

Abstract
Potent inhibitors of Trypanosoma brucei methionyl-tRNA synthetase were previously designed using a structure-guided approach. Compounds 1 and 2 were the most active compounds in the cyclic and linear linker series, respectively. To further improve cellular potency, SAR investigation of a binding fragment targeting the “enlarged methionine pocket” (EMP) was performed. The optimization led to the identification of a 6,8-dichloro-tetrahydroquinoline ring as a favorable fragment to bind the EMP. Replacement of 3,5-dichloro-benzyl group (the EMP binding fragment) of inhibitor 2 using this tetrahydroquinoline fragment resulted in compound 13, that exhibited an EC50 of 4 nM.
Keywords: Human African trypanosomiasis, Methionyl-tRNA synthetase, Structure-based design, Trypanosoma brucei
Graphical abstract

Human African trypanosomiasis (HAT), commonly known as sleeping sickness, is a neglected tropical disease caused by the protozoan parasite Trypanosoma brucei.1 The parasite is transmitted to humans through the bite of the tsetse fly. The disease progresses in two distinct stages: an initial acute stage where the parasitic infection is restricted to the hemolymphatic system and a late stage where the parasites cross the blood-brain barrier and reside in brain tissue.2 Current treatment options are severely inadequate for this disease.1,3 For the treatment of early stage infection, the two drugs, pentamidine and suramin, have toxicity and require injection.4 The late stage infection is particularly difficult to treat, as drugs must cross the blood-brain barrier to be effective. The two drugs available for the late stage infection, melarsoprol and eflornithine, are toxic, have limited ability to cross the blood-brain barrier, and require injection.4–6 New drugs that are safe and easy to administer are urgently needed for both stages of HAT.
We recently reported on structure-guided design of Trypanosoma brucei methionyl-tRNA synthetase (TbMetRS) inhibitors.7 Two series of compounds were designed and demonstrated to be potent TbMetRS inhibitors. The most potent compound in each series is shown in Figure 1. Compound 1 is a cyclic linker inhibitor with an EC50 of 39 nM and compound 2 is a linear linker inhibitor with an EC50 of 22 nM. In the previous study, the 3,5-dichlorophenyl moiety was fixed as the fragment to fill the so-called “enlarged methionine pocket” (EMP)8 and the investigation was mainly focused on the linker part. Here, we report on the optimization of the EMP binding fragment based on the cyclic and linear linker compounds 1 and 2 in order to identify the preferred moiety for binding the EMP. This led to the identification of inhibitors with significantly enhanced potency.
Figure 1.

Structures of compounds 1 and 2.
Analogues of compound 1 in which the 3,5-dichlorophenyl moiety was replaced by various 3,5-disubstituted phenyl or 2,3,5-trisubstituted phenyl ring were prepared as shown in Scheme 1. The synthesis was following previously reported procedures.7 In brief, (S)-tert-butyl piperidin-3-ylcarbamate reacted with 2-bromo-5-chloro-3H-imidazo[4,5-b]pyridine through nucleophilic substitution reaction, and the following Boc removal provided intermediate 4. Reductive amination of 4 with various substituted benzaldehydes afforded the final products 5a-5p.
Scheme 1.

Reagents and conditions: (a) (S)-tert-butyl piperidin-3-ylcarbamate, pyridine, MW, 100 °C, 30 min; (b) TFA, DCM, r.t. overnight; (c) Substituted benzaldehyde, DIPEA, NaBH3CN, AcOH, CH3OH, r.t., overnight.
An analogue of compound 2 in which the 3,5-dichlorophenyl moiety was replaced by 3,5-dichloro-2-ethoxy phenyl was also designed, and synthesized as shown in Scheme 2. Compound 7 was synthesized following the same procedure used for synthesizing compound 5. Additional analogues of compound 2 were prepared through introducing substituents onto the benzylic α-position of the 3,5-dichlorophenyl ring (Scheme 3). Reagent 2,2-dimethoxy-N-methylethanamine reacted with 2-bromo-5-chloro-3H-imidazo[4,5-b]pyridine under the same microwave assisted nucleophilic substitution reaction, but with extended reaction time. The resulted intermediate containing an acetal group was hydrolyzed under acidic condition to produce the aldehyde intermediate 8, which underwent reductive amination with methyl 2-amino-2-(3,5-dichlorophenyl)acetate to generate compound 9a. The methyl ester group of 9a was reduced by lithium aluminum hydride to generate compound 9b, while ammonolysis of the ester group produced compound 9c.
Scheme 2.

