

Abstract
Aims
Heart failure with preserved ejection fraction (HFpEF) accounts for 30–50% of patients with heart failure (HF). A major obstacle in HF management is the difficulty in differentiating between HFpEF and heart failure with reduced ejection fraction (HFrEF) using conventional clinical and laboratory investigations. The aim of this study is to develop robust transcriptomic and proteomic biomarker signatures that can differentiate HFpEF from HFrEF.
Methods and results
A total of 210 HF patients were recruited in participating institutions from the Alberta HEART study. An expert clinical adjudicating panel differentiated between patients with HFpEF and HFrEF. The discovery cohort consisted of 61 patients, and the replication cohort consisted of 70 patients. Transcriptomic and proteomic data were analysed to find panels of differentiating HFpEF from HFrEF. In the discovery cohort, a 22‐transcript panel was found to differentiate HFpEF from HFrEF in male patients with a cross‐validation AUC of 0.74, as compared with 0.70 for N‐terminal pro‐B‐type natriuretic peptide (NT‐proBNP) in those same patients. An ensemble of the transcript panel and NT‐pro‐BNP yielded a cross‐validation AUC of 0.80. This performance improvement was also observed in the replication cohort. An ensemble of the transcriptomic panel with NT‐proBNP produced a replication AUC of 0.90, as compared with 0.74 for NT‐proBNP alone and 0.73 for the transcriptomic panel.
Conclusions
We have identified a male‐specific transcriptomic biomarker panel that can differentiate between HFpEF and HFrEF. These biosignatures could be further replicated on other patients and potentially be developed into a blood test for better management of HF patients.
Keywords: Heart failure, Heart failure with preserved ejection fraction, Heart failure with reduced ejection fraction, Transcriptomics, Proteomics, Biomarkers
Introduction
Heart failure (HF) is a global epidemic with an increasing economic burden on healthcare utilization.1, 2 The prevalence of HF with preserved ejection fraction (HFpEF), most commonly defined as HF with an ejection fraction (EF) equal to or greater than 40–50%, is progressively increasing and now accounts for approximately 30–50% of patients with HF.1, 3, 4, 5 HFpEF, characterized by stiffness of the left ventricle (LV) and an associated increase in LV filling pressures, is commonly associated with other comorbidities including older age, female sex, and hypertension.3 The signs and symptoms of HFpEF are similar to those of HF with reduced ejection fraction (HFrEF), and thus, guideline recommendations encourage identification of the overall HF syndrome, then proceeding to an imaging modality such as echocardiography to assess LV systolic and diastolic function.
The correct initial diagnosis of HFpEF is paramount because its management differs considerably from HFrEF. According to current recommendations, a diagnosis of HFpEF requires the presence of signs and symptoms of HF, objective evidence of a normal or near normal EF in the absence of significant valvular abnormalities, evidence of abnormal LV relaxation or elevated BNP.3 Unfortunately, such diagnoses often require invasive measurements or cardiac imaging modalities that may not be readily available. Although conventional biomarkers such as B‐type natriuretic peptide (BNP) have demonstrated minimal clinical utility when used as a single biomarker to distinguish between HFpEF and HFrEF (AUC 0.65–0.69),6, 7, 8, 9 multiple biomarker panels utilizing different peptides, micro RNA, metabolomites, and clinical features have yielded improved discriminatory ability.6, 8, 10, 11 However, given that the risk factors for and symptoms of heart failure often differ between males and females, previous work using biomarker panels has not explicitly examined the applicability of biomarkers panels based on sex. Thus, there remains a significant unmet need for readily available and inexpensive diagnostic tests for application in patients with HFpEF.
Interest in the potential use of ‘‐omic’ biomarker panels as diagnostic, predictive, monitoring and prognostic tools in HF has grown.6, 12, 13 While most initial work in this area have focused on diseased cardiac tissue samples, subsequent studies have demonstrated that informative biomarkers can be found in peripheral blood and that blood and cardiac tissue may show comparable transcriptomic (gene expression) and proteomic profiles.14, 15 Because the pathobiology of HF reflects a complex array of underlying molecular perturbations, ‘multimarker’ approaches such as measurement of both the whole blood transcriptome and plasma proteome as a comprehensive strategy may yield deeper insights into HF while revealing new signatures that reflect cardiac function. Hence, the current study is intended to assess the potential for blood‐based ‘‐omic’ biomarkers to differentiate HFpEF and HFrEF in a clinical setting, and to determine whether there are sex‐specific differences in these biomarker panels.
Methods
Patient population
Following informed consent, patients were recruited into the study from two tertiary referral centres in Alberta, Canada as part of the Alberta Heart Failure Etiology and Analysis (Alberta HEART) initiative.16 Ethics approval was obtained from the local ethics boards of the respective study institutions. Exclusion criteria in the trial included age <18 years, known malignancy with expected survival <1 year, current pregnancy or recent pregnancy <6 months prior, recent hospitalization (<2 weeks after acute coronary syndrome, HF or other admission) and severe aortic or mitral stenosis. Using the 2013 ACCF/AHA HF guidelines, an expert clinical adjudication panel classified patients into either HFpEF and HFrEF groups.1 Specifically from an echocardiographic standpoint, patients with EF ≥ 50% were labelled HFpEF, and patients with EF ≤40% were labelled HFrEF. All EF measurements were visually estimated by qualified echocardiographers. Data were collected and stored in the Alberta HEART Registry. The first 61 participants in the study comprised the discovery cohort, while the next 70 participants comprised the replication cohort. The remaining 79 participants were enrolled in the Alberta HEART Registry had an intermediate EF (>40%–<50%), were healthy age‐matched controls or were at high risk for developing HF but had no history of clinically relevant HF.
Proteomic
Blood samples were collected in P100 plasma tubes (BD, Franklin Lake, NJ) and stored on ice until processing. Blood was spun down within 2 hours of collection, and plasma was collected and stored at −80°C until selected for proteomic analysis. Discovery and replication patient plasma samples were analysed by multiple reaction monitoring mass spectrometry (MRM‐MS) at the University of Victoria Genome BC Proteomic Centre (Victoria, BC, Canada). The semi‐targeted MRM‐MS assay measures 306 peptides corresponding to 132 proteins. These were proteins found to be related to HF, HFpEF or HFrEF from literature review17 or from a pilot study in which we analysed the discovery samples using a non‐targeted iTRAQ‐LC‐MALDI‐TOF/TOF mass spectrometry method (data not shown).
Transcriptomic
Gene expression was measured in whole blood of the discovery cohort samples and the replication samples. PAXgene (PreAnalytiX, Hombrechtikon, Switzerland) whole blood samples were collected, stored at −80°C and subsequently shipped on dry ice to Scripps Research Institute (La Jolla, CA) for analysis. Microarray analyses were performed using Affymetrix GeneChip Human Gene 1.1 ST Chips (Santa Clara, CA).
B‐type natriuretic peptide measurement
N‐terminal proBNP (NT‐proBNP) from plasma samples was analysed at the Prevention of Organ Failure Centre of Excellence in Vancouver, BC using the point‐of‐care RAMP® assay (Response Biomedical Corp, Vancouver, BC). This assay has been validated against the Roche Diagnostics Elecsys NT‐ProBNP assay with 99% concordance (95% CI 97–100%) in subjects with HF.18
Statistical analyses
Transcriptomic and proteomic data were analysed using R and Bioconductor.
For transcriptomic data, Factor Analysis for Robust Microarray Summarization was applied to filter out transcripts with inconsistent relative probe intensity levels and summarize probe intensities into transcript expression data. These transcripts were analysed for differential expression between HFpEF and HFrEF, using a moderated t‐test (limma). Probe sets with false discovery rate (FDR) < 0.05 were selected, and a biomarker panel was then identified by performing an elastic net classification on the selected transcripts (glmnet).
Proteomic data were pre‐processed to remove peptides that did not pass quality control metrics or had >25% missing values. Protein expression data were created by taking the log 2 of the relative ratios of sample peptide abundance to SIS peptide abundance and summarizing these at the protein‐level. Significantly, differentially expressed proteins (P < 0.1) were analysed, selected and combined in a biomarker panel, in a similar fashion as the transcriptomic data.
The performance of each classifier was estimated using repeated 10‐fold cross validation (CV). The proteomic classifier with the best CV area under the ROC curve (AUC) was tested in the replication cohort.
Transcriptomic and proteomic biomarker panels were also combined with NT‐proBNP in ensemble classifiers. Ensembling of classifiers was done by taking averages of the probabilities predicted by different models.19
Effect of cellular composition on peripheral whole blood gene expression
A large proportion of the variation observed in whole blood gene expression data can be accounted for by differences in the cellular composition of the underlying samples,20 and it is therefore desirable to quantify this heterogeneity. In order to assess the effect of cellular composition of the underlying samples on our transcriptomic signature, we first used a statistical model to impute the cell proportions of all samples from their gene expression data. We assessed whether significant differences in cell proportions existed between HFpEF and HFrEF groups, and quantified the extent to which the observed variation in gene expression could be accounted for by cellular composition of the samples (Supporting Information Figure S1 ).21 We also evaluated whether any blood cell‐specific gene sets were over‐represented in any of our identified transcriptomic signatures using data derived from public gene expression datasets. Finally, we generated biomarker panels, using the process outlined above, using the cell proportion data only to demonstrate that the transcriptomic signature provides additional information beyond that provided by the cellular composition alone. In addition, we also combined into an ensemble, the cell proportion‐ and transcriptomics‐based panels.
Functional enrichment
To identify key biological processes represented by the biomarker panels, functional enrichment of significant genes and proteins was performed using MetaCore (Thomson Reuters). Process networks with a FDR < 0.05 were considered enriched.
Results
Patient population
Baseline demographics for the discovery and replication cohorts are summarized in Table 1. There were a total of 61 patients in the discovery cohort (34 HFrEF, 27 HFpEF) and 69 patients in the replication cohort (48 HFrEF, 21 HFpEF).
Table 1.
Baseline demographics—discovery and replication cohorts
| Discovery | Replication | ||||
|---|---|---|---|---|---|
| HFpEF (n=27) | HFrEF (n=34) | HFpEF (n=21) | HFrEF (n=48) | P‐value | |
| Age (years) | 71.0 [61.5, 75.0] | 63.0 [56.0, 69.0] | 70.0 [63.0, 79.0] | 66.0 [58.8, 72.5] | 0.012 |
| Sex (male) | 21 (77.8%) | 23 (67.6%) | 10 (47.6%) | 35 (72.9%) | 0.124 |
| BMI | 33.8 [29.9, 36.7] | 29.7 [26.6, 34.4] | 32.0 [27.6, 34.8] | 29.4 [26.3, 34.1] | 0.08 |
| Ethnicity | 0.72 | ||||
| Aboriginal | 3 (11.1%) | 2 (5.9%) | 1 (4.8%) | 1 (2.1%) | |
| Caucasian | 22 (81.5%) | 28 (82.4%) | 17 (81%) | 43 (89.6%) | |
| Other | 2 (7.4%) | 4 (11.8%) | 3 (14.3%) | 4 (8.3%) | |
| Aetiology | 0.001 | ||||
| Alcoholic | 1 (3.7%) | 0 (0%) | 1 (4.8%) | 3 (6.2%) | |
| Diabetic | 2 (7.4%) | 1 (2.9%) | 1 (4.8%) | 0 (0%) | |
| Idiopathic | 14 (51.9%) | 12 (35.3%) | 16 (76.2%) | 24 (50%) | |
| Ischaemic | 6 (22.2%) | 20 (58.8%) | 3 (14.3%) | 21 (43.8%) | |
| Other | 4 (14.8%) | 1 (2.9%) | 0 (0%) | 0 (0%) | |
| Smoker | 0.087 | ||||
| Current | 5 (18.5%) | 2 (5.9%) | 1 (4.8%) | 3 (6.2%) | |
| Former | 16 (59.3%) | 17 (50%) | 12 (57.1%) | 17 (35.4%) | |
| Never | 4 (14.8%) | 13 (38.2%) | 8 (38.1%) | 27 (56.2%) | |
| Unknown | 2 (7.4%) | 2 (5.9%) | 0 (0%) | 1 (2.1%) | |
| Diabetes | 10 (37%) | 8 (23.5%) | 10 (47.6%) | 21 (43.8%) | 0.264 |
| Dyslipidemia | 14 (51.9%) | 15 (44.1%) | 7 (33.3%) | 21 (43.8%) | 0.637 |
| Hypertension | 20 (74.1%) | 19 (55.9%) | 15 (71.4%) | 23 (47.9%) | 0.064 |
| Atrial fibrillation | 12 (44.4%) | 9 (26.5%) | 11 (52.4%) | 25 (52.1%) | 0.106 |
| Systolic BP (mmHg) | 135 [124, 146] | 113 [100, 124] | 126 [120, 131] | 112.0 [104, 1312] | <0.001 |
| Diastolic BP (mmHg) | 75 [61, 83] | 70 [64, 77] | 70 [70, 77] | 71 [62, 79] | 0.633 |
| LVEF (%) | 62 [58, 66] | 28 [26, 33] | 60 [56, 62] | 30 [23, 36] | <0.001 |
