

Abstract
CLN diseases are rare lysosomal storage diseases characterized by progressive axonal degeneration and neuron loss in the CNS, manifesting in disability, blindness, and premature death. We have previously demonstrated that, in animal models of infantile and juvenile forms of CLN disease (CLN1 and CLN3, respectively), secondary neuroinflammation in the CNS substantially amplifies neural damage, opening the possibility that immunomodulatory treatment might improve disease outcome. First, we recapitulated the inflammatory phenotype, originally seen in mice in autopsies of CLN patients. We then treated mouse models of CLN1 and CLN3 disease with the clinically approved immunomodulatory compounds fingolimod (0.5 mg/kg/day) and teriflunomide (10 mg/kg/day) by consistent supply in the drinking water for 5 months. The treatment was well tolerated and reduced T cell numbers and microgliosis in the CNS of both models. Moreover, axonal damage, neuron loss, retinal thinning, and brain atrophy were substantially attenuated in both models, along with reduced frequency of myoclonic jerks in Ppt1−/− mice. Based on these findings, and because side effects were not detected, we suggest that clinically approved immune modulators such as fingolimod and teriflunomide may be suitable to attenuate progression of CLN1 and CLN3 disease and, possibly, other orphan diseases with pathogenically relevant neuroinflammation.
Keywords: CLN disease, Batten disease, axon degeneration, neurodegeneration, inflammation, immune modulation, treatment
Graphical Abstract
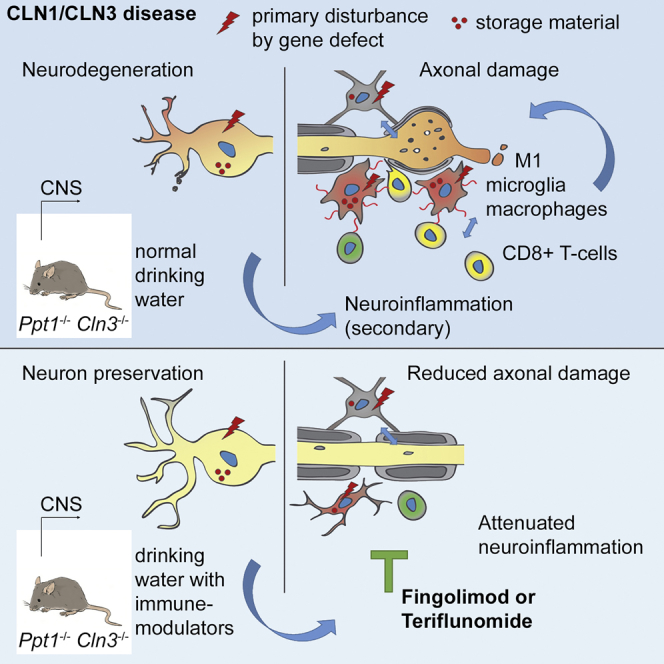
Neuronal ceroid lipofuscinoses (CLNs) are fatal storage diseases causing CNS damage with blindness and disability. An important disease amplifier is neuroinflammation. Groh et al. show that two clinically approved immune modulators, fingolimod and teriflunomide, mitigate neuroinflammation and disease hallmarks in mouse models, suggesting their suitability for treating CLN patients.
Introduction
Neuronal ceroid lipofuscinoses (NCLs, CLN diseases) are rare, genetically mediated lysosomal storage disorders characterized by substantial neurodegeneration in the CNS, leading to blindness, seizures, disability, and early death.1, 2 Apart from neuronal degeneration, a characteristic histopathological feature is the accumulation of lipofuscin-like autofluorescent storage material of uncertain pathogenetic relevance in the cytoplasm of neurons, macroglia, microglia, and other cell types.1, 3 13 distinct CLN genes functionally related to lysosomal and cellular homeostasis have been identified as primary disease causes.4 In spite of the progress in molecular genetics of the respective orphan disorders, there is presently no cure available to mitigate the severe clinical burden, resulting in an often desperate and hopeless situation for the patients and their relatives, particularly because many CLN forms manifest in early childhood and progress rapidly.
One possible therapeutic target of CLN disease is neuroinflammation. We have previously demonstrated that, in two mouse models of CLN disease, secondary inflammation in the CNS amplifies axonal perturbation, neurodegeneration, and clinical outcome.5, 6 One of these models is Ppt1−/− mice, which are deficient in the soluble lysosomal enzyme palmitoyl protein thioesterase 1.7 Ppt1−/− mice represent infantile CLN (CLN1) disease in humans, which is characterized by an early onset at around 1 year of age and a premature death at around age 10.8 The other model is deficient in the lysosomal integral membrane protein CLN39 and mimics the most frequent, juvenile form: CLN3 disease. In humans, this disease variant starts around 6 years of age with rapidly progressing visual loss and many other neurological symptoms. The patients mostly die in their mid-20s.4
The abovementioned pathogenetic relevance of neuroinflammation in these models was demonstrated by cross-breeding the mouse models with either lymphocyte-deficient mice5 or with mice deficient in the microglia/macrophage-related cell adhesion and pro-inflammatory signaling molecule sialoadhesin.6 Both approaches identified CD8+ effector T lymphocytes as substantial culprit cells in the two disease situations, demonstrating a surprising, but unequivocal disease convergence in both models. The present study was designed to target these effector cells in both models with two pharmacological compounds of approved clinical safety already established for much more frequent neuroinflammatory diseases, such as multiple sclerosis. One substance, fingolimod (or FTY720), a modulator of the sphingosine-1-phosphate (S1P) receptor, impairs lymphocyte emigration from the lymph nodes and infiltration into the brain.10, 11, 12 The other compound, teriflunomide, inhibits the de novo synthesis of pyrimidine nucleotides and reduces the proliferation and function of activated but not resting cells of the adaptive immune system.12 In the present study, we demonstrate that both orally applied substances robustly attenuate neuroinflammation in the CNS of CLN1 and CLN3 models. Moreover, features indicative of neural perturbation in the retinotectal system were strongly reduced/prevented, as identified by repetitive optical coherence tomography in vivo and, eventually, by histopathological post-mortem analysis using immunohistochemistry and electron microscopy to quantify axonal spheroids and histology to quantify neuronal cells. Last, total brain atrophy was attenuated, and the frequency of myoclonic jerks as a clinical readout in the CLN1 disease model was strongly reduced upon pharmacological immune modulation. Based on these results and our concomitant observation that, in CNS autopsies of CLN disease patients, both microgliosis and accumulation of CD8+ T lymphocytes were detectable, we suggest that fingolimod and teriflunomide could serve as clinically safe and approved immune modulators to mitigate the disease burden in at least two forms of presently untreatable CLN diseases.
