

Abstract
Studies in rodents highlight a role for leptin in stimulation of pituitary growth hormone (GH) secretion, with an impact on body composition regulation. We have reported that maternal obesity (MO) during ovine pregnancy results in hyperphagia, glucose-insulin dysregulation, increased adiposity, hypercortisolemia and hyperleptinemia in mature offspring subjected to a bout of ad libitum feeding. We hypothesized that MO reduces leptin signaling in the pituitary and down regulates the GH/IGF1 axis and increases circulating cortisol leading to increased adiposity in their adult offspring. Male lambs born to MO (n = 6) or control (CON, n = 6) ewes were fed only to requirements until placed on a 12 week ad libitum feeding trial at maturity. The pituitary, hypothalamic arcuate nucleus, and liver were collected at necropsy and mRNA and protein expression determined. Plasma cortisol concentrations were increased (P<0.05) in MO vs. CON offspring at the end of the feeding trial. Further, serum concentrations of IGF1 decreased (P<0.01) and GH tended to decrease (P<0.08) in MO vs. CON offspring. Pituitary mRNA and leptin receptor protein expression were decreased in MO vs. CON offspring in association with decreased GH mRNA expression, and decreased IGF1 mRNA and protein expression in liver. Liver 11β-hydroxysteroid dehydrogenase 1 (11βHSD1) expression was increased (P<0.01) and its cofactor hexose-6-phosphate dehydrogenase tended to increase (P<0.06) in MO vs. CON offspring. 11βHSD2 expression remained unchanged. These data indicate that MO induced an increase in liver conversion of cortisone to cortisol in adult offspring and support a role for leptin signaling in the pituitary in mediating offspring adiposity.
Introduction
Obesity is a major human health concern. The current global obesity epidemic, together with its associated chronic diseases, represents a significant economic cost [1,2]. In a survey carried out between 2003 and 2006, the US obesity rate in women of child bearing age (20–44 years old) was estimated at 32% [3]. Additionally, there is strong evidence that maternal obesity and overnutrition during human pregnancy is associated with an increased incidence of insulin resistance and obesity later in life [2,4,5]. There is also compelling evidence from animal studies that maternal obesity alters offspring phenotype in a similar manner [6–8].
Leptin is produced and secreted by adipocytes and circulates to the hypothalamus where it binds to leptin receptor (OB-Rb), signaling the level of adiposity and suppressing appetite [9–11]. Rodents with leptin or OB-Rb mutations show several phenotypes including infertility, hyperphagia, obesity, and reduced energy expenditure as well as reduced somatotrope numbers [12,13]. There is now strong evidence that OB-Rb is also localized to pituitary somatotropes [14–16] and exerts an important regulatory role on the maintenance of growth hormone (GH) stores in somatotropes [15,17]. Recently, Childs et al. [18] reported that a pituitary specific OB-Rb mutation in mice resulted in severe GH deficiency and significantly increased adiposity. Further, an in vivo study showed that exogenous leptin infusion in ob/ob mice restored GH secretion, but hypothalamic growth hormone releasing hormone (GHRH) mRNA expression was not altered [19]. Collectively, these data highlight the importance of leptin in modulating somatotrope GH secretion and thus regulation of hepatic IGF1 secretion.
The GH/IGF1 axis is an important regulator of growth and metabolism through effects on carbohydrate, fat and protein metabolism [20–22]. IGF1 infusion in healthy humans reduces blood insulin and lipid levels [23], and circulating GH levels are negatively correlated with BMI [19,24,25]. Further, GH deficiency is associated with an increased risk of obesity, insulin resistance and diabetes mellitus [26], and these adverse metabolic profiles and increased adiposity in GH deficient adults are improved by GH treatment [27]. Either GH [28] or IGF1 [23] treatments are effective in reducing hyperphagia, obesity, hyperinsulinemia and hypertension in adult rat offspring that were exposed to sub-optimal nutrition in utero.
We have developed and characterized a sheep model of maternal overnutrition/obesity [6,29,30]. In this model newborn lambs born to MO ewes have a markedly increased adiposity compared to lambs born to lean ewes fed only to requirements [29]. Further, when adult offspring of MO and CON ewes are subjected to an ad libitum feeding challenge, MO offspring exhibited hyperphagia, leading to increased weight gain, glucose dysregulation and hyperleptinemia when compared to offspring born to CON ewes. The increased weight gain of MO offspring in response to the feeding challenge was predominantly due to increased fat accumulation both viscerally and subcutaneously [6,31]. We hypothesized that MO programs reduced pituitary sensitivity to leptin, down regulating development of the GH/IGF1 axis and leading to an increase in adiposity in adult offspring.
Materials and methods
Animals
All animal procedures were approved by the University of Wyoming Animal Care and Use Committee. Blood and tissues utilized in the present study were obtained at necropsy from adult male offspring in a study previously reported by Long et al [31]. Offspring were born to either overfed/obese ewes (MO ewes) who exhibited an 85% increase in body weight when overfed from 60 days before conception through late gestation, or lean ewes fed only to requirements (CON ewes) who gained only 15.8% over the same time period. As reported by Long et al. [31] male lambs born to MO (n = 6) or CON (n = 6) ewes were housed together and fed only to requirements until being placed on an 12 week ad libitum feeding trial at maturity (2.5 years of age). Weights and blood samples were obtained throughout the feeding trial. At necropsy, the animals were euthanized with an overdose of sodium pentobarbital (Beuthanasia-D Special; Schering-Plough Animal Health, Union, NJ). Organs and tissues were removed and weighed. For the present study, the pituitary, the arcuate nucleus of the hypothalamus, and the right lobe of the liver were collected and portions snap frozen in liquid nitrogen and stored at –80°C. Pituitary tissue was also fixed in paraformaldehyde and paraffin embedded for immunohistochemical evaluation.