| NT‐proBNP (ng/L) | 416 [130, 1327] | 1422 [637, 1992] | 295 [143, 1550] | 1174 [401, 2516] | 0.09 |
| BNP (pg/mL) | 72 [42, 281] | 199 [94, 417] | 75 [48, 200] | 202 [81, 385] | 0.017 |
| Creatinine (umol/L) | 102 [78‐141] | 97.0 [84.0, 116.0] | 93.0 [69.0, 123.0] | 89.5 [78.0, 113.0] | 0.407 |
| NYHA class | 0.609 | ||||
| I | 4 (14.8%) | 5 (14.7%) | 3 (14.3%) | 8 (16.7%) | |
| II | 14 (51.9%) | 15 (44.1%) | 13 (61.9%) | 27 (56.2%) | |
| III | 9 (33.3%) | 11 (32.4%) | 5 (23.8%) | 13 (27.1%) | |
| IIII | 0 (0%) | 2 (5.9%) | 0 (0%) | 0 (0%) | |
| Unknown | 0 (0%) | 1 (2.9%) | 0 (0%) | 0 (0%) | |
| Pharmacotherapy | |||||
| Ace‐inhibitor | 16 (59.3%) | 24 (70.6%) | 12 (57.1%) | 31 (64.6%) | 0.666 |
| ARB | 8 (29.6%) | 8 (23.5%) | 10 (47.6%) | 14 (29.2%) | 0.324 |
| Beta Blocker | 20 (74.1%) | 32 (94.1%) | 12 (57.1%) | 46 (95.8%) | <0.001 |
| MRA | 3 (11.1%) | 13 (38.2%) | 0 (0%) | 26 (54.2%) | <0.001 |
| Diuretic | 20 (74.1%) | 15 (44.1%) | 17 (81%) | 39 (81.2%) | 0.002 |
| Digoxin | 4 (14.8%) | 4 (11.8%) | 1 (4.8%) | 6 (12.5%) | 0.716 |
| ASA | 17 (63%) | 22 (64.7%) | 11 (52.4%) | 28 (58.3%) | 0.693 |
| Anticoagulant | 11 (40.7%) | 12 (35.3%) | 11 (52.4%) | 21 (43.8%) | 0.714 |
| Statin | 20 (74.1%) | 20 (58.8%) | 16 (76.2%) | 31 (64.6%) | 0.443 |
| Cardiac Device | 0.528 | ||||
| Brady | 0 (0%) | 0 (0%) | 0 (0%) | 1 (2.1%) | |
| ICD | 1 (3.7%) | 9 (26.5%) | 0 (0%) | 7 (14.6%) | |
| ICD‐CRT | 0 (0%) | 2 (5.9%) | 0 (0%) | 0 (0%) | |
| None | 26 (96.3%) | 23 (67.6%) | 21 (100%) | 40 (83.3%) | |
| Cell Proportions | |||||
| Granulocytes | 65.72 | 62.23 | 67.15 | 62.13 | 0.064 |
| Monocytes | 8.00 | 8.22 | 7.61 | 8.33 | 0.511 |
| B Lymphocytes | 5.61 | 6.25 | 5.85 | 6.22 | 0.301 |
| CD4+ T Lymphocytes | 11.93 | 14.15 | 11.85 | 13.77 | 0.007 |
| CD8+ T Lymphocytes | 4.71 | 5.56 | 4.69 | 5.64 | 0.212 |
| NK Lymphocytes | 7.14 | 7.27 | 6.32 | 7.53 | 0.764 |
ARB, angiotensin receptor blockers; ASA, acetylsalicylic acid; BMI, body mass index; BNP, B‐type natriuretic peptide; CRT, cardiac resynchronization therapy; HFpEF, heart failure with preserved ejection fraction; HFrEF, heart failure with reduced ejection fraction; ICD, implantable cardioverter defibrillator; LVEF, left ventricular ejection fraction; MRA, mineralocorticoid receptor antagonists; NT‐proBNP, N‐terminal proBNP.
Numeric variables are reported as median and interquartile range.
P‐values are calculated based on 2‐way ANOVA with interaction for numeric variables, and a chi‐square test for categorical variables. Cell proportions were imputed from gene expression profiles, where available (discovery 33 HFrEF vs. 27 HFpEF; replication 35 HFrEF vs. 9 HFpEF).
In the discovery cohort, HFpEF patients were older (70 vs. 65 years, P = 0.047), more likely to have diabetes (19% vs. 3%, P = 0.039), and less likely to have an ischaemic aetiology for HF (22% vs. 62%, P = 0.009) compared with the HFrEF patients. HFpEF patients had higher systolic BP (130 mmHg vs. 100 mmHg, P = 0.006) and LVEF (61% vs. 30%, P < 0.001) and lower NT‐proBNP (416 ng/L vs. 1422 ng/L, P = 0.006) and BNP (72 pg/mL vs. 199 pg/mL, P = 0.029) compared with HFrEF patients. HFrEF patients were more likely to be on beta blockers (BB) (94.1% vs. 74.1%, P < 0.001) and mineralocorticoid receptor antagonists (MRA) (38.2% vs. 11.1%, P < 0.001) than HFpEF patients, whereas the use of angiotensin converting enzyme inhibitor (ACEi) and angiotensin receptor blockers (ARB) was similar.
In the replication cohort, HFpEF patients tended to be older (71 vs. 67 years, P = 0.11) and were less likely to have ischaemic aetiology for HF (19% vs. 45%, P = 0.11) compared with HFrEF patients. There was no significant difference in systolic BP between the two groups but the HFpEF group had higher LVEF (60% vs. 29%, P < 0.001) and lower NT‐proBNP (538 ng/L vs. 1224 ng/L, P = 0.018), BNP (82 pg/mL vs. 210 pg/mL, P = 0.049), and CD4+ T lymphocyte proportions, compared with HFrEF patients. HFrEF patients were more likely to be on BB (95.8% vs. 57.1.1%, P < 0.001) and MRA (54.2% vs. 0.0%, P < 0.001) than HFpEF patients, whereas the use of ACEi and ARB was similar.
Discovery analysis
Transcriptomic
To find discriminative markers, we focused biomarker discovery efforts on patients with EF ≥ 50% (HFpEF) and with EF ≤ 40% (HFrEF), and patients with intermediate EF (>40% to <50%) were excluded. In the discovery cohort comprising of 61 patients with HFrEF or HFpEF, there were 61 differentially expressed transcripts with FDR < 0.05. T lymphocyte‐specific genes were significantly over‐represented in these transcripts (Figure S2A ). These transcripts were subjected to an elastic net analysis which subsequently yielded a 26 transcript diagnostic classifier for distinguishing between HFpEF and HFrEF. When compared with HFrEF, 10 transcripts were up‐regulated in HFpEF, and 16 were down‐regulated (Table 2). The cross‐validation AUC of this classifier was 0.62. The best performing model derived using cell proportion data alone achieved equivalent cross‐validation AUC of 0.62. To identify enriched biological pathways, the differentially expressed transcripts were also subjected to gene enrichment analyses using MetaCore (Thomson Reuters). The enriched pathways are highlighted in Figure 1. Key pathways upregulated in HFpEF as compared with HFrEF involve Notch signalling, the Sin3 and NuRD histone dacetylase complexes, and the epigenetic gene silencing protein N‐Cor/SMRT. Downregulated pathways include well‐known signalling pathways involving the chemokine G‐protein coupled receptor CXCR‐4, and cell growth, survival and differentiation regulators such as Signal Transduction and Transcription proteins, phospholipase C (PLC), PI3K/AKT and mitogen‐activated protein kinase.
