

Abstract
Drugs that allosterically modulate G protein‐coupled receptor (GPCR) activity display higher specificity and may improve disease treatment. However, the rational design of compounds that target the allosteric site is difficult, as conformations required for receptor activation are poorly understood. Guided by photopharmacology, a set of prearranged positive allosteric modulators (PAMs) with restricted degrees of freedom was designed and tested against the glucagon‐like peptide‐1 receptor (GLP‐1R), a GPCR involved in glucose homeostasis. Compounds incorporating a trans‐stilbene comprehensively outperformed those with a cis‐stilbene, as well as the benchmark BETP, as GLP‐1R PAMs. We also identified major effects of ligand conformation on GLP‐1R binding kinetics and signal bias. Thus, we describe a photopharmacology‐directed approach for rational drug design, and introduce a new class of stilbene‐containing PAM for the specific regulation of GPCR activity.
Keywords: allosterism, BETP, glucagon-like peptides, glucagon-like peptide-1 receptors, G protein-coupled receptors, stilbene
G protein‐coupled receptors (GPCRs) are critical for organism homeostasis by converting signals encoded by extracellular molecules into an appropriate cell response.1 Classically, ligands bind the extracellularly located orthosteric face of the GPCR, leading to conformational changes and activation of second messenger pathways.1 GPCRs are also subject to allosteric regulation, whereby small molecular entities influence orthosteric activation.2 Such allosteric modulators are therapeutically desirable, as they demonstrate excellent specificity and selectivity.2a
Recently, we described allosteric optical control of the glucagon‐like peptide‐1 receptor (GLP‐1R),3 a prototypical class B GPCR involved in the maintenance of blood glucose levels and a blockbuster target for type 2 diabetes treatment.4 As peptide ligands that target the GLP‐1R must be injected, the development of orally available small‐molecule GLP‐1R activators is a priority. In our previous work, an azobenzene‐containing molecular photoswitch termed PhotoETP was designed and synthesized,3 based upon an “azologue”5 of the known GLP‐1R positive allosteric modulator (PAM) 4‐(3‐(benzyloxy)phenyl)‐2‐(ethylsulfinyl)‐6‐(trifluoro‐methyl)pyrimidine (BETP).6 Notably, signaling responses to glucagon‐like peptide‐1 (GLP‐1) degradation products could be sculpted by the application of blue light to induce formation of the trans‐isomer.3 We hypothesized that maximal GLP‐1R activation by PAMs requires a molecule with fewer degrees of freedom. As the molecular conformations/interactions required for allosteric activation are largely unknown, we thought that this may be achievable by “prearranging” the molecule.
To produce PAMs with improved activity at the GLP‐1R, the PhotoETP azobenzene diazene unit, which exhibits non‐binary photostationary states,5 was replaced with a C=C fragment to introduce a stilbene that displays single isomers. A range of prearranged molecules were synthesized and tested for their ability to potentiate GLP‐1R signaling responses. In all cases, compounds incorporating the trans‐stilbene outperformed those with cis‐stilbene, in addition to native BETP. As such, we introduce a photopharmacology‐based strategy to direct the rational design of prearranged PAMs, with broad applicability to the allosteric regulation of GPCRs involved in health and disease.
