts (p<0.05, fold change>1.5) (Supplemental Figure 9A). Pathway analysis identified the seven most significantly upregulated pathways in HCM, including ECM-receptor interaction, focal adhesion, regulation of actin cytoskeleton, hypertrophic cardiomyopathy, dilated cardiomyopathy, TGF-β signaling pathway, and arrhythmogenic right ventricular cardiomyopathy (Supplemental Figure 9B). Notably, this HCM-specific signature was downregulated in cardiac fibroblasts after p38 inhibitor treatment (Supplemental Figure 9C/D). 63 DEGs were transcriptionally regulated in the same direction (43 DEGs were upregulated, and 20 DEGs were downregulated in each of the two groups), and 121 DEGs in opposite directions (97 DEGs were upregulated and 24 DEGs were downregulated genes in opposite patterns) after p38 inhibition (Supplemental Figure 9E). Moreover, pathway analysis revealed that downregulation of 97 overlapped DEGs after p38 inhibition was enriched in the 7 signature HCM pathways (Supplemental Figure 9F-H).
Discussion
We have made two novel findings in this study. Firstly, by utilizing discovery driven/nonbiased approaches, we have demonstrated that hypertensive stimuli selectively upregulated ATF3 expression in cardiac fibroblast cells, not in cardiac myocytes. Given recent recognition that the same molecule expressed in different cell types may have different, or even opposite, impacts upon global organ function26,27. Secondly, cardiac fibroblast-specific ATF3 protects against hypertensive remodeling and heart failure by suppressing Map2K3 transcription and p38 signaling.
Although it is well-recognized that ATF3 is encoded by an immediate early gene with quickly upregulated expression in response to various stress28, the physiologic/pathologic consequence of ATF3 upregulation remains largely unknown29. Recent studies demonstrate that cardiac ATF3 expression is increased in response to pathologic stress30,31. However, the currently available information regarding the functional importance of ATF3 in ventricular remodeling is limited and controversial. Specifically, Zhou et al first reported that global ATF3 deficiency promotes cardiac hypertrophy and fibrosis in pressure overload-induced heart failure, suggesting a cardioprotective function of ATF331. However, another study published 2 years later reported an exactly opposite effect, noting cardiac myocyte-specific ATF3 overexpression promotes ventricular hypertrophy and fibrosis32. Our study demonstrated that ATF3 expression is selectively increased in cardiac fibroblasts in both Ang II infusion-induced and TAC-induced hypertensive remodeling models, challenging the pathologic relevance and subsequent conclusions concerning cardiac myocyte-specific ATF3 overexpression. To obtain the most reliable evidence possible to clarify the origin of ATF3 pertaining to cardiac remodeling, we employed three separate approaches. We first utilized a loss-of-function approach, demonstrating significantly enhanced Ang II -induced cardiac remodeling in ATF3KO mice. We then established a miRNA-aided ATF3-carring lentivirus system preventing cardiomyocyte ATF3 overexpression, demonstrating that non-cardiac myocyte ATF3 overexpression rescued the pro-fibrotic/hypertrophic ATF3KO phenotype. Most importantly, we generated a tamoxifen-inducing cardiac fibroblast cell-specific ATF3 transgenic mouse line, demonstrating cardiac fibroblast-specific ATF3 overexpression attenuated Ang II -induced cardiac remodeling and improved cardiac function. Collectively, we have provided clear evidence that cardiac fibroblast expression of ATF3 is a self-protective mechanism that counteracts harmful cardiac TFs, protecting the heart against hypertensive remolding. Our results not only provide a likely explanation regarding the recently reported opposite effects of ATF3 in cardiac remodeling31,32, but also significantly aid our understanding of the Yin-and- Yang balance underlying hypertensive cardiac remolding regulation.