Table 2.
Annotated differentially expressed transcripts (FDR < 0.05) for HFpEF vs. HFrEF
| Gene symbol | Gene name | Fold change | Direction | FDR |
|---|---|---|---|---|
| TMEM204 | transmembrane protein 204 | 1.26 | down | 0.04 |
| PBX1 | pre‐B‐cell leukaemia homeobox 1 | 1.42 | up | 0.04 |
| SPOCK2 | sparc/osteonectin, cwcv and kazal‐like domains proteoglycan (testican) 2 | 1.26 | down | 0.04 |
| FAM102A | family with sequence similarity 102, member A | 1.28 | down | 0.04 |
| CD5 | CD5 molecule | 1.22 | down | 0.04 |
| LEF1 | lymphoid enhancer‐binding factor 1 | 1.37 | down | 0.04 |
| ITK | IL2‐inducible T‐cell kinase | 1.26 | down | 0.04 |
| RNF11 | ring finger protein 11 | 1.29 | up | 0.04 |
| PIK3IP1 | phosphoinositide‐3‐kinase interacting protein 1 | 1.16 | down | 0.04 |
| NELL2 / LOC100653255 / LOC100653018 | NEL‐like 2 (chicken) / uncharacterized LOC100653255 / uncharacterized LOC100653018 | 1.39 | down | 0.04 |
| PDK4 | pyruvate dehydrogenase kinase, isozyme 4 | 1.32 | up | 0.04 |
| SERINC5 | serine incorporator 5 | 1.18 | down | 0.05 |
| C6orf25 | chromosome 6 open reading frame 25 | 1.36 | up | 0.05 |
| TCF7 | transcription factor 7 (T‐cell specific, HMG‐box) | 1.25 | down | 0.05 |
| HIST2H4B / HIST4H4 / HIST2H4A / HIST1H4L / HIST1H4E / HIST1H4B / HIST1H4H / HIST1H4C / HIST1H4J / HIST1H4K / HIST1H4F / HIST1H4D / HIST1H4A / HIST1H4I | histone cluster 2, H4b / histone cluster 4, H4 / histone cluster 2, H4a / histone cluster 1, H4l / histone cluster 1, H4e / histone cluster 1, H4b / histone cluster 1, H4h / histone cluster 1, H4c / histone cluster 1, H4j / histone cluster 1, H4k / histone cluster 1, H4f / histone cluster 1, H4d / histone cluster 1, H4a / histone cluster 1, H4i | 1.13 | up | 0.05 |
| IL7R | interleukin 7 receptor | 1.26 | down | 0.05 |
| CD6 | CD6 molecule | 1.26 | down | 0.05 |
| RIOK3 | RIO kinase 3 (yeast) | 1.29 | up | 0.05 |
| IGF2BP2 | insulin‐like growth factor 2 mRNA binding protein 2 | 1.44 | up | 0.05 |
| C6orf25 | chromosome 6 open reading frame 25 | 1.35 | up | 0.05 |
| TESPA1 | thymocyte expressed, positive selection associated 1 | 1.20 | down | 0.05 |
| CD3G | CD3g molecule, gamma (CD3‐TCR complex) | 1.32 | down | 0.05 |
| VSIG1 | V‐set and immunoglobulin domain containing 1 | 1.28 | down | 0.05 |
| KAT2B | K(lysine) acetyltransferase 2B | 1.19 | up | 0.05 |
| TCP11L2 | t‐complex 11 (mouse)‐like 2 | 1.18 | up | 0.05 |
| RCAN3 | RCAN family member 3 | 1.22 | down | 0.05 |
| BNIP3L | BCL2/adenovirus E1B 19kDa interacting protein 3‐like | 1.09 | up | 0.05 |
| LDLRAP1 | low density lipoprotein receptor adaptor protein 1 | 1.17 | down | 0.05 |
| MXI1 | MAX interactor 1 | 1.29 | up | 0.05 |
| TRABD2A | TraB domain containing 2A | 1.25 | down | 0.05 |
| CCR7 | chemokine (C‐C motif) receptor 7 | 1.42 | down | 0.05 |
| MMP8 | matrix metallopeptidase 8 (neutrophil collagenase) | 1.74 | up | 0.05 |
| CCNDBP1 | cyclin D‐type binding‐protein 1 | 1.09 | up | 0.05 |
| RMND5A | required for meiotic nuclear division 5 homolog A (S. cerevisiae) | 1.12 | up | 0.05 |
| ABCD2 | ATP‐binding cassette, sub‐family D (ALD), member 2 | 1.17 | down | 0.05 |
| ZDHHC2 | zinc finger, DHHC‐type containing 2 | 1.20 | up | 0.05 |
| STOM | stomatin | 1.14 | up | 0.05 |
| DGKA | diacylglycerol kinase, alpha 80kDa | 1.14 | down | 0.05 |
| EIF4B / LOC100653227 | eukaryotic translation initiation factor 4B / uncharacterized LOC100653227 | 1.13 | down | 0.05 |
| GLTSCR2 | glioma tumour suppressor candidate region gene 2 | 1.11 | down | 0.05 |
| ARHGEF12 | Rho guanine nucleotide exchange factor (GEF) 12 | 1.31 | up | 0.05 |
| CD27 | CD27 molecule | 1.39 | down | 0.05 |
| YOD1 | YOD1 OTU deubiquinating enzyme 1 homolog (S. cerevisiae) | 1.36 | up | 0.05 |
| ST6GAL1 | ST6 beta‐galactosamide alpha‐2,6‐sialyltranferase 1 | 1.11 | down | 0.05 |
| PLCG1 | phospholipase C, gamma 1 | 1.10 | down | 0.05 |
| ELOVL7 | ELOVL fatty acid elongase 7 | 1.26 | up | 0.05 |
| IGHV3‐33 | immunoglobulin heavy variable 3‐33 | 1.42 | down | 0.05 |
| TMEM63A | transmembrane protein 63A | 1.09 | down | 0.05 |
| SIT1 | signalling threshold regulating transmembrane adaptor 1 | 1.18 | down | 0.05 |
FDR, false discovery rate; HFpEF, heart failure with preserved ejection fraction; HFrEF, heart failure with reduced ejection fraction.
Figure 1.

The 10 most statistically significant pathway maps identified by MetaCore, based on all differentially expressed transcripts [false discovery rate < 0.05] for heart failure with preserved ejection fraction vs. heart failure with reduced ejection fraction.