Results
Based on our previous studies identifying neuroinflammation as a robust amplifier of CLN1 and CLN3 disease in mouse models, we investigated whether, in human CLN disorders, inflammation might be a disease-modifying candidate as well. For this purpose, we investigated frozen brain autopsy sections (thalamus and occipital cortex) from CLN2 and CLN3 disease patients for evidence for inflammation (Table 1). At first, as expected, we identified a high amount of autofluorescent material in neuronal and non-neuronal cells in the brain sections of CLN disease but not in those of control patients (data not shown). In comparison with the sections from control patients, we found significantly increased numbers of CD4+ and CD8+ T lymphocytes in sections from CLN patients (Figure 1). Similar to our observations in the mouse models, CD8+ T-lymphocytes outnumbered the CD4+ ones. Numbers of CD68+ and CD11b+ microglial cells/macrophages were also elevated, as well as cells positive for the pro-inflammatory microglial/macrophage cell signaling molecule sialoadhesin (Sn; Figure S1). In line with these findings was the rounded or bushy morphology of CD68+ and CD11b+ microglial cells/macrophages, indicating their activated state. One control autopsy from a patient with a “congenital abnormality of the CNS” was included and did not display signs of CNS inflammation. These observations corroborate our assumption that, not only in mouse models but also in human CLN patients, secondary inflammation might play a detrimental role.
Table 1.
Brain Autopsies
| Autopsy No. | MRC BBN-ID | Sex | Age | Diagnosis |
|---|---|---|---|---|
| A137/12 | BBN_4576 | F | 10 | CLN2 |
| A114/08 | BBN_16328 | F | 11 | CLN3 |
| A091/07 | BBN_16363 | M | 23 | CLN3 |
| A206/03 | BBN_15741 | F | 10 | congenital abnormality of nervous system, control |
| A113/09 | BBN_16281 | M | 18 | no specific features, control |
| A002/94 | n/a | M | 25 | normal adult brain, control |
The frozen samples of thalamus and occipital cortex of the corresponding patients were obtained by the London Neurodegenerative Disease Brain Bank and Brains for Dementia Research and analyzed by immunohistochemistry. F, female; M, male.
Figure 1.

Increased Numbers of T Lymphocytes in the CNS of CLN Disease Patients
(A) Representative light microscopy of immunohistochemically labeled CD8+ T lymphocytes (arrows) in thalamic brain autopsy sections from a CLN3 patient (BBN_16328) and a control patient (BBN_15741). Right: higher magnification of the inset. Scale bars, 30 μm. (B and C) Quantification of CD8+ (B) and (C) CD4+ T lymphocytes. The numbers of CD8+ and CD4+ T cells were significantly increased in the thalamus and occipital cortex of CLN disease patients (pooled data of two CLN3 cases and one CLN2 case) compared with controls (n = 3 patients/group). Student’s t test. *p < 0.05, **p < 0.01.
In a subsequent attempt to treat detrimental inflammation in CLN, we used fingolimod and teriflunomide as preventive (before the appearance of significant degenerative alterations) pharmacological intervention in Ppt1−/− and Cln3−/− mice, mimicking CLN1 and CLN3 disease, respectively. Based on our previous work regarding onset of neuroinflammation and neurodegeneration in the models,5, 6 1-month-old Ppt1−/− mice and 12-month-old Cln3−/− mice were consistently supplied for 5 months, respectively, with drinking water containing either fingolimod (0.5 mg/kg/day) or teriflunomide (10 mg/kg/day). Control mice were supplied with normal drinking water lacking the immune modulators. All treated mice were generally undistinguishable from the nontreated controls regarding fur and body weight, reflecting that both treatments were well tolerated. At the end of the respective treatment periods, 6-month-old Ppt1−/− mice and 17-month-old Cln3−/− mice were analyzed regarding the systemic effect of treatment on distinct immune cell populations of peripheral blood. Based on flow cytometry, we detected a significant elevation of leukocytes in peripheral blood in untreated Ppt1−/− (Figure S2A) and Cln3−/− mice (data not shown). Using antibodies against CD4, CD8, and CD11b, we identified T lymphocytes and myeloid cells, respectively, as contributing populations of increased leukocyte numbers in the untreated mutants. Interestingly, in both models, fingolimod, but not teriflunomide, significantly reduced leukocyte numbers by strongly depleting T lymphocytes, but not myeloid cells, from the circulation (Figures S2A and S2B). This lymphopenic effect was independent of the mouse genotype (data not shown). Reflecting the lymphopenic effects, a reduction in spleen weight was typical for fingolimod- but not teriflunomide-treated mutants (Figure S3).
As a next step, we quantified the number of CD4+ and CD8+ T lymphocytes and CD11b+ microglia/macrophages in the CNS fiber tracts, as represented by longitudinal sections of the optic nerves. In accordance with our previous studies,5, 6 the respective cell types were elevated in number in both untreated models compared with wild-type (WT) mice. However, in the fingolimod- and teriflunomide-treated mutants, the elevation of T lymphocyte numbers was prevented (Figures 2A and 2B; Figure S4). Interestingly, both compounds also reduced the increase in numbers of CD11b+ cells in CNS tissue (Figure S5). Also, activated microglia/macrophages positive for the cell recognition molecule Sn were less frequent in the treated mutants in comparison with the untreated ones (Figure 3). Similarly, the numbers of CD8+ T lymphocytes (Figure 2C) and Sn+ microglia/macrophages (Figure 3C) were also attenuated in gray matter brain regions such as the somatosensory barrel field cortex of Ppt1−/− and Cln3−/− mice after treatment with fingolimod or teriflunomide. Thus, both immune modulators clearly mitigate neuroinflammation in the CLN models investigated.
Figure 2.

Fingolimod and Teriflunomide Prevent the Elevation of CD8+ T Lymphocyte Numbers in CLN1 and CLN3 Disease Models
(A) Representative immune fluorescence microscopy of CD8+ T lymphocytes (arrows) in longitudinal optic nerve sections from 6-month-old Ppt1+/+, Ppt1−/−, fingolimod-treated Ppt1−/− (+ FTY720), and teriflunomide-treated Ppt1−/− (+ Teriflunomide) mice (top) and 17-month-old Cln3+/+, Cln3−/−, fingolimod-treated Cln3−/− (+ FTY720), and teriflunomide-treated Cln3−/− (+ Teriflunomide) mice (bottom). Scale bar, 20 μm. (B) Quantification of CD8+ T lymphocytes in optic nerve sections. The numbers of CD8+ T cells were significantly increased in the untreated CLN1 and CLN3 disease models compared with WT mice. This increase was prevented in both models by treatment with fingolimod as well as with teriflunomide for 150 days (n = 5 mice/group). One-way ANOVA and Tukey’s post hoc tests. ***p < 0.001. (C) Representative light microscopy of CD8+ T lymphocytes (circles) in coronal sections of S1BF cortex from 6-month-old Ppt1+/+, Ppt1−/−, fingolimod-treated Ppt1−/− (+ FTY720), and teriflunomide-treated Ppt1−/− (+ Teriflunomide) mice (top) and 17-month-old Cln3+/+, Cln3−/−, fingolimod-treated Cln3−/− (+ FTY720), and teriflunomide-treated Cln3−/− (+ Teriflunomide) mice (bottom). Note the reduction of cortical atrophy in treated Ppt1−/− mice. Scale bar, 50 μm. (D) Higher magnification of CD8+ T lymphocytes. Scale bar, 20 μm.