Cardiac fibroblasts have central and dynamic roles in modulating cardiac function33. During physiologic conditions, cardiac fibroblasts provide the mechanical scaffold for cardiac myocytes, and coordinate cardiac pump function. Under pathologic conditions, cardiac fibroblasts are preferentially activated by stress signals34,35. Activated cardiac fibroblasts produce an array of biologically active molecules, including growth factors and cytokines, which may either contribute to or protect against cardiac myocyte injury via paracrine-mediated cell-cell communications36. However, the precise molecular mechanisms rendering cardiac fibroblasts more sensitive to stress (e.g. selective ATF3 overexpression in response to Ang II and TAC stimuli observed in the present study) remain unclear, and warrant direct investigation.
Second, we combined both in vitro and in vivo models, performed bioinformatics analysis, and integrated transcriptomics in ATF3-depleted fibroblasts and Chip-sequencing for ATF3 binding sites. We identified Map2K3 as a novel ATF3 target. Interestingly, ATF3-deficient mice exhibited increased Map2K3 expression, whereas ATF3 overexpression inhibited Map2K3 expression. Contrary to expectations given its nomenclature, binding of the Map2K3 promoter by ATF3 suppresses, not activates, Map2K3 mRNA expression. Functionally, inhibition of Map2K3 signaling ameliorated the remodeling phenotype observed in ATF3-deficient animals, indicating augmented cardiac fibroblast ATF3 expression in response to hypertensive stress impedes Map2K3 expression. Given the well-recognized pro-fibrotic/hypertrophic effect of Map2K3 signaling37,38, transcriptional suppression of Map2K3 expression is a novel mechanism by which ATF3 functionally protects against hypertensive remodeling.
When co-expressed with heterodimeric partners, or expressed as an alternative splicing product, ATF3 activates transcription. However, as a homodimer, native ATF3 represses transcription39. The molecular mechanisms responsible for such contradictory function remain unidentified. Gene expression is regulated at multiple levels, including epigenetic regulation by DNA methylation and histone acetylation. In particular, HDAC-mediated histone deacetylation tightens chromatin structure and inhibits gene expression. In this regard, we demonstrate for the first time that ATF3 not only directly binds the Map2K3 promoter, but additionally recruits HDAC1. More importantly, the levels of Map2K3 promoter-bound histone acetylation in cardiac fibroblasts significantly increased after Ang II treatment. ATF3 overexpression blocked the effects of Ang II, and significantly increased Map2K3 promoter-bound histone acetylation levels. To the best of our knowledge, this is the first study providing explanation for ATF3-mediated suppression, not activation, of Map2K3 gene transcription. Moreover, our current work further underscores the important and unique regulatory function of ATF3, as it activates select target genes but represses others, depending upon both promoter and cellular context.
We demonstrated p38, not JNK or ERK, is activated in ATK3KO cardiac fibroblasts. Moreover, in similar fashion to Map2K3 inhibition, p38 inhibition ameliorated the exacerbated remodeling phenotype observed in ATF3-deficient cells. These results indicate that p38 activation in cardiac fibroblasts is responsible for Map2K3-mediated pro-fibrotic/hypertrophic effects in ATF3KO animals. These observations are supported by a previous study demonstrating that TNF-α administration to cardiac fibroblasts promotes Map2K3 expression, and predominantly activates p38-mediated inflammatory response40. Interestingly, pharmacological inhibition of p38 is cardioprotective in diabetic mice or mice subjected to myocardial ischemia41,42. However, cardiac myocyte-specific attenuation of p38α results in enhanced cardiac myocyte hypertrophy, apoptosis, and cardiac fibrosis43. Therefore, the origin of p38 (from cardiac myocyte versus cardiac fibroblast) bears influence upon cardiac remodeling and overall cardiac function. This very phenomenon plausibly explains the discrepancy between cardiomyocytes-expressed ATF3 and cardiac fibroblast-expressed ATF3 in concerning their hypertrophic/pro-fibrotic (as previously reported31,32) and anti-hypertrophic/anti-fibrotic effect (as observed in our study).