Proteomic
The plasma protein levels in the 61 discovery samples analysed using a semi‐targeted multiple‐reaction monitoring mass spectrometry assay consisted of 306 peptides that corresponded to 132 proteins. These proteins consisted of relevant HF proteins identified from either the literature or from a non‐targeted proteomic17 study on this discovery cohort (data not shown). Of the 306 peptides, 123 passed quality control. These corresponded to 73 unique proteins. There were 10 proteins statistically different between HFpEF and HFrEF patients (Table S1 ). These were subjected to elastic net analysis, which yielded a 9 protein classifier, and the cross‐validation AUC of this classifier was 0.58.
Natriuretic peptides
NT‐proBNP had a cross‐validation AUC of 0.69 in differentiating between HFpEF and HFrEF subjects. Ensembles of NT‐proBNP with transcriptomic (cross‐validation AUC = 0.61) or proteomic (cross‐validation AUC = 0.56) panels did not improve performance compared with NT‐proBNP alone.
Sex‐specific biomarker panels
Given that the risks, associations and symptoms for HF often differ between males and females, we performed discovery analyses in males and females separately in both the transcriptomic and proteomic data. Since the transcript biomarker panel was not discriminatory between HFrEF and HFpEF in the female‐specific discovery cohort, we focused on proteomic data for the female‐specific analyses.
An analysis of only male patients yielded a 22‐transcript biomarker panel (Table S2 ) with a cross‐validation AUC of 0.74. Comparatively, the best performing model derived using cell proportion data only achieved a cross‐validation AUC of 0.63. Of the 22 transcripts, 8 were up‐regulated, and 14 were down‐regulated in HFpEF compared with HFrEF. T lymphocyte‐specific genes were significantly over‐represented in these transcripts (Figure S2B ). MetaCore analysis was performed on the list of transcripts that were differentially expressed in the male‐only analysis, to look for unique pathways (Figure 2). No proteomic panel with an AUC > 0.70 was found.
Figure 2.

The three pathway maps identified as statistically significant (false discovery rate < 0.05) by MetaCore, based on differentially expressed transcripts (false discovery rate < 0.05) for heart failure with preserved ejection fraction vs. heart failure with reduced ejection fraction in male patients that are not differentially expressed in all patients.
When ‘omics’‐based classifiers were ensembled with NT‐proBNP, performance improvements were seen in the male‐specific 22‐transcript transcriptomic based biomarker panel, with a cross‐validation AUC of 0.80. In comparison, NT‐proBNP alone had an AUC of 0.70 in these same patients. The ensemble of the cell proportion‐based and transcriptomics‐based panels had a 0.70 cross‐validation AUC.
In females, a panel of seven proteins (Table S3 ) was found with a cross‐validation AUC of 0.71. Comparatively, the best performing model derived using cell proportion data only achieved a cross‐validation AUC of 0.44. Of the seven proteins, six were up‐regulated and one was down‐regulated in HFpEF compared with HFrEF.
NT‐proBNP alone had an AUC of 0.50 in these patients. The ensemble of the female‐specific proteomic panel with NT‐proBNP resulted in a performance improvement to a cross‐validation AUC of 0.69, but when combined with the cell proportion panel, the AUC decreased to 0.48.
Replication analysis
In the replication cohort comprised of 70 subjects, we tested the male‐specific NT‐proBNP and transcriptomic ensemble, and the female‐specific protein classifier, both of which had good discovery cross‐validation AUCs. The male‐specific proteomic panel and female‐specific transciptomic panel were not pursued given their lack of discriminatory ability in the discovery cohort.
The male‐specific transcriptomic panel had a replication AUC of 0.73 compared with NT‐proBNP at 0.74 (10 HFpEF vs. 35 HFrEF male patients) and cell proportions alone at 0.65. The ensemble of cell proportion and transcriptomic panel had a test AUC of 0.67. However, the ensemble of NT‐proBNP with the transcriptomic panel yielded a replication AUC of 0.90 (Figure 3).
Figure 3.
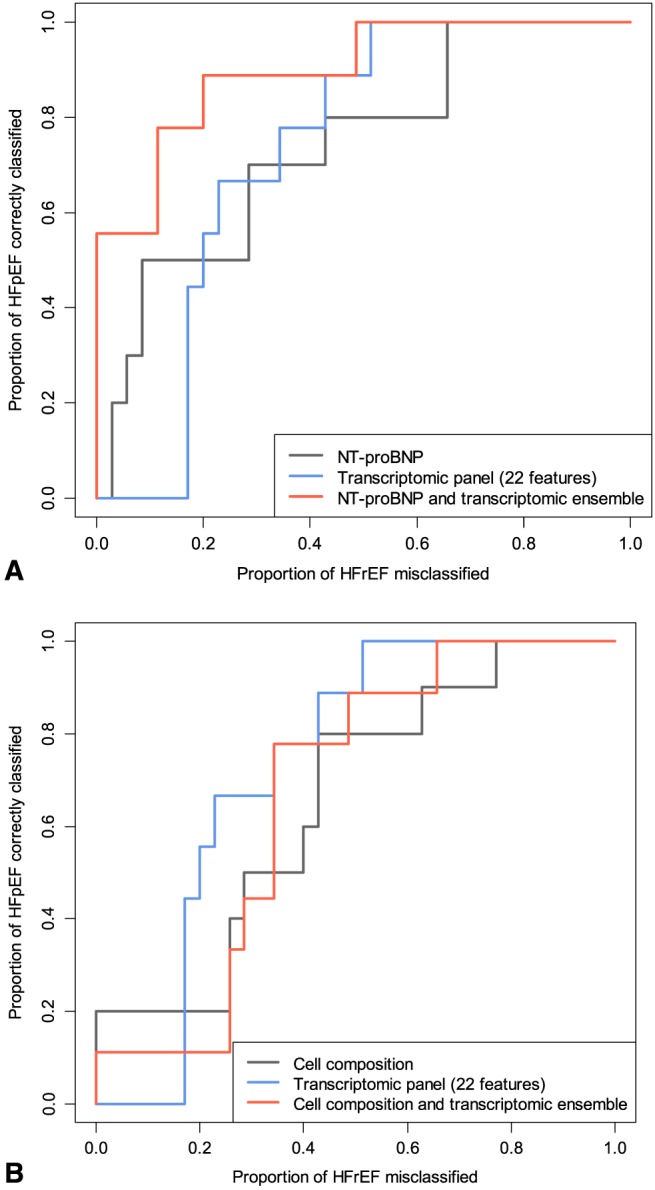
Replication receiver operating characteristic curves of male‐specific classifiers. (A) Performance of the transcriptomic panel, N‐terminal proBN (NT‐proBNP), and their ensemble; (B) Performance of the transcriptomic panel, cell proportion model, and their ensemble. HFpEF, heart failure with preserved ejection fraction; HFrEF, heart failure with reduced ejection fraction.
The female‐specific proteomic panel had a replication AUC of 0.66 in 11 HFpEF vs. 13 HFrEF female patients, compared with the AUC of 0.44 for NT‐proBNP alone (Figure 4). The ensemble of NT‐proBNP with the proteomic panel yielded a replication AUC of 0.62.
Figure 4.
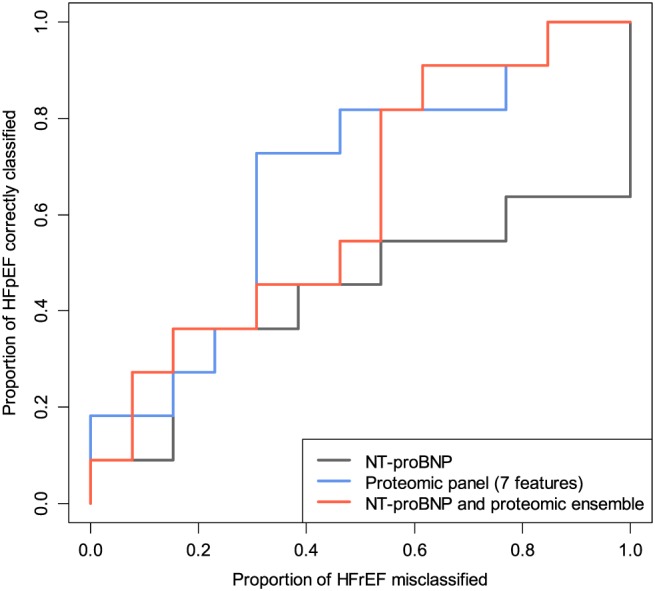
Replication receiver operating characteristic curves of female‐specific classifiers. HFpEF, heart failure with preserved ejection fraction; HFrEF, heart failure with reduced ejection fraction; NT‐proBNP, N‐terminal proBN.
Discussion
In this study, we examined the potential for blood‐based mRNA and protein multi‐marker panels in improving the differentiation of patients with HFpEF from those with HFrEF. The findings indicate that transcriptomic classifier panels could reasonably differentiate between HFpEF and HFrEF, yet, they did not enhance the diagnostic ability of the natriuretic peptide, NT‐proBNP, in the full discovery cohort. Although proteomic panels did not achieve a strong diagnostic performance in differentiating HFpEF from HFrEF in the overall discovery cohort, we identified a male‐specific transcriptomic panel that performed well in the replication cohort. A male‐specific ensemble classifier consisting of the transcriptomic signatures and NT‐proBNP was also found to have strong performance in differentiating between HFpEF and HFrEF.
Moreover, while systematic differences in CD4+ T lymphocyte abundances between HFpEF and HFrEF patients may have impacted both differential expression and biomarker signature discovery, the male‐specific transcriptomic classifier outperformed a cellular composition classifier, suggesting that the component features of the transcriptomic classifier are more than simple surrogates of cell proportions, at least in males.
HFpEF and HFrEF are typically difficult to differentiate in a clinical setting without the assistance of imaging modalities. Further, it has also been noted that natriuretic peptides, a mainstay of the diagnostic armamentarium of HF, is not useful in differentiating between HFpEF and HFrEF. By analysing differentially expressed transcriptomic and proteomic biosignatures, the current study provides insight into the biological mechanisms and pathways which make each heart failure phenotype its own distinct clinical entity.
A number of different proteins and transcripts were noted to be differentially expressed in HFpEF and HFrEF (Table S4 ), including modulators of inflammation.22 This is consistent with prior evidence highlighting that inflammatory pathways play an important role in the progression of HF.23 Furthermore, a number of components from the complement pathway were also expressed in HFpEF vs. HFrEF. An increasing body of evidence supports a functional role for complement activation in the pathogenesis of cardiovascular disease through pleiotropic effects on endothelial and haematopoietic cell function and haemostasis.24 These effects in particular, influence the development of atherosclerosis, thrombosis and inflammation.24
Another protein found to be differentially expressed in our cohort was galectin‐3 binding protein. Galectin‐3 has been studied extensively as a biomarker in HF and has an important role in the profibrotic process and diastolic dysfunction of the ventricle.25 It is uncertain why galectin‐3 was downregulated instead of being upregulated in female HFpEF subjects in our discovery cohort but possible reasons could include concomitant use of pharmacological agents with antifibrotic properties such as spironolactone, which may suppress Galectin‐3.25
It is not surprising that there was a sex‐related difference in both the transcriptomic and proteomic marker panels. Other biomarkers have also been shown to have different discriminatory power in men and women.26 The distribution of BNP has been shown to be higher in women compared with men, in both healthy subjects as well as those with heart failure.27 Also, concentrations of galactin‐3 levels have been shown to be higher in women compared with men in population‐based studies, whereas soluble ST2 and growth differentiation factor‐15 have been shown to be lower in women compared with men.28 Although the exact role sex plays in cardiac diseases remains to be fully understood, what is known is that there are fundamental intrinsic sex differences in cardiac tissue.29 These differences may include variable ion channel expression and diverse responses to sex hormonal regulations via long term genomic and acute non‐genomic pathways.29, 30 Other explanations for these sex‐related differences may be derived from previous studies which have proposed that sex hormones may induce epigenetic change, thereby predisposing males and females differently to non‐Mendelian complex diseases.31
Overall, our data have identified a blood‐based transcriptomic and proteomic panel classifiers in separating HFpEF and HFrEF. Separate panels could be considered for each sex in view of the differences seen in this study. The ultimate validation and introduction of these panels in health care could be particularly useful in the setting of primary care or where technical/imaging expertise in diagnosing HFpEF are not readily available. However, further transcriptomic replication work is required and validation in a bigger population sample before clinical implementation is considered.
Limitations in the study include the fact that sample sizes were comparatively small. However, in order to accommodate the smaller sample size, robust statistical techniques were applied in the discovery of discriminative transcriptomic and proteomic marker sets. Our discovery observations were supported by correspondent replication data, and thus reproducible classifier panels. Another potential limitation is that the recruited population may not be reflective of a ‘true’ population‐based HFpEF cohort as there were more males than females with relatively low BNP/NT‐proBNP in our study. Hence, it is important that the panels discovered in this work are validated prospectively in a larger cohort of patients, which reflect a ‘real world’ HFpEF population. The analysis performed and presented focused on the two ‘extreme’ phenotypes of interest, as per ESC guidelines. Given we would expect more overlap in the intermediate EF group, biomarker discovery could be confounded in this group of patients. Future analyses will focus on assessing the ability of a biomarker panel to assess or predict LVEF as a continuous variable. Further, the cohort of patients included in this analysis was all stable ambulatory HF patients. The results may not apply to those with acute decompensated HF. Also, it is well established that there is overlap between the HFrEF and HFpEF; however, we tried to address this issue by excluding the intermediate EF patients to identify more discrete HF phenotypes with HFpEF and HFrEF. Additionally, while some of these biological pathways may be relevant in the development of HFpEF, it is important to take into account that there was a trend towards a higher lymphocyte count in the HFrEF group which may affect the validity of our findings.
In conclusion, this study provides provisional indications of a viable diagnostic approach using blood‐based ‘‐omic’ biomarker panels in combination with natriuretic peptides in differentiating between HFpEF and HFrEF. We showed specific sex differences in transcriptomic and proteomic panels in patients with these distinct HF phenotypes. In particular, transcriptomic panels had better diagnostic ability in males in differentiating between HFpEF and HFrEF and these results were replicated in our analysis. Further transcriptomic replication work and validation in larger observational trials will be required prior to consideration for implementation in a clinical setting.
Conflict of interest
None declared.
Supporting information
Table S1. Differentially expressed proteins between HFpEF and HFrEF.
Table S2. Annotated transcripts in the male‐specific transcriptomic biomarker panel for HFpEF versus HFrEF.
Table S3. Female‐specific proteomic biomarker panel for HFpEF versus HFrEF.
Table S4. Molecular and Clinical effects of transcripts and protiens that were noted to be differentially expressed in HFpEF and HFrEF.
Figure S1. Eigen‐R2 analysis reveals strong association between transcriptomic data and cellular composition
Figure S2. Evidence of cell‐specificity in all differentially expressed transcripts (FDR <0.05; A), or the male‐specific transcriptomic panel (B), for HFpEF versus HFrEF
Toma, M. , Mak, G. J. , Chen, V. , Hollander, Z. , Shannon, C. P. , Lam, K. K. Y. , Ng, R. T. , Tebbutt, S. J. , Wilson‐McManus, J. E. , Ignaszewski, A. , Anderson, T. , Dyck, J. R. B. , Howlett, J. , Ezekowitz, J. , McManus, B. M. , and Oudit, G. Y. (2017) Differentiating heart failure phenotypes using sex‐specific transcriptomic and proteomic biomarker panels. ESC Heart Failure, 4: 301–311. doi: 10.1002/ehf2.12136.
Institutions where work was performed: University of Alberta, Univerity of British Columbia, and Centre of Excellence for Prevention of Organ Failure (PROOF Centre)
Contributor Information
Justin Ezekowitz, Email: justin.ezekowitz@ualberta.ca.
Bruce M. McManus, Email: bruce.mcmanus@hli.ubc.ca
Gavin Y. Oudit, Email: gavin.oudit@ualberta.ca
References
- 1. Writing Committee M , Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE, Jr. , Drazner MH, Fonarow GC, Geraci SA, Horwich T, Januzzi JL, Johnson MR, Kasper EK, Levy WC, Masoudi FA, McBride PE, McMurray JJ, Mitchell JE, Peterson PN, Riegel B, Sam F, Stevenson LW, Tang WH, Tsai EJ, Wilkoff BL, American College of Cardiology Foundation/American Heart Association Task Force on Practice G . 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation 2013; 128: e240–327. [DOI] [PubMed] [Google Scholar]
- 2. Kaul P, Reed SD, Hernandez AF, Howlett JG, Ezekowitz JA, Li Y, Zheng Y, Rouleau JL, Starling RC, O'Connor CM, Califf RM, Armstrong PW. Differences in treatment, outcomes, and quality of life among patients with heart failure in Canada and the United States. JACC Heart Fail 2013; 1: 523–530. [DOI] [PubMed] [Google Scholar]
- 3. Paulus WJ, Tschope C, Sanderson JE, Rusconi C, Flachskampf FA, Rademakers FE, Marino P, Smiseth OA, De KG, Leite‐Moreira AF, Borbely A, Edes I, Handoko ML, Heymans S, Pezzali N, Pieske B, Dickstein K, Fraser AG, Brutsaert DL. How to diagnose diastolic heart failure: a consensus statement on the diagnosis of heart failure with normal left ventricular ejection fraction by the Heart Failure and Echocardiography Associations of the European Society of Cardiology. Eur Heart J 2007; 28: 2539–2550. [DOI] [PubMed] [Google Scholar]
- 4. Brouwers FP, de Boer RA, van der Harst P, Voors AA, Gansevoort RT, Bakker SJ, Hillege HL, van Veldhuisen DJ, van Gilst WH. Incidence and epidemiology of new onset heart failure with preserved vs. reduced ejection fraction in a community‐based cohort: 11‐year follow‐up of PREVEND. Eur Heart J 2013; 34: 1424–1431. [DOI] [PubMed] [Google Scholar]
- 5. Owan TE, Hodge DO, Herges RM, Jacobsen SJ, Roger VL, Redfield MM. Trends in prevalence and outcome of heart failure with preserved ejection fraction. N Engl J Med 2006; 355: 251–259. [DOI] [PubMed] [Google Scholar]
- 6. Watson CJ, Gupta SK, O'Connell E, Thum S, Glezeva N, Fendrich J, Gallagher J, Ledwidge M, Grote‐Levi L, McDonald K, Thum T. MicroRNA signatures differentiate preserved from reduced ejection fraction heart failure. Eur J Heart Fail 2015; 17: 405–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Krishnaswamy P, Lubien E, Clopton P, Koon J, Kazanegra R, Wanner E, Gardetto N, Garcia A, DeMaria A, Maisel AS. Utility of B‐natriuretic peptide levels in identifying patients with left ventricular systolic or diastolic dysfunction. Am J Med 2001; 111: 274–279. [DOI] [PubMed] [Google Scholar]
- 8. Sanders‐van Wijk S, van Empel V, Davarzani N, Maeder MT, Handschin R, Pfisterer ME, Brunner‐La Rocca HP, investigators T‐C. Circulating biomarkers of distinct pathophysiological pathways in heart failure with preserved vs. reduced left ventricular ejection fraction. Eur J Heart Fail 2015; 17: 1006–1014. [DOI] [PubMed] [Google Scholar]
- 9. Putko BN, Wang Z, Lo J, Anderson T, Becher H, Dyck JR, Kassiri Z, Oudit GY, Alberta HI. Circulating levels of tumor necrosis factor‐alpha receptor 2 are increased in heart failure with preserved ejection fraction relative to heart failure with reduced ejection fraction: evidence for a divergence in pathophysiology. PLoS One 2014; 9: e99495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chan MM, Santhanakrishnan R, Chong JP, Chen Z, Tai BC, Liew OW, Ng TP, Ling LH, Sim D, Leong KT, Yeo PS, Ong HY, Jaufeerally F, Wong RC, Chai P, Low AF, Richards AM, Lam CS. Growth differentiation factor 15 in heart failure with preserved vs. reduced ejection fraction. Eur J Heart Fail 2016; 18: 81–88. [DOI] [PubMed] [Google Scholar]
- 11. Cheng ML, Wang CH, Shiao MS, Liu MH, Huang YY, Huang CY, Mao CT, Lin JF, Ho HY, Yang NI. Metabolic disturbances identified in plasma are associated with outcomes in patients with heart failure: diagnostic and prognostic value of metabolomics. J Am Coll Cardiol. 2015; 65: 1509–1520. [DOI] [PubMed] [Google Scholar]
- 12. Allen LA, Felker GM. Multi‐marker strategies in heart failure: clinical and statistical approaches. Heart Fail Rev 2010; 15: 343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. D'Elia E, Vaduganathan M, Gori M, Gavazzi A, Butler J, Senni M. Role of biomarkers in cardiac structure phenotyping in heart failure with preserved ejection fraction: critical appraisal and practical use. Eur J Heart Fail 2015; 17: 1231–1239. [DOI] [PubMed] [Google Scholar]
- 14. Liew CC. Expressed genome molecular signatures of heart failure. Clin Chem Lab Med 2005; 43: 462–469. [DOI] [PubMed] [Google Scholar]
- 15. Liew CC, Ma J, Tang HC, Zheng R, Dempsey AA. The peripheral blood transcriptome dynamically reflects system wide biology: a potential diagnostic tool. J Lab Clin Med 2006; 147: 126–132. [DOI] [PubMed] [Google Scholar]
- 16. Ezekowitz JA, Becher H, Belenkie I, Clark AM, Duff HJ, Friedrich MG, Haykowsky MJ, Howlett JG, Kassiri Z, Kaul P, Kim DH, Knudtson ML, Light PE, Lopaschuk GD, McAlister FA, Noga ML, Oudit GY, Paterson DI, Quan H, Schulz R, Thompson RB, Weeks SG, Anderson TJ, Dyck JR. The Alberta Heart Failure Etiology and Analysis Research Team (HEART) study. BMC Cardiovasc Disord 2014; 14: 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cohen Freue GV, Meredith A, Smith D, Bergman A, Sasaki M, Lam KK, Hollander Z, Opushneva N, Takhar M, Lin D, Wilson‐McManus J, Balshaw R, Keown PA, Borchers CH, McManus B, Ng RT, McMaster WR. Computational biomarker pipeline from discovery to clinical implementation: plasma proteomic biomarkers for cardiac transplantation. PLoS Comput Biol 2013; 9: e1002963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lee‐Lewandrowski E, Januzzi JL, Green SM, Tannous B, Wu AH, Smith A, Wong A, Murakami MM, Kaczmarek J, Apple FS, Miller WL, Hartman K, Jaffe AS. Multi‐center validation of the Response Biomedical Corporation RAMP NT‐proBNP assay with comparison to the Roche Diagnostics GmbH Elecsys proBNP assay. Clin Chim Acta 2007; 386: 20–24. [DOI] [PubMed] [Google Scholar]
- 19. Gunther OP, Chen V, Freue GC, Balshaw RF, Tebbutt SJ, Hollander Z, Takhar M, McMaster WR, McManus BM, Keown PA, Ng RT. A computational pipeline for the development of multi‐marker bio‐signature panels and ensemble classifiers. BMC Bioinformatics 2012; 13: 326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xu Q, Ni S, Wu F, Liu F, Ye X, Mougin B, Meng X, Du X. Investigation of variation in gene expression profiling of human blood by extended principle component analysis. PLoS One 2011; 6: e26905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Benita Y, Cao Z, Giallourakis C, Li C, Gardet A, Xavier RJ. Gene enrichment profiles reveal T‐cell development, differentiation, and lineage‐specific transcription factors including ZBTB25 as a novel NF‐AT repressor. Blood 2010; 115: 5376–5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Frey A, Ertl G, Angermann CE, Hofmann U, Stork S, Frantz S. Complement c3c as a biomarker in heart failure. MediatorsInflamm 2013; 2013: 716902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Heymans S, Hirsch E, Anker SD, Aukrust P, Balligand JL, Cohen‐Tervaert JW, Drexler H, Filippatos G, Felix SB, Gullestad L, Hilfiker‐Kleiner D, Janssens S, Latini R, Neubauer G, Paulus WJ, Pieske B, Ponikowski P, Schroen B, Schultheiss HP, Tschope C, Van Bilsen M, Zannad F, McMurray J, Shah AM. Inflammation as a therapeutic target in heart failure? A scientific statement from the Translational Research Committee of the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail 2009; 11: 119–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Carter AM. Complement activation: an emerging player in the pathogenesis of cardiovascular disease. Scientifica(Cairo) 2012; 2012: 402783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. de Boer RA, Edelmann F, Cohen‐Solal A, Mamas MA, Maisel A, Pieske B. Galectin‐3 in heart failure with preserved ejection fraction. Eur J Heart Fail 2013; 15: 1095–1101. [DOI] [PubMed] [Google Scholar]
- 26. Hollander Z, Dai DL, Putko BN, Yogasundaram H, Wilson‐McManus JE, Thompson RB, Khan A, West ML, McManus BM, Oudit GY. Gender‐specific plasma proteomic biomarkers in patients with Anderson‐Fabry disease. Eur J Heart Fail 2015; 17: 291–300. [DOI] [PubMed] [Google Scholar]
- 27. Redfield MM, Rodeheffer RJ, Jacobsen SJ, Mahoney DW, Bailey KR, Burnett JC Jr. Plasma brain natriuretic peptide concentration: impact of age and gender. J Am Coll Cardiol 2002; 40: 976–982. [DOI] [PubMed] [Google Scholar]
- 28. Motiwala SR, Sarma A, Januzzi JL, O'Donoghue ML. Biomarkers in ACS and heart failure: should men and women be interpreted differently? Clin Chem 2014; 60: 35–43. [DOI] [PubMed] [Google Scholar]
- 29. Xiao L, Zhang L, Han W, Wang Z, Nattel S. Sex‐based transmural differences in cardiac repolarization and ionic‐current properties in canine left ventricles. Am J Physiol Heart Circ Physiol 2006; 291: H570–HH80. [DOI] [PubMed] [Google Scholar]
- 30. Furukawa T, Kurokawa J. Regulation of cardiac ion channels via non‐genomic action of sex steroid hormones: implication for the gender difference in cardiac arrhythmias. Pharmacol Ther 2007; 115: 106–115. [DOI] [PubMed] [Google Scholar]
- 31. Kaminsky Z, Wang SC, Petronis A. Complex disease, gender and epigenetics. Ann Med 2006; 38: 530–544. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Differentially expressed proteins between HFpEF and HFrEF.
Table S2. Annotated transcripts in the male‐specific transcriptomic biomarker panel for HFpEF versus HFrEF.
Table S3. Female‐specific proteomic biomarker panel for HFpEF versus HFrEF.
Table S4. Molecular and Clinical effects of transcripts and protiens that were noted to be differentially expressed in HFpEF and HFrEF.
Figure S1. Eigen‐R2 analysis reveals strong association between transcriptomic data and cellular composition
Figure S2. Evidence of cell‐specificity in all differentially expressed transcripts (FDR <0.05; A), or the male‐specific transcriptomic panel (B), for HFpEF versus HFrEF