Biochemical assays
Plasma IGF1 and ACTH concentrations were determined using an Immulite 1000 immunoassay analyzer with a sensitivity of 9 pg/mL (Siemens Medical Solutions, Malvern, PA, USA). IGF1 measurements were completed in a single assay, with an intra-assay coefficient of variation of 5.5%. All ACTH measurements were completed in a single assay, which resulted in intra-assay coefficient of variation of 5.5%. Plasma cortisol was determined in duplicate as previously described [21], using a commercial cortisol RIA kit with a sensitivity of 0.5 μg/dL (Siemens Healthcare Diagnostics, Deerfield, IL, USA). All cortisol measurements were also completed in a single assay and the intra-assay CV for cortisol was 4.5%. Serum GH was quantified in the laboratory of Dr. Dennis Hallford (New Mexico State University) using a double antibody RIA as described by and Hoefler and Hallford [32], and using primary antisera and purified ovine GH provided by the National Hormone and Peptide Program. Further, GH was determined in a single assay and the within assay coefficient of variation was 6.1%.)
Immunohistochemistry
Immunostaining of GH and OB-Rb in 5μm pituitary tissue sections were accomplished following the general immunohistochemistry protocols described by Ma et al [30]. Briefly, paraffin embedded sections were deparaffinized and rehydrated by routine methods, and then antigen retrieval was accomplished by boiling the sections in citrate buffer. Sections were then probed with rabbit anti-GH (Geneway, San Diego, CA) and goat anti-OB-Rb (Abbiotec, San Diego, CA) primary antibodies followed by fluorescence-labeled anti-rabbit (green) and anti-goat (red) secondary antibodies. Images were visualized using an Olympus BS50 fluorescence microscope and captured digitally using a Retiga ExiFast camera. Pictures at 40 X magnification were taken using QED Imaging software (Media Cybernetics, Silver Spring, MD).
RNA extraction and real-time PCR
Pituitary and liver tissues were pulverized in liquid nitrogen. Total RNA was extracted from 50 mg of each sample using Trizol reagent (Invitrogen Corp., Carlsbad, CA) treated with DNase I (QIAGEN Inc. Valencia, CA) and then purified by RNeasy mini column (QIAGEN Inc. Valencia, CA) according to the manufacturer’s corresponding protocols. Two μg of purified RNA was used for cDNA synthesis using Promega ImProm-II™ Reverse Transcription System (Promega BioSiences, San Luis Obispo, CA) according to the kit protocol. The genes and the primers in real-time PCR experiments are provided in Table 1. All Real-time PCR reactions were conducted using a Bio-Rad IQ5 Realtime-PCR Reaction System (Bio-Rad Laboratories Inc., Hercules, CA) as previously utilized in our laboratory [30]. Final data was analyzed by the Ct method relative to 18s rRNA expression.
Table 1. Primer sequences of genes evaluated by qPCR.
| Gene* | Sense | Anti-sense |
|---|---|---|
| OB-Rb | GAT GAG ATG GTG CCA ACA ACT A | TGG GTT TCT ATT TCC CAT GAT C |
| Leptin | ATC TCA CAC ACG CAG TCC GT | CCA GCA GGT GGA GAA GGT C |
| GH | TCC AGA ACA CCC AGG TTG CC | CAT CTT CCA GCT CCC GCA TC |
| IGF1 | GAC AGG AAT CGT GGA TGA GTG | AAC AGG TAA CTC GTG CAG AGC |
| 11βHSD1 | GCG CCA GAT CCC TGT CTG AT | AGC CCC ATA CCA CCT TCT TT |
| 11βHSD2 | AGC AGG AGA CAT GCC GTT TC | GCA ATG CCA AGG CTG CTT |
| IGFBP3 | CAG AAC TTC TCC TCC GAG TCC | CCA CAC ACC AGC AGA AAC C |
| GHR | ATG AAC CCA TCT GCA TGT GA | TTC AGT CTT CTC ATC AGG GTC A |
*Leptin receptor (OB-Rb), growth hormone (GH), insulin-like growth factor-1 (IGF1), 11β-hydroxysteroid dehydrogenase-1 (11β HSD1), 11β-hydroxysteroid dehydrogenase-2 (11β HSD2), growth hormone receptor (GHR), insulin-like growth factor binding protein-3 (IGFBP3).
Protein extraction and western blotting
Protein was extracted by homogenizing ~100 mg of pulverized pituitary, arcuate nucleus of the hypothalamus and liver tissues in ice-cold lysis buffer using a polytron homogenizer. Homogenates were then centrifuged and the supernatants mixed with 2 × standard SDS sample loading buffer and then boiled at 95°C for 5 minutes. Approximately, 50 μg of protein extracts were separated in SDS-PAGE gels and transferred to nitrocellulose membranes for immunoblotting with primary antibodies for leptin, OB-Rb, GH releasing hormone (GHRH), GH inhibiting hormone (GHIH, somatostatin), GH receptor (GHR), IGF1, IGF-binding protein 3 (IGFBP3), IGF1 receptor(IGF1R), 11 β hydroxysteroid dehydrogenase (HSD)1, 11-β HSD2, Hexose-6-phosphate dehydrogenase (H6PD) and β-actin. The membranes were incubated with appropriate secondary antibodies and then visualized using enhanced chemiluminescence (Amersham Biosciences) and exposure to an X-ray film. The target protein band density was analyzed by ImageQuant TL software (Amersham Biosciences) and normalized according to the density of β-actin as previously described [30].
Statistical analysis
Repeated variables were analyzed using PROC MIXED procedures of SAS (SAS Institute Inc., Cary, NC) with repeated measures including leptin, ACTH and cortisol analysis from the different time points. The model contained time, treatment and their interaction for repeated measurements. PROC CORR was used for correlation analysis for ACTH and cortisol. Differences in non-repeated variables were determined using analysis of variance by PROC GLM in SAS with treatment in the model statement. Differences were considered significant at P≤0.05, tendencies at P≤0.10. Data are presented as mean ± SEM.
Results
Plasma ACTH and cortisol and liver expression of enzymes that regulate cortisol metabolism
Plasma concentrations of ACTH were similar between the CON and MO offspring both at the beginning and the end of the feeding trial (Fig 1A). However, there was a marked increase (P < 0.05) in plasma cortisol concentration in both CON and MO offspring at necropsy compared to the concentrations at initiation of the feeding trial (Fig 1B). Further, while cortisol concentrations were similar between CON and MO offspring at the initiation of the feeding trial, cortisol concentrations were markedly higher in MO than CON offspring at the end of the trial. A significant correlation (P<0.01) between ACTH and cortisol levels was found at the initiation of the feeding trial, but no significant correlation (P>0.10) was found at necropsy.
Fig 1. Plasma ACTH (A) and cortisol (B) levels of CON (open bars; n = 6) and MO (solid bars; n = 6) adult male F1 offspring at the initiation and the end of the feeding trial.
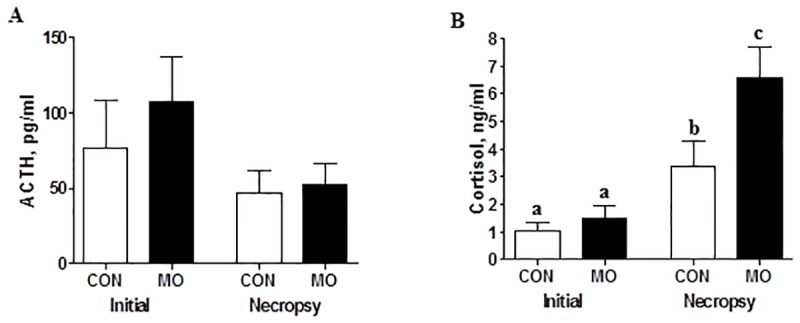
a,b,cMeans ± SEM with different superscripts differ, (P < 0.05).
Expression of 11βHSD1, its co-factor H6PD and 11βHSD2 in the livers of adult male offspring was determined to evaluate the potential contribution of peripheral tissues to cortisol production. Expression of 11βHSD1 mRNA and protein was increased (P<0.01) and H6PD protein tended to increase (P<0.06) in MO vs. CON offspring, but there was no difference in 11βHSD2 mRNA and protein expression between the two groups (Fig 2A–2E).
Fig 2. Gene and protein expression of 11βHSD1 (A,C), 11βHSD2 (B,D) and protein expression of H6PD (E) in the livers of CON (open bars, n = 6) and MO (solid bars; n = 6) adult male F1 offspring.
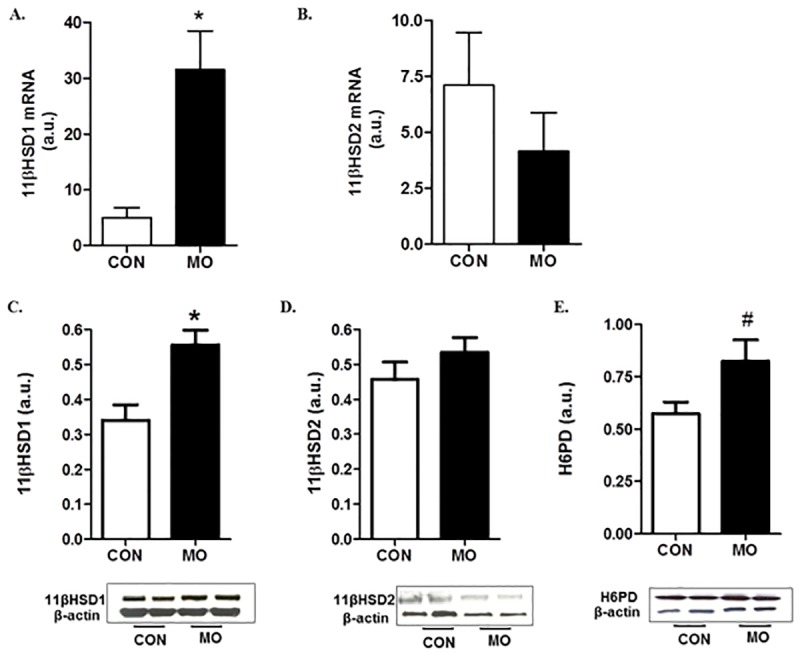
#Means ± SEM tended to differ, (P < 0.06). *Means ± SEM differ, (P < 0.01).
Serum GH and IGF1
Serum concentrations of IGF1 were decreased (P<0.01) in MO compared to CON offspring at necropsy (327.0 ± 31.0 vs. 443.0 ± 28.0 ng/mL, respectively). There was a tendency for reduced serum concentrations of GH (P<0.08) in MO compared with CON offspring at the end of the feeding trial (6.80 ± 0.52 vs. 9.44 ± 0.68 ng/mL, respectively).
Co-localization of GH and OB-Rb in the pituitary somatotropes
Both GH and OB-Rb were extensively expressed in the ovine pituitary (Fig 3A & 3B) with many somatotropes co-expressing GH and OB-Rb (Fig 3C).
Fig 3. Localization of GH and leptin receptor (OB-Rb) on paraffin embedded pituitary sections using double-labeled immunofluorescence microscopy.
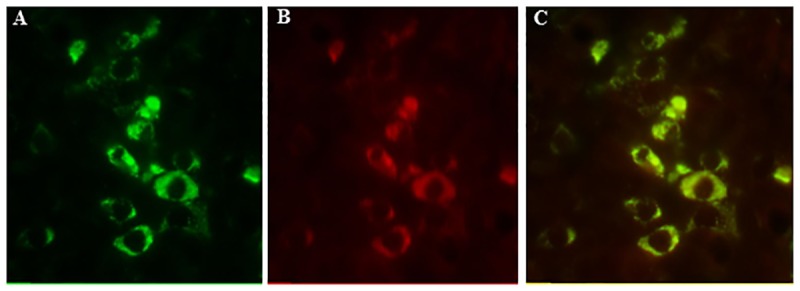
GH (green) positive cells (A); OB-Rb (red) positive cells (B); and merged images for GH and OB-Rb expressing cells (C) showing co-localization in somatotropes.
Expression of the components and the regulators of the GH/IGF1 axis
Protein expression of hypothalamic GHRH and GHIH peptides were determined, and there were no significant differences (P>0.10) found in protein expression in either of these peptides in CON and MO adult offspring. Further, the ratio of GHRH/GHIH did not differ between the MO and CON groups (data not shown). However, both mRNA and protein expression of OB-Rb in the pituitary were decreased (P<0.05) in MO adults compared with CON adults (Figs 4A & 5A). Pituitary GH mRNA expression was decreased (P<0.05) in MO offspring (Fig 4B) but there was only a trend for a decrease (P<0.10) in GH protein expression in MO offspring compared with CON offspring (Fig 5B). Further, liver GHR mRNA and protein expression were lower (P<0.05) in MO offspring than in CON offspring (Figs 4C & 5C). Liver IGF1 mRNA and protein expression were also lower in MO offspring compared to CON offspring (Figs 4D & 5D). However, no significant differences (P>0.05) were found in liver IGFBP3 mRNA or protein expression (Figs 4E & 5E).
Fig 4. Gene expression of OB-Rb (A), GH (B) in pituitary, and GH receptor (GHR, C), IGF1(D), and IGF binding protein 3 (IGFBP3, E) in liver samples of CON (open bars, n = 6) and MO (solid bars; n = 6) adult male F1 offspring.
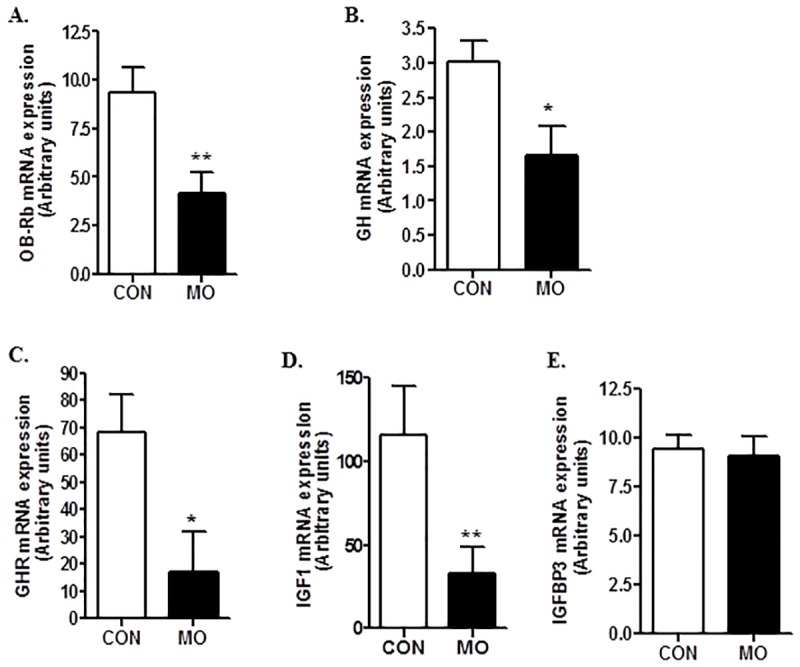
*Means ± SEM differ, (P < 0.05). **Means ± SEM differ, (P < 0.01).
Fig 5. Protein expression of OB-Rb (A), and GH (B) in pituitary, and GHR (C), IGF1 (D), and IGFBP3 (E) in liver samples of CON (open bars, n = 6) and MO (solid bars; n = 6) adult male F1 offspring.
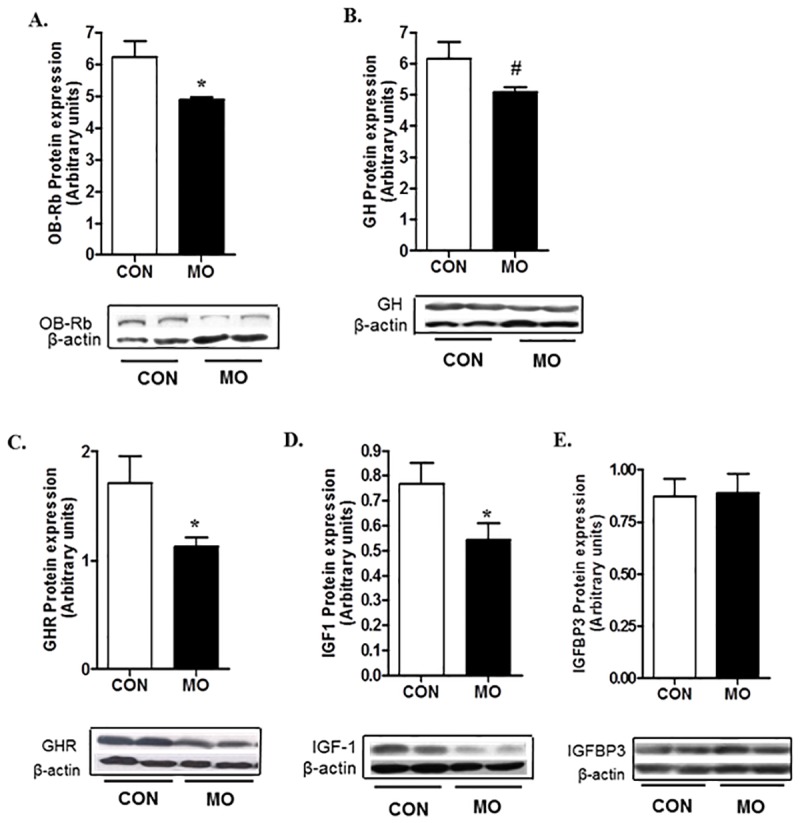
*Means ± SEM differ, (P < 0.05). #Means ± SEM tended to differ, (P < 0. 10).
Discussion
It is well-documented that maternal overnutrition/obesity during pregnancy can increase the risk of insulin resistance and obesity in offspring postnatally [33–35]. Further, there is strong evidence that production of leptin, a regulatory hormone mainly produced by adipocytes, can be altered by the maternal nutritional environment and is involved in prenatal or early postnatal programming of increased offspring appetite and obesity [36–38]. The data presented in this study support a novel hypothesis that leptin signaling in the pituitary plays a role in programming of increased adult offspring adiposity by maternal overnutrition/obesity. We have previously shown that lambs born to MO ewes had similar body weights to lambs of CON mothers, but they had an increased percentage of body fat at birth when compared to CON lambs [29]. Further, we reported that the neonatal leptin peak was eliminated by maternal overnutrition/obesity in MO lambs [37,39]. The neonatal leptin peak is present across species, and we have demonstrated that the neonatal leptin peak demonstrated in altricial species is also present in the sheep, a precocial species [37,38,40], and evidence supports the concept that this surge of leptin during the early neonatal period is responsible for development of normal hypothalamic neuronal trajectories that regulate appetite [9,41]. As predicted, the absence of a leptin peak in these MO offspring during the neonatal period [37,39] predisposed them to become hyperphagic in adulthood when they were subjected to an ad libitum feeding trial despite having elevated circulating leptin concentration.
Further, we have previously demonstrated [6,31] that male offspring born to MO mothers gain more weight and accumulate more fat when subjected to a postnatal ad libitum feeding challenge at maturity than offspring from mothers fed only to requirements. In that study, plasma leptin concentrations were elevated in MO offspring compared with CON offspring, yet MO offspring had a greater feed intake, suggesting that the MO male offspring were leptin resistant. Nevertheless, increased appetite alone cannot explain why the MO adult offspring only increased fat mass rather than lean tissue mass. Rodents with mutations in leptin receptor have low numbers of somatotropes [42,43]. Further, exogenous leptin injection to ob/ob mice restores GH secretion [44]. Recently, Childs et al. [18] demonstrated that a pituitary specific deletion of OB-Rb in mouse caused obesity and dramatic decreases in both somatotrope numbers and GH secretion. The group later reported more extreme cases of GH deficiency and abdominal obesity in male mice that had selective ablation of exon 1 of OB-Rb in somatotropes [45]. This excision of OB-Rb exon 1 resulted in the loss of all isoforms of the leptin receptor and uncovered a broader role for somatotropes as metabolic sensors including sex-specific responses to leptin [45,46].
In our study, we report reduced pituitary OB-Rb expression in MO offspring in addition to co-localization of GH and OB-Rb in the same population of anterior pituitary cells demonstrating a probable functional relationship. This decrease in OB-Rb expression in somatotropes could compromise the function of the GH/IGF1 axis, leading to increased adiposity due to a reduced lipolytic action of GH and IGF1 [18,47,48]. We found that the mRNA expression of both pituitary GH and liver IGF1 were reduced in MO offspring compared to CON offspring. Further, there was a trend towards a decrease in protein expression of GH in the pituitary, and a trend for a decrease in serum GH levels in MO vs. CON offspring. The serum IGF1 concentration was significantly lower in MO offspring than CON offspring, consistent with a decrease in liver IGF1 secretion. It is well established that the hypothalamus has an important role in regulation of the somatotrope axis through GH releasing hormone (GHRH) and somatostatin (GH inhibiting hormone, GHIH) secretion from the arcuate nucleus [49], and that the ratio of these two antagonistic peptides modulate growth hormone secretion [50]. However, as we found no differences in hypothalamic expression of these two peptide hormones, differences in hypothalamic function may not be the driving force for the difference observed in GH and IGF1 levels of MO and CON offspring. Collectively, these results are consistent with the concept that the GH/IGF1 axis in MO offspring was down-regulated in association with reduced leptin signaling in the pituitary.
The GH/ IGF1 axis is one of the most important regulators of growth and metabolism in mammals, due to its regulation of carbohydrate, fat and protein metabolism [51]. With prolonged exposure to GH, protein anabolism, organ growth and lean body mass increases and body fat mass decreases [52,53]. The level of visceral obesity is strongly and negatively associated with both circulating GH and IGF1 levels, and treatment of GH deficient young adults with GH and IGF1 reduces the percentage fat mass and increases the rate of protein synthesis, which contributes to elevated lean muscle mass [54]. GH treatment reduces abdominal obesity in women [55]. Further, greater visceral adiposity and insulin resistance in overweight girls were associated with lower GH and higher cortisol levels [56]. These data indicate that lower GH and/or IGF1 levels are associated with increased visceral adiposity and insulin resistance. The lack of hepatic expression differences in IGFBP-3 in MO vs. CON offspring suggests no role for IGFBP-3 in altering half-life of IGF1 in the blood stream. IGFBP-3 is produced by the liver and released into the systemic circulation, binding to and transporting 70–90% of all circulating IGF [57].
We have reported increased circulating glucose and visceral adiposity as well as reduced pancreatic insulin release in these MO offspring [31], in addition to the lower circulating IGF1 levels and higher cortisol levels at the end of an ad libitum feeding trial reported here. The elevated cortisol level was not correlated with ACTH level in MO offspring suggesting either altered adrenocortical responsiveness to ACTH or cortisol production that we and others have shown in the peripheral tissues [58]. Further, Moore et al. [59] showed an inhibitory role for IGF1 on 11βHSD1 activity the key enzyme that converts cortisone to cortisol. Transgenic mice studies show that 11βHSD1 overexpression causes increased visceral adiposity and profound insulin resistance [60]. In contrast, overexpressing 11βHSD2, the enzyme that inactivates cortisol by converting it to cortisone in peripheral tissues protects against obesity and improves insulin resistance [61]. Protein expression of 11βHSD1 and H6PD were increased in MO offspring vs. CON offspring, while no changes were found in 11βHSD2 expression in the liver. Elevation of H6PD expression in the liver could enhance intracellular NADPH availability that drives 11βHSD1 reductase activity resulting in increased local cortisol production from the conversion of cortisone to cortisol. We suggest that the reduced IGF1 level in MO offspring in this study is associated with enhanced 11βHSD1 activity, thus stimulating increased cortisol production. Taken together, the reduction in IGF1 and increased cortisol levels may explain the elevated adiposity in MO offspring. In conclusion, the data presented here provide evidence for the first time that leptin signaling in the pituitary plays a role in programming of adiposity in offspring born to overnourished/obese mothers.
Supporting information
(PDF)
Acknowledgments
The authors would like to thank Dr. Dennis Hallford, Department of Animal and Range Sciences, New Mexico State University for conducting the radioimmunoassay for growth hormone in his laboratory, and Dr. A.F. Parlow, Pituitary Hormones and Antisera Center, Harbor/UCLA medical Center, Torrance, CA for supplying assay materials. The authors thank the students of the Center for the Study of Fetal Programming for their assistance in animal care and data collection on the farm, and Mr. Adam Uthlaut and Robert Cordery-Cotter, DVM for animal care and management.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by the University of Wyoming National Institutes of Health (NIH) Grant INBRE #P20 RR016474 and NIH Grant 1 R01 HD070096-01A1. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Must A, Spadano J, Coakley EH, Field AE, Colditz G, Dietz WH. The disease burden associated with overweight and obesity. JAMA 1999; 282: 1523–1529. [DOI] [PubMed] [Google Scholar]
- 2.Nelson SM, Matthews P, Poston L. Maternal metabolism and obesity: modifiable determinants of pregnancy outcome. Hum Reprod Update 2010; 16: 255–275. doi: 10.1093/humupd/dmp050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Flegal KM, Caroll MD, Ogden CL, Curtin LR. Prevalence and Trends in Obesity Among US Adults, 1999–2008 (NHANES). JAMA 2010; 303: 235–241. doi: 10.1001/jama.2009.2014 [DOI] [PubMed] [Google Scholar]
- 4.Narayan KM, Boyle JP, Geiss LS, Saaddine JB, Thompson TJ. Impact of recent increase in incidence on future diabetes burden: U.S., 2005–2050. Diabetes Care 2006; 29: 2114–2116. doi: 10.2337/dc06-1136 [DOI] [PubMed] [Google Scholar]
- 5.Whitaker RC, Wright JA, Pepe MS, Seidel KD, Dietz WH. Predicting obesity in young adulthood from childhood and parental obesity. N Engl J Med 1997; 337: 869–873. doi: 10.1056/NEJM199709253371301 [DOI] [PubMed] [Google Scholar]
- 6.Long NM, George LA, Uthlaut AB, Smith DT, Nijland MJ, Nathanielsz PW, et al. Maternal obesity and increased nutrient intake before and during gestation in the ewe results in altered growth, adiposity, and glucose tolerance in adult offspring. J Anim Sci 2010; 88: 3546–3553. doi: 10.2527/jas.2010-3083 [DOI] [PubMed] [Google Scholar]
- 7.McCurdy CE, Bishop JM, Williams SM, Grayson BE, Smith MS, Friedman JE, et al. Maternal high-fat diet triggers lipotoxicity in the fetal livers of nonhuman primates. J Clin Invest 2009; 119: 323–335. doi: 10.1172/JCI32661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Samuelsson AM, Matthews PA, Argenton M, Christie MR, McConnell JM, Jansen EH, et al. Diet-induced obesity in female mice leads to offspring hyperphagia, adiposity, hypertension, and insulin resistance: a novel murine model of developmental programming. Hypertension 2008; 51: 383–392. doi: 10.1161/HYPERTENSIONAHA.107.101477 [DOI] [PubMed] [Google Scholar]
- 9.Bouret SG, Draper SJ, Simerly RB. Trophic action of leptin on hypothalamic neurons that regulate feeding. Science 2004; 304: 108–110. doi: 10.1126/science.1095004 [DOI] [PubMed] [Google Scholar]
- 10.McGarry JD. Appetite control: Does leptin lighten the problem of obesity? Curr Biol 1995; 5: 1342–1344. [DOI] [PubMed] [Google Scholar]
- 11.Mercer JG, Moar KM, Findlay PA, Hoggard N, Adam CL. Association of leptin receptor (OB-Rb), NPY and GLP-1 gene expression in the ovine and murine brainstem. Regul Pept 1998; 75–76: 271–278. [DOI] [PubMed] [Google Scholar]
- 12.Lloyd RV, Jin L, Tsumanuma I, Vidal S, Kovacs K, Horvath E, et al. Leptin and leptin receptor in anterior pituitary function. Pituitary 2001; 4: 33–47. [DOI] [PubMed] [Google Scholar]
- 13.Urbanski HF. Leptin and puberty. Trends Endocrinol Metab 2001; 12: 428–429. [DOI] [PubMed] [Google Scholar]
- 14.Cai A, Hyde JF. The human growth hormone-releasing hormone transgenic mouse as a model of modest obesity: differential changes in leptin receptor (OBR) gene expression in the anterior pituitary and hypothalamus after fasting and OBR localization in somatotrophs. Endocrinology 1999; 140: 3609–3614. doi: 10.1210/endo.140.8.6925 [DOI] [PubMed] [Google Scholar]
- 15.Iqbal J, Pompolo S, Considine RV, Clarke IJ. Localization of leptin receptor-like immunoreactivity in the corticotropes, somatotropes, and gonadotropes in the ovine anterior pituitary. Endocrinology 2000; 141: 1515–1520. doi: 10.1210/endo.141.4.7433 [DOI] [PubMed] [Google Scholar]
- 16.Jin L, Zhang S, Burguera BG, Couce ME, Osamura RY, Kulig E, et al. Leptin and leptin receptor expression in rat and mouse pituitary cells. Endocrinology 2000; 141: 333–339. doi: 10.1210/endo.141.1.7260 [DOI] [PubMed] [Google Scholar]
- 17.Sone M, Nagata H, Takekoshi S, Osamura RY. Expression and localization of leptin receptor in the normal rat pituitary gland. Cell Tissue Res 2001; 305: 351–356. [DOI] [PubMed] [Google Scholar]
- 18.Childs GV, Akhter N, Haney A, Syed M, Odle A, Cozart M, et al. The somatotrope as a metabolic sensor: deletion of leptin receptors causes obesity. Endocrinology 2011; 152: 69–81. doi: 10.1210/en.2010-0498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luque RM, Gahete MD, Cordoba-Chacon J, Childs GV, Kineman RD. Does the pituitary somatotrope play a primary role in regulating GH output in metabolic extremes? Ann N Y Acad Sci 2011; 1220: 82–92. doi: 10.1111/j.1749-6632.2010.05913.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baker J, Liu JP, Robertson EJ, Efstratiadis A. Role of insulin-like growth factors in embryonic and postnatal growth. Cell 1993; 75: 73–82. [PubMed] [Google Scholar]
- 21.Dunger DB, Acerini CL. Does recombinant human insulin-like growth factor-1 have a role in the treatment of diabetes? Diabet Med 1997; 14: 723–731. doi: 10.1002/(SICI)1096-9136(199709)14:9<723::AID-DIA480>3.0.CO;2-S [DOI] [PubMed] [Google Scholar]
- 22.Powell-Braxton L, Hollingshead P, Warburton C, Dowd M, Pitts-Meek S, Dalton D, et al. IGF-I is required for normal embryonic growth in mice. Genes Dev 1993; 7: 2609–2617. [DOI] [PubMed] [Google Scholar]
- 23.Vickers MH, Ikenasio BA, Breier BH. IGF-I treatment reduces hyperphagia, obesity, and hypertension in metabolic disorders induced by fetal programming. Endocrinology 2001; 142: 3964–3973. doi: 10.1210/endo.142.9.8390 [DOI] [PubMed] [Google Scholar]
- 24.Pombo M, Maccario M, Seoane LM, Tovar S, Micic D, Ghigo E, et al. Control and function of the GH-IGF-I axis in obesity. Eat Weight Disord 2001; 6: 22–27. [PubMed] [Google Scholar]
- 25.Scacchi M, Pincelli AI, Cavagnini F. Growth hormone in obesity. Int J Obes Relat Metab Disord 1999; 23: 260–271. [DOI] [PubMed] [Google Scholar]
- 26.Abs R, Mattsson AF, Thunander M, Verhelst J, Góth MI, Wilton P, et al. Prevalence of diabetes mellitus in 6050 hypopituitary patients with adult-onset GH deficiency before GH replacement: a KIMS analysis. Eur J Endocrinol 2013; 168: 297–305. doi: 10.1530/EJE-12-0807 [DOI] [PubMed] [Google Scholar]
- 27.Monson JP. Long-term experience with GH replacement therapy: efficacy and safety. Eur J Endocrinol 2003; 148 (Suppl 2): S9–14. [DOI] [PubMed] [Google Scholar]
- 28.Vickers MH, Ikenasio BA, Breier BH. Adult growth hormone treatment reduces hypertension and obesity induced by an adverse prenatal environment. J Endocrinol 2002; 175: 615–623. [DOI] [PubMed] [Google Scholar]
- 29.Ford SP, Zhang L, Zhu M, Miller MM, Smith DT, Hess BW, et al. Maternal obesity accelerates fetal pancreatic beta-cell but not alpha-cell development in sheep: prenatal consequences. Am J Physiol Regul Integr Comp Physiol 2009; 297: R835–843. doi: 10.1152/ajpregu.00072.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ma Y, Zhu MJ, Zhang L, Hein SM, Nathanielsz PW, Ford SP. Maternal obesity and overnutrition alter fetal growth rate and cotyledonary vascularity and angiogenic factor expression in the ewe. Am J Physiol Regul Integr Comp Physiol 2010; 299: R249–258. doi: 10.1152/ajpregu.00498.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Long NM, Rule DC, Tuersunjiang N, Nathanielsz PW, Ford SP. Maternal obesity in sheep increases fatty acid synthesis, upregulates nutrient transporters, and increases adiposity in adult male offspring after a feeding challenge. PLoS One 2015; e-pub ahead of print 15 April, 2015; doi: 10.1371/journal.pone.0122152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hoefler WC, Hallford DM. Influence of suckling status and type of birth on serum hormone profiles and return to estrus in early postpartum spring-lambing ewes. Theriogenology 1987; 27: 887–895. [DOI] [PubMed] [Google Scholar]
- 33.Catalano PM, Presley L, Minium J, Hauguel-de Mouzon S. Fetuses of obese mothers develop insulin resistance in utero. Diabetes Care 2009; 32: 1076–1080. doi: 10.2337/dc08-2077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fernandez-Twinn DS, Ozanne SE. Mechanisms by which poor early growth programs type-2 diabetes, obesity and the metabolic syndrome. Physiol Behav 2006; 88: 234–243. doi: 10.1016/j.physbeh.2006.05.039 [DOI] [PubMed] [Google Scholar]
- 35.Vickers MH, Breier BH, Cutfield WS, Hofman PL, Gluckman PD. Fetal origins of hyperphagia, obesity, and hypertension and postnatal amplification by hypercaloric nutrition. Am J Physiol Endocrinol Metab 2000; 279: E83–87. [DOI] [PubMed] [Google Scholar]
- 36.Attig L, Larcher T, Gertler A, Abdennebi-Najar L, Djiane J. Postnatal leptin is necessary for maturation of numerous organs in newborn rats. Organogenesis 2011; 7: 88–94. doi: 10.4161/org.7.2.14871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Long NM, Ford SP, Nathanielsz PW. Maternal obesity eliminates the neonatal lamb plasma leptin peak. J Physiol 2011; 589: 1455–1462. doi: 10.1113/jphysiol.2010.201681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vickers MH, Gluckman PD, Coveny AH, Hofman PL, Cutfield WS, Gertler A, et al. Neonatal leptin treatment reverses developmental programming. Endocrinology 2005; 146: 4211–4216. doi: 10.1210/en.2005-0581 [DOI] [PubMed] [Google Scholar]
- 39.Shasa DR, Odhiambo JF, Long NM, Tuersunjiang N, Nathanielsz PW, Ford SP. Multigenerational impact of maternal overnutrition/obesity in the sheep on the neonatal leptin surge in granddaughters. Int J Obes (Lond) 2015; 39: 695–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ahima RS, Prabakaran D, Flier JS. Postnatal leptin surge and regulation of circadian rhythm of leptin by feeding. Implications for energy homeostasis and neuroendocrine function. J Clin Invest 1998; 101: 1020–1027. doi: 10.1172/JCI1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McMillen IC, Edwards LJ, Duffield J, Muhlhausler BS. Regulation of leptin synthesis and secretion before birth: implications for the early programming of adult obesity. Reproduction 2006; 131: 415–427. doi: 10.1530/rep.1.00303 [DOI] [PubMed] [Google Scholar]
- 42.Isozaki O, Tsushima T, Miyakawa M, Demura H, Seki H. Interaction between leptin and growth hormone (GH)/IGF-I axis. Endocr J 1999; 46 (Suppl): S17–24. [DOI] [PubMed] [Google Scholar]
- 43.Popovic V, Damjanovic S, Dieguez C, Casanueva FF. Leptin and the pituitary. Pituitary 2001; 4: 7–14. [DOI] [PubMed] [Google Scholar]
- 44.Luque RM, Park S, Kineman RD. Role of endogenous somatostatin in regulating GH output under basal conditions and in response to metabolic extremes. Mol Cell Endocrinol 2008; 286: 155–168. doi: 10.1016/j.mce.2007.12.005 [DOI] [PubMed] [Google Scholar]
- 45.Allensworth-James ML, Odle A, Haney A, Childs G. Sex differences in somatotrope dependency on leptin receptors in young mice: Ablation of lepr causes severe growth hormone deficiency and abdominal obesity in males. Endocrinol 2015; 156:3253–3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Odle AK, Allensworth-James ML, Akhter N, Syed M, Haney AC, MacNicol M, et al. A sex-dependent, tropic role for leptin in the somatotrope as a regulator of POU1F1 and POU1F1-dependent hormones. Endocrinol 2016; 157: 3958–3971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Agha A, Monson JP. Modulation of glucocorticoid metabolism by the growth hormone—IGF-1 axis. Clin Endocrinol (Oxf) 2007; 66: 459–465. [DOI] [PubMed] [Google Scholar]
- 48.Asada N, Takahashi Y, Wada M, Naito N, Uchida H, Ikeda M, et al. GH induced lipolysis stimulation in 3T3-L1 adipocytes stably expressing hGHR: analysis on signaling pathway and activity of 20K hGH. Mol Cell Endocrinol 2000; 162: 121–129. [DOI] [PubMed] [Google Scholar]
- 49.Ceda GP, Davis RG, Rosenfeld RG, Hoffman AR. The growth hormone (GH)-releasing hormone (GHRH)-GH-somatomedin axis: evidence for rapid inhibition of GHRH-elicited GH release by insulin-like growth factors I and II. Endocrinology 1987; 120: 1658–1662. doi: 10.1210/endo-120-4-1658 [DOI] [PubMed] [Google Scholar]
- 50.Alvarez P, Isidro L, Leal-Cerro A, Casanueva FF, Dieguez C, Cordido F. Effect of withdrawal of somatostatin plus GH-releasing hormone as a stimulus of GH secretion in obesity. Clin Endocrinol (Oxf) 2002; 56: 487–492. [DOI] [PubMed] [Google Scholar]
- 51.Breier BH. Regulation of protein and energy metabolism by the somatotropic axis. Domest Anim Endocrinol 1999; 17: 209–218. [DOI] [PubMed] [Google Scholar]
- 52.Djurhuus C, Gravholt C, Nielsen S, Pedersen S, Moller N, Schmitz O. Additive effects of cortisol and growth hormone on regional and systemic lipolysis in humans. American Journal of Physiology-Endocrinology and Metabolism 2004; 286: E488–E494. doi: 10.1152/ajpendo.00199.2003 [DOI] [PubMed] [Google Scholar]
- 53.Møller N, Jørgensen JO. Effects of growth hormone on glucose, lipid, and protein metabolism in human subjects. Endocr Rev 2009; 30: 152–177. doi: 10.1210/er.2008-0027 [DOI] [PubMed] [Google Scholar]
- 54.Mauras N, O'Brien K, Welch S, Rini A, Helgeson K, Vieira N, et al. Insulin-like growth factor I and growth hormone (GH) treatment in GH-deficient humans: Differential effects on protein, glucose, lipid, and calcium metabolism. Journal of Clinical Endocrinology & Metabolism 2000; 85: 1686–1694. [DOI] [PubMed] [Google Scholar]
- 55.Bredella MA, Lin E, Brick DJ, Gerweck AV, Harrington LM, Torriani M, et al. Effects of GH in women with abdominal adiposity: a 6-month randomized, double-blind, placebo-controlled trial. Eur J Endocrinol 2012; 166: 601–611. doi: 10.1530/EJE-11-1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Misra M, Bredella MA, Tsai P, Mendes N, Miller KK, Klibanski A. Lower growth hormone and higher cortisol are associated with greater visceral adiposity, intramyocellular lipids, and insulin resistance in overweight girls. Am J Physiol Endocrinol Metab 2008; 295: E385–392. doi: 10.1152/ajpendo.00052.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Blum WF, Ranke MB. Insulin-like growth factor binding proteins (IGFBPs) with special reference to IGFBP-3. Acta Paediatr Scand 1990; 367 (Suppl): 55–62. [DOI] [PubMed] [Google Scholar]
- 58.Guo C, Li C, Myatt L, Nathanielsz PW, Sun K. Sexually dimorphic effects of maternal nutrient reduction on expression of genes regulating cortisol metabolism in fetal baboon adipose and liver tissues. Diabetes 2013; 62: 1175–85. doi: 10.2337/db12-0561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moore J, Monson J, Kaltsas G, Putignano P, Wood P, Sheppard M, et al. Modulation of 11 beta-hydroxysteroid dehydrogenase isozymes by growth hormone and insulin-like growth factor: In vivo and in vitro studies. Journal of Clinical Endocrinology & Metabolism 1999; 84: 4172–4177. [DOI] [PubMed] [Google Scholar]
- 60.Masuzaki H, Paterson J, Shinyama H, Morton N, Mullins J, Seckl J, et al. A transgenic model of visceral obesity and the metabolic syndrome. Science 2001; 294: 2166–2170. doi: 10.1126/science.1066285 [DOI] [PubMed] [Google Scholar]
- 61.Kershaw E, Morton N, Dhillon H, Ramage L, Seckl J, Flier J. Adipocyte-specific glucocorticoid inactivation protects against diet-induced obesity. Diabetes 2005; 54: 1023–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.