Figure 3.

Fingolimod and Teriflunomide Attenuate the Elevation of Sn+ Microglia/Macrophage Numbers in CLN1 and CLN3 Disease Models
(A) Representative immune fluorescence microscopy of Sn+ microglia/macrophages in longitudinal optic nerve sections from 6-month-old Ppt1+/+, Ppt1−/−, fingolimod-treated Ppt1−/− (+ FTY720), and teriflunomide-treated Ppt1−/− (+ Teriflunomide) mice (top) and 17-month-old Cln3+/+, Cln3−/−, fingolimod-treated Cln3−/− (+ FTY720), and teriflunomide-treated Cln3−/− (+ Teriflunomide) mice (bottom). Scale bar, 20 μm. (B) Quantification of Sn+ microglia/macrophages (percentage of CD11b+) in optic nerve sections. The numbers of Sn+ cells were significantly increased in the CLN1 and CLN3 disease models compared with WT mice. This increase was attenuated in both models by treatment with fingolimod as well as teriflunomide for 150 days (n = 5 mice/group). One-way ANOVA and Tukey’s post hoc tests. *p < 0.05, **p < 0.01, ***p < 0.001. (C) Representative light microscopy of Sn+ microglia/macrophages in coronal sections of S1BF cortex from 6-month-old Ppt1+/+, Ppt1−/−, fingolimod-treated Ppt1−/− (+ FTY720), and teriflunomide-treated Ppt1−/− (+ Teriflunomide) mice (top) and 17-month-old Cln3+/+, Cln3−/−, fingolimod-treated Cln3−/− (+ FTY720) and teriflunomide-treated Cln3−/− (+ Teriflunomide) mice (bottom). Note the reduction of cortical atrophy in treated Ppt1−/− mice. Scale bar, 50 μm. (D) Higher magnification of Sn+ microglia/macrophages. Scale bar, 20 μm.
We also scored the effect of therapeutic intervention on disease severity. In living mutants, we first analyzed retinopathic changes using non-invasive optical coherence tomography (OCT) for live imaging of the retina13 and assessed clinical features in the CLN1 disease model by quantifying myoclonic jerks. For OCT, Ppt1−/− mice were analyzed at 4, 5, and 6 months of age regarding total retinal and composite layer thickness. Corroborating previous findings,6, 13 a constant decrease in the thickness of the innermost composite layers (nerve fiber layer [NFL], ganglion cell layer [GCL], and inner plexiform layer [IPL]) was detected, reaching highly significant levels at 6 months (Figure 4A). Strikingly, both immune modulators prevented reduction of the retinal layer thickness. This preservation of retinal integrity in the CLN1 disease model after fingolimod and teriflunomide treatment was accompanied by an ameliorated clinical outcome in the form of reduced occurrence of myoclonic jerks at 6 months of age (Figure 4B).
Figure 4.

Fingolimod and Teriflunomide Attenuate Retinal Thinning in CLN1 and CLN3 Disease Models and Myoclonic Jerks in the CLN1 Model
(A) Longitudinal analysis of NFL/GCL/IPL thickness at 4, 5, and 6 months of age in Ppt1+/+, Ppt1−/−, fingolimod-treated Ppt1−/− (FTY720), and teriflunomide-treated Ppt1−/− (Teri) mice (top) and at 12, 15, and 17 months of age in Cln3+/+, Cln3−/−, fingolimod-treated Cln3−/− (FTY720), and teriflunomide-treated Cln3−/− (Teri) mice (bottom). Inner retinal thickness progressively decreased with age in the CLN1 and CLN3 disease models compared with WT mice. This decrease was attenuated in both models by treatment with fingolimod as well as teriflunomide for 150 days (n = 5 mice/group). One-way ANOVA and Tukey’s post hoc tests. (B) Quantification of myoclonic jerks in Ppt1+/+, Ppt1−/−, fingolimod-treated Ppt1−/− (FTY720), and teriflunomide-treated Ppt1−/− (Teri) mice. The frequency of myoclonic jerks was robust in the untreated CLN1 disease model but substantially decreased by treatment with fingolimod as well as with teriflunomide for 150 days (n = 5 mice/group). One-way ANOVA and Tukey’s post hoc tests. *p < 0.05, **p < 0.01, ***p < 0.001.
Retinopathy of Cln3−/− mice was scored at 12, 15, and 17 months with OCT. Extending previous results,13 a decrease in the thickness of the innermost composite layer was detected, with a highly significant degree of retinal thinning at 15 and 17 months of age. Treatment with each of the immune modulators reduced the retinal thinning but did not completely prevent it (Figure 4A).
Last, the effects of the distinct treatment approaches on histopathological changes in the retinotectal system were quantified. SMI32+ axonal spheroids, as representatives of axonal perturbation, were amply detectable in optic nerve sections of 6- and 17-month-old CLN1 and CLN3 disease models, respectively. Both fingolimod and teriflunomide substantially suppressed their formation in Ppt1−/− mice, whereas, in Cln3−/− mice, this ameliorating effect only reached significance after teriflunomide treatment (Figure 5). However, electron microscopic quantification demonstrated a significant attenuation of axonal spheroid formation after both treatment approaches (Figure 6). Quantification of retinal ganglion cells in flat mount preparations recapitulated previous observations5, 6, 13 in that cell numbers were significantly reduced in 6- and 17-month-old Ppt1−/− and Cln3−/− mice, respectively. Strikingly, treatment with fingolimod and teriflunomide had a strong beneficial effect and prevented significant loss of retinal ganglion cells in both models, corroborating our observations in living mice using OCT (Figure 7). Similarly, total brain atrophy, as reflected by reduced brain weights of Ppt1−/− and Cln3−/− mice, was significantly attenuated after treatment with fingolimod and teriflunomide, arguing for a generalized effect of immunomodulatory therapy extending beyond the retinotectal system (Figure 8A). This beneficial effect on brain atrophy was also obvious in coronal brain sections of Ppt1−/− mice (Figure 8B; see also Figures 2C, 3C, and 8C). In contrast, the accumulation of autofluorescent storage material appeared not to be prominently affected by immunomodulation (Figure 8C), as similarly observed with our previous cross-breeding approaches to genetically eliminate cytotoxic T lymphocytes.5 This indicates that immunomodulation has no effect on primary disease-related abnormalities and that inflammation is a secondary modifier of disease outcome.
Figure 5.