In summary, we have provided the first evidence that cardiac fibroblast upregulation of ATF3 is a compensatory cardiac protective mechanism activated by hypertensive stimuli. Mechanistically, we demonstrated for the first time that ATF3 binds to the Map2K3 promotor, recruits HDAC1, and results in histone deacetylation, blocking Map2K3 expression and subsequent p38-TGFβ signaling. Our data suggest positive modulation of cardiac fibroblast ATF3, a “hub” of the cellular adaptive-response network, may represent a novel therapeutic approach against hypertensive cardiac remodeling.
Supplementary Material
Clinical Perspective.
What is new?
We have identified both a novel transcription factor (Activating Transcription Factor 3) and the mechanism by which it protects against the development of heart remodeling.
ATF3 protected against cardiac hypertensive remodeling, heart failure, and fibrosis, via suppression of Map2K3 expression and p38 MAPK signaling.
What are the clinical implications?
Identifying molecules mimicking endogenous ligands or inhibiting miRNA that downregulate ATF3 expression may represent a novel therapeutic approach against hypertensive cardiac remodeling.
Acknowledgments
We thank Professor Yongqiang Lai for provision of human myocardial samples, and Professor Hongliang Li for graciously providing the ATF3-deficient mice. We appreciate Dr. Wanshi Cai and Dr. Fenbiao Mao for providing assistance in CHIP-sequencing and data analysis. We are grateful to Dr. Jian Cui (Shanghai BioGenius Biotechnology Co., Ltd) and Weiping Tang (Cnkingbio Company Ltd, Beijing, China) for bioinformatics assistance.
Sources of Funding: This study was supported by grants from the National Science Foundation of China (81230006, 81470428, 91539121 and 81500265), Key Laboratory of Remodeling-Related Cardiovascular Diseases, Ministry of Education, China (PXM2014-014226-000012), Beijing collaborative innovative research center for cardiovascular diseases, Beijing Natural Science Foundation (7132043 and 7142050); and NIH HL-096686, HL-123404, American Diabetes Association 1-15-BS-122 (to XLM).
Footnotes
Disclosures: None.
References
- 1.Drazner MH. The progression of hypertensive heart disease. Circulation. 2011;123:327–334. doi: 10.1161/CIRCULATIONAHA.108.845792. [DOI] [PubMed] [Google Scholar]
- 2.Burchfield JS, Xie M, Hill JA. Pathological ventricular remodeling: Mechanisms: Part 1 of 2. Circulation. 2013;128:388–400. doi: 10.1161/CIRCULATIONAHA.113.001878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chobanian AV, Bakris GL, Black HR, Cushman WC, Green LA, Izzo JL, Jr, Jones DW, Materson BJ, Oparil S, Wright JT, Jr, Roccella EJ. The seventh report of the joint national committee on prevention, detection, evaluation, and treatment of high blood pressure: The jnc 7 report. JAMA. 2003;289:2560–2572. doi: 10.1001/jama.289.19.2560. [DOI] [PubMed] [Google Scholar]
- 4.Feron O, Salomone S, Godfraind T. Action of the calcium channel blocker lacidipine on cardiac hypertrophy and endothelin-1 gene expression in stroke-prone hypertensive rats. Br J Pharmacol. 1996;118:659–664. doi: 10.1111/j.1476-5381.1996.tb15451.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Makuc DM, Marcus GM, Marelli A, Matchar DB, Moy CS, Mozaffarian D, Mussolino ME, Nichol G, Paynter NP, Soliman EZ, Sorlie PD, Sotoodehnia N, Turan TN, Virani SS, Wong ND, Woo D, Turner MB. Heart disease and stroke statistics--2012 update: A report from the american heart association. Circulation. 2012;125:e2–e220. doi: 10.1161/CIR.0b013e31823ac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Judd SE, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Mackey RH, Magid DJ, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER, 3rd, Moy CS, Mussolino ME, Neumar RW, Nichol G, Pandey DK, Paynter NP, Reeves MJ, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Wong ND, Woo D, Turner MB. Heart disease and stroke statistics--2014 update: A report from the american heart association. Circulation. 2014;129:e28–e292. doi: 10.1161/01.cir.0000441139.02102.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heineke J, Ritter O. Cardiomyocyte calcineurin signaling in subcellular domains: From the sarcolemma to the nucleus and beyond. J Mol Cell Cardiol. 2012;52:62–73. doi: 10.1016/j.yjmcc.2011.10.018. [DOI] [PubMed] [Google Scholar]
- 8.Bujak M, Ren G, Kweon HJ, Dobaczewski M, Reddy A, Taffet G, Wang XF, Frangogiannis NG. Essential role of smad3 in infarct healing and in the pathogenesis of cardiac remodeling. Circulation. 2007;116:2127–2138. doi: 10.1161/CIRCULATIONAHA.107.704197. [DOI] [PubMed] [Google Scholar]
- 9.van Oort RJ, van Rooij E, Bourajjaj M, Schimmel J, Jansen MA, van der Nagel R, Doevendans PA, Schneider MD, van Echteld CJ, De Windt LJ. Mef2 activates a genetic program promoting chamber dilation and contractile dysfunction in calcineurin-induced heart failure. Circulation. 2006;114:298–308. doi: 10.1161/CIRCULATIONAHA.105.608968. [DOI] [PubMed] [Google Scholar]
- 10.Oka T, Maillet M, Watt AJ, Schwartz RJ, Aronow BJ, Duncan SA, Molkentin JD. Cardiac-specific deletion of gata4 reveals its requirement for hypertrophy, compensation, and myocyte viability. Circ Res. 2006;98:837–845. doi: 10.1161/01.RES.0000215985.18538.c4. [DOI] [PubMed] [Google Scholar]
- 11.Hai T, Hartman MG. The molecular biology and nomenclature of the activating transcription factor/camp responsive element binding family of transcription factors: Activating transcription factor proteins and homeostasis. Gene. 2001;273:1–11. doi: 10.1016/s0378-1119(01)00551-0. [DOI] [PubMed] [Google Scholar]
- 12.Hartman MG, Lu D, Kim ML, Kociba GJ, Shukri T, Buteau J, Wang X, Frankel WL, Guttridge D, Prentki M, Grey ST, Ron D, Hai T. Role for activating transcription factor 3 in stress-induced beta-cell apoptosis. Mol Cell Biol. 2004;24:5721–5732. doi: 10.1128/MCB.24.13.5721-5732.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li Y, Wu Y, Zhang C, Li P, Cui W, Hao J, Ma X, Yin Z, Du J. γδT cell-derived interleukin-17a via an interleukin-1β-dependent mechanism mediates cardiac injury and fibrosis in hypertension. Hypertension. 2014;64:305–314. doi: 10.1161/HYPERTENSIONAHA.113.02604. [DOI] [PubMed] [Google Scholar]
- 14.Li Y, Zhang C, Wu Y, Han Y, Cui W, Jia L, Cai L, Cheng J, Li H, Du J. Interleukin-12p35 deletion promotes CD4 t-cell-dependent macrophage differentiation and enhances angiotensin ii-induced cardiac fibrosis. Arterioscler Thromb Vasc Biol. 2012;32:1662–1674. doi: 10.1161/ATVBAHA.112.249706. [DOI] [PubMed] [Google Scholar]
- 15.da Costa Martins PA, Salic K, Gladka MM, Armand AS, Leptidis S, el Azzouzi H, Hansen A, Coenen-de Roo CJ, Bierhuizen MF, van der Nagel R, van Kuik J, de Weger R, de Bruin A, Condorelli G, Arbones ML, Eschenhagen T, De Windt LJ. Microrna-199b targets the nuclear kinase dyrk1a in an auto-amplification loop promoting calcineurin/nfat signalling. Nat Cell Biol. 2010;12:1220–1227. doi: 10.1038/ncb2126. [DOI] [PubMed] [Google Scholar]
- 16.Sassi Y, Ahles A, Truong DJ, Baqi Y, Lee SY, Husse B, Hulot JS, Foinquinos A, Thum T, Muller CE, Dendorfer A, Laggerbauer B, Engelhardt S. Cardiac myocyte-secreted camp exerts paracrine action via adenosine receptor activation. J Clin Invest. 2014;124:5385–5397. doi: 10.1172/JCI74349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Collins CA, Kretzschmar K, Watt FM. Reprogramming adult dermis to a neonatal state through epidermal activation of beta-catenin. Development. 2011;138:5189–5199. doi: 10.1242/dev.064592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brown BD, Venneri MA, Zingale A, Sergi Sergi L, Naldini L. Endogenous microrna regulation suppresses transgene expression in hematopoietic lineages and enables stable gene transfer. Nat Med. 2006;12:585–591. doi: 10.1038/nm1398. [DOI] [PubMed] [Google Scholar]
- 19.Santulli G, Wronska A, Uryu K, Diacovo TG, Gao M, Marx SO, Kitajewski J, Chilton JM, Akat KM, Tuschl T, Marks AR, Totary-Jain H. A selective microrna-based strategy inhibits restenosis while preserving endothelial function. J Clin Invest. 2014;124:4102–4114. doi: 10.1172/JCI76069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jayawardena TM, Egemnazarov B, Finch EA, Zhang L, Payne JA, Pandya K, Zhang Z, Rosenberg P, Mirotsou M, Dzau VJ. Microrna-mediated in vitro and in vivo direct reprogramming of cardiac fibroblasts to cardiomyocytes. Circ Res. 2012;110:1465–1473. doi: 10.1161/CIRCRESAHA.112.269035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ubil E, Duan J, Pillai IC, Rosa-Garrido M, Wu Y, Bargiacchi F, Lu Y, Stanbouly S, Huang J, Rojas M, Vondriska TM, Stefani E, Deb A. Mesenchymal-endothelial transition contributes to cardiac neovascularization. Nature. 2014;514:585–590. doi: 10.1038/nature13839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wong VW, Rustad KC, Akaishi S, Sorkin M, Glotzbach JP, Januszyk M, Nelson ER, Levi K, Paterno J, Vial IN, Kuang AA, Longaker MT, Gurtner GC. Focal adhesion kinase links mechanical force to skin fibrosis via inflammatory signaling. Nat Med. 2012;18:148–152. doi: 10.1038/nm.2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Berlo JH, Maillet M, Molkentin JD. Signaling effectors underlying pathologic growth and remodeling of the heart. J Clin Invest. 2013;123:37–45. doi: 10.1172/JCI62839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kakkar R, Lee RT. Intramyocardial fibroblast myocyte communication. Circ Res. 2010;106:47–57. doi: 10.1161/CIRCRESAHA.109.207456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sporn MB. The early history of tgf-beta, and a brief glimpse of its future. Cytokine Growth Factor Rev. 2006;17:3–7. doi: 10.1016/j.cytogfr.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 27.Dotto GP. Notch tumor suppressor function. Oncogene. 2008;27:5115–5123. doi: 10.1038/onc.2008.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hai T, Wolfgang CD, Marsee DK, Allen AE, Sivaprasad U. Atf3 and stress responses. Gene Expr. 1999;7:321–335. [PMC free article] [PubMed] [Google Scholar]
- 29.Hai T, Wolford CC, Chang YS. Atf3, a hub of the cellular adaptive-response network, in the pathogenesis of diseases: Is modulation of inflammation a unifying component? Gene Expr. 2010;15:1–11. doi: 10.3727/105221610x12819686555015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hasin T, Elhanani O, Abassi Z, Hai T, Aronheim A. Angiotensin ii signaling up-regulates the immediate early transcription factor atf3 in the left but not the right atrium. Basic Res Cardiol. 2011;106:175–187. doi: 10.1007/s00395-010-0145-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou H, Shen DF, Bian ZY, Zong J, Deng W, Zhang Y, Guo YY, Li H, Tang QZ. Activating transcription factor 3 deficiency promotes cardiac hypertrophy, dysfunction, and fibrosis induced by pressure overload. PLoS One. 2011;6:e26744. doi: 10.1371/journal.pone.0026744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koren L, Elhanani O, Kehat I, Hai T, Aronheim A. Adult cardiac expression of the activating transcription factor 3, atf3, promotes ventricular hypertrophy. PLoS One. 2013;8:e68396. doi: 10.1371/journal.pone.0068396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Furtado MB, Nim HT, Boyd SE, Rosenthal NA. View from the heart: Cardiac fibroblasts in development, scarring and regeneration. Development. 2016;143:387–397. doi: 10.1242/dev.120576. [DOI] [PubMed] [Google Scholar]
- 34.Souders CA, Bowers SL, Baudino TA. Cardiac fibroblast: The renaissance cell. Circ Res. 2009;105:1164–1176. doi: 10.1161/CIRCRESAHA.109.209809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takeda N, Manabe I, Uchino Y, Eguchi K, Matsumoto S, Nishimura S, Shindo T, Sano M, Otsu K, Snider P, Conway SJ, Nagai R. Cardiac fibroblasts are essential for the adaptive response of the murine heart to pressure overload. J Clin Invest. 2010;120:254–265. doi: 10.1172/JCI40295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Manabe I, Shindo T, Nagai R. Gene expression in fibroblasts and fibrosis: Involvement in cardiac hypertrophy. Circ Res. 2002;91:1103–1113. doi: 10.1161/01.res.0000046452.67724.b8. [DOI] [PubMed] [Google Scholar]
- 37.Streicher JM, Ren S, Herschman H, Wang Y. Mapk-activated protein kinase-2 in cardiac hypertrophy and cyclooxygenase-2 regulation in heart. Circ Res. 2010;106:1434–1443. doi: 10.1161/CIRCRESAHA.109.213199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liao P, Georgakopoulos D, Kovacs A, Zheng M, Lerner D, Pu H, Saffitz J, Chien K, Xiao RP, Kass DA, Wang Y. The in vivo role of p38 map kinases in cardiac remodeling and restrictive cardiomyopathy. Proc Natl Acad Sci U S A. 2001;98:12283–12288. doi: 10.1073/pnas.211086598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen BP, Liang G, Whelan J, Hai T. Atf3 and atf3 delta zip. Transcriptional repression versus activation by alternatively spliced isoforms. J Biol Chem. 1994;269:15819–15826. [PubMed] [Google Scholar]
- 40.Wysk M, Yang DD, Lu HT, Flavell RA, Davis RJ. Requirement of mitogen-activated protein kinase kinase 3 (mkk3) for tumor necrosis factor-induced cytokine expression. Proc Natl Acad Sci U S A. 1999;96:3763–3768. doi: 10.1073/pnas.96.7.3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Westermann D, Rutschow S, Van Linthout S, Linderer A, Bucker-Gartner C, Sobirey M, Riad A, Pauschinger M, Schultheiss HP, Tschope C. Inhibition of p38 mitogen-activated protein kinase attenuates left ventricular dysfunction by mediating pro-inflammatory cardiac cytokine levels in a mouse model of diabetes mellitus. Diabetologia. 2006;49:2507–2513. doi: 10.1007/s00125-006-0385-2. [DOI] [PubMed] [Google Scholar]
- 42.Liu YH, Wang D, Rhaleb NE, Yang XP, Xu J, Sankey SS, Rudolph AE, Carretero OA. Inhibition of p38 mitogen-activated protein kinase protects the heart against cardiac remodeling in mice with heart failure resulting from myocardial infarction. J Card Fail. 2005;11:74–81. doi: 10.1016/j.cardfail.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 43.Braz JC, Bueno OF, Liang Q, Wilkins BJ, Dai YS, Parsons S, Braunwart J, Glascock BJ, Klevitsky R, Kimball TF, Hewett TE, Molkentin JD. Targeted inhibition of p38 mapk promotes hypertrophic cardiomyopathy through upregulation of calcineurin-nfat signaling. J Clin Invest. 2003;111:1475–1486. doi: 10.1172/JCI17295. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.