Reagents and conditions: (a) tert-butyl (2-(methylamino)ethyl) carbamate, pyridine, MW, 100 °C, 30 min; (b) TFA, DCM, r.t. overnight; (c) 3,5-dichloro-2-ethoxybenzaldehyde, DIPEA, NaBH3CN, AcOH, CH3OH, r.t., overnight.
Scheme 3.

Reagents and conditions: (a) 2,2-dimethoxy-N-methylethanamine, pyridine, MW, 100 °C, 60 min; (b) HCl (2 M), acetone, reflux, 60 min; (c) methyl 2-amino-2-(3,5-dichlorophenyl)acetate (HCl salt), DIPEA, NaBH3CN, AcOH, CH3OH, r.t., overnight; (d) LiAlH4, THF, 0 °C, 1h; (e) NH3·H2O, r.t. overnight.
Inspired by the superior potency of bacterial MetRS and TbMetRS inhibitors that contained a tetrahydroquinoline group reported previously,9–11 a 6,8-dichloro-tetrahydroquinoline group was employed to replace the 3,5-dichlorophenyl moiety in compounds 1 and 2 to generate compounds 11 and 13. Their synthesis is shown in Schemes 4 and 5. For compound 11, amine and ketone were pre-reacted with Ti(OEt)4 as catalyst, and the reductant NaBH3CN was added 30 min later followed by an 8 h reaction under microwave conditions (Scheme 4). Compound 13 was synthesized through reductive amination using intermediates 8 and 12 (Scheme 5). Intermediate 12 was prepared following previously published procedures.9
Scheme 4.

Reagents and conditions: Ti(OEt)4, EtOH, MW, 100 °C, 30 min; then NaBH3CN, MW, 100 °C, 8 h.
Scheme 5.

Reagents and conditions: DIPEA, NaBH3CN, AcOH, CH3OH, r.t., overnight.
All the compounds were first evaluated for enzymatic potency against TbMetRS using an ATP depletion assay as described previously.7,12 As shown in Table 1, most of the compounds are very potent inhibitors of TbMetRS, exhibiting IC50s below 50 nM (the enzyme concentration used in the assay). Compounds 5k and 5l were found to have significantly reduced inhibitory potency compared to 1, with IC50s >300 nM.
Table 1.
Inhibitory activities of compounds against TbMetRS enzyme and T. brucei cell growth

| ||||
|---|---|---|---|---|
| Compound number | R1/R2 | Structure of R1/R2 | IC50 (nM)b | EC50 (nM)c |
| 1a | R1 |

|
< 50 | 39 |
| 2a | R2 |
|
< 50 | 22 |
| 5a | R1 |

|
< 50 | 111 |
| 5b | R1 |

|
< 50 | 71 |
| 5c | R1 |

|
< 50 | 85 |
| 5d | R1 |
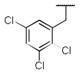
|
< 50 | 354 |
| 5e | R1 |