Fingolimod and Teriflunomide Attenuate the Formation of SMI32+ Axonal Spheroids in CLN1 and CLN3 Disease Models
(A) Representative light microscopy of immunohistochemically labeled SMI32+ axonal spheroids (arrows) in longitudinal optic nerve sections from 6-month-old Ppt1+/+, Ppt1−/−, fingolimod-treated Ppt1−/− (+ FTY720), and teriflunomide-treated Ppt1−/− (+ Teriflunomide) mice (top) and 17-month-old Cln3+/+, Cln3−/−, fingolimod-treated Cln3−/− (+ FTY720), and teriflunomide-treated Cln3−/− (+ Teriflunomide) mice (bottom). Scale bar, 20 μm. (B) Quantification of SMI32+ axonal spheroids in optic nerves. The numbers of SMI32+ axonal spheroids were significantly increased in the CLN1 and CLN3 disease models compared with WT mice. This increase was attenuated in the CLN1 model by treatment with fingolimod and in both models by treatment with teriflunomide for 150 days (n = 5 mice/group). One-way ANOVA and Tukey’s post hoc tests. *p < 0.05, **p < 0.01, ***p < 0.001.
Figure 6.

Fingolimod and Teriflunomide Attenuate the Formation of Axonal Spheroids in CLN1 and CLN3 Disease Models
(A) Representative electron micrographs of axonal spheroids (red arrows) in optic nerve cross-sections from 6-month-old Ppt1−/−, fingolimod-treated Ppt1−/− (+ FTY720), and teriflunomide-treated Ppt1−/− (+ Teriflunomide) mice (top) and 17-month-old Cln3−/−, fingolimod-treated Cln3−/− (+ FTY720), and teriflunomide-treated Cln3−/− (+ Teriflunomide) mice (bottom). Scale bar, 2 μm. (B) Electron microscopy-based quantification of axonal spheroids (percentage of all analyzed axons) in optic nerves. Axonal spheroids were prominent in the untreated CLN1 and CLN3 disease models compared with WT mice. Axonal spheroid formation was attenuated in both models by treatment with fingolimod as well as with teriflunomide for 150 days (n = 5 mice/group). Kruskal-Wallis test with Bonferroni-Holm correction. *p < 0.05, **p < 0.01, ***p < 0.001.
Figure 7.

Fingolimod and Teriflunomide Prevent Loss of Retinal Ganglion Cells in CLN1 and CLN3 Disease Models
(A) Representative light microscopy of cresyl violet-stained ganglion cells in retinal flat mount preparations from 6-month-old Ppt1+/+, Ppt1−/−, fingolimod-treated Ppt1−/− (+ FTY720), and teriflunomide-treated Ppt1−/− (+ Teriflunomide) mice (top) and 17-month-old Cln3+/+, Cln3−/−, fingolimod-treated Cln3−/− (+ FTY720), and teriflunomide-treated Cln3−/− (+ Teriflunomide) mice (bottom). Scale bar, 20 μm. (B) Quantification of retinal ganglion cells. The numbers of cells were significantly reduced in the CLN1 and CLN3 disease models compared with WT mice. This reduction was prevented in both models by treatment with fingolimod as well as with teriflunomide for 150 days (n = 5 mice/group). One-way ANOVA and Tukey’s post hoc tests. *p < 0.05, **p < 0.01.
Figure 8.

Fingolimod and Teriflunomide Attenuate Brain Atrophy in CLN1 and CLN3 Disease Models
(A) Total brain weights of 6-month-old Ppt1+/+, Ppt1−/−, fingolimod-treated Ppt1−/− (+ FTY720), and teriflunomide-treated Ppt1−/− (+ Teriflunomide) mice and 17-month-old Cln3+/+, Cln3−/−, fingolimod-treated Cln3−/− (+ FTY720), and teriflunomide-treated Cln3−/− (+ Teriflunomide) mice. Brain atrophy was attenuated in both models by treatment with fingolimod as well as with teriflunomide for 150 days (n = 5 mice/group). One-way ANOVA and Tukey’s post hoc tests. *p < 0.05, ***p < 0.001. (B) Representative light microscopy of cresyl violet-stained coronal brain sections of 6-month-old Ppt1+/+, Ppt1−/−, fingolimod-treated Ppt1−/− (+ FTY720), and teriflunomide-treated Ppt1−/− (+ Teriflunomide) mice. Note the reduction of brain atrophy in treated Ppt1−/− mice. Scale bar, 1 mm. (C) Representative microscopy of autofluorescence (Af) in coronal sections of somatosensory cortex from 6-month-old Ppt1+/+, Ppt1−/−, fingolimod-treated Ppt1−/− (+ FTY720), and teriflunomide-treated Ppt1−/− (+ Teriflunomide) mice. There was no obvious difference regarding autofluorescence in treated Ppt1−/− mice. Scale bar, 50 μm.
Discussion
Previous proof-of-principle investigations from our laboratory showed that CLN1 and CLN3 disease models with genetically attenuated immune activation display strongly ameliorated histopathological changes in the CNS and improved disease outcome.5, 6 The respective studies are, therefore, suggestive of a potential anti-inflammatory treatment option for the corresponding human diseases. However, until now, two significant drawbacks may have limited the enthusiasm to consider immune modulation as a therapeutic approach for CLN diseases: in spite of some hints from the literature,14 it is not known whether neuroinflammation is a typical feature in CLN disease patients, and suppression of parts of the immune system with an intended block of auto-antibody deposition with the cytostatic mycophenolate mofetil only very moderately improved disease outcome in a relatively young and, therefore, only mildly affected mouse model for CLN3 disease.15
The purpose of our present study was to shift focus again on the potential therapeutic effect of immune modulation in CLN diseases. Along these lines, we could verify that, in autopsies of CLN disease patients, including both CLN2 and CLN3, robust inflammatory features implicating both the adaptive and the innate immune system are detectable. A striking feature was the predominance of CD8+ over CD4+ T lymphocytes, resembling the situation in the CLN3 mouse model.6 It is interesting in this context that the neuroinflammatory features of the CLN3 patients were indistinguishable from the inflammatory hallmarks seen in the CLN2 autopsies. Because of the scarcity of brain autopsy samples from CLN1 patients, we were not able to investigate whether similar neuroinflammatory reactions occur as in the mouse model. However, based on the pathogenic effect of neuroinflammation in CLN1 model mice and the similarities (except the much later onset in CLN3) of neuroinflammatory features in the distinct mouse models and the CLN3 patients, we assume that CLN1 patients likely show comparable inflammatory reactions as CLN3 (and CLN2) patients. Of note, we found that, in one of our control autopsies from a patient with a congenital abnormality of the CNS (Table 1), no signs of inflammation were detectable, suggesting that not every abnormality leads to inflammation.
All in all, we found that secondary neuroinflammation with a predominance of CD8+ T cells is not confined to CLN mouse models but appears to be a typical histopathological feature of CLN patients. It is, therefore, of particular relevance that clinically approved and well tolerated immune modulators, initially designed for long-term therapy of more frequent, neuroinflammatory disorders such as MS, robustly improved histopathological changes and disease outcome in CLN1 and CLN3 models. The compounds were applied at doses based on established protocols regarding the treatment of neuroinflammation-associated mouse models.30, 31 Because of the compounds’ stability in neutral solutions, fingolimod and teriflunomide were consistently supplied ad libitum in weekly renewed drinking water, which is the least stressful delivery method for long-term application.
Along these lines, both fingolimod and teriflunomide treatment ameliorated retinal thinning, neuron loss, and other histopathological features of the retinotectal system as well as total brain atrophy in both mutants. Moreover, in the severe CLN1 disease model, both drugs significantly reduced clinical features in the form of myoclonic jerks, which is in line with our previous proof-of-principle studies.5, 6 Also, in a recent pharmacological approach using the anti-inflammatory small molecule MW151, seizure frequency was significantly reduced in the CLN1 model,16 supporting our finding that development of seizures is amplified by neuroinflammation. These observations, made in two different laboratories, are of particular relevance because CLN disease patients often respond only poorly to conventional anti-epileptic drugs or even react with deterioration of the disease course.17 Taken together, degenerative changes in the retinotectal system and other brain areas of both models and the development of seizures in the severe model appear to be strongly co-mediated or amplified by the neuroinflammatory aspect of the primarily genetically caused diseases. This might be of pivotal relevance for treatment strategies regarding the presently non-curable CLN1 and CLN3 disease and possibly related disorders in humans, such as other CLNs, immune-related leukodystrophies, or hereditary spastic paraplegia.18, 19, 20
The similar efficacy of both drugs is remarkable and impressively mimics the ameliorating effects of genetic impairment of inflammation in the CLN1 and CLN3 disease models. Fingolimod impairs lymphocyte emigration from the lymph nodes,10, 11, 12 likely reflected by lymphopenia and reduced spleen weights in both treated models. This was not seen in teriflunomide-treated models, in line with the drug being predominantly a cytostatic for proliferation of activated but not resting cells of the adaptive immune system, thus preserving the protective function of the immune system.12, 21 Interestingly, in a rat model of Alzheimer disease, fingolimod improved spatial22 and aversive learning.23 Also, the drug mitigated the outcome of ischemia, although the respective mechanisms are controversially discussed.24, 25, 26 Further pleiotropic mechanisms independent of the immune system are protection of oligodendrocytes, remyelination, and astrocyte migration.21 A previous study in a Rett syndrome model displayed a direct neuroprotective effect because of fingolimod-induced brain-derived neurotrophic factor (BDNF) expression.27 It is therefore conceivable that the ameliorated phenotype of our CLN models may also be influenced by pleiotropic effects not related to the immune system. Two observations argue against this possibility. First, recent studies from our laboratory not only displayed the pathogenetic effect of cytotoxic CD8+ T cells in CLN disease models5 but also identified intrinsic immune-regulatory mechanisms implicating CD8+CD122+ T lymphocytes.5, 6 These regulatory cells are negatively controlled by the microglia/macrophage-related cell signaling molecule Sn. Consequently, absence of Sn caused a robust mitigation of disease in both CLN1 and CLN3 models,6 closely resembling the presently reported improvement by the immune modulators, supporting their immune-directed effect in these models. Second, teriflunomide- and fingolimod-treated CLN models not only strikingly resemble CLN models with genetically impaired immune activation but also resemble each other. This is remarkable because the non-immune effects of these drugs differ considerably.21 Thus, it is more likely that the improved histopathological and clinical changes by fingolimod and teriflunomide are the predominant result of immune modulation.
The scope of the present study was to open opportunities to increase the quality of life for CLN1 and CLN3 patients by reducing damage of the visual system, seizures, and possibly other consequences of brain atrophy. We think there is much hope to achieve these goals with use of the two immune modulators because particularly seizure frequency and the retinotectal system strongly “respond” not only to the genetic immune ablation but also to the pharmacological immune modulation. Early application of the drugs, as in the present approaches, should be possible in most cases because diagnosis of the common CLN disease forms improved significantly over the last years. Moreover, younger siblings, being pre-symptomatic disease carriers, will likely profit most. Furthermore, immunomodulation could be combined with future gene therapeutic approaches or enzyme replacement therapies with possible synergistic effects.16, 28 Because impaired vision leading to eventual blindness is one of the very first and likely the most disabling and insulating symptoms in CLN1 and CLN3 disease,17 stalling and/or ameliorating these symptoms would be of outstanding significance. Future treatment studies should expand the current findings and additionally focus on the effect of immune modulation on neuronal function in both models and on the longevity of Ppt1−/− mice. Ideally, clinical trials using fingolimod or teriflunomide should be performed in the future to assess their efficacy in CLN1 and CLN3 disease. However, because both drugs are relatively safe and already “established” in clinical use for more frequent chronic neuroinflammatory disorders, at the moment, off-label use of the immune modulators may help to make the orphan CLN diseases more bearable for patients and their relatives, at least until safe causal therapies may become available.
Materials and Methods
Animals
Mice were kept at the animal facility of the Department of Neurology, University of Würzburg, under barrier conditions and at a constant cycle of 12 hr in the light (<300 lux) and 12 hr in the dark. All animal experiments were approved by the government of Lower Franconia, Germany. Ppt1-deficient (Ppt1−/−) mice with disruption of exon 97 and age-matched WT (Ppt1+/+) littermates were on a uniform C57BL/6 genetic background. Cln3-deficient (Cln3−/−) mice with disruption of exons 2–6 and most of exon 19 and age-matched WT littermates (Cln3+/+) were on a uniform Sv/129 genetic background. Genotypes were determined by conventional PCR using isolated DNA from ear punch biopsies following previously published protocols. Mice were screened for the confounding Rd8 mutation in the Crb1 gene as described previously.29 Neither Ppt1−/− nor Cln3−/− mice carried the respective mutation.
Immunomodulatory Treatment
Fingolimod (FTY720, Sigma-Aldrich, SML0700) was dissolved in autoclaved drinking water at 3 μg/mL and provided ad libitum. With an approximate consumption of 5 mL/day and 30-g body weight, this corresponds to a dose of 0.5 mg/kg body weight/day. Teriflunomide (Biorbyt, orb146201) was dissolved in autoclaved drinking water containing 0.6% Tween 80 at 60 μg/mL, corresponding to a dose of 10 mg/kg body weight/day. These concentrations are based on previous animal experiments in other laboratories30, 31 and nearly correspond to doses used for human multiple sclerosis patients when dose conversion scaling is applied.32 Non-treated controls received autoclaved drinking water without the compounds (but with or without 0.6% Tween 80), and the water with or without the compounds was changed weekly. Water containing only 0.6% Tween 80 had no effect on neuroinflammation and neural damage in Ppt1−/− or Cln3−/− mice (data not shown). Mice were treated for 150 days and monitored daily regarding defined burden criteria and phenotypic abnormalities. No obvious side effects or significant changes in body weight were detected with both treatment approaches. The frequency of myoclonic jerks in treated and untreated Ppt1−/− mice was determined as described previously.5 At the end of the treatment, mice were euthanized with CO2 (according to guidelines by the State Office of Health and Social Affairs Berlin), blood was rinsed with PBS containing heparin, and mice were transcardially perfused with 2% paraformaldehyde (PFA) in PBS. Tissue was harvested, post-fixed, dehydrated, and processed as described previously.5 Before embedding of the brains, olfactory bulbs and medullae were separated at defined positions, and total brains, including pontes, were weighed using an analytical balance (ABT 220-5DM, Kern).
