

Abstract
Chimeric antigen receptor (CAR)-redirected T lymphocytes are a promising immunotherapeutic approach and object of pre-clinical evaluation for the treatment of acute myeloid leukemia (AML). We developed a CAR against CD123, overexpressed on AML blasts and leukemic stem cells. However, potential recognition of low CD123-positive healthy tissues, through the on-target, off-tumor effect, limits safe clinical employment of CAR-redirected T cells. Therefore, we evaluated the effect of context-dependent variables capable of modulating CAR T cell functional profiles, such as CAR binding affinity, CAR expression, and target antigen density. Computational structural biology tools allowed for the design of rational mutations in the anti-CD123 CAR antigen binding domain that altered CAR expression and CAR binding affinity without affecting the overall CAR design. We defined both lytic and activation antigen thresholds, with early cytotoxic activity unaffected by either CAR expression or CAR affinity tuning but later effector functions impaired by low CAR expression. Moreover, the anti-CD123 CAR safety profile was confirmed by lowering CAR binding affinity, corroborating CD123 is a good therapeutic target antigen. Overall, full dissection of these variables offers suitable anti-CD123 CAR design optimization for the treatment of AML.
Keywords: immunotherapy, CAR, AML, affinity, CD123
Graphical Abstract
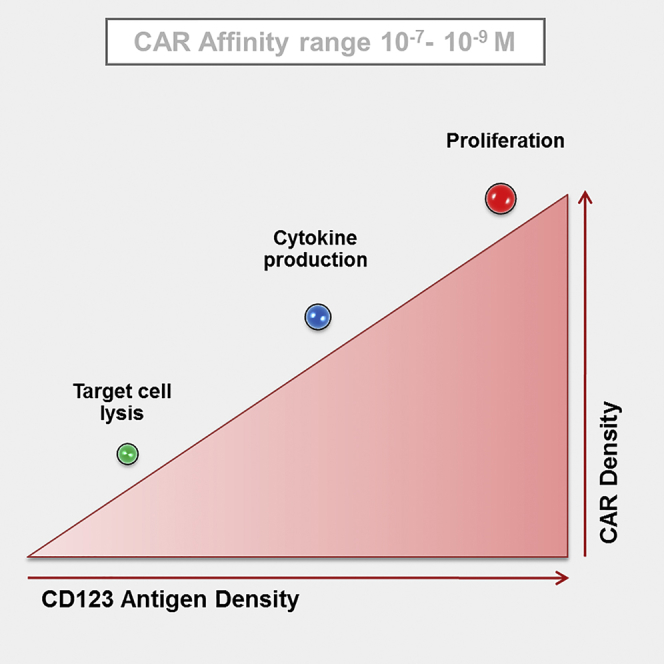
In the context of CAR T cell immunotherapy, a proper balance between safety and efficacy should be evaluated before reaching the clinic. Arcangeli et al. demonstrated how context-dependent variables, i.e., CAR binding affinity, CAR expression, and target antigen density, modulate CAR T cell functionality in the context of AML targeting by an anti-CD123 CAR.
Introduction
Adoptive cellular immunotherapy (ACT) employing T lymphocytes engineered with chimeric antigen receptors (CARs) has demonstrated impressive antitumor effects in relapsed or refractory patients affected by B cell neoplasms,1, 2, 3, 4 providing ACT with the possibility of being translated to other aggressive hematological malignancies, such as acute myeloid leukemia (AML), which is still associated with high relapse rates.5 CARs are artificial molecules comprising an extracellular antigen binding domain, usually derived from a monoclonal antibody (mAb) in the form of a single-chain fragment variable (scFv) and an intracellular signaling moiety, such as the CD3-ζ complex, together with one or more co-stimulatory signaling modules.6, 7, 8
CAR T cell technology is an extremely sensitive approach, because the minimum number of surface target molecules that can be recognized is markedly low compared to the mAb from which the same CAR has been derived due to avidity effects arising from the presence of multiple CARs on the same T cell.9 Therefore, the target antigen choice for CAR-based therapies should be carefully evaluated, because drawbacks have emerged in the clinical translation of CARs. Serious side effects led in the worst cases to the death of the treated patients because of CAR-mediated recognition of low expressed tumor-associated antigens (TAAs) on normal tissues, a problem known as “on target, off tumor.”10, 11 Thus, strategies aimed at ameliorating the CAR-based approach should focus on an off-tumor target antigen expression at very low levels or be limited to healthy tissues whose depletion could be well tolerated by the organism without causing major morbidity (i.e., CD19 targeting in B cell malignancies).
In the context of AML, the disease heterogeneity and similarities between hematopoietic stem cells (HSCs) and leukemia stem cells (LSCs) make the identification of a good target antigen a challenging aspect. In this scenario, our attention has been focused on the interleukin-3 receptor alpha (IL-3RA; CD123), which is a poor-prognosis overexpressed marker on AML blasts and LSCs.12 An anti-CD123 CAR previously generated by our group was able to eradicate AML blasts in vitro and in vivo, showing a safer profile toward normal hematopoietic stem or progenitor cells (HSPCs) compared to anti-CD33 CAR.13, 14 However, a moderate in vitro toxicity was observed toward CD123-low-expressing endothelium and monocytes, suggesting a higher caution level for future clinical translation to avoid potential on-target, off-tumor effects.
Although the exact structural and signaling features for an ideal CAR design are still undefined, it has been demonstrated in several tumor models that it is possible to affect the functional activity of engineered T cells by tuning the CAR-TAA binding properties.15, 16, 17, 18 However, further investigation of CAR binding affinity in a more context-dependent manner is warranted given the differences in each pathological situation in terms of percentage and density of antigen-positive cells, antigen localization, accessibility of the targeted epitope, and CAR design. In a model of anti-CD20 CAR, both a lytic and an activation threshold have been defined as the number of cell surface antigens required to produce full cytotoxic activity and activation or expansion of CAR+ T cells, respectively.9 Therefore, we aimed to unravel how the interplay among CAR affinity, antigen density, and CAR expression could affect anti-CD123 CAR-redirected effector cell efficacy and safety profiles.
We generated and tested anti-CD123 lower-affinity CAR mutants by single-residue substitution on the wild-type (WT) CAR scFv. In this way, the affinity should be the only variable in the system while the same epitope binding site and the overall features within the CAR structure are maintained. Mutation of these residues resulted in lower-affinity antibodies, with Koff (off-rate constant) identical to the original molecule but a slower Kon (on-rate constant). This is expected to decrease CAR sensitivity toward low antigen concentrations, as it does in off-tumor organs with low target antigen expression, thus favoring the recognition of overexpressing tumor cells.
The CAR-redirected effector T cell population has been represented by cytokine-induced killer (CIK) cells, an ex vivo, easily expandable population that is greatly heterogeneous, displaying natural killer-like properties and showing powerful cytolytic activity against both solid and hematological tumors with no graft versus host disease in the allogeneic clinical setting.18, 19, 20 We show that the CAR expression profile can strongly influence CAR-CIK cell effector functions, particularly their later properties, such as proliferation and cytokine production. In addition, our set of anti-CD123 CAR affinity mutants (CAMs) allowed us to define in vitro antigen-specific lytic and activation functional thresholds, influenced by CAR binding kinetics.
Results
Structure-Based Design of CAR Mutants with Lower Affinity for the CD123 Antigen
To explore the role of the sole CAR binding affinity, we used a combination of computational tools and structural analysis to design mutants of the anti-CD123 antibody that would (1) equally recognize the same epitope within the CD123 antigen and (2) have reduced binding affinity due to slower association rates. We first obtained an atomic model of the 7G3 antibody (previously used to design our anti-CD123 CAR)13 and of its complex with the CD123 target antigen by computational simulations, validated by available experimental information regarding the antibody binding site.21, 22 Computational and visual analysis allowed us to identify single antibody residues in the antigen binding loops that, once mutated, could decrease, but not abrogate, the antibody binding. We then produced the mutated antibodies as scFv versions and measured their binding properties for the CD123 antigen with surface plasmon resonance (SPR).
In addition to the WT antibody, four mutants named CAMs (CAR affinity mutants) were selected and incorporated in a CAR to engineer CIK cells for biological characterization. Two antibody mutants had an affinity approximately 10- and 100-fold weaker than the WT antibody (KD: 3 × 10−9 M for WT, 3 × 10−7 M for CAM-L [CAM low affinity], and 3 × 10−8 M for CAM-M [CAM medium affinity]). Two further mutants with binding properties similar to the WT molecule were generated as controls (1 × 10−9 M for CAM-H1 [CAM high affinity mutant 1] and 2 × 10−9 M for CAM-H2 [CAM high affinity mutant 2]). All mutants had Koff rates similar to the WT molecule but different Kon rates (Kon: 1 × 105 M−1s−1 for WT, 8 × 102 M−1s−1 for CAM-L, and 4 × 104 M−1s−1 for CAM-M; Koff: 2 × 10−4 s−1 for WT, 2 × 10−4 s−1 for CAM-L, and 9 × 10−4 s−1 for CAM-M) (Figure 1). The mutations did not alter the binding site and cross-competed with the WT (data not shown).
Figure 1.

Three-Dimensional Model of the Anti-CD123/CD123 Complex and SPR Binding Analysis
(Top) Cartoon visualization of CD123 domain 1 (blue, with the region important for anti-CD123 binding in cyan) and anti-CD123 antibody (heavy and light chain in dark and light gray, respectively). Single mutations, shown as stick, were introduced in the antigen binding loops to affect CAR affinity. (Bottom) SPR sensogram showing the binding of immobilized WT single-chain antibody, CAM-L, and CAM-M to CD123. Raw data are shown in gray, whereas the fit used to calculate the binding properties is in color, with gradation indicating different concentrations. Association, dissociation, and binding affinity are shown.
Reduced CAR Expression Impairs the Anti-CD123 CAR-CIK Cell’s Later Efficacy Profile
The typical CIK cell phenotype was minimally affected by CAR engineering for all tested conditions (WT CAR and CAMs), being comparable to the unmanipulated NO DNA control (Figure S1). The percentage of CAR expression at the end of culture accounted for 61% ± 13%, for WT CAR, 62% ± 14% for CAM-H1, and 20% ± 6% for CAM-H2 (Figure 2A). We also observed a reduction of CAM-H2 mean fluorescence intensity (MFI) in comparison to the other constructs (Figure 2B).
Figure 2.

Later Effector Functions, and Not Early Cytotoxic Profile, Are Impaired in CAR-CIK Cells Displaying Identical Binding Affinity but Reduced Expression
(A and B) CAR expression (A) and MFI values (B) at day 21 of anti-CD123 CAR-CIK cells and NO DNA control. Data represented are the result of mean ± SEM. (A) n = 6 (**p < 0.01; one-way ANOVA, Bonferroni test); (B) n = 9 (**p < 0.01; ***p < 0.001; one-way ANOVA, Dunn’s multiple comparison test). (C–F) Short-term AnnV-7AAD assay by means of the cytotoxic double-target challenge at an effector-target (E:T) ratio of 5:1 after a 4 hr co-culture with both THP-1 and MHH-CALL-4 cell lines (C). Long-term proliferation assay after a 72 hr co-culture of the effector cells with THP-1 and MHH-CALL-4 cell lines (D). Intracellular cytokine staining of IFN-γ (E) and IL-2 (F) after a 5 hr co-culture between effector cells and both THP-1 and MHH-CALL-4 targets. Data represented are the result of mean ± SEM. n = 4 (***p < 0.001; two-way ANOVA, Bonferroni test).
Cytotoxic activity, cytokine production, and proliferation ability of CAR-redirected CIK cells against two cell targets were evaluated: THP-1 (AML cell line, CD123-positive control) and MHH-CALL-4 (B-cell acute lymphoblastic leukemia [B-ALL] cell line, CD123-negative control). We applied a variant to the canonical cytotoxic assay by performing a double-target challenge through co-culture of both target cell lines, together with CAR-redirected CIK cells. This allowed evaluation of their ability to specifically recognize the CD123+ AML cells in the presence of a second, unintended target (negative control or low antigen-positive target cells).
CAR-engineered CIK cell lytic activity against the MHH-CALL-4-negative control was comparable to NO DNA, indicating that the mutations introduced did not render them unspecifically active against a CD123-negative target. Furthermore, CAR-engineered CIK cells showed similar and higher killing ability against the THP-1-positive target, compared to NO DNA, despite the markedly reduced expression of CAM-H2 (Figure 2C). By contrast, upon stimulation with THP-1 target cells, the CAM-H2-CIK cell variant proliferated less than the others, with levels comparable to the NO DNA control (Figure 2D). This suggests that the low expression of CAM-H2, and not the affinity for its target, has a strong impact on later effector functions (Figure 2D).
CAR-redirected CIK cells were also able to produce cytokines, such as interleukin-2 (IL-2) and interferon gamma (IFN-γ), in response to the THP-1 cell line, and not to MHH-CALL-4. However, CAM-H2-CIK cells stimulated with the THP-1 target produced statistically significant lower levels of IFN-γ and IL-2 in comparison to WT- and CAM-H1-redirected CIK cells (Figures 2E and 2F). These results suggest that, at comparable binding affinities (CAM-H1 and CAM-H2), an optimal CAR expression is required for later effector functions, such as proliferation and cytokine production, in contrast to the more immediate cytotoxic activity.
CAR Expression Affects the Anti-CD123 CAR-CIK Cell’s Later Efficacy Compared to CAR Affinity
To characterize the biological effects of the affinity tuning on CAR-CIK cell functional properties, we focused on CAM-L and CAM-M mutants with binding affinity approximately 100- and 10-fold weaker than the WT (KD: 3 × 10−9 M for WT, 3 × 10−8 for CAM-M, and 3 × 10−7 for CAM-L). Both the genetic manipulation and the affinity tuning minimally affected the phenotype of engineered CIK cells, being comparable to the NO DNA control (Figure S2). WT CAR, CAM-H1, and CAM-L had similar expression profiles, whereas both CAR expression and MFI values appeared lower for CAM-M (CAM-M = 41% ± 7%; WT CAR = 70% ± 5%; CAM-H1 = 74% ± 5%; CAM-L = 62% ± 5%) (Figures 3A and 3B).
Figure 3.

Reduced CAR Binding Affinity Does Not Affect CIK Cell Cytotoxic Functions in Response to Highly CD123+ Target Cells
(A and B) CAR expression (A) and MFI values (B) at day 21 of anti-CD123 CAR-CIK cells and NO DNA control. Data represented are the result of mean ± SEM. (A) WT CAR, CAM-H1, and CAM-L: n = 18; CAM-3: n = 7; (B) n = 7 (one-way ANOVA, Bonferroni test). (C–F) AnnV-7AAD assay through double-target challenge after 4 hr co-culture with both THP-1 and MHH-CALL-4 cell lines and primary AML and healthy bone marrow samples (C). E:T ratio 5:1, n = 4. Long-term proliferation assay after the co-culture of the effector cells with THP-1 and MHH-CALL-4 cell lines for 72 hr (D). Intracellular cytokine staining of IFN-γ (E) and IL-2 (F) after 5 hr co-culture between effector cells and THP-1, MHH-CALL-4 targets, primary AML, and healthy BM samples. Data represented are the result of mean ± SEM. WT CAR, CAM-H1, and CAM-L: n = 7; CAM-M: n = 4 (*p < 0.05, ***p < 0.001; two-way ANOVA, Bonferroni test).
The double-target challenge cytotoxic assay showed that CIK cells engineered with the lower-affinity CAM-M and CAM-L receptors maintained significant killing activity for highly CD123+ cells in comparison to NO DNA. All tested CAM-CIK cells showed comparable killing capacity against primary AML cells and no activity toward the co-cultured healthy bone marrow cells (Figure 3C). Similarly, the highly CD123-positive THP-1 cells were lysed by the engineered CIK cells, whereas the co-cultured CD123-negative MHH-CALL-4 cells were not.
The proliferation assay confirmed CAM-L- and CAM-M-redirected CIK cell specificity and ability to respond to the high CD123+ target compared to the negative control. If CAM-L had a response comparable to WT- and CAM-H1-CIK cells, proliferation induced by CAM-M was lower. This highlights that CAR expression profile is more relevant than affinity in determining later effector functions (Figure 3D). CAM-L expression appeared sufficiently adequate to grant redirected CIK cells a proliferative advantage with respect to CAM-M and CAM-H2, even if its binding affinity is one and two orders of magnitude lower, respectively.
The cytokine production profile after stimulation with CD123+ target cells further showed that CAM-M-CIK cells, like CAM-H2, produced significantly less IFN-γ and IL-2 than the other constructs (Figures 3E and 3F). Cytokine production was more abundant in the presence of CAM-L, strengthening the more prominent role of CAR expression over target affinity for later cellular functions. All these assays confirmed that CAM-L is equally effective in redirecting CIK cells as WT CAR and CAM-H1, despite having a 100-fold lower binding affinity for its target antigen.
We also measured the killing of CD123-positive cells over time in a long-term cytotoxic assay for 1 week (Figure S3A). CAM-L-, WT CAR-, and CAM-H1-CIK cells were all found to be effective in killing THP-1 cells, while sparing CD123-negative MHH-CALL-4 cells, in comparison to NO DNA. Altogether, the preceding observations indicate that a 10−7 M binding affinity for the target antigen is sufficient to ensure significant and robust functional responses in vitro against high CD123+ leukemic conditions in both THP-1 and primary AML cells.
Lowering CAR Binding Affinity Confirms CD123 as a Safe Target Antigen for CAR-CIK Cell Approach
With the aim of studying the role played by CAR binding affinity in the recognition of cells with lower CD123 expression, we investigated the functional effects of CAM-L-CIK cells on cell lines with a progressive CD123 expression and density (Figure S4). We chose THP-1 (97% of CD123+ cells, with 7,435 ± 1,986 CD123 molecules/cell) and KG-1 (81% of CD123+ cells, with 4,550 ± 559 CD123 molecules/cell) AML cell lines as high CD123+ target cells. The AML U937 cell line and healthy telomerase immortalized human microvascular endothelium (TIME) cells were representative of CD123 low-expressing cells, with a CD123 expression of 13% for U937 and 9% for TIME and comparable numbers of CD123 surface molecules (U937 = 1,603 ± 215; TIME = 1,688 ± 328).
As before, we confirmed both the efficacy and the specificity of CAR-CIK cells when co-cultured with CD123+ THP-1 and CD123− MHH-CALL-4 (Figure 4A). Co-culture of the THP-1 cell line with low CD123-positive conditions showed preferential killing of THP-1 cells (Figures 4B and 4C), highlighting that the presence of a second target, low CD123+, does not inhibit the killing of a first intended highly CD123+ target. Although CAM-L-CIK cells were as effective as WT- and CAM-H1-CIK cells in killing highly CD123+ cells, they showed a trend of lower killing activity in comparison to the other constructs when challenged with low CD123+ cells (Figure 4D).
Figure 4.

Short-Term Cytotoxic Double-Target Challenge
AnnV-7AAD assay, E:T ratio 5:1, (A) THP-1/MHH-CALL-4 (n = 5; CAM-H1: n = 4), (B) THP-1/U937 (n = 5; CAM-H1: n = 4), (C) THP-1/TIME (n = 6), (D) U937/MHH-CALL-4 (n = 5; CAM-H1: n = 4), and (E) TIME/MHH-CALL-4 (n = 6) double-target combinations. Data represented are the result of mean ± SEM (*p < 0.05; **p < 0.01; ***p < 0.001; two-way ANOVA, Bonferroni test).
To characterize the safety profile of CAM-L-CIK cells against the TIME healthy tissue, we also performed a 72 hr long-term cytotoxicity assay (Figure S3B). Results supported the observation that CAM-L-CIK cells had cytotoxic effects against THP-1 cells, but not against the MHH-CALL-4-negative control or the TIME endothelial cells. This strengthens the idea that a mutant with 10−7 M affinity, 100-fold lower than the WT, is able to retain an optimal effector profile, carrying a safe profile against the healthy tissue.
Furthermore, because the killing activity exerted by CAR-CIK cells against the low CD123+ cells was higher than the negative control, but significantly lower compared to the high CD123+ leukemic target, it appeared that the presence of approximately 1,600 CD123 molecules/cell is sufficient to induce the activation of the lytic cell machinery by CAR-redirected CIK cells, in line with the presence of a lytic threshold suggested by other literature.9
By contrast, no proliferation activity was detected against the low CD123+ cells (Figure 5A), while there was a strong and equal proliferative response obtained against the high CD123+ targets for all CARs studied. According to these findings, ∼1,600 CD123 molecules are sufficient to trigger the lytic activity, but not to induce the proliferation of CAR-redirected CIK cells.
Figure 5.

Target Antigen Density, and Not CAR Binding Affinity, Affects Later Effector Functions
(A) Long-term proliferation assay after the co-culture of the effector cells with the indicated cell lines for 72 hr. (B–E) Intracellular cytokine staining of IFN-γ (B), TNF-α (C), IL-2 (D), and IL-6 (E). Effector cells were co-cultured for 5 hr with THP-1, KG-1, U937, TIME, and MHH-CALL-4 targets. Data represented are the result of mean ± SEM. n = 12 (ALONE condition); n = 9 (THP-1, U937, and TIME experimental cell conditions); n = 3 for all CARs tested against the KG-1 cell line (*p < 0.05; **p < 0.01; ***p < 0.001; two-way ANOVA, Bonferroni test).
Concerning the cytokine production, CAR-CIK cell sensitivity to the targets appeared to be clearly influenced by their CD123 positivity. A hierarchical cytokine production, from the highly CD123+ THP-1 cells to the lower and lowest CD123 levels of KG-1 and U937 cells, was found. This was particularly evident in the case of IFN-γ and IL-2 production. Instead, a significant amount of tumor necrosis factor alpha (TNF-α) and interleukin-6 (IL-6) was only produced in the presence of THP-1 cells (Figure 5). No activation was induced against the CD123-negative control and the healthy tissue by all CAR-CIK cells tested and for all cytokines analyzed.
Moreover, with the aim of evaluating the response of anti-CD123 CAR-redirected CIK cells against the endothelium in a more physiological context, we set up a co-culture of Matrigel-embedded endothelial cells and CIK cells. Endothelial TIME cells are known to undergo tubule formation when cultured on Matrigel.23 Thus, the impact on vessel formation induced by unmodified and CAR-redirected CIK cells was monitored after 4 hr co-culture. Branching points were counted to have a measure of the potential impairment on endothelial network stability and spreading. The same and limited impairment of vessel formation was observed, with no differences encountered between NO DNA and CAR-redirected CIK cells (Figures 6A and 6B). Therefore, in this model of endothelial vessel formation, anti-CD123 CAR CIK cells were not activated more than the unmanipulated counterpart, independently of CAR binding affinity.
Figure 6.

TIME Cell Vessels Are Not Hampered by CAR-CIK Cells
(A) Representative 5× and 10× magnifications of TIME cell vessels after a 4 hr culture on Matrigel (TIME cells alone) or with NO DNA and CAR-CIK cells (WT CAR and CAM-L), respectively. (B) Analysis of TIME cell branching point number, alone or after a 4 hr co-culture with unmanipulated and CAR-CIK cells, as a measure of TIME cell tissue integrity and vessel spreading. Data represented are the result of mean ± SEM. n = 3 (***p < 0.001; one-way ANOVA, Bonferroni test).
Later Effector Functions, and Not Early Cytotoxic Activity, Are Proportional to CAR Downmodulation
After antigen engagement by T cells with canonical T cell receptors (TCRs), a serial triggering process leads to the internalization of surface TCR molecules until the activation threshold is overcome. This process is due to the low affinity of TCRs, which can be sequentially engaged and thus serially downmodulated. According to this model, the signaling strength is proportional to the rate of receptor internalization and thus to the potency of T cell activation.24, 25, 26, 27, 28 Similarly, CAR+ T cell activation can be described as a specific case of TCR-antigen engagement in which serial CAR triggering is abrogated due to the high affinity of CARs compared to TCRs.29 Therefore, to better understand the mechanisms at the basis of the lower CIK cell functional later responses encountered with both CAM-M and CAM-H2 receptors, we analyzed the CAR downmodulation in relation to both later cellular responses and lytic activity.
The amount of CAR downmodulation was found to be independent of binding affinity for the target antigen. CAM-L, for instance, had the same amount of downmodulation as WT CAR and CAM-H1, despite a 100-fold lower binding affinity. By contrast, the lower-expressing CAMs, CAM-M and CAM-H2, had progressively lower amounts of downmodulation and lower IFN-γ production while having higher affinity than CAM-L (Figures 7A and 7B).
Figure 7.

CAR Downmodulation in CAR-CIK Cells
(A and B) Percentage of CAR-CIK cell downmodulation (A) and percentage of IFN-γ production by CAR-CIK cells (B) in response to THP-1 cells. (A) WT CAR, CAM-H1, and CAM-L: n = 11; CAM-M: n = 9; CAM-H2: n = 7 (*p < 0.05, **p < 0.01, ***p < 0.001; one-way ANOVA, Bonferroni test). (B) WT CAR, CAM-H1, and CAM-L: n = 11; CAM-H2 and CAM-M: n = 4. (C) Plot of CAR downmodulation as a function of killing activity against both THP-1 and MHH-CALL-4 targets. (D) CAR-CIK cell downmodulation as a function of target antigen density. Data represented are the result of mean ± SEM, n = 11 for the stimulation with MHH-CALL-4, TIME, U937, and THP-1 cell lines; n = 7 for the stimulation with KG-1 cells.
By plotting the percentage of the killing activity detected against THP-1 cells as a function of CAR downmodulation, we found no apparent relation between the cytotoxic activity on highly CD123-positive THP-1 cell lines and the amount of CAR downmodulation. The killing response of CAM-M- and CAM-H2-CIK cells was similar to that of the other CARs, even with a lower CAR downmodulation pattern (Figure 7C).
The amount of CAR downmodulation was found to be related to CD123 antigen density (Figure 7D), accounting for a different strength of CAR+ T cell activation and thus functional related responses (Figures 5 and 7D). Increasing numbers of CD123 molecules per cell lead to a larger amount of CAR downmodulation, resulting in escalating CAR+ T cell activation. The results are comparable among WT CAR, CAM-H1, and CAM-L and thus independent of binding affinity, reflecting the hierarchical trend previously observed with cytokine production.
Discussion
In search for a CAR-mediated targeting optimization, we investigated the effect of three of the main variables affecting CAR T cell activity: (1) CAR binding affinity for the target antigen, reported to modulate CAR T cell effector functions;15, 16, 17, 18 (2) CAR T cell expression levels; and (3) target antigen density.
Anti-CD123 CAMs were rationally designed on the basis of computational and structural investigation of CD123 antigen-anti-CD123 antibody interaction to produce antibody mutants with slower association rate in comparison to the WT. Experimental SPR analysis confirmed that two anti-CD123 mutants had 100-fold (CAM-L) and 10-fold (CAM-M) lower affinity in comparison to the WT. Introduction of single amino acid substitutions in the antigen binding loops allows evaluating the real effect of CAR affinity tuning without simply comparing antibodies targeting different binding epitopes.
Altogether, computational simulations allowed rapid and inexpensive design of a limited set of mutants, bypassing the need to generate and screen large number of mutations in randomized sequence searches. This integrated approach connects the biochemical aspects of in silico selection of antibody mutants with the biological relevance given by their subsequent experimental validation.
We found that all CAMs were specific for CD123+ AML cells, including AML primary samples. Unlike other published contexts,15, 16, 18, 30 even the weaker binders CAM-L and CAM-M showed enhanced redirected-CIK cell effector properties. However, CAM-H2 and CAM-M had lower CAR expression and MFI, which strongly affected the later functional cell properties, independently of CAR binding affinity. CAM-M-CIK cells showed lower proliferation and cytokine production than CAM-L-CIK cells, despite a 10-fold higher binding affinity. At the same time, the low CAM-M- and CAM-H2-CAR expression profile was not a limiting factor in determining a powerful cytotoxic activity against a highly CD123+ leukemic target, similar to WT. These results are in sharp contrast with previous findings suggesting the presence of a functional balance between TAA densities and CAR expression profiles. Accordingly, a lower CAR density was equally able to induce both target cell lysis and production of pro-inflammatory cytokines against a high TAA+ target cell.31
In our model, a CAR expression ceiling of ∼40% CAR+ cells, together with lower CAR MFI values, was observed. Below this threshold, no satisfactory cell activation was detected, even in the presence of high CAR affinity and high TAA density, leading to decreased later cellular effector functions.
We then investigated the effect of CAR binding affinity and target antigen density on the efficacy and safety profiles of CAR-redirected CIK cells. No reduction of cytotoxic activity against a first intended target (THP-1), highly CD123+, in the presence of a second unintended target (U937-AML or TIME-endothelium), low CD123+, was detected. This is in contrast to what observed in an anti-CD20 CAR model described by James et al.29
A value of approximately 1,600 CD123 molecules/cell (in U937 and TIME) was found sufficient to produce a detectable lytic activity, albeit significantly lower than the response to highly CD123+ THP-1 cells. Watanabe et al. described this feature as the CAR lytic threshold.9 The double-target challenge cytotoxic assay, which is to our knowledge the first attempt in challenging CAR T cells with multiple targets, allowed us to better assess this threshold.
Moreover, CAM-L-CIK cells were as able to kill THP-1 cells as WT CAR- and CAM-H1-CIK cells, despite having a 100-fold lower binding affinity. Therefore, binding affinity above 10−7 M is sufficient to achieve a satisfactory CAR T cell lytic activation. In contrast, CAM-L showed a trend of inferior killing levels in comparison to CAM-H1- and WT CAR-redirected cells, against the low CD123+ U937 and TIME cells, pointing out that 10−7 M binding affinity could offer a better safety profile.
In a model of anti-ErbB2 targeting, Chmielewski et al. identified an affinity ceiling of 10−8 M, below which there is no additional improvement of receptor-mediated cellular activation.16 However, differences in CAR affinities arose from the dissociation rate. In our case, by contrast, the lower affinity depends on the association rate. This could allow a long-lasting interaction between CAR and antigen, which might be important to induce potent CAR-CIK cell later effector functions.
Furthermore, we found later T cell effector properties being affected by target antigen density. An antigen escalation seems to govern CAR-CIK cell-dependent cytokine production, particularly concerning IFN-γ and IL-2. In contrast, CAR-CIK cell proliferative capability was no longer sensitive to an antigen density above ∼5,000 antigen molecules/cell. We observed that the triggering of cytokine production and T cell proliferation depends on different antigen thresholds, whose definition is of fundamental importance to propose proper and specific target antigens in any CAR-based adoptive cell immunotherapy approach.
Caruso et al. proposed the possibility of limiting the recognition of healthy tissues by lowering anti-epidermal growth factor receptor (EGFR) CAR binding affinity. However, similar later effector functions by low-affinity CARs were detected against the low EGFR+ healthy tissue (∼15,000 molecules/cell) and the U87 tumor cell line (engineered with ∼30,000 antigens/cell). Thus, the lytic and activation thresholds might have been exceeded,30 limiting the proper interpretation of a potential on-target, off-tumor effect.
Anti-CD123 CAR-CIK cells could recognize healthy targets, such as HSPCs, monocytes, and endothelium, reaching the antigen activation thresholds. In a previous publication, we employed colony assays and in vivo secondary transplantation experiments to demonstrate that anti-CD123 WT CAR was able to spare the different CD34 subpopulations at levels similar to the unmanipulated CIK cells and was safer than an anti-CD33 CAR.13, 14 In the present work, we performed cytotoxic assays against healthy bone marrow (BM) cells and observed that the CAM mutants had the same low toxicity as the previously characterized anti-CD123 WT CAR (Figure 3C), strongly suggesting that we are in a safe window in regards to the potential on-target, off-tumor effect against the normal hematopoietic compartment. Therefore, we investigated CAR affinity-tuned targeting of low CD123+ endothelium by co-culturing differentiated endothelial cells and CIK cells on Matrigel. The antigen presentation within the fully differentiated endothelial network has been shown to be different from monolayer plating on plastic wells.23 Same and limited impairment of vessel formation was found, with no differences between NO DNA and CAR-CIK cells, suggesting lower CD123 antigen detection in this more physiological context. The lack of homology of human and mouse CD123 molecules represents the major limitation for performing in vivo experiments on safety profile evaluation. Gene-editing approaches might deserve further consideration to generate a humanized mouse model to confirm the safety profile of our CD123 CAR mutants against the endothelial tissue and are under evaluation for future studies.
TCR downmodulation was reported to be proportional to antigen-TCR binding strength and thus to T cell activation.24, 25, 26, 27, 28 Similarly, CAR-antigen engagement can trigger CAR internalization at a specific rate, leading to reduced CAR availability on the engineered T cell surface.29, 30 We noted that CAR affinity has no impact on CAR downmodulation when CAR-CIK cells are challenged with highly antigen-positive THP-1 cells. The lytic activity is not related to CAR downmodulation, because equal killing activity was detected for mutants with different downmodulation levels. Instead, mutants with higher downmodulation appeared to have increased IFN-γ production, strengthening the idea that downmodulation is mainly responsible for later effector functions rather than early lytic activity.
Finally, there was a clear correlation between antigen density and amount of CAR downmodulation, suggesting that the number of CARs internalized (downmodulation) depends on the number of interactions with antigen molecules. According to this functional profile, there would be a threshold of CAR downmodulation able to trigger the cytotoxic activity of CAR T cells. Internalization of CARs above such threshold would represent accessory signaling events that can be required to sustain different and later effector functions.
In conclusion, low CAR expression levels resulted in impaired CAR-CIK cell activity, especially regarding later functions. This seems to arise from decreased CAR internalization or downmodulation levels upon antigen binding, which has no impact on early lytic activity but leads to decreased cytokine production. As a consequence, when designing a new CAR within the specific tumor context, efforts should be directed toward the screening of optimal CAR expression profiles, and not exclusively toward the hunt for a high-affinity antibody-derived CAR. CIK cells redirected with the lowest-affinity mutant showed a trend of decreased killing of low CD123+ targets and were as active as high-affinity CAR-CIK cells against highly antigen-positive cells. In conclusion, a combination of computational structural biology and cellular assays allowed us to characterize the role and interplay of CAR binding affinity and CAR expression in the efficacy and safety profiles of anti-CD123 CAR-redirected CIK cells toward AML cells and healthy tissues, expressing different target antigen levels. Overall, the pre-clinical identification of a proper balance between efficacy and safety of CAR therapy will help the improvement of the therapeutic index of such innovative treatments, particularly when dealing with highly aggressive malignancies not responsive to available standard treatments.
Materials and Methods
Cell Lines and AML Primary Cells
THP-1, U937, KG-1 (AML cell lines), MHH-CALL-4 (B-ALL cell line), and TIME (ATCC CRL-4025, dermal micro-vascular endothelial cell line) have been obtained from the American Type Culture Collection (ATCC). The THP-1, U937, and KG-1 cell lines were maintained in culture with Advanced RPMI medium (Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 2 mM L-glutamine, 25 IU/mL of penicillin and 25 mg/mL of streptomycin (Lonza). The MHH-CALL-4 cell line was maintained in culture with 20% FBS Advanced RPMI complete medium. The TIME cell line was maintained in culture with Vascular Cell Basal Medium (ATCC), supplemented with the Microvascular Endothelial Cell Growth Kit-VEGF, containing several purified human recombinant (rh) growth factors (rh_VEGF [vascular endothelial growth factor], rh_EGF [epidermal growth factor], rh_FGF [fibroblast growth factor] basic, and rh IGF-1 [insulin growth factor 1]) and combined with 10 mM L-glutamine, 0.75 U/mL of heparin sulfate, 1 μg/mL of hydrocortisone hemisuccinate, 5% FBS, and 50 μg/mL of ascorbic acid (ATCC).
Bone marrow and peripheral blood cells were collected from non-leukemic controls and children with AML at diagnosis. The Institutional Review Board approved this study, and informed consent was obtained from patients or their guardians.
Protein Modeling of Anti-CD123 and CD123
Structural analysis of anti-CD123 antibody in complex with its antigen was performed using a computational approach. CD123 experimental structure is available at PDB: 4JZJ, while anti-CD123 antibody was obtained using RosettaAntibody software. The templates are coded as follows (sequence identity is indicated in parentheses)—PDB: 1MVU for the light-chain framework (96.77%), PDB: 1MJ8 for the heavy-chain framework (91%); PDB: 1MVU for L1 (100%) and for L3 (88.89%), PDB: 1Q9R for L2 (100%), PDB: 1EGJ for H1 (100%) and for H2 (82.35%), and PDB: 12E8 for H3 (same length, no identity). The best 10 of 2,100 antibody models were selected because of their RosettaAntibody algorithm score.
Computational Modeling of Anti-CD123-CD123 Complex
Complex models were predicted using Rosetta Docking 2.3, a computational docking software. The starting structures were visually oriented with the antibody (Ab) complementarity-determining region (CDR) loops facing the N-terminal part of the CD123 protein, because it is known that domain 1 of CD123 is fundamental for anti-CD123 binding,32 and then separated by 25 Å. Docking runs of ten antibody models with its antigen were conducted as previously described.33 Best models of each simulation, selected because of their energetic score and by visual analysis after discarding complexes with an interface far from the first 30 residues of N-terminal CD123, were then subjected to clustering and contact map analysis. This allowed us to select residues showing interaction in several models likely to be important in mediating binding.
Antigen, Antibody, and Variant Protein Production
The human CD123 domain 1+2 nucleotide sequence was cloned in frame into pET21a plasmid and expressed using the E. coli system (Rosetta DE2 cells). Single-clone cells were grown in Luria-Bertani broth ampicillin+cyclohexylammonium salt+ (LB AMP+CHA+) media until 0.6 optical density (OD) and then induced with 1 mM isopropil-β-D-1-tiogalattopiranoside (IPTG) and harvested after 3 hr. CD123 domain 1+2 was found in the insoluble fraction, so the protein pellet was washed and solubilized using mild-denaturing buffer (100 mM Tris [pH 12.5], 2 M urea, 5 mM β-mercaptoethanol [B-ME]). Protein was loaded into an anion exchange column (HiPrep Q FF 16/10, GE Healthcare) pre-equilibrated with solubilization buffer and eluted with NaCl gradient, starting from 0 to 1M. Fractions containing CD123 1+2 protein were then loaded into size exclusion column (HiLoad 16/60 Superdex 75, GE Healthcare) pre-equilibrated with 50 mM Tris (pH 8.5), 150 mM NaCl, and 1% PEG3350. CD123 domain 1+2 elutes at 68 mL according to its monomeric molecular weight.
The anti-CD123 single-chain antibody nucleotide sequence was cloned in frame into pET21a plasmid and expressed using the E. coli system (Rosetta DE2 cells). Single-clone cells were grown in LB AMP+CHA+ media until 0.6 OD and then induced with 1 mM IPTG and harvested after 3 hr. Single-chain antibody was found in the insoluble fraction, so the protein pellet was washed and solubilized using denaturing buffer (50 mM MES [pH 6.5], 1 M NaCl, 6 M guanidinium-HCl). Protein was loaded into a HiTrap column (GE Healthcare) pre-equilibrated with solubilization buffer and eluted with same buffer plus 500 mM imidazole. Fractions containing antibody were refolded using direct dilution into 20 mM NaP (pH 10), 150 mM NaCl, 200 mM arginine, and 1 mM Glut Red 0.1 Glut Ox. Protein was concentrated by centrifugation (2,000 rpm, 4°C) with 10 kDa Vivaspin (Sartorius) and loaded on a size exclusion column (HiLoad 16/60 Superdex 75, GE Healthcare) pre-equilibrated with 50 mM Tris (pH 9) and 50 mM NaCl. Anti-CD123 elutes at 64 mL according to its monomeric molecular weight.
Mutagenesis of Anti-CD123 Single-Chain Ab
Mutagenesis was performed using the QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent Technologies). Specific mutated primers were synthesized by Microsynth and used to generate protein variants.
Site-directed mutagenesis PCRs were performed according to the protocol kit and following the cycling parameters listed below. After the PCR reaction, the mixtures were treated with DpnI restriction enzyme at 37°C for 5 min to remove the parental (i.e., the nonmutated), supercoiled, double-stranded DNA (dsDNA). The DpnI-treated DNAs (mutated plasmids) were used to transform XL10-Gold Ultracompetent Cells; the colonies obtained from the transformations were used for DNA amplification and extraction with the QIAGEN Maxi or Mini Prep Kit.
The sequence of each mutated plasmid was verified by DNA sequencing (Microsynth); the verified plasmids were used for protein expression and stored in aliquots at −80°C. Antibody variants were tested for protein purification, showing no difference in yield and stability in comparison to WT.
PCR cycling parameters were as follows:
SPR
SPR experiments were performed to validate computational results. The scFvs were immobilized on the surface of a GLC chip (a thin alginate layer for amine coupling) at 500 nM in 10 mM NaOAc (pH 4.0). CD123 domain 1+2 was used as analyte (protein and running buffer: 20 mM HEPES (pH 7.4), 150 mM NaCl, 3 mM EDTA, 0.005% Tween 20). The injection of the antigen spanned a concentration range between 200–12.5 nM at flow rate of 70 μL/min. Data were fit using the Langmuir equation.
Transposons Plasmids
The WT anti-CD123/pTMNDU3 Sleeping Beauty (SB) transposon expresses the human third-generation anti-CD123-CD28-OX40-CD3z CAR under pTMNDU3 promoter. The construct has been derived as a SB expression plasmid, replacing the EGFP sequence from pT-MNDU3-EGFP with the scFv CD123 (7G3 clone) previously cloned in frame with CH2CH3-CD28-OX40-ζ from SFG.aGD2 (provided by Dr. Martin Pule, University College of London). The DNA sequences of each anti-CD123 affinity mutant scFv were cloned in place of the anti-CD123 WT scFv. The plasmid pCMV-SB11 encodes for the SB11X transposase (from the University of Minnesota).
Generation of CIK Cells Genetically Modified for the Expression of the Anti-CD123 CARs
CIK cells were generated starting from peripheral blood mononuclear cells (PBMCs) from healthy subjects, obtained after centrifugation of fresh blood on a density gradient using Ficoll-Hypaque (Pharmacia). Once collected, PBMCs were resuspended in Amaxa Nucleofector solution, provided with the P3 Primary Cell 4D-Nuceofector X kit (Lonza), together with SB11X transposase and DNA plasmid encoding for one of the anti-CD123 CAR mutants, and were transfected using the 4D-Amaxa Nucleofector device (Lonza). Our controls included unmodified cells, which were nucleofected with no DNA, and EGFP episomal transfected cells (with 2 μg of Amaxa control plasmid) to assess the gene transfer efficiency. After nucleofection, the cells were transferred into a 6-well plate containing 4 mL of pre-warmed medium (advanced RPMI supplemented with 20% of heat-inactivated FBS and 1% of L-glutamine), and 1,000 U/mL of IFN-γ (Dompè Biotec) were added to each well.34 Twenty-four hours later, IL-2 (Chiron) and OKT-3 (Janssen-Cilag) were added at 300 U/mL and at 50 ng/mL, respectively. Fresh medium and IL-2 were added twice a week, and cell concentration was maintained around 0.75 × 106 cells/mL. Cells were then cultured for 21 days.
Flow Cytometry
Immunostaining and flow cytometric analysis were performed with the following antibodies: allophycocyanin (APC)-anti-CD123 (Becton Dickinson [BD]), phycoerythrin (PE)-anti-CD123 (BD), v-500-anti-CD45 (BD), phycoerythrin-cyanine 7 (Pe-Cy7)-anti-CD34 (BD), PE-anti-CD38 (BD), peridinin-chlorophyll-protein complex (PerCP)-anti-CD3, PE-anti-CD56 (BD), fluorescein isothiocyanate (FITC)-anti-CD8 (BD), PE-anti-CD4 (BD), PE-anti-CD62L (BD), FITC-anti-CD45RO (BD), Alexa Fluor 647-F(ab′)2-anti-immunoglobulin G (IgG) (H+L) (anti-Fc), PE-anti-IL-2, FITC-anti-IFN-γ, Pe-Cy7-anti-TNF-α, Horizon BV421-anti-IL-6, Horizon BV421-anti-Ki-67, FITC-anti-CD19 (BD), and PE-anti-CD144 (BD). Cell death and apoptosis were detected using the GFP-Certified Apoptosis/Necrosis detection kit (Enzo Life Sciences), according to the manufacturer’s instructions. Cell membrane labeling was also performed using two lipophilic fluorescent dyes: FITC- and PE-Cell Tracker (Invitrogen).
To quantify the number of CD123 molecules on the surface of the target cell lines and primary AML samples, we used the QuantiBRITE PE fluorescence quantitation kit (BD), which allows the conversion of cell fluorescence intensity values into absolute numbers of receptors per cell through the creation of a calibration curve.35 Flow cytometry was performed on a FACSCanto II flow cytometer (BD), and data were analyzed using BD FACSDiva software v.6.1.3.
Short- and Long-Term Cytotoxicity Assays
To evaluate the killing ability of both unmodified and CAR-redirected CIK cells, short- and long-term cytotoxicity assays were performed. In the short-term cytotoxic assay assessed by means of the double-target challenge, CIK cells were co-cultured for 4 hr with the CD123-positive targets (THP-1, primary AML cells, U937, and TIME cell line), together with the CD123-negative control (MHH-CALL-4 cell line), at an effector-target (E:T) ratio of 5:1. Target cells were previously labeled with FITC- and PE-Cell Trackers. At the end of the incubation, target cell killing was measured through apoptosis detection by flow cytometry, after annexin V and 7-amino-actinomycin D (7-AAD) (AnnV-7AAD) staining, gating in the Cell Tracker PE+ and FITC+ cells. The percentage of killed cells was calculated according to the following formula.
Two long-term cytotoxicity assays were conducted at an E:T ratio of 1:5. The first was performed by co-culturing CIK cells with THP-1 and MHH-CALL-4 cell lines for 1 week, and the second was performed by co-culturing CIK cells with THP-1, MHH-CALL-4, and TIME cell lines for 72 hr. Target cell survival was then evaluated after surface staining with PerCP-anti-CD3 (BD) antibody, to detect CIK cells, and by comprising the following antibodies: APC-anti-CD123 (BD), FITC-anti-CD19 (BD), and PE-anti-CD144 (BD), for THP-1, MHH-CALL-4, and TIME cell line detection, respectively. The percentage of target cell survival was calculated according to the following formula.
Proliferation Assay
The proliferation ability of CAR-CIK cells was evaluated after co-culture with the various Cell Tracker-labeled targets, irradiated at 100 Gy γ-radiations at an E:T ratio of 1:1. After a 72 hr co-culture, the cells were collected, immunostained for intracellular Ki-67, and then analyzed by flow cytometry by performing detection of Cell Tracker negative-Ki-67+ cells.
Intracellular Cytokine Staining
CAR-CIK cell ability to produce cytokines was evaluated following a stimulation with the various target cell conditions at an E:T ratio of 1:3. After a 2 hr and 30 min co-culture, BD GolgiStop was added. The co-culture was then maintained for an additional period of 2 hr and 30 min, after which the cells were collected and stained for anti-CD3 and anti-Fc surface molecule detection. Finally, intracellular cytokine staining (ICS) for IL-2, IFN-γ, IL-6, and TNF-α was performed using the BD Cytofix/Cytoperm kit, according to the manufacturer’s protocol. Specimens were then analyzed by flow cytometry.
Matrigel-Embedded Endothelium and CIK Cell Assay
250 μL of Matrigel (Corning) were plated on the surface of a 24-well plate and allowed to solidify by incubation at 37°C for 30 min. Subsequently, a number of 60,000 TIME cells and CIK cells (both unmodified and CAR redirected) were plated together, at an E:T ratio of 1:1, and left for additional 2 hr at 37°C.
After this time interval, real-time monitoring of vessel formation was performed by microscope observation to assess the state of tubule formation of each sample, duplicated in every independent experiment. Four hours after plating, picture acquisition was performed by collecting nine pictures per well at 10× magnification. Branching point count through ImageJ software was then performed in each of the nine pictures per well to obtain a sufficient representation of the entire well surface.
CAR Downmodulation Quantitation
Following the ICS protocol previously described, after the CIK cell surface staining of CAR expression, we analyzed the CAR MFI values with no stimulation and in response to different targets. The CAR downmodulation percentage was calculated according to the following formula, as described by James et al.29
Statistical Analysis
Data were analyzed using GraphPad Prism 5 software (GraphPad). One-way ANOVA and two-way ANOVA statistical tests were performed for column statistics and grouped statistics, respectively, using no matching and repeated-measures criteria according to the type of dataset analyzed and presence or absence of matched values, as indicated in the figure legends. Reported values of the statistical analyses are the result of the evaluation of mean ± SEM. All p values are provided in the figure legends.
Author Contributions
S.A., M.C.R., M.B., and S.T. designed the research, performed the experiments, analyzed the data, and wrote the paper. L.S. contributed in performing the experiments. C.F.M. offered technical support and optimized the non-viral gene transfer protocol for CIK cell CAR engineering. S.T. and L.V. designed and coordinated the research. E.B., A.B., and L.V. revised the data and performed a final revision of the paper.
Conflicts of Interest
The authors have no potential conflict of interest.
Acknowledgments
The authors thank Dr. Daniela Zanta and Dr. Nice Turazzi for technical support and Dr. Gino Vairo for providing 7G3 cDNA on behalf of CSL Research, CSL. The authors also thank Prof. Olivier Michielin and Dr. Mattia Pedotti for scientific discussions and Dr. Roberto Giovannoni and Dr. Stefania Galimberti for giving precious suggestions in planning the experiments and during paper writing. The authors thank the parent committees “Quelli che…con LUCA onlus,” “Comitato Maria Letizia Verga,” and “Stefano Verri” for their generous and constant support. This work was supported by grants from AIRC, Molecular Clinical Oncology 5 per mille 2010, “Innate immunity in cancer: Molecular targeting and cellular therapy” (grant 9962), and IG 2015, “Novel leukemia treatment by the use of chimeric antigen receptors (CARs)” (grant 17248); “Libera Le Ali” 2011 project, Fondazione Just Italia; SNF (grant 310030 166445); and Oncosuisse (grant KFS-3728-08-2015 to L.V. S.A. is a fellow of the University of Milano-Bicocca, Milan, Doctoral Program in Molecular and Translational Medicine (DIMET). Gratitude goes to “Quelli che…con LUCA onlus,” which generously supported S.T.’s fellowship and funded the project.
Footnotes
Supplemental Information includes four figures and can be found with this article online at http://dx.doi.org/10.1016/j.ymthe.2017.04.017.
Contributor Information
Andrea Biondi, Email: abiondi.unimib@gmail.com.
Ettore Biagi, Email: e.biagi@hsgerardo.org.
Supplemental Information
References
- 1.Gill S., Tasian S.K., Ruella M., Shestova O., Li Y., Porter D.L., Carroll M., Danet-Desnoyers G., Scholler J., Grupp S.A. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor-modified T cells. Blood. 2014;123:2343–2354. doi: 10.1182/blood-2013-09-529537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brentjens R.J., Rivière I., Park J.H., Davila M.L., Wang X., Stefanski J., Taylor C., Yeh R., Bartido S., Borquez-Ojeda O. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118:4817–4828. doi: 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee D.W., Kochenderfer J.N., Stetler-Stevenson M., Cui Y.K., Delbrook C., Feldman S.A., Fry T.J., Orentas R., Sabatino M., Shah N.N. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385:517–528. doi: 10.1016/S0140-6736(14)61403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maude S.L., Frey N., Shaw P.A., Aplenc R., Barrett D.M., Bunin N.J., Chew A., Gonzalez V.E., Zheng Z., Lacey S.F. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014;371:1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Robak T., Wierzbowska A. Current and emerging therapies for acute myeloid leukemia. Clin. Ther. 2009;31:2349–2370. doi: 10.1016/j.clinthera.2009.11.017. [DOI] [PubMed] [Google Scholar]
- 6.Eshhar Z., Waks T., Gross G., Schindler D.G. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc. Natl. Acad. Sci. USA. 1993;90:720–724. doi: 10.1073/pnas.90.2.720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gross G., Levy S., Levy R., Waks T., Eshhar Z. Chimaeric T-cell receptors specific to a B-lymphoma idiotype: a model for tumour immunotherapy. Biochem. Soc. Trans. 1995;23:1079–1082. doi: 10.1042/bst0231079. [DOI] [PubMed] [Google Scholar]
- 8.Sadelain M., Rivière I., Brentjens R. Targeting tumours with genetically enhanced T lymphocytes. Nat. Rev. Cancer. 2003;3:35–45. doi: 10.1038/nrc971. [DOI] [PubMed] [Google Scholar]
- 9.Watanabe K., Terakura S., Martens A.C., van Meerten T., Uchiyama S., Imai M., Sakemura R., Goto T., Hanajiri R., Imahashi N. Target antigen density governs the efficacy of anti-CD20-CD28-CD3 ζ chimeric antigen receptor-modified effector CD8+ T cells. J. Immunol. 2015;194:911–920. doi: 10.4049/jimmunol.1402346. [DOI] [PubMed] [Google Scholar]
- 10.Morgan R.A., Yang J.C., Kitano M., Dudley M.E., Laurencot C.M., Rosenberg S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010;18:843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lamers C.H., Sleijfer S., van Steenbergen S., van Elzakker P., van Krimpen B., Groot C., Vulto A., den Bakker M., Oosterwijk E., Debets R., Gratama J.W. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol. Ther. 2013;21:904–912. doi: 10.1038/mt.2013.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vergez F., Green A.S., Tamburini J., Sarry J.-E., Gaillard B., Cornillet-Lefebvre P., Pannetier M., Neyret A., Chapuis N., Ifrah N. High levels of CD34+CD38low/-CD123+ blasts are predictive of an adverse outcome in acute myeloid leukemia: a Groupe Ouest-Est des Leucemies Aigues et Maladies du Sang (GOELAMS) study. Haematologica. 2011;96:1792–1798. doi: 10.3324/haematol.2011.047894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tettamanti S., Marin V., Pizzitola I., Magnani C.F., Giordano Attianese G.M.P., Cribioli E., Maltese F., Galimberti S., Lopez A.F., Biondi A. Targeting of acute myeloid leukaemia by cytokine-induced killer cells redirected with a novel CD123-specific chimeric antigen receptor. Br. J. Haematol. 2013;161:389–401. doi: 10.1111/bjh.12282. [DOI] [PubMed] [Google Scholar]
- 14.Pizzitola I., Anjos-Afonso F., Rouault-Pierre K., Lassailly F., Tettamanti S., Spinelli O., Biondi A., Biagi E., Bonnet D. Chimeric antigen receptors against CD33/CD123 antigens efficiently target primary acute myeloid leukemia cells in vivo. Leukemia. 2014;28:1596–1605. doi: 10.1038/leu.2014.62. [DOI] [PubMed] [Google Scholar]
- 15.Hudecek M., Lupo-Stanghellini M.T., Kosasih P.L., Sommermeyer D., Jensen M.C., Rader C., Riddell S.R. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T cells. Clin. Cancer Res. 2013;19:3153–3164. doi: 10.1158/1078-0432.CCR-13-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chmielewski M., Hombach A., Heuser C., Adams G.P., Abken H. T cell activation by antibody-like immunoreceptors: increase in affinity of the single-chain fragment domain above threshold does not increase T cell activation against antigen-positive target cells but decreases selectivity. J. Immunol. 2004;173:7647–7653. doi: 10.4049/jimmunol.173.12.7647. [DOI] [PubMed] [Google Scholar]
- 17.Chmielewski M., Hombach A.A., Abken H. CD28 cosignalling does not affect the activation threshold in a chimeric antigen receptor-redirected T-cell attack. Gene Ther. 2011;18:62–72. doi: 10.1038/gt.2010.127. [DOI] [PubMed] [Google Scholar]
- 18.Liu X., Jiang S., Fang C., Yang S., Olalere D., Pequignot E.C., Cogdill A.P., Li N., Ramones M., Granda B. Affinity-tuned ErbB2 or EGFR chimeric antigen receptor T cells exhibit an increased therapeutic index against tumors in mice. Cancer Res. 2015;75:3596–3607. doi: 10.1158/0008-5472.CAN-15-0159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu P.H., Negrin R.S. A novel population of expanded human CD3+CD56+ cells derived from T cells with potent in vivo antitumor activity in mice with severe combined immunodeficiency. J. Immunol. 1994;153:1687–1696. [PubMed] [Google Scholar]
- 20.Baker J., Verneris M.R., Ito M., Shizuru J.A., Negrin R.S. Expansion of cytolytic CD8(+) natural killer T cells with limited capacity for graft-versus-host disease induction due to interferon gamma production. Blood. 2001;97:2923–2931. doi: 10.1182/blood.v97.10.2923. [DOI] [PubMed] [Google Scholar]
- 21.Pedotti M., Simonelli L., Livoti E., Varani L. Computational docking of antibody-antigen complexes, opportunities and pitfalls illustrated by influenza hemagglutinin. Int. J. Mol. Sci. 2011;12:226–251. doi: 10.3390/ijms12010226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Simonelli L., Pedotti M., Beltramello M., Livoti E., Calzolai L., Sallusto F., Lanzavecchia A., Varani L. Rational engineering of a human anti-dengue antibody through experimentally validated computational docking. PLoS ONE. 2013;8:e55561. doi: 10.1371/journal.pone.0055561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Venetsanakos E., Mirza A., Fanton C., Romanov S.R., Tlsty T., McMahon M. Induction of tubulogenesis in telomerase-immortalized human microvascular endothelial cells by glioblastoma cells. Exp. Cell Res. 2002;273:21–33. doi: 10.1006/excr.2001.5424. [DOI] [PubMed] [Google Scholar]
- 24.Valitutti S., Müller S., Cella M., Padovan E., Lanzavecchia A. Serial triggering of many T-cell receptors by a few peptide-MHC complexes. Nature. 1995;375:148–151. doi: 10.1038/375148a0. [DOI] [PubMed] [Google Scholar]
- 25.Cai Z., Kishimoto H., Brunmark A., Jackson M.R., Peterson P.A., Sprent J. Requirements for peptide-induced T cell receptor downregulation on naive CD8+ T cells. J. Exp. Med. 1997;185:641–651. doi: 10.1084/jem.185.4.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sykulev Y., Joo M., Vturina I., Tsomides T.J., Eisen H.N. Evidence that a single peptide-MHC complex on a target cell can elicit a cytolytic T cell response. Immunity. 1996;4:565–571. doi: 10.1016/s1074-7613(00)80483-5. [DOI] [PubMed] [Google Scholar]
- 27.Valitutti S., Müller S., Dessing M., Lanzavecchia A. Different responses are elicited in cytotoxic T lymphocytes by different levels of T cell receptor occupancy. J. Exp. Med. 1996;183:1917–1921. doi: 10.1084/jem.183.4.1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Viola A., Lanzavecchia A. T cell activation determined by T cell receptor number and tunable thresholds. Science. 1996;273:104–106. doi: 10.1126/science.273.5271.104. [DOI] [PubMed] [Google Scholar]
- 29.James S.E., Greenberg P.D., Jensen M.C., Lin Y., Wang J., Budde L.E., Till B.G., Raubitschek A.A., Forman S.J., Press O.W. Mathematical modeling of chimeric TCR triggering predicts the magnitude of target lysis and its impairment by TCR downmodulation. J. Immunol. 2010;184:4284–4294. doi: 10.4049/jimmunol.0903701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Caruso H.G., Hurton L.V., Najjar A., Rushworth D., Ang S., Olivares S., Mi T., Switzer K., Singh H., Huls H. Tuning sensitivity of CAR to EGFR density limits recognition of normal tissue while maintaining potent antitumor activity. Cancer Res. 2015;75:3505–3518. doi: 10.1158/0008-5472.CAN-15-0139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weijtens M.E.M., Hart E.H., Bolhuis R.L.H. Functional balance between T cell chimeric receptor density and tumor associated antigen density: CTL mediated cytolysis and lymphokine production. Gene Ther. 2000;7:35–42. doi: 10.1038/sj.gt.3301051. [DOI] [PubMed] [Google Scholar]
- 32.Sun Q., Woodcock J.M., Rapoport A., Stomski F.C., Korpelainen E.I., Bagley C.J., Goodall G.J., Smith W.B., Gamble J.R., Vadas M.A., Lopez A.F. Monoclonal antibody 7G3 recognizes the N-terminal domain of the human interleukin-3 (IL-3) receptor alpha-chain and functions as a specific IL-3 receptor antagonist. Blood. 1996;87:83–92. [PubMed] [Google Scholar]
- 33.Daily M.D., Masica D., Sivasubramanian A., Somarouthu S., Gray J.J. CAPRI rounds 3–5 reveal promising successes and future challenges for RosettaDock. Proteins. 2005;60:181–186. doi: 10.1002/prot.20555. [DOI] [PubMed] [Google Scholar]
- 34.Magnani C.F., Turazzi N., Benedicenti F., Calabria A., Tenderini E., Tettamanti S., Giordano Attianese G.M., Cooper L.J., Aiuti A., Montini E. Immunotherapy of acute leukemia by chimeric antigen receptor-modified lymphocytes using an improved Sleeping Beauty transposon platform. Oncotarget. 2016;7:51581–51597. doi: 10.18632/oncotarget.9955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haso W., Lee D.W., Shah N.N., Stetler-Stevenson M., Yuan C.M., Pastan I.H., Dimitrov D.S., Morgan R.A., FitzGerald D.J., Barrett D.M. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood. 2013;121:1165–1174. doi: 10.1182/blood-2012-06-438002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.