BETP adopts a conformationally free benzyl ether when bound,6b suggesting that, upon activation of the GLP‐1R by orthosteric ligands, it is able to rearrange its shape to fully engage the allosteric site (Figure 1 a). This motion is restricted by light in PhotoETP, which possesses a photoisomerizable azobenzene in place of the O‐benzyl group (Figure 1 b). We reasoned that replacement of the diazene bridge in PhotoETP by a C=C moiety to introduce a stilbene would allow production of prearranged PAMs where the cis‐ and trans‐states are mimicked, but without complications arising from photostationary states. Stilbenes have the added advantage of being more drug‐like than azobenzenes and may demonstrate better metabolic stability in terms of double‐bond cleavage in the intestine and the possible liberation of reactive anilines. Furthermore, BETP uses an ethyl sulfoxide as a leaving group that is replaced by covalent cysteine attachment on the receptor to give ethyl sulfenic acid as a leaving group,6b and other electrophilic moieties, mainly aryl chlorides and aryl sulfones, have also been reported to undergo covalent labelling toward the GLP‐1R.7 We, therefore, wanted to explore whether a methyl sulfoxide would be tolerated (Figure 1 c), as this moiety would: 1) be easier and faster to introduce synthetically from commercially‐available substrates, giving cheaper access to PAMs that target the GLP‐1R (Figure 1 d); and 2) exhibit methyl sulfenic acid as an unstable and, therefore, quickly‐cleared leaving group.8
Figure 1.
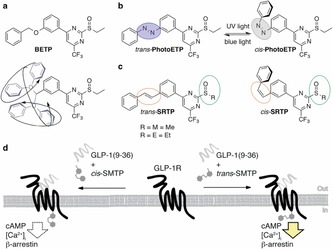
Logic and design of prearranged GLP‐1R positive allosteric modulators. a) BETP possesses an O‐benzyl ether that can freely rotate and adopt various conformations, occupying a large space, as denoted in the lower drawing. b) PhotoETP 3 endows GLP‐1R with light sensitivity when the azobenzene is locked in its active trans‐state under blue light illumination (blue circle), which is interchangeable with UV light to its inactive cis‐state (gray circle). c) Novel stilbene congeners are constantly locked (orange circle) in their respective state and do not exhibit photostationary states. The functional group for displacement can be a methyl (Me) or ethyl (Et) sulfoxide (green circle), giving pure trans‐ and cis‐isomers of the prearranged PAMs SMTP and SETP. d) Schematic representation of GLP‐1R activation with GLP‐1(9–36)NH2 in the presence of either trans‐ or cis‐SMTP.
To obtain BMTP, that is, the methyl analogue of the lead compound BETP, as a first model compound, boronic ester 1 was coupled under Suzuki–Miyaura conditions with chloropyrimidine 2 to biaryl 3 in quantitative yield,9 which was monooxidized with one equivalent of mCPBA to give BMTP in 94 % yield (Scheme 1 a). SMTP and SETP were synthesized in a comparable synthetic sequence. Commencing with benzyltriphenylphosphonium bromide (4) and 3‐bromobenzaldehyde (5) that underwent a non‐stereoselective Wittig reaction at room temperature in THF with LiHMDS as base, cis‐ and trans‐bromo stilbene 6 could be separated through flash column chromatography (FCC) and isolated in 79 % overall yield (Scheme 1 b). Single crystals of trans‐6 were obtained, which unambiguously provided the isomeric identity, and this was further confirmed by the coupling constant 3 J HH of the olefinic protons through 1H NMR spectroscopy, being approximately 12 and 16 Hz for cis‐ and trans‐isomers, respectively. Next, Miyaura coupling with B2pin2 using PdCl2(dppf) under standard conditions, employing DMSO as the solvent and KOAc as the base,10 gave access to cis‐ and trans‐boronic ester 7 in good yields (78 and 77 %, respectively), which were subsequently subjected to Suzuki–Miyaura Pd cross‐coupling with chloropyrimidine 2 to yield the cis‐ and trans‐thioether precursor 8 in good 76 % and excellent 95 % yields, respectively. Progressing towards SMTP, thioether 6 was monooxidized with one equivalent mCPBA to give cis‐ and trans‐SMTP in 91 and 89 %, respectively.
Scheme 1.