Histochemistry and Immunofluorescence
Immunohistochemistry was performed on 10-μm-thick longitudinal optic nerve cryo-sections after post-fixation in 4% PFA in PBS or ice-cold acetone for 10 min. Sections were blocked using 5% BSA in PBS and incubated overnight at 4°C with one or an appropriate combination of up to three of the following antibodies: rat anti-CD4 (1:1,000, Bio-Rad AbD Serotec), rat anti-CD8 (1:500, Bio-Rad AbD Serotec), rat anti-CD11b (1:100, Bio-Rad AbD Serotec), rat anti-CD169 (1:300, Bio-Rad AbD Serotec), and mouse anti-SMI32 (1:1,000, BioLegend). Immune reactions were visualized using fluorescently labeled (1:300, Dianova) secondary antibodies or biotinylated secondary antibodies (1:100, Vector Laboratories) and streptavidin-biotin-peroxidase (Vector Laboratories) complex using diaminobenzidine-HCl and H2O2, and nuclei were stained with DAPI (Sigma-Aldrich). Moreover, 40-μm-thick coronal brain sections were used for free-floating immunohistochemistry using the same antibodies. Light and fluorescence microscopic images were acquired using an Axiophot 2 microscope (Zeiss) with an attached charge-coupled device (CCD) camera (SPOT Imaging, Diagnostic Instruments). Images were minimally processed for generation of figures using Photoshop CS6 (Adobe). For quantification, immunoreactive profiles were counted in at least three non-adjacent sections for each animal and related to the total area of these sections using the cell counter plugin in ImageJ (NIH). For quantification of retinal ganglion cells, eyes were enucleated and post-fixed in 4% PFA in PBS for 15 min, and retinal flat mounts were prepared. Cresyl violet staining and quantification of Nissl-positive cells in the ganglion cell layer were performed according to previously published protocols in three images of the middle retinal region per flat mount.5, 6, 13 Air-dried, 40-μm-thick coronal brain sections were similarly stained using cresyl violet.
Electron Microscopy
For electron microscopy, optic nerves were dissected after transcardial perfusion and post-fixed with 4% PFA and 2% glutaraldehyde in cacodylate buffer overnight. Tissue was osmicated, dehydrated, and embedded in Spurr’s medium. Semi-thin (0.5 μm) and ultra-thin (70 nm) optic nerve cross-sections were prepared and stained with methylene blue or lead citrate, respectively. Micrographs were acquired using a Leo 906 E electron microscope (Zeiss) with a ProScan Slow Scan CCD camera. Axonal spheroids were quantified as previously described in 10 images/section.5, 6
Flow Cytometry of Blood Leukocytes
Before transcardial perfusion of the euthanized mice, blood was collected from the right atrium of the heart, and coagulation was prevented by adding PBS containing heparin. Erythrocytes were lysed, and the remaining cells were washed and analyzed by flow cytometry as described previously.33, 34 Total leukocytes were gated based on forward and side scatter, myeloid cells were stained using phycoerythrin (PE)-conjugated antibodies against CD11b (1:50, BD Biosciences), and T lymphocytes were stained using antibodies against CD4 and CD8 (1:50, BD Biosciences). At least 1 × 105 leukocytes/mouse were analyzed using a FACSCalibur with CellQuest software (BD Biosciences) and their numbers per microliter of blood were calculated.
Spectral Domain OCT
Mice were subjected to OCT imaging with a commercially available device (Spectralis OCT, Heidelberg Engineering) and additional lenses as described previously.6, 13 Mice were measured at different ages for longitudinal analysis, and the thickness of the innermost retinal composite layer comprising the NFL, GCL, and IPL was measured in high-resolution peripapillary circle scans (at least 10 measurements/scan) by an investigator unaware of the genotype of the mice.
Human Brain Autopsy Samples
Frozen brain autopsy samples (Table 1) of thalamus and occipital cortex were provided by the London Neurodegenerative Disease Brain Bank and Brains for Dementia Research. The local Ethical Committee had no concerns regarding the use of autopsy material in this study and dispensed the authors from a corresponding application. Samples from two CLN3 patients (aged 11 and 23 years) and one CLN2 patient (aged 10 years) were pooled into a CLN disease group to allow statistical comparison of the tissue available from the rare disorders. Samples from two normal adults (aged 18 and 25 years) and one patient with a congenital abnormality of the nervous system (aged 10 years) served as the control group. Samples were cut into 10-μm-thick cryo-sections and post-fixed in 4% PFA in PBS or ice-cold acetone for 10 min. Sections were blocked using 5% BSA in PBS and incubated overnight at 4°C with one of the following antibodies: mouse anti-CD4 (1:100, BD Biosciences), mouse anti-CD8 (1:100, BD Biosciences), mouse anti-CD68 (1:500, Dako, Agilent Technologies), rat anti-CD11b (1:100, Bio-Rad AbD Serotec), or mouse anti-CD169 (1:100, Bio-Rad AbD Serotec). Immune reactions were visualized using biotinylated (1:100, Vector Laboratories) secondary antibodies and diaminobenzidine. Light microscopic images were acquired using an Axiophot 2 microscope (Zeiss) with an attached CCD camera (SPOT Imaging, Diagnostic Instruments). For quantification, immunoreactive profiles were counted in at least five images per section in three non-adjacent sections for each sample using the cell counter plugin in ImageJ (NIH) and related to the area of the images. Samples from CLN2 and CLN3 disease patients showed comparable numbers of immune cells that were increased in comparison with control samples.
Experimental Design and Statistical Analysis
All quantifications and behavioral analyses were performed by investigators unaware of the genotypes and mode of treatment of the respective mice after concealment of genotypes and treatment groups with individual, uniquely coded labels. Animals were randomly placed into experimental or control groups according to genotyping results using a random generator (https://www.randomizer.org). For biometrical sample size estimation, the program G*Power (version 3.1.3) was used.35 Calculation of appropriate sample size groups was performed in a priori power analysis by comparing the mean of two groups with a defined adequate power of 0.8 (1 − β-error) and an α-error of 0.05. To determine the prespecified effect size d, previously published data obtained in Ppt1−/−Rag1+/+ and Ppt1−/−Rag1−/− mice were considered comparable reference values.5 This resulted in prespecified effect sizes of 2.07 for numbers of cresyl violet-stained retinal ganglion cells, 4.30 for numbers of SMI32+ axonal spheroids, 4.18 for quantification of axonal spheroids by electron microscopy, and 2.12 for myoclonic jerks. Statistical analysis was performed using PASW Statistics 18 (SPSS, IBM) software. Shapiro-Wilk test was used to check for normal distribution of data. For multiple comparisons, one-way ANOVA followed by Tukey’s post hoc tests (parametric data) or Kruskal-Wallis tests with Bonferroni correction (non-parametric data) was applied. Values from brain autopsy samples of CLN patients and controls were compared using unpaired two-tailed Student’s t test. p Values considered significant are indicated by asterisks according to the following scheme: *p < 0.05, **p < 0.01, ***p < 0.001. Significant differences of a respective genotype group in comparison with WT mice are indicated above the corresponding bar.
Author Contributions
J.G. and R.M. designed the experiments and wrote the paper. J.G. and K.B. performed the experiments and analyzed the data.
Acknowledgments
The authors are grateful to Guido Stoll and Robert Steinfeld for reading the manuscript and helpful advice and Jon Cooper for valuable discussions. We thank the London Neurodegenerative Disease Brain Bank and Brains for Dementia Research for providing autopsy samples. We thank Heinrich Blazyca, Silke Loserth, and Bettina Meyer for expert technical assistance and Helga Brünner, Jacqueline Schreiber, Anja Weidner, Jennifer Bauer, and Thomas Bimmerlein for attentive care of mice. The work was supported by the Interdisciplinary Centre for Clinical Research (IZKF) of the University of Würzburg (A-302) and the Gemeinnützige Hertie Stiftung (P1150084 to J.G.).
Footnotes
Supplemental Information includes five figures and can be found with this article online at http://dx.doi.org/10.1016/j.ymthe.2017.04.021.
Contributor Information
Janos Groh, Email: groh_j@ukw.de.
Rudolf Martini, Email: rudolf.martini@uni-wuerzburg.de.
Supplemental Information
References
- 1.Radke J., Stenzel W., Goebel H.H. Human NCL Neuropathology. Biochim. Biophys. Acta. 2015;1852(10 Pt B):2262–2266. doi: 10.1016/j.bbadis.2015.05.007. [DOI] [PubMed] [Google Scholar]
- 2.Cooper J.D., Tarczyluk M.A., Nelvagal H.R. Towards a new understanding of NCL pathogenesis. Biochim. Biophys. Acta. 2015;1852(10 Pt B):2256–2261. doi: 10.1016/j.bbadis.2015.05.014. [DOI] [PubMed] [Google Scholar]
- 3.Palmer D.N., Barry L.A., Tyynelä J., Cooper J.D. NCL disease mechanisms. Biochim. Biophys. Acta. 2013;1832:1882–1893. doi: 10.1016/j.bbadis.2013.05.014. [DOI] [PubMed] [Google Scholar]
- 4.Mole S.E., Cotman S.L. Genetics of the neuronal ceroid lipofuscinoses (Batten disease) Biochim. Biophys. Acta. 2015;1852(10 Pt B):2237–2241. doi: 10.1016/j.bbadis.2015.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Groh J., Kühl T.G., Ip C.W., Nelvagal H.R., Sri S., Duckett S., Mirza M., Langmann T., Cooper J.D., Martini R. Immune cells perturb axons and impair neuronal survival in a mouse model of infantile neuronal ceroid lipofuscinosis. Brain. 2013;136:1083–1101. doi: 10.1093/brain/awt020. [DOI] [PubMed] [Google Scholar]
- 6.Groh J., Ribechini E., Stadler D., Schilling T., Lutz M.B., Martini R. Sialoadhesin promotes neuroinflammation-related disease progression in two mouse models of CLN disease. Glia. 2016;64:792–809. doi: 10.1002/glia.22962. [DOI] [PubMed] [Google Scholar]
- 7.Gupta P., Soyombo A.A., Atashband A., Wisniewski K.E., Shelton J.M., Richardson J.A., Hammer R.E., Hofmann S.L. Disruption of PPT1 or PPT2 causes neuronal ceroid lipofuscinosis in knockout mice. Proc. Natl. Acad. Sci. USA. 2001;98:13566–13571. doi: 10.1073/pnas.251485198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Santavuori P., Haltia M., Rapola J. Infantile type of so-called neuronal ceroid-lipofuscinosis. Dev. Med. Child Neurol. 1974;16:644–653. doi: 10.1111/j.1469-8749.1974.tb04183.x. [DOI] [PubMed] [Google Scholar]
- 9.Mitchison H.M., Bernard D.J., Greene N.D., Cooper J.D., Junaid M.A., Pullarkat R.K., de Vos N., Breuning M.H., Owens J.W., Mobley W.C., The Batten Mouse Model Consortium Targeted disruption of the Cln3 gene provides a mouse model for Batten disease. The Batten Mouse Model Consortium [corrected] Neurobiol. Dis. 1999;6:321–334. doi: 10.1006/nbdi.1999.0267. [DOI] [PubMed] [Google Scholar]
- 10.Brinkmann V., Billich A., Baumruker T., Heining P., Schmouder R., Francis G., Aradhye S., Burtin P. Fingolimod (FTY720): discovery and development of an oral drug to treat multiple sclerosis. Nat. Rev. Drug Discov. 2010;9:883–897. doi: 10.1038/nrd3248. [DOI] [PubMed] [Google Scholar]
- 11.Chun J., Brinkmann V. A mechanistically novel, first oral therapy for multiple sclerosis: the development of fingolimod (FTY720, Gilenya) Discov. Med. 2011;12:213–228. [PMC free article] [PubMed] [Google Scholar]
- 12.Melzer N., Meuth S.G. Disease-modifying therapy in multiple sclerosis and chronic inflammatory demyelinating polyradiculoneuropathy: common and divergent current and future strategies. Clin. Exp. Immunol. 2014;175:359–372. doi: 10.1111/cei.12195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Groh J., Stadler D., Buttmann M., Martini R. Non-invasive assessment of retinal alterations in mouse models of infantile and juvenile neuronal ceroid lipofuscinosis by spectral domain optical coherence tomography. Acta Neuropathol. Commun. 2014;2:54. doi: 10.1186/2051-5960-2-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lim M.J., Alexander N., Benedict J.W., Chattopadhyay S., Shemilt S.J., Guérin C.J., Cooper J.D., Pearce D.A. IgG entry and deposition are components of the neuroimmune response in Batten disease. Neurobiol. Dis. 2007;25:239–251. doi: 10.1016/j.nbd.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 15.Seehafer S.S., Ramirez-Montealegre D., Wong A.M., Chan C.H., Castaneda J., Horak M., Ahmadi S.M., Lim M.J., Cooper J.D., Pearce D.A. Immunosuppression alters disease severity in juvenile Batten disease mice. J. Neuroimmunol. 2011;230:169–172. doi: 10.1016/j.jneuroim.2010.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Macauley S.L., Wong A.M., Shyng C., Augner D.P., Dearborn J.T., Pearse Y., Roberts M.S., Fowler S.C., Cooper J.D., Watterson D.M., Sands M.S. An anti-neuroinflammatory that targets dysregulated glia enhances the efficacy of CNS-directed gene therapy in murine infantile neuronal ceroid lipofuscinosis. J. Neurosci. 2014;34:13077–13082. doi: 10.1523/JNEUROSCI.2518-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schulz A., Kohlschütter A., Mink J., Simonati A., Williams R. NCL diseases - clinical perspectives. Biochim. Biophys. Acta. 2013;1832:1801–1806. doi: 10.1016/j.bbadis.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moser H.W. Adrenoleukodystrophies. In: Lazzarini R.A., Griffin J.W., Lassmann H., Nave K.A., Miller R.H., Trapp B.D., editors. Volume 2. Elsevier Academic Press; Amsterdam: 2004. pp. 807–839. (Myelin Biology and Disorders). [Google Scholar]
- 19.Barrette B., Nave K.A., Edgar J.M. Molecular triggers of neuroinflammation in mouse models of demyelinating diseases. Biol. Chem. 2013;394:1571–1581. doi: 10.1515/hsz-2013-0219. [DOI] [PubMed] [Google Scholar]
- 20.Klebe S., Depienne C., Gerber S., Challe G., Anheim M., Charles P., Fedirko E., Lejeune E., Cottineau J., Brusco A. Spastic paraplegia gene 7 in patients with spasticity and/or optic neuropathy. Brain. 2012;135:2980–2993. doi: 10.1093/brain/aws240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bar-Or A., Pachner A., Menguy-Vacheron F., Kaplan J., Wiendl H. Teriflunomide and its mechanism of action in multiple sclerosis. Drugs. 2014;74:659–674. doi: 10.1007/s40265-014-0212-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Asle-Rousta M., Kolahdooz Z., Oryan S., Ahmadiani A., Dargahi L. FTY720 (fingolimod) attenuates beta-amyloid peptide (Aβ42)-induced impairment of spatial learning and memory in rats. J. Mol. Neurosci. 2013;50:524–532. doi: 10.1007/s12031-013-9979-6. [DOI] [PubMed] [Google Scholar]
- 23.Hemmati F., Dargahi L., Nasoohi S., Omidbakhsh R., Mohamed Z., Chik Z., Naidu M., Ahmadiani A. Neurorestorative effect of FTY720 in a rat model of Alzheimer’s disease: comparison with memantine. Behav. Brain Res. 2013;252:415–421. doi: 10.1016/j.bbr.2013.06.016. [DOI] [PubMed] [Google Scholar]
- 24.Wei Y., Yemisci M., Kim H.H., Yung L.M., Shin H.K., Hwang S.K., Guo S., Qin T., Alsharif N., Brinkmann V. Fingolimod provides long-term protection in rodent models of cerebral ischemia. Ann. Neurol. 2011;69:119–129. doi: 10.1002/ana.22186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kraft P., Göb E., Schuhmann M.K., Göbel K., Deppermann C., Thielmann I., Herrmann A.M., Lorenz K., Brede M., Stoll G. FTY720 ameliorates acute ischemic stroke in mice by reducing thrombo-inflammation but not by direct neuroprotection. Stroke. 2013;44:3202–3210. doi: 10.1161/STROKEAHA.113.002880. [DOI] [PubMed] [Google Scholar]
- 26.Schuhmann M.K., Krstic M., Kleinschnitz C., Fluri F. Fingolimod (FTY720) Reduces Cortical Infarction and Neurological Deficits During Ischemic Stroke Through Potential Maintenance of Microvascular Patency. Curr. Neurovasc. Res. 2016;13:277–282. doi: 10.2174/1567202613666160823152446. [DOI] [PubMed] [Google Scholar]
- 27.Deogracias R., Yazdani M., Dekkers M.P., Guy J., Ionescu M.C., Vogt K.E., Barde Y.A. Fingolimod, a sphingosine-1 phosphate receptor modulator, increases BDNF levels and improves symptoms of a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. USA. 2012;109:14230–14235. doi: 10.1073/pnas.1206093109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Katz M.L., Tecedor L., Chen Y., Williamson B.G., Lysenko E., Wininger F.A., Young W.M., Johnson G.C., Whiting R.E., Coates J.R., Davidson B.L. AAV gene transfer delays disease onset in a TPP1-deficient canine model of the late infantile form of Batten disease. Sci. Transl. Med. 2015;7:313ra180. doi: 10.1126/scitranslmed.aac6191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mattapallil M.J., Wawrousek E.F., Chan C.C., Zhao H., Roychoudhury J., Ferguson T.A., Caspi R.R. The Rd8 mutation of the Crb1 gene is present in vendor lines of C57BL/6N mice and embryonic stem cells, and confounds ocular induced mutant phenotypes. Invest. Ophthalmol. Vis. Sci. 2012;53:2921–2927. doi: 10.1167/iovs.12-9662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Metzler B., Gfeller P., Wieczorek G., Li J., Nuesslein-Hildesheim B., Katopodis A., Mueller M., Brinkmann V. Modulation of T cell homeostasis and alloreactivity under continuous FTY720 exposure. Int. Immunol. 2008;20:633–644. doi: 10.1093/intimm/dxn023. [DOI] [PubMed] [Google Scholar]
- 31.Merrill J.E., Hanak S., Pu S.F., Liang J., Dang C., Iglesias-Bregna D., Harvey B., Zhu B., McMonagle-Strucko K. Teriflunomide reduces behavioral, electrophysiological, and histopathological deficits in the Dark Agouti rat model of experimental autoimmune encephalomyelitis. J. Neurol. 2009;256:89–103. doi: 10.1007/s00415-009-0075-3. [DOI] [PubMed] [Google Scholar]
- 32.Nair A.B., Jacob S. A simple practice guide for dose conversion between animals and human. J. Basic Clin. Pharm. 2016;7:27–31. doi: 10.4103/0976-0105.177703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ip C.W., Kroner A., Bendszus M., Leder C., Kobsar I., Fischer S., Wiendl H., Nave K.A., Martini R. Immune cells contribute to myelin degeneration and axonopathic changes in mice overexpressing proteolipid protein in oligodendrocytes. J. Neurosci. 2006;26:8206–8216. doi: 10.1523/JNEUROSCI.1921-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kroner A., Ip C.W., Thalhammer J., Nave K.A., Martini R. Ectopic T-cell specificity and absence of perforin and granzyme B alleviate neural damage in oligodendrocyte mutant mice. Am. J. Pathol. 2010;176:549–555. doi: 10.2353/ajpath.2010.090722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Faul F., Erdfelder E., Lang A.G., Buchner A. G*Power 3: a flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav. Res. Methods. 2007;39:175–191. doi: 10.3758/bf03193146. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.