|
< 50 | 241 |
| 5f | R1 |

|
< 50 | 357 |
| 5g | R1 |

|
< 50 | 111 |
| 5h | R1 |

|
< 50 | 326 |
| 5i | R1 |

|
< 50 | 827 |
| 5j | R1 |

|
< 50 | 511 |
| 5k | R1 |

|
389 | 3115 |
| 5l | R1 |

|
305 | 1703 |
| 5m | R1 |

|
< 50 | 359 |
| 5n | R1 |

|
< 50 | 230 |
| 5o | R1 |

|
< 50 | 40 |
| 5p | R1 |

|
< 50 | 40 |
| 7 | R2 |
|
< 50 | 314 |
| 9a | R2 |

|
86 | 5273 |
| 9b | R2 |

|
101 | 995 |
| 9c | R2 |

|
75 | 4488 |
| 11 | R1 |

|
89 | 57 |
| 13 | R2 |

|
< 50 | 4 |
Data was published previously, included here for comparison;
The values are averages of triplicate data, control for TbMetRS IC50 assay: Met-SA 17 (± 10.1%; n = 11 assays);
The values are averages of triplicate date, control for TbEC50 assay : Pentamidine (± 15.5%; n=9).
All the compounds were also tested for potency against T. brucei parasites using a growth inhibition assay as previously described.7,9,13 The results are shown in Table 1 and the inhibition curves of compounds with EC50 < 100 nM are shown in Figure s1 (Supporting Information). Compounds with larger groups at the 3- and 5-positions, such as bromo and cyano (5a-5c), were tolerated by the enzyme, but had higher EC50 values by 2–3 folds. All compounds that contain the 2,3,5-trisubstituted phenyl ring except 5p were less potent in the parasite growth inhibition assay. The ethoxy group was the best 2-position substituent as evidenced by 5g and 5p being more potent than their counterparts with other substituents. In fact the cellular potency of compound 5p was nearly identical to the EC50 of 1 (39 nM). Larger alkoxy groups at 2-position resulted in significant reductions in cellular activity, especially for branched alkoxy groups. In general, the larger the alkoxy group at the 2-position, the less cellular potency the compound exhibited (5g<5h<5j<5i). Compounds 5k and 5l that contain branched alkoxy groups at 2-position lost cellular potency significantly. Compounds 9a-9c that contain substitutions at the benzylic α-position in the case of the linear linker series exhibited weak cellular potency. The 6,8-dichlorotetrahydroquinoline moiety was a good match for the linear linker series, but not in the cyclic linker series as indicated by compounds 11 and 13. In the cyclic linker series, compound 11 was less potent than 1, while in the linear linker series, compound 13 was more potent than 2. Compound 13 was the most potent compound found in this study, exhibiting an EC50 of 4 nM. The calculated physicochemical properties of compounds with EC50 <100 nM against T. brucei are presented in Table S1 (Supporting Information). All these potent compounds possess Ligand Efficiency (LE) values > 0.3, indicating a good balance of size and lipophilicity. It is noteworthy that compounds 2 and 13 show the highest LE of 0.43, and the Ligand-Lipophilicity Efficiency (LLE) of 2.96 and 4.38, respectively. This indicates the optimization of 2 to 13 not only improved potency but also maintained LE and increased LLE.
To check for unexpected changes in the binding mode, we obtained crystal structure of TbMetRS in complex with compounds 13 at 2.3Å resolution (Figure 2A). The binding mode of compound 13 was compared to compound 2 bound to TbMetRS (Figure 2B). The 5-chloro-imidazopyridine group of compound 13 bound in the same manner to the auxiliary pocket (AP) as the corresponding part in compound 2, forming a hydrogen bond interaction with the catalytic residue Asp287 (Figures 2C, 2D). The linkers of both compounds superimposed almost perfectly (Figure 2B). The 6,8-dichlorotetrahydroquinoline ring of compound 13 also bound similarly to the EMP compared to the 3,5-dichloro-benzyl group in compound 2, as observed before for analogs with longer linkers.9 The dichloro benzene group of compound 2 was in essentially the same position as the corresponding part in compound 13. The availability of compound 13 bound crystal structure could help to explain the structure-activity relationship (SAR) data generated in Table 1 and guide future inhibitor design.
Figure 2.

Binding of compound 13 to TbMetRS. (A) TbMetRS•Compound 13 complex structure (PDB: 5V49). The protein surface and the two pockets, EMP and AP, where the inhibitor is bound are shown. Protein carbon atoms are colored grey, nitrogens blue and oxygens red. (B) Superposition of TbMetRS structures bound to compounds 13 and 2 (PDB: 5J59)7. Chlorine atoms are colored green, carbon atoms of compounds 13 and 2 are colored orange and cyan, respectively. (C and D) Hydrogen bond interactions in TbMetRS•compound 2 and TbMetRS•compound 13 structures are shown with dotted lines and labels for interacting residues are underlined.
Potent compounds with EC50 <100 nM against T. brucei were selected to examine the host cell toxicity. The compounds shown in Table 2 were tested using a human lymphoblast cell line (CRL-8155) and a hepatocellular carcinoma cell line (Hep G2) following procedures described previously.7,14 Compounds 5b and 5c had CC50s (concentration to cause 50% cytotoxicity) close to or greater than 40 μM, whereas compounds 11 and 13 had CC50s of ~20 μM, and compound 5p had CC50 of ~10 μM. Overall, these compounds exhibited low toxicities to mammalian cell lines.
Table 2.
Toxicity, oral PK, and brain penetration data of select compounds.
| Cpd. | CRL-8155 (μM)b | Hep G2 (μM)c | Oral PK (50 mg/kg)d | Brain/Plasma Ratio (%)d | |
|---|---|---|---|---|---|
| AUC (min·μM)b | Cmax (μM) | ||||
| 1a | 29.3 | 49.0 | 6223±2160 | 37.6±22.1 | 0 |
| 2a | 22.6 | 39.2 | 952±331 | 9.7±4.5 | 27.2±7.1 |
| 5b | 43.0 | >50 | 4688±2123 | 19.2±16.4 | 0 |
| 5c | 47.6 | 39.1 | Not done | Not done | Not done |
| 5p | 10.5 | 18.7 | 182±43 | 2.1±1.5 | 4.4±0.6 |
| 11 | 19.0 | 40.2 | 947±354 | 14.7±4.3 | Not done |
| 13 | 18.5 | 30.6 | 140±40 | 1.3±0.7 | 19.1±7.5 |
Data was published previously, included here for comparison;
The values are averages of quadruplicate, control for CRL-8155 EC50 assay: Quinacrine (± 12.5%; n=2);
The values are averages of quadruplicate, control for HepG2 EC50 assay: Quinacrine (± 14.9%; n=3);
The values are from one experiment with 3 mice per group.
Compounds with good cellular potency were further tested for oral pharmacokinetic (PK) properties and/or brain penetration in mice (Table 2). The PK studies were performed following published procedures with compounds being administered by oral gavage at 50 mg/kg.7,9,15–17 The brain permeability was tested at a dose of 5 mg/kg IP as described previously.7,9 Compound 5b exhibited high plasma exposure comparable to compound 1 with a Cmax at 19.2 μM, and an AUC of 4687.5 min·μM. Unfortunately, like compound 1, the brain permeability of 5b was poor with undetectable brain levels at 60 min after IP injection. Compound 5p, the 2,3,5-trisubstituted analogue of 1, showed slightly improved brain/plasma ratio but less favorable oral PK compared to 1. Compound 11 had promising PK properties with a Cmax at 14.7 μM and AUC of 947.4 min·μM, whereas compound 13 showed low plasma exposure. Both the plasma and brain exposure of 13 are lower than those of 2, but its improvement in EC50 over 2 may compensate for the lower exposure in an in vivo efficacy model.
In summary, a series of 3,5-disubstitued and 2,3,5-trisubstituted benzyl groups, as well as a 6,8-dichlorotetrahydroquinoline moiety were used to replace the 3,5-dichlorobenzyl group as the EMP binding moiety based on previously discovered cyclic and linear linker TbMetRS inhibitors. It was found that substituents larger than chloro at 3-and/or 5-positions do not improve potency while 2,3,5-trisubstituted benzyl groups generally resulted in decreased potency. Substituents at the benzylic μ-position in the case of the linear linker series also led to a loss of potency. The 6,8-dichlorotetrahydroquinoline moiety however, afforded compound 13 in the linear linker series with improved potency against T. brucei parasites. We obtained the crystal structure of TbMetRS in complex with compound 13 which will help guide future inhibitor design. Compound 13 also had moderately good brain penetration in mice. This study identified potentially better fragments for binding the EMP than the 3,5-dichloro benzyl group in the cyclic and linear linker series of TbMetRS inhibitors.
Supplementary Material
Acknowledgments
Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award numbers R01AI084004 and R01AI097177. We acknowledge the support of a Fulbright Fellowship to X.B.-A. We thank Stewart Turley and Robert Steinfeldt for providing support for the X-ray data collection and computing environment at the Biomolecular Structure Center of the University of Washington. Crystallography performed in support of the work benefitted from remote access to resources at the Stanford Synchrotron Radiation Lightsource supported by the U.S. Department of Energy Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515 and by the National Institutes of Health (P41GM103393).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Brun R, Blum J, Chappuis F, Burri C. Human African trypanosomiasis. Lancet. 2010;375:148. doi: 10.1016/S0140-6736(09)60829-1. [DOI] [PubMed] [Google Scholar]
- 2.Grab DJ, Kennedy PGE. Traversal of human and animal trypanosomes across the blood-brain barrier. Journal of Neurovirology. 2008;14:344. doi: 10.1080/13550280802282934. [DOI] [PubMed] [Google Scholar]
- 3.Rodgers J. Human African trypanosomiasis, chemotherapy and CNS disease. J Neuroimmunol. 2009;211:16. doi: 10.1016/j.jneuroim.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 4. [accessed 12.06.16];Trypanosomiasis, human African (sleeping sickness) 2016 http://www.who.int/mediacentre/factsheets/fs259/en/
- 5.Croft SL, Barrett MP, Urbina JA. Chemotherapy of trypanosomiases and leishmaniasis. Trends Parasitol. 2005;21:508. doi: 10.1016/j.pt.2005.08.026. [DOI] [PubMed] [Google Scholar]
- 6.Wilkinson SR, Kelly JM. Trypanocidal drugs: mechanisms, resistance and new targets. Expert Rev Mol Med. 2009:11. doi: 10.1017/S1462399409001252. [DOI] [PubMed] [Google Scholar]
- 7.Huang W, Zhang Z, Barros-Álvarez X, Koh CY, Ranade RM, Gillespie JR, Creason SA, Shibata S, Verlinde CLM, Hol WGJ, Buckner FS, Fan E. Structure-guided design of novel Trypanosoma brucei Methionyl-tRNA synthetase inhibitors. Eur J Med Chem. 2016;124:1081. doi: 10.1016/j.ejmech.2016.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koh CY, Kim JE, Shibata S, Ranade RM, Yu M, Liu J, Gillespie JÂR, Buckner FS, Verlinde CLMJ, Fan E, Hol WGJ. Distinct States of Methionyl-tRNA Synthetase Indicate Inhibitor Binding by Conformational Selection. Structure. 2012;20:1681. doi: 10.1016/j.str.2012.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Z, Koh CY, Ranade RM, Shibata S, Gillespie JR, Hulverson MA, Huang W, Nguyen J, Pendem N, Gelb MH, Verlinde CLMJ, Hol WGJ, Buckner FS, Fan E. 5-Fluoroimidazo[4,5-b]pyridine Is a privileged fragment that conveys bioavailability to potent Trypanosomal methionyl-tRNA synthetase inhibitors. ACS Infec Dis. 2016;2:399. doi: 10.1021/acsinfecdis.6b00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jarvest RL, Berge JM, Berry V, Boyd HF, Brown MJ, Elder JS, Forrest AK, Fosberry AP, Gentry DR, Hibbs MJ, Jaworski DD, O’Hanlon PJ, Pope AJ, Rittenhouse S, Sheppard RJ, Slater-Radosti C, Worby A. Nanomolar inhibitors of Staphylococcus aureus methionyl tRNA synthetase with potent antibacterial activity against Gram-positive pathogens. J Med Chem. 2002;45:1959. doi: 10.1021/jm025502x. [DOI] [PubMed] [Google Scholar]
- 11.Jarvest RL, Armstrong SA, Berge JM, Brown P, Elder JS, Brown MJ, Copley RCB, Forrest AK, Hamprecht DW, O’Hanlon PJ, Mitchell DJ, Rittenhouse S, Witty DR. Definition of the heterocyclic pharmacophore of bacterial methionyl tRNA synthetase inhibitors: potent antibacterially active non-quinolone analogues. Bioorg Med Chem Lett. 2004;14:3937. doi: 10.1016/j.bmcl.2004.05.070. [DOI] [PubMed] [Google Scholar]
- 12.Pedró-Rosa L, Buckner FS, Ranade RM, Eberhart C, Madoux F, Gillespie JR, Koh CY, Brown S, Lohse J, Verlinde CLM, Fan E, Bannister T, Scampavia L, Hol WGJ, Spicer T, Hodder P. Identification of potent inhibitors of the Trypanosoma brucei methionyl-tRNA synthetase via high throughput orthogonal screening. J Biomol Screening. 2015;20:122. doi: 10.1177/1087057114548832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shibata S, Gillespie JR, Kelley AM, Napuli AJ, Zhang Z, Kovzun KV, Pefley RM, Lam J, Zucker FH, Van Voorhis WC, Merritt EA, Hol WG, Verlinde CL, Fan E, Buckner FS. Selective inhibitors of methionyl-tRNA synthetase have potent activity against Trypanosoma brucei infection in mice. Antimicrob Agents Chemother. 2011;55:1982. doi: 10.1128/AAC.01796-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shibata S, Gillespie JR, Ranade RM, Koh CY, Kim JE, Laydbak JU, Zucker FH, Hol WG, Verlinde CL, Buckner FS, Fan E. Urea-based inhibitors of Trypanosoma brucei Methionyl-tRNA synthetase: selectivity and in vivo characterization. J Med Chem. 2012;55:6342. doi: 10.1021/jm300303e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kraus JM, Verlinde CLMJ, Karimi M, Lepesheva GI, Gelb MH, Buckner FS. Rational modification of a candidate cancer drug for use against Chagas disease. J Med Chem. 2009;52:1639–1647. doi: 10.1021/jm801313t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Suryadevara PK, Olepu S, Lockman JW, Ohkanda J, Karimi M, Verlinde CLMJ, Kraus JM, Schoepe J, Van Voorhis WC, Hamilton AD, Buckner FS, Gelb MH. Structurally simple inhibitors of Lanosterol 14α-demethylase are efficacious in a rodent model of acute Chagas disease. J Med Chem. 2009;52:3703. doi: 10.1021/jm900030h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tatipaka HB, Gillespie JR, Chatterjee AK, Norcross NR, Hulverson MA, Ranade RM, Nagendar P, Creason SA, McQueen J, Duster NA, Nagle A, Supek F, Molteni V, Wenzler T, Brun R, Glynne R, Buckner FS, Gelb MH. Substituted 2-phenylimidazopyridines: A new class of drug leads for human African Trypanosomiasis. J Med Chem. 2014;57:828. doi: 10.1021/jm401178t. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.