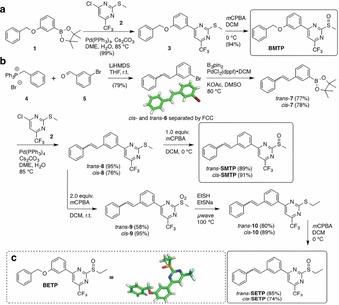
Synthesis of GLP‐1R prearranged PAMs. a) BMTP is obtained after a two‐step synthetic sequence from commercially available boronic ester 1 and chloropyrimidine 2 after Pd cross‐coupling and subsequent monooxidation. b) Wittig transformation gives access to trans‐ and cis‐stilbenes that can be separated by FCC and independently processed towards thioether 8. trans‐ and cis‐SMTP are obtained as the monooxidized products from 8, whereas bisoxidation, aromatic substitution, and monooxidation yield the prearranged positive allosteric modulators trans‐ and cis‐SETP. c) Crystal structure of BETP.
To progress to the ethyl sulfoxide, thioether 6 was oxidized with two equivalents of mCPBA to give the corresponding sulfone 9 (yield for cis: 95 %; trans: 58 %), which was subjected to aromatic substitution with a mixture of ethylsulfide/sodium thioethanolate to ethyl thioether 10 (cis: 89 %; trans: 80 %) in a microwave reactor before final installation of the ethyl sulfoxide with one equivalent of mCPBA to obtain cis‐ and trans‐SETP in 74 and 85 % yields, respectively. Lastly, we obtained a crystal structure of BETP by leaving a DMSO solution open to the atmosphere for 2 weeks (Scheme 1 c), providing atomic coordinates and showing that BETP is stable for this timeframe in solution at room temperature and under benchtop light.
The GLP‐1R signals primarily through the generation of cyclic adenosine monophosphate (cAMP).11 As expected, BETP potently augmented the cAMP responses to the GLP‐1 breakdown product and weak agonist GLP‐1(9–36)NH2 (Figure 2 a), but not fully active GLP‐1(7–36)NH2 (Figure S1 in the Supporting Information). BETP also conferred agonist activity on the GLP‐1R orthosteric antagonist exendin(9–39) (Figure 2 b). Although cis‐SETP allosterically enhanced cAMP responses, it was outperformed by trans‐SETP, which displayed a threefold increase in potency as a GLP‐1R PAM (as indicated by EC50 values in Table S1). Similar results were seen with BMTP and SMTP, suggesting that the replacement of ethyl sulfoxide with its methyl counterpart is well tolerated. Potentiation of GLP‐1(9–36)NH2‐induced β‐arrestin2 recruitment to the GLP‐1R was also greatest with trans‐isoforms (Figure 2 c).
Figure 2.
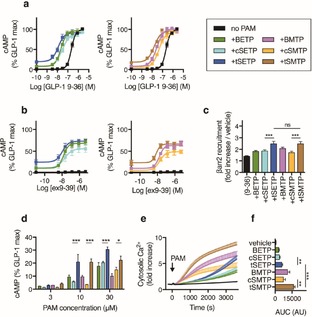
Prearranged PAMs potently enhance GLP‐1R signaling. a) Allosteric enhancement of GLP‐1(9–36)NH2 (“9–36”) cAMP responses (30 min incubation; n=4) (4‐parameter logistic fit shown). b) As for (a), but with exendin(9–39) (ex9‐39) (n=5). c) Allosteric enhancement of 10 μμ GLP‐1(9–36)NH2‐induced β‐arrestin2 (βarr2) recruitment (30 min incubation; n=5). d) cAMP responses to indicated PAM or prearranged PAM concentration in the absence of orthosteric ligand (30 min incubation; n=3). e) Cytosolic Ca2+ responses in Calcium 6 dye‐loaded cells, expressed relative to baseline fluorescent signal (60 min incubation; n=7). f) Area under curve (AUC) determined from (e). *P<0.05, **P<0.01, ***P<0.001; one‐ or two‐way randomized block ANOVA followed by either Sidak's or Tukey's post‐hoc test. Values represent the mean + or ± SEM. Except where indicated, PAMs or prearranged PAMs were applied at 10 μμ.
PAMs stabilize active receptor conformations and can behave as agonists, even in the absence of the orthosteric ligand. When BETP, BMTP, and related prearranged PAMs were applied in pure agonist mode, cAMP responses were greatest with trans‐diastereomers (Figure 2 d). There was, however, no detectable increase in β‐arrestin2 recruitment with any PAM (Figure S2). We also investigated the ability of these compounds to liberate Ca2+ from internal stores. In the absence of GLP‐1, BMTP induced strong cytosolic Ca2+ rises, more so than BETP (Figure 2 e, f). SETP and SMTP trans‐isomers were again most effective. The effects of BETP and BMTP were likely to be GLP‐1R mediated, as they were almost absent in CHO cells without GLP‐1R overexpression (Figure S3 a) and, like GLP‐1(7–36)NH2‐induced responses, could be reduced by inhibiting G‐protein signaling intermediates including Epac2 (ESI09) and phospholipase C (U73122) (Figure S3 b–e). Surprisingly, BETP was the only PAM that enhanced the Ca2+ response to GLP‐1(7–36)NH2 when pre‐incubated, as previously described.2a This may reflect depletion of accessible intracellular Ca2+ stores at a rate dependent on the intrinsic activity of the PAM (Figure S4).
PAMs increase receptor affinity for extracellular ligand, coupling to intracellular effectors, or both.12 We employed a time‐resolved Förster resonance energy transfer (TR‐FRET) approach to measure ligand‐receptor binding in real‐time, whereby FITC‐conjugated orthosteric ligand binding to SNAP‐GLP‐1R self‐ labeled with Lumi4‐Tb leads to an increase in FRET.13 For further details of this technique, see Section 6.6 in the Supporting Information. We determined that GLP‐1R PAMs substantially slow down dissociation of GLP‐1(7–36)NH2‐FITC, increasing residence time14 (Figure 3 a), and also decreasing the rate of association (Figure 3 b). The overall effect was to increase binding affinity of GLP‐1(7–36)NH2‐FITC approximately fivefold (Figure 3 c). trans‐SETP and trans‐SMTP increased the residence time and binding affinity more than their cis‐counterparts.
Figure 3.
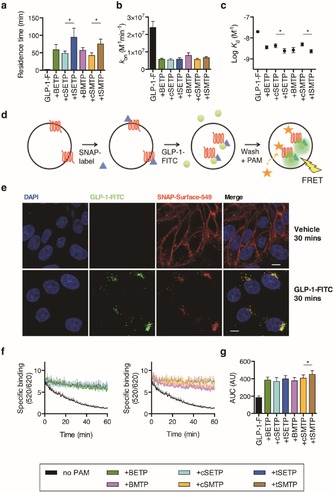
Prearranged PAMs modify GLP‐1 binding kinetics. a) GLP‐1‐FITC residence time (=1/ dissociation rate constant k off) at surface SNAP‐GLP‐1R after pre‐incubation with PAM or prearranged PAM, determined by non‐linear regression of GLP‐1‐FITC binding data15 (n=7). b) As for (a), but association rate constant (k on). c) Effect of PAMs and prearranged PAMs on affinity constants (K d) for GLP‐1‐FITC (n=7). d) Schematic showing the endosomal binding protocol. e) Representative images demonstrating complete internalization of surface‐labeled SNAP‐GLP‐1R by, and co‐localization with, 100 nμ GLP‐1‐FITC (n=2) (scale bars; 8 μm). f) Effect of PAMs and prearranged PAMs on endosomal GLP‐1‐FITC dissociation after complete SNAP‐GLP‐1R internalization, agonist washout, and compound application in the presence of exendin(9–39) (n=5). g) AUC from (f) relative to zero baseline. *P<0.05; one‐way randomized block ANOVA followed by Sidak's post‐hoc test. Values represent the mean + or ± SEM. PAMs or prearranged PAMs were applied at 10 μμ.
The GLP‐1R undergoes extensive internalization after agonist stimulation, and ongoing cAMP generation by internalized receptors may play a role in GLP‐1R signaling.16 We hypothesized that the PAMs described here, which unlike peptide ligands are membrane‐permeating, might access and modulate the behavior of pre‐internalized ligand–receptor complexes (Figure 3 d). After complete GLP‐1R internalization with 100 nμ GLP‐1(7–36)NH2‐FITC (Figure 3 e) and washout of remaining extracellular ligand, a steady reduction in FRET was detected, indicative of ligand–receptor dissociation within endosomes. However, when BETP, BMTP, and prearranged PAMs were applied to the cell immediately post‐washout, marked reductions in dissociation from endosomal receptors were observed (Figure 3 f, g). This effect was most marked with trans‐SMTP.
In the present study, we describe a photopharmacology‐inspired strategy for the rational design of potent GLP‐1R PAMs based upon the allosteric photoswitch PhotoETP.3 By restricting degrees of freedom using stilbenes, it could be shown that compounds prearranged as their trans‐isomer were more potent than other PAMs, including BETP.6 Such findings likely reflect the requirement for bound PAMs to undergo fine conformational changes at the allosteric site for full activation in response to orthosteric binding.6b Prearrangement may circumvent this by stabilizing molecule dynamics, allowing PAMs to adopt an allosteric ′ON′ state when unbound. Moreover, replacement of the ethyl sulfoxide moiety with a methyl sulfoxide maintained full activity, whilst promoting cheaper and faster access to PAMs and prearranged PAMs from commercially available chemicals in high yields. Although the differences in signaling described here for the prearranged PAMs may seem small, it should be noted that GLP‐1R PAMs allow less active GLP‐1 metabolites to signal at the orthosteric site,2b and resulting small alterations in cell signaling are expected to translate into large functional differences.
Intriguingly, intracellular Ca2+ fluxes induced by BMTP exceeded those of BETP, despite equivalent cAMP responses. As such, the prearranged PAMs may provide useful tools to tease apart the receptor conformations required for biased signaling and, more widely, the impact of second messenger preference on relevant biological endpoints. Although our studies implicated classical GLP‐1R pathways, the precise mechanism(s) underlying differences in Ca2+ responses remain unclear. The different leaving groups for BMTP versus BETP (methyl versus ethyl sulfenate) are potential candidates, as sulfenic acids are known signaling intermediaries in the context of cysteine modification.17 In addition, further studies using recently described conformational FRET sensors,18 or concentration responses to calculate alpha values,12, 19 are required to unambiguously demonstrate the differential actions of the prearranged PAMs at the GLP‐1R. It should be noted that the herein‐described prearranged PAMs, as well as BETP, exist as racemates, and in the future it will be interesting to study the labeling kinetics of the separate enantiomers before chirality is lost through covalent modification of GLP‐1R.
Finally, we provide the first demonstration that GLP‐1R PAMs can markedly increase agonist residence time, proposed as a therapeutic strategy to drive sustained responses in vitro and in vivo.20 These molecules can also directly access pre‐internalized GLP‐1Rs to modulate ligand–receptor binding within endosomes. They could, therefore, be used to prolong non‐canonical cAMP signaling from internalized GLP‐1Rs,16 delineate effects of membrane versus endosomal GLP‐1R signaling, or study the influence of occupancy on post‐endocytic receptor trafficking.21
In summary, we unveil a new class of positive allosteric modulator PAMs that are prearranged. These compounds perform better than their compacted stablemates (i.e. those incorporating a cis‐stilbene), as well as benchmark PAM, BETP, and also display signal bias. Thus, prearranged PAMs provide a template for the production of newer and more potent allosteric modulators of the GLP‐1R, with broad‐applicability to GPCR research and drug discovery.
Experimental Section
Experimental details including synthesis, spectroscopic and spectrometric characterization, and biology can be found in the Supporting Information. CCDC 1530565 and 1530566 contain the supplementary crystallographic data for this paper. The data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/structures.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
B.J.J. was funded by an MRC Clinical Research Training Fellowship. S.R.B. was funded by grants from the MRC, BBSRC, NIHR, an Integrative Mammalian Biology (IMB) Capacity Building Award, an FP7‐HEALTH‐2009–241592 EuroCHIP grant and is supported by the NIHR Imperial Biomedical Research Centre Funding Scheme. A.T. was supported by an MRC Project Grant (MR/M012646/1) and a Diabetes UK Early‐Career Small Grant (16/0005441). D.J.H. was supported by Diabetes UK R. D. Lawrence (12/0004431), EFSD/Novo Nordisk Rising Star and Birmingham Fellowships, a Wellcome Trust Institutional Support Award, and an MRC Project Grant (MR/N00275X/1). This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (Starting Grant 715884 to D.J.H.). J.B. acknowledges support from the ‘EPFL Fellows’ fellowship programme co‐funded by Marie Skłodowska‐Curie, Horizon 2020 Grant agreement no. 665667. We are grateful to a reviewer of Broichhagen et al. 2016 3 for hinting at the potential of prearranged PAMs, and thank Prof. Dr. Kai Johnsson for kindly providing reagents and helpful discussions.
B. J. Jones, R. Scopelliti, A. Tomas, S. R. Bloom, D. J. Hodson, J. Broichhagen, ChemistryOpen 2017, 6, 501.
Contributor Information
Prof. Dr. David J. Hodson, Email: d.hodson@bham.ac.uk.
Dr. Johannes Broichhagen, Email: johannes.broichhagen@mpimf-heidelberg.mpg.de.
References
- 1. Katritch V., Cherezov V., Stevens R. C., Annu. Rev. Pharmacol. Toxicol. 2013, 53, 531–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.
- 2a. Wootten D., Savage E. E., Valant C., May L. T., Sloop K. W., Ficorilli J., Showalter A. D., Willard F. S., Christopoulos A., Sexton P. M., Mol. Pharmacol. 2012, 82, 281–290; [DOI] [PubMed] [Google Scholar]
- 2b. Koole C., Savage E. E., Christopoulos A., Miller L. J., Sexton P. M., Wootten D., Mol. Endocrinol. 2013, 27, 1234–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Broichhagen J., Johnston N. R., von Ohlen Y., Meyer-Berg H., Jones B. J., Bloom S. R., Rutter G. A., Trauner D., Hodson D. J., Angew. Chem. Int. Ed. 2016, 55, 5865–5868; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 5961–5965. [Google Scholar]
- 4. Campbell J. E., Drucker D. J., Cell Metab. 2013, 17, 819–837. [DOI] [PubMed] [Google Scholar]
- 5. Broichhagen J., Frank J. A., Trauner D., Acc. Chem. Res. 2015, 48, 1947–1960. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Willard F. S., Wootten D., Showalter A. D., Savage E. E., Ficorilli J., Farb T. B., Bokvist K., Alsina-Fernandez J., Furness S. G., Christopoulos A., Sexton P. M., Sloop K. W., Mol. Pharmacol. 2012, 82, 1066–1073; [DOI] [PubMed] [Google Scholar]
- 6b. Nolte W. M., Fortin J. P., Stevens B. D., Aspnes G. E., Griffith D. A., Hoth L. R., Ruggeri R. B., Mathiowetz A. M., Limberakis C., Hepworth D., Carpino P. A, Nat. Chem. Biol. 2014, 10, 629–631. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Bueno A. B., Showalter A. D., Wainscott D. B., Stutsman C., Marin A., Ficorilli J., Cabrera O., Willard F. S., Sloop K. W., J. Biol. Chem. 2016, 291, 10700–10715; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b. Knudsen L. B., Kiel D., Teng M., Behrens C., Bhumralkar D., Kodra J. T., Holst J. J., Jeppesen C. B., Johnson M. D., de Jong J. C., Jorgensen A. S., Kercher T., Kostrowicki J., Madsen P., Olesen P. H., Petersen J. S., Poulsen F., Sidelmann U. G., Sturis J., Truesdale L., May J., Lau J., Proc. Natl. Acad. Sci. USA 2007, 104, 937–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Penn R. E., Block E., Revelle L. K., J. Am. Chem. Soc. 1978, 100, 3622–3623. [Google Scholar]
- 9. Eng H., Sharma R., McDonald T. S., Edmonds D. J., Fortin J. P., Li X., Stevens B. D., Griffith D. A., Limberakis C., Nolte W. M., Price D. A., Jackson M., Kalgutkar A. S., Drug Metab. Dispos. 2013, 41, 1470–1479. [DOI] [PubMed] [Google Scholar]
- 10. Ishiyama T., Murata M., Miyaura N., J. Org. Chem. 1995, 60, 7508–7510. [Google Scholar]
- 11.
- 11a. Leech C. A., Dzhura I., Chepurny O. G., Kang G., Schwede F., Genieser H. G., Holz G. G., Prog. Biophys. Mol. Biol. 2011, 107, 236–247; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11b. MacDonald P. E., El-Kholy W., Riedel M. J., Salapatek A. M., Light P. E., Wheeler M. B., Diabetes 2002, 51, S434–442. [DOI] [PubMed] [Google Scholar]
- 12. May L. T., Leach K., Sexton P. M., Christopoulos A., Annu. Rev. Pharmacol. Toxicol. 2007, 47, 1–51. [DOI] [PubMed] [Google Scholar]
- 13. Emami-Nemini A., Roux T., Leblay M., Bourrier E., Lamarque L., Trinquet E., Lohse M. J., Nat. Protoc. 2013, 8, 1307–1320. [DOI] [PubMed] [Google Scholar]
- 14. Tautermann C. S., Curr. Opin. Pharmacol. 2016, 30, 22–26. [DOI] [PubMed] [Google Scholar]
- 15. Klein Herenbrink C., Sykes D. A., Donthamsetti P., Canals M., Coudrat T., Shonberg J., Scammells P. J., Capuano B., Sexton P. M., Charlton S. J., Javitch J. A., Christopoulos A., Lane J. R., Nat. Commun. 2016, 7, 10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kuna R. S., Girada S. B., Asalla S., Vallentyne J., Maddika S., Patterson J. T., Smiley D. L., DiMarchi R. D., Mitra P., Am. J. Physiol. Endocrinol. Metab. 2013, 305, E161–170. [DOI] [PubMed] [Google Scholar]
- 17. Paulsen C. E., Carroll K. S., ACS Chem. Biol. 2010, 5, 47–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lecat-Guillet N., Monnier C., Rovira X., Kniazeff J., Lamarque L., Zwier J. M., Trinquet E., Pin J. P., Rondard P., Cell Chem. Biol 2017, 24, 229–245. [DOI] [PubMed] [Google Scholar]
- 19. Conn P. J., Christopoulos A., Lindsley C. W., Nat. Rev. Drug Discovery 2009, 8, 41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.
- 20a. Guo D., Heitman L. H., AP I. J., ACS Med. Chem. Lett. 2016, 7, 819–821; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20b. de Witte W. E., Danhof M., van der Graaf P. H., de Lange E. C., Trends Pharmacol. Sci. 2016, 37, 831–842. [DOI] [PubMed] [Google Scholar]
- 21. Yeatman H. R., Lane J. R., Choy K. H., Lambert N. A., Sexton P. M., Christopoulos A., Canals M., J. Biol. Chem. 2014, 289, 15856–15866. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary