

Supplemental Digital Content is available in the text.
Keywords: acetylation, heart failure, mitochondrial dynamics, molecular dynamics simulation, tumor necrosis factor-alpha
Abstract
Rationale:
Mitochondria are important cellular organelles and play essential roles in maintaining cell structure and function. Emerging evidence indicates that in addition to having proinflammatory and proapoptotic effects, TNFα (tumor necrosis factor α) can, under certain circumstances, promote improvements in mitochondrial integrity and function, phenomena that can be ascribed to the existence of TNFR2 (TNFα receptor 2).
Objective:
The present study aimed to investigate whether and how TNFR2 activation mediates the effects of TNFα on mitochondria.
Methods and Results:
Freshly isolated neonatal mouse cardiac myocytes treated with shRNA targeting TNFR1 were used to study the effects of TNFR2 activation on mitochondrial function. Neonatal mouse cardiac myocytes exhibited increases in mitochondrial fusion, a change that was associated with increases in mitochondrial membrane potential, intracellular ATP levels, and oxygen consumption capacity. Importantly, TNFR2 activation–induced increases in OPA1 (optic atrophy 1) protein expression were responsible for the above enhancements, and these changes could be attenuated using siRNA targeting OPA1. Moreover, both Stat3 and RelA bound to the promoter region of OPA1 and their interactions synergistically upregulated OPA1 expression at the transcriptional level. Stat3 acetylation at lysine 370 or lysine 383 played a key role in the ability of Stat3 to form a supercomplex with RelA. Meanwhile, p300 modulated Stat3 acetylation in HEK293T (human embryonic kidney 293T) cells, and p300-mediated Stat3/RelA interactions played an indispensable role in OPA1 upregulation. Finally, TNFR2 activation exerted beneficial effects on OPA1 expression in an in vivo transverse aortic constriction model, whereby TNFR1-knockout mice exhibited better outcomes than in mice with both TNFR1 and TNFR2 knocked out.
Conclusions:
TNFR2 activation protects cardiac myocytes against stress by upregulating OPA1 expression. This process was facilitated by p300-mediated Stat3 acetylation and Stat3/RelA interactions, leading to improvements in mitochondrial morphology and function.
TNFα (tumor necrosis factor α) is a cell signaling protein (cytokine) that participates in systemic inflammation, and increased serum TNFα levels are observed in patients with advanced heart failure.1 TNFα transmits its signals through 2 receptors, and a large body of evidence indicates that TNFR1 (TNFα receptor 1)–dependent pathways mediate adverse cardiac remodeling and impairments in cardiac function,2,3 whereas TNFR2 (TNFα receptor 2) signaling pathways counter cardiac injury and ameliorate heart failure progression.4–6 TNFα can protect the heart against apoptosis in the setting of ischemia-reperfusion injury. Specifically, administering low concentrations of exogenous TNFα (0.5 ng/mL, in vitro) before ischemia-reperfusion enhances cell survival in a dose-dependent manner, whereas administering higher concentrations (10–20 ng/mL, in vitro) leads to dose-dependent toxic effects on cells.7–9 Given that TNFR2 exhibits a higher affinity for TNFα than TNFR1,10,11 we propose that the cardioprotective effects of low concentrations of TNFα are attributable to TNFR2 activation and that the toxic effects of higher concentrations of TNFα are attributable to TNFR1 activation. In addition, the failure of previous clinical trials evaluating anti-TNFα therapies, such as RENAISSANCE (Randomised Etanercept North American Strategy to Study Antagonism of Cytokines), RECOVER (Research Into Etanercept Cytokine Antagonism in Ventricular Dysfunction Trial), and ATTACH (Anti-Tnf Alpha Therapy Against Chronic Heart Failure; where etanercept and infliximab were used to inhibit TNFα), may be attributable to the blockade of TNFR2 signaling.12–14
Mitochondria serve as key regulators of cellular metabolic activity. In the heart, mitochondrial fitness is required for the maintenance of cardiac myocyte homeostasis. Mitochondrial fitness is facilitated largely by the following 2 distinct phenomena: mitochondrial biogenesis, a process that controls mitochondrial mass and copy number,15,16 and mitochondrial dynamics, which comprises mitochondrial fission and fusion.17 Mitochondrial biogenesis is modulated mainly by several key transcription factors, such as PGC-1α (peroxisome proliferator–activated receptor gamma coactivator 1-alpha), which regulates the de novo generation of mitochondrial proteins, whereas mitochondrial dynamics is regulated mainly by a family of dynamin-related large GTPases. One of the members of this family, Drp1 (dynamin-related protein 1), is largely localized in the cytoplasm and can translocate to the mitochondria to participate in fission of the outer mitochondrial membrane.18 Two other members of this family, Mfn1 and Mfn2 (mitofusin), are located in the outer mitochondrial membrane, and another member of the family, OPA1 (optic atrophy 1), is located in the inner mitochondrial membrane; Mfn1 (mitofusin 1) and Mfn2 control outer mitochondrial membrane, whereas OPA1 controls inner mitochondrial membrane fusion.19,20 The boundary membrane and the cristae are 2 inner mitochondrial membrane subcompartments connected via tubular structures known as cristae junctions, which are characterized by relatively small diameters.21 Cristae are studded with proteins, including respiratory chain supercomplexes and ATP synthase and provide a sufficient surface area for chemical reactions to occur, a feature that enables them to serve as the major sites of aerobic cellular respiration and ATP synthesis.22 OPA1 comprises at least 8 isoforms, which exist as a long OPA1 form and a short OPA1 form. The oligomers formed by long OPA1 form and short OPA1 form tighten cristae junctions to stabilize respiratory chain supercomplexes, thereby increasing mitochondrial respiration efficiency.23–25
TNFα can cause mitochondrial fragmentation26 and decrease both mitochondrial metabolism-related gene expression27 and respiratory chain supercomplex activity28,29; however, studies have also shown that TNFα can enhance mitochondrial integrity30 and stimulate mitochondrial metabolism.31 These seemingly contradictory data support the notion that TNFR2 can compromise the therapeutic effects of the anti-TNFα agents used to treat patients with heart failure. Moreover, TNFR2 activation has been demonstrated previously to protect the heart against ischemia.5,6 Given that mitochondria play a significant role in maintaining normal cellular structure and function, investigating whether TNFR2 activation can protect cardiac myocytes by regulating mitochondrial morphology and function is worthwhile. In this study, we used cultured neonatal mouse cardiac myocytes (NMCMs) in which TNFR1 was knocked down using siRNA to study the effects of TNFR2 activation at the cellular level. We also subjected both TNFR1-knockout mice and TNFR1 and TNFR2-double (TNFR1/2)-knockout mice to transverse aortic constriction (TAC)–induced stress to determine whether TNFR2 activation can protect against cardiac remodeling. Our data demonstrated that TNFR2 activation protected cardiac myocytes by modulating p300/Stat3/RelA/OPA1 signaling, resulting in cristae junction tightening and significant improvements in mitochondrial integrity, adaptive responses that enabled the indicated cells to withstand the indicated stressors.
Methods
Generation of Stat3-Knockdown and Put-Back Cells
Lentiviruses containing shRNA targeting mouse Stat3 (Hanheng Biotechnology, Shanghai, China) were transfected into NMCMs to knock down endogenous Stat3. To render the put-back different exogenous Stat3 resistant to siRNA, 2 silent nucleotide substitutions (the siRNA-resistant nucleotide sequence 5′-CTACATTGGTGTTTCATAA-3′ was used to replace the original nucleotide sequence 5′-CCACGTTGGTGTTTCATAA-3′) were introduced into Flag-tagged mouse wild-type (WT) Stat3 and 2 other Flag-tagged mutant forms of Stat3 for which we either generated a lysine (K)-to-glutamine (Q) mutation at residue 370 (K370Q) to mimic constitutive acetylation or introduced lysine (K)-to-arginine (R) mutations at residues 370 and 383 (K370/383R) to mimic acetylation resistance. Adenoviruses containing the sequences of the Flag-tagged WT Stat3 and Stat3 mutants as described above were produced by Hanheng Biotechnology for transfection into Stat3-knockdown NMCMs. Additional information on this procedure is available in the Online Data Supplement.
Immunoprecipitation and Western Blotting
Cultured cells were lysed in immunoprecipitation lysis buffer, after which the cell lysates were incubated in immunoprecipitation washing buffer with the appropriate antibodies overnight at 4°C, with gentle agitation. The resultant protein–antibody immunocomplexes were precipitated after 1 hour of incubation with protein A/G plus agarose (Santa Cruz Biotechnology, Shanghai, China). Protein A/G plus agarose absorbed with protein–antibody immunocomplexes were washed 5× in lysis buffer before SDS–PAGE and immunoblotting with the indicated antibodies. Additional information on this procedure is available in the Online Data Supplement.
ATP Measurements
Cellular ATP content was measured using a luciferin/luciferase-based kit (Beyotime Biotechnology, Shanghai, China), according to the manufacturer’s instructions.
Measurement of Cardiac Myocyte Oxygen Consumption
An Oxygraph-2k (O2k; OROBOROS Instruments, Innsbruck, Austria) was used to measure respiration in intact cells.32,33 Approximately 1×106 cells were suspended in MiR05 (respiration media containing 0.5 mmol/L EGTA, 3 mmol/L MgCl2·6H2O, 60 mmol/L potassium lactobionate, 20 mmol/L taurine, 10 mmol/L KH2PO4, 20 mmol/L HEPES, 110 mmol/L sucrose, and 1 g/L fatty acid-free BSA, pH 7.1) and then added to the chamber with 2 mL in volume (37°C). The basal oxygen consumption rate (OCR) of the above cells was measured in MiR05 containing no substrates, and the maximum OCR of the cells was measured in MiR05 when adding 1 μmol/L titration of carbonyl cyanide m-chlorophenylhydrazone.
Transmission Electron Microscopy.
Murine heart specimens of the indicated genotypes were fixed in glutaraldehyde, post-fixed in osmium tetroxide, stained with ranyl acetate, dehydrated by ethanol solutions, and embedded in epoxy resin. Thereafter, the blocks were trimmed and cut into ultrathin sections (120 nm) of the indicated thickness. The sections were subsequently observed under a transmission electron microscope. We randomly captured images to measure mitochondrial area, circularity and cristae width using the multi-measure region of interest tool developed by the manufacturers of ImageJ. Additional information on this procedure is available in the Online Data Supplement.
p300-Induced Stat3 Acetylation Site Prediction
Protein–protein docking was performed using ZDOCK, and the indicated acetylation residues and their adjacent residues in Stat3 were set as the receptor binding sites.34 Any one of the following residues in p300 could serve as a donor binding site, as each residue in question was within the indicated distance (15 Å) of the receptor binding sites: ILE1456 (isoleucine1456), ARG1461 (arginine1461), and TRP1465 (tryptophan1465). The binding status of each docking model was evaluated using ZRANK scores.35
Protein Docking and Molecular Dynamics Simulation
Mutated Stat3 (ie, S1, S2, S3, and S4)-RelA protein docking was performed using ZDOCK.34 For this procedure, the angular step size was set at 15, the residues between LEU358 and ILE368 served as the receptor binding residues, and the docking models were ranked using ZRANK scores.35 For each of the docking complexes, that is, S1-RelA, S2-RelA, S3-RelA, and S4-RelA, the top-ranked structure was selected for subsequent molecular dynamics simulations (MDSs) and binding free energy calculations. To conduct the MDS, we first optimized each docking complex. We then heated the entire system to 300 K in 50 ps. A weak restraint force constant of k=10 kcal mol−1 Å−2 was applied to the protein atoms. The time for this step of the procedure was 1 fs. The complex was then simulated for 5 ns with a time step of 2 fs at 300 K. A quadratic constraint force constant of k=1 kcal mol−1 Å−2 was applied to RelA during the simulation. The SHAKE algorithm36 was used to constrain all the chemical bonds containing hydrogen atoms, and the particle-mesh Ewald method37 was used to quantify long-range electrostatic interactions. The nonbonded cutoff was 8 Å.
Binding Free Energy Calculation
The binding free energy (ΔG) of each complex was calculated using the molecular mechanics/Poisson−Boltzmann surface area equation,
| (1) |
where ΔEele and ΔEvdw are the electrostatic and van der Waals interaction energies, respectively, characterizing the interactions between the receptor and ligand; ΔGpB and ΔGnonpolar are the polar and nonpolar components of the desolvation energy based on the PB model, respectively; and TΔS is the change in conformational entropy induced by protein binding. For the binding free energy calculation, 20 snapshots of the 5-ns trajectory were taken for analysis. The value of the exterior dielectric constant was set to 80, and the value of the solute dielectric constant was set to 1. The nonpolar solvation term was calculated from the solvent-accessible surface area38 using the following equation: 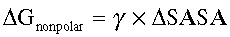 (where γ=0.0072 kcal/(mol·Å2), and the unit of
(where γ=0.0072 kcal/(mol·Å2), and the unit of 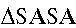 is Å2).
is Å2).
Additional information on the antibodies, chemicals, vectors, viruses, cell culture and transfection methods, western blot analyses, real-time polymerase chain reaction analyses, chromatin immunoprecipitation (ChIP) assays, immunofluorescence assays, mitochondrial DNA quantification experiments, mitochondrial flow cytometry analyses, mitochondrial respiration measurements, in silico structure optimization for protein optimization experiments, echocardiography studies, animal models, and histological analyses performed in the study is available in the Online Data Supplement.
Statistical Analysis
All values are presented as the mean±SE. After confirming that all variables were normally distributed using the Kolmogorov–Smirnov test, we evaluated the significance of the differences between groups by Student t test or 2-way ANOVA followed by Bonferroni multiple comparison test. P<0.05 was considered statistically significant.
Results
TNFR2 Activation Increased Mitochondrial Fusion and Function in NMCMs
To quantitatively assess the protective effects of TNFR2 activation on mitochondrial homeostasis, we transduced freshly isolated NMCMs and H9C2 cells with lentiviruses containing TNFR1-shRNAm (shRNA targeting mouse TNFR1) and TNFR1-shRNAr (shRNA targeting rat TNFR1), respectively. TNFR1 silencing efficiency was prevalidated in experiments involving NMCMs and H9C2 cells transfected with negative control shRNA (NC-shRNA; Online Figure IA and IB). Twelve hours after transduction, NMCMs with TNFR1 knocked down (NMCMs-TNFR1-KD) were treated with TNFα (0.5 ng/mL) for different time periods and then harvested for transmission electron microscopic examination to analyze the mitochondrial morphology. Twelve hours after TNFα treatment, NMCMs-TNFR1-KD featured mitochondria exhibiting a tubular morphology (Figure 1A). We quantified the changes in mitochondria morphology induced by TNFα treatment and found that TNFR2 activation in NMCMs-TNFR1-KD resulted in a 76% increase in mitochondrial size in the corresponding cells compared with placebo-treated cells, a change that was associated with a decrease in the circularity index (from 0.83±0.01–0.73±0.01) and a 45% increase in mitochondrial density (Figure 1B). These data indicate that treatment with TNFα enhanced mitochondrial fusion in NMCMs-TNFR1-KD.
Figure 1.
TNFR2 (TNFα receptor 2) activation increased mitochondrial fusion and function in NMCMs (neonatal mouse cardiac myocytes). A, NMCMs were transfected with shRNA targeting mouse TNFR1 (NMCMs-TNFR1-KD) and treated with either TNFα (0.5 ng/mL) or placebo for 12 h. Cells obtained before and after TNFα exposure were subjected to transmission electron microscopy (TEM) examination. Representative images are shown (bar=1 μm). B, The average mitochondrial area (μm2), circularity index, and number of mitochondria per μm2 were quantified via TEM analysis (n=3 separate studies, 100 mitochondria per group). C, Intracellular ATP levels were determined via luciferin/luciferase-based assays (n=3) involving NMCMs-TNFR1-KD. D, Oxygen consumption rates in NMCMs-TNFR1-KD were assayed under both basal and maximal conditions (ie, under carbonyl cyanide m-chlorophenylhydrazone [CCCP] treatment; n=3). E, Mitochondrial membrane potential (Δψm) was measured in H9C2 cells transfected with TNFR1-shRNAr and treated with either CCCP (50 μmol/L) or oligomycin (10 μmol/L), which served as negative and positive controls, respectively (n=3). The results are shown in the bar graph. F, Real-time polymerase chain reaction was performed to quantify mitochondrial cytochrome B (cytoB as mtDNA) expression levels, which were normalized to β-actin expression levels in NMCMs-TNFR1-KD to assess the total mitochondrial mass. G, MitoTracker Red staining was performed on H9C2 cells transfected with TNFR1-shRNAr to stain the mitochondria for flow cytometric analysis (n=3). *P<0.05, **P<0.01.
Previous studies have shown that mitochondrial fusion is associated with increased mitochondrial oxidative phosphorylation.25,39 To confirm this finding, we studied the effects of treatment with TNFα on ATP levels. Treatment with TNFα resulted in a 35% increase in ATP levels in NMCMs-TNFR1-KD compared with placebo-treated cells (Figure 1C). Moreover, treatment with TNFα resulted in a 35% increase in maximal OCRs (under carbonyl cyanide m-chlorophenylhydrazone treatment) but not basal OCRs in NMCMs-TNFR1-KD compared with placebo-treated cells (Figure 1D). In addition, TNFR2 activation increased the Δψm (+18%) in TNFα-treated H9C2 cells with TNFR1-shRNAr compared with placebo-treated cells at 12 hours post-treatment (Figure 1E).
TNFR2 Activation Did Not Promote Mitochondrial Biogenesis in NMCMs
We observed that TNFR2 activation induced significant changes in mitochondrial morphology and function. We subsequently aimed to determine whether these changes were attributable to enhanced mitochondrial biogenesis. We assessed cytoB expression levels (representing mtDNA levels), which were normalized to β-actin levels, by quantitative real-time polymerase chain reaction to measure the total mitochondrial mass in NMCMs-TNFR1-KD. Treatment with TNFα induced no changes in the above parameters in the corresponding cells compared with placebo-treated cells, indicating that TNFR2 activation does not stimulate enhancements of mitochondrial biogenesis (Figure 1F). We also performed flow cytometry analysis, in which MitoTracker Deep Red was used to stain the mitochondria (Figure 1G). The flow cytometry results also showed that treatment with TNFα elicited no significant changes in mitochondrial mass in the corresponding cells compared with placebo-treated cells. Moreover, we quantified the expression levels of the 3 core proteins involved in oxidative phosphorylation (ie, ATP5A, MTCO1, and SDHB) by Western blotting, with both HSP60 (heat shock protein 60) and β-actin serving as loading controls. Treatment with TNFα did not induce significant changes in the expression levels of the indicated proteins (Figure 2A) in NMCMs-TNFR1-KD compared with placebo-treated cells. In addition, we noted no changes in the protein expression levels of PGC1α, a central regulator of mitochondrial biogenesis, in TNFα-treated cells compared with placebo-treated cells (Figure 2A).
Figure 2.
TNFR2 (TNFα receptor 2) activation enhanced mitochondrial fusion by upregulating OPA1 (optic atrophy 1) expression. A, Neonatal mouse cardiac myocytes (NMCMs)-TNFR1-KD were treated with TNFα (tumor necrosis factor α; 0.5 ng/mL) for the indicated times and then harvested, after which the total protein was obtained. The protein expression levels of PGC-1α (peroxisome proliferator–activated receptor gamma coactivator 1-alpha), a transcriptional factor that regulates mitochondrial biogenesis, and selected proteins that participate in mitochondrial oxidative phosphorylation (OXPHOS; including ATP5A, MTC01, and SDHB) were determined by Western blotting (n=3) and did not change after TNFR2 activation. Both HSP60 (heat shock protein 60; a mitochondrial marker protein) and β-actin (a cytosolic marker protein) served as loading controls. B and C, The expression levels of mitochondrial dynamic-related proteins, including OPA1 (optic atrophy 1), Drp1 (dynamin-related protein 1), Mfn1 (mitofusin 1), and Mfn2 (mitofusin 2), were determined by Western blotting. Representative western blots are shown (n=3). Two antibodies against OPA1, including a polyclonal antibody (the first lane in C) and a monoclonal antibody (the fourth lane in C), were used to detect OPA1. The densitometry of each protein was quantified and normalized to that of either β-actin or GAPDH, and the differences in protein expression between the groups are shown in the relevant bar graphs. D–G, To determine whether upregulated OPA1 was responsible for enhancements of mitochondrial fusion, we cotransfected NMCMs-TNFR1-KD with OPA1-siRNA before treating them with TNFα for 12 h. Transmission electron microscopic examinations were performed, and representative images for each group are shown (D, bar=1 μm). The average mitochondrial area (μm2), circularity index and number of mitochondria per μm2 were quantified and are shown in a bar graph (E, n=3, 100 mitochondria per group). Intracellular ATP levels (F) and oxygen consumption rates (G) were also determined (n=3, respectively). **P<0.01.
TNFR2 Activation Modulated OPA1 Expression in NMCMs
To further evaluate the changes in mitochondrial fusion induced by TNFR2 activation in NMCMs-TNFR1-KD, we measured the expression levels of the key proteins involved in mitochondrial dynamics40–42 by Western blot analysis. Mfn1, Mfn2, and Drp1 protein expression levels did not change significantly in TNFα-treated cells compared with placebo-treated cells (Figure 2B); however, surprisingly, total OPA1 expression levels were significantly increased by TNFR2 activation (Figure 2C). The results of the Western blot analysis, which was performed using 2 different antibodies specific for OPA1, showed that the expression levels of five OPA1 isoforms (5 bands) recognized by polyclonal antibodies increased by 1.52±0.1-fold in NMCMs in response to TNFR2 stimulation. Moreover, the expression levels of the long and short isoforms of OPA1 (2 bands), which were detected using monoclonal antibodies, increased by 1.73±0.2-fold (Figure 2C). Thus, our data strongly suggested that the improvements in mitochondrial fusion and function induced by TNFR2 activation are because of enhancements of OPA1-dependent mitochondrial fusion.
TNFR2 Activation-Induced OPA1 Upregulation Was Responsible for Enhancing Mitochondrial Fusion and Function in NMCMs.
We subsequently assessed whether enhancements of OPA1 expression were responsible for improving mitochondrial fusion and function after TNFR2 stimulation. We transfected NMCMs-TNFR1-KD with either NC-siRNA or siRNA targeting mouse OPA1 (OPA1-siRNA). The efficiency of the siRNAs was tested previously (Online Figure IC). Consistent with the results shown in Figure 1, the results of this experiment showed that NMCMs-TNFR1-KD transfected with NC-siRNA exhibited a 56% increase in mitochondrial area, a 10% decrease in the circularity index, and a 42% decrease in mitochondrial density compared with untransfected cells after TNFR2 stimulation. All of these changes were attenuated in NMCMs-TNFR1-KD transfected with OPA1-siRNA (Figure 2D and 2E). Similar results were observed in the experiments in which intracellular ATP levels (Figure 2F) and maximum oxygen consumption capacity (Figure 2G) were quantified, indicating that OPA1 upregulation contributed to enhancements of mitochondrial function. Interestingly, we also observed that Drp1 mitochondrial localization was slightly decreased in OPA1-siRNA–transfected cells compared with NC-siRNA–transfected cells (Figure II).
Both Stat3 and NF-kB Signaling Promoted OPA1 Expression After TNFR2 Activation
To identify the specific signaling pathway that regulates OPA1 expression, we used specific inhibitors to block the signaling pathways downstream of TNFR2. Specifically, we pretreated NMCMs-TNFR1-KD with the following agents: pyrrolidine dithiocarbamate (at 50 µmol/L), which inhibits the NF-kB (nuclear factor-kappa B) signaling pathway43; bisindolylmaleimide I (at 50 µmol/L), which inhibits the PKC signaling pathway44; wortmannin (500 nmol/L), which inhibits the PI3K signaling pathway45; ruxolitinib (250 nmol/L), which inhibits the Stat3 signaling pathway5,6; or KN-62 (3 µmol/L), which inhibits the CaMKII (Ca2+/calmodulin-dependent protein kinase II) signaling pathway.46 We found that the enhancements of OPA1 expression that were induced by TNFR2 activation were largely abrogated by pretreatment with either pyrrolidine dithiocarbamate or ruxolitinib however were unaffected by treatment with bisindolylmaleimide I, wortmannin or KN-62 (Figure 3A), suggesting that Stat3 and NF-kB both participate in regulating OPA1 expression.
Figure 3.
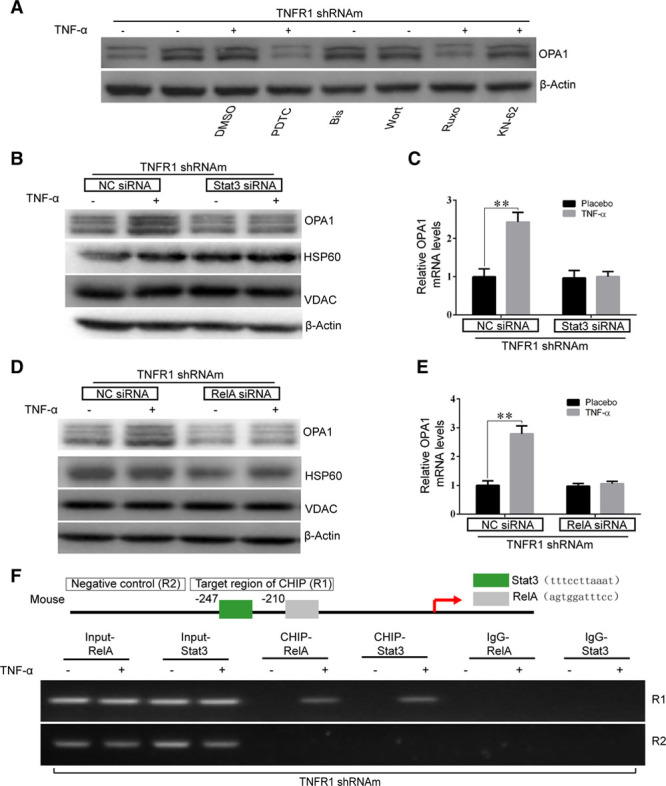
Stat3 and NF-kB (nuclear factor-kappa B) signaling pathways participated in TNFR2 (TNFα receptor 2) activation-induced OPA1 (optic atrophy 1) upregulation in neonatal mouse cardiac myocytes (NMCMs). A, Various pharmacological inhibitors of different signaling pathways, including bisindolylmaleimide I (Bis I, PKC inhibitor), ruxolitinib (Ruxo, Stat3 inhibitor), pyrrolidine dithiocarbamate (PDTC, NF-kB inhibitor), KN-62 (CaMKII [Ca2+/calmodulin-dependent protein kinase II] inhibitor), and wortmannin (Wort, PI3K inhibitor), were pre-administered to NMCMs-TNFR1-KD. TNFR2 activation-induced OPA1 expression levels were then quantified. B–E, Stat3 (B and C) or RelA (D and E) was knocked down by siRNA against Stat3 or RelA, respectively, in NMCMs-TNFR1-KD, after which the cells were treated with TNFα (tumor necrosis factor α). OPA1 protein (B and D) and mRNA expression levels (C and E) were quantified. F, Chromatin immunoprecipitation (ChIP)-polymerase chain reaction assay was performed to identify the binding sites for Stat3 and RelA in the promoter region of OPA1. **P<0.01.
To confirm that Stat3 and NF-kB participate in regulating OPA1 expression in response to TNFR2 activation, we used siRNA specifically targeting either mouse Stat3 (Stat3-siRNA) or mouse RelA (RelA-siRNA). The silencing efficiency of the indicated siRNAs was tested previously (Online Figure ID and IE). Treatment with either Stat3-siRNA or RelA-siRNA significantly attenuated the indicated enhancements of OPA1 expression in the corresponding cells compared with NC-siRNA–treated cells after TNFR2 stimulation, as shown by the results of experiments in which protein (by Western blotting) and mRNA expression (by polymerase chain reaction) levels in the indicated cells were assessed (Figure 3B through 3E). As Stat3 and RelA are both well-known important transcription factors, we aimed to determine whether Stat3 and RelA regulated OPA1 expression at the transcriptional level after TNFR2 activation. Analysis of the OPA1 promoter using Jaspar software (http://jaspar.genereg.net/) identified 2 putative binding sequences for Stat3 and RelA (as indicated in Figure 3F) that were adjacent to each other. To directly determine how Stat3 and RelA mediated OPA1 expression at the transcriptional level in NMCMs-TNFR1-KD, we performed ChIP assays. We designed a pair of primers that targeted the R1 region covering the 2 putative binding sequences (Online Table I). Primers targeting the R2 region were used as negative controls. The results of the ChIP analysis, in which antibodies targeting either mouse Stat3 or mouse RelA were used, showed that TNFR2 stimulation resulted in the binding of both Stat3 and RelA to the OPA1 promoter (Figure 3F). These data implied that Stat3 and RelA can synergistically enhance OPA1 transcription by binding to adjacent regions of the OPA1 promoter.
TNFR2 Activation–Mediated Increases in Stat3 Acetylation Enhanced Stat3/RelA Interactions and Thus Upregulated OPA1 Expression
The roles of Stat3/RelA interactions in the nuclear retention of both proteins and the modulation of the expression of prosurvival genes have been studied previously.47,48 In the present study, we tested whether TNFR2 activation also facilitated Stat3/RelA interactions to increase OPA1 expression. We designed TNFR1-shRNAr (shRNA targeting rat TNFR1) and TNFR2-shRNAr (shRNA targeting rat TNFR2) and a negative control shRNA (NC-shRNA) and then transfected them into H9C2 cells. The gene silencing efficiency of each siRNA was tested before the experiment (Online Figure IB and IF). We examined post-TNFα treatment Stat3/RelA colocalization in the nucleus by confocal immunofluorescence assay. Stat3/RelA clearly colocalized in the nuclei of H9C2 cells infected with NC-shRNA and TNFR1-shRNAr; however, only RelA localized in the nuclei of H9C2 cells infected with TNFR2-shRNAr (Figure 4A). To confirm that TNFR2 activation modulates the interactions between Stat3 and RelA in mice, we performed an experiment involving NMCMs. We transfected TNFR2-shRNAm (shRNA targeting mouse TNFR2) into NMCMs (NMCMs-TNFR2-KD). The silencing efficiency of the indicated siRNA was tested previously (Online Figure IG). We then performed a co-immunoprecipitation (co-IP) assay to test the interactions between endogenous Stat3 and RelA in NMCMs-TNFR2-KD and NRCMs-TNFR1-KD after the cells were stimulated by TNFα. As expected, Stat3 and RelA interacted in NRCMs-TNFR1-KD (Figure 4B) but not in NRCMs-TNFR2-KD (Figure 4C).
Figure 4.
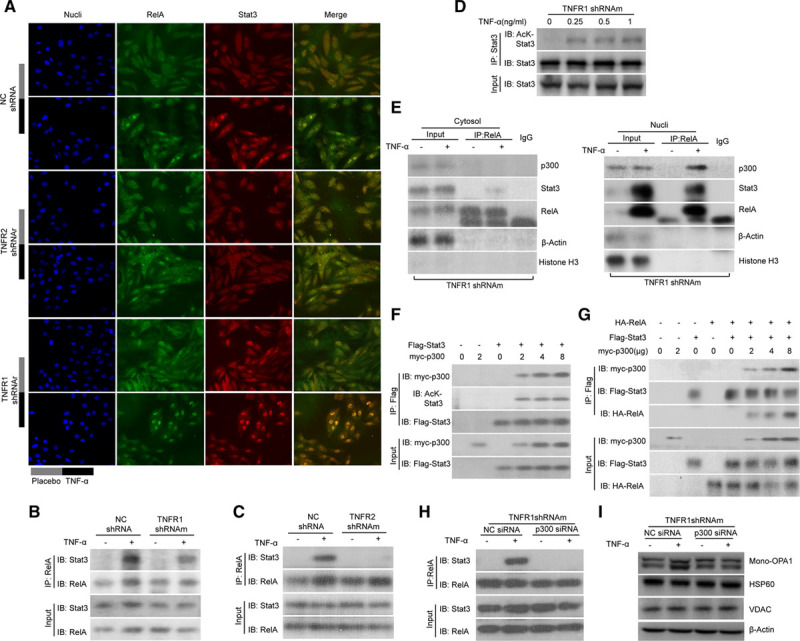
p300 was essential for Stat3–NF-kB (nuclear factor-kappa B) interactions and, consequently, OPA1 (optic atrophy 1) expression on TNFR2 (TNFα receptor 2) activation. A, H9C2 cells were transfected with shRNA targeting rat TNFR1 (TNFR1-shRNAr), shRNA targeting rat TNFR2 (TNFR2-shRNAr), or negative control (NC)-shRNA. These cells were then treated with TNFα (tumor necrosis factor α; 0.5 ng/mL) or placebo, which served as a control, for 3 h. Confocal microscopy examination was performed. Fluorescence staining was performed to detect RelA (in green) and Stat3 (in red), and DAPI (4',6-diamidino-2-phenylindole) used to identify the nucleus. B and C, Co-immunoprecipitation (Co-IP) experiments were performed to assess the interactions between Stat3 and RelA in neonatal mouse cardiac myocytes (NMCMs)-TNFR1-KD and NMCMs-TNFR2-KD after TNFα treatment. D, Stat3 was immunoprecipitated in NMCMs-TNFR1-KD treated with incremental doses of TNFα and then immunoblotted using pan-antiacetyl lysine antibodies (a-Ac). E, Cytosolic (left) and nuclear (right) protein extracts were prepared from the same cells described in D. The proteins were then precipitated with antibodies against RelA followed by immunoblotting (IB) detecting RelA, Stat3, and p300. F, Plasmids containing myc-p300 and Flag-Stat3 were cotransfected into HEK293T (human embryonic kidney 293T) cells. Co-IP was performed again using antibodies against Flag to pull down Stat3, IB was then performed using a-Ac. G, Plasmids containing myc-p300, Flag-Stat3, and HA-RelA were then cotransfected into HEK293T cells, as indicated, followed by co-IP to detect the interactions of Stat3 with myc-p300 and HA-RelA. H and I, NMCMs-TNFR1-KD were transfected with p300-siRNA and then treated with TNFα. The interactions between endogenous Stat3 and RelA were subsequently assessed by co-IP (H), as described above. I, OPA1, HSP60 (heat shock protein 60), and VDAC (voltage-dependent anion-selective channel) protein expression levels were determined by Western blot analysis.
Protein acetylation has been demonstrated to play essential roles in protein–protein interactions,49,50 and the acetylation status of Stat3 has also been thoroughly investigated.51–54 Moreover, the cross talk and interactions between Stat3 and NF-kB signaling have been described in previous studies.48,55,56 In the present study, we sought to determine whether Stat3 acetylation could also affect Stat3/RelA interactions after TNFR2 activation. We thus evaluated the acetylation status of Stat3 in TNFα-treated NMCMs-TNFR1-KD. We immunoprecipitated total endogenous Stat3 and then assayed the expression of the protein by Western blotting using antiacetyl-lysine antibodies. We observed that TNFR2 activation resulted in a dose-dependent increase in acetylated Stat3 levels (Figure 4D). Additionally, p300-mediated modulation of Stat3 acetylation has also been described in previous studies.57–59 In the present study, we determined whether p300 participated in the enhancements of Stat3 acetylation induced by TNFR2 activation. Indeed, TNFR2 activation in NMCMs-TNFR1-KD significantly increased the interactions between p300 and the Stat3/RelA complex in the nucleus but not in the cytosol (Figure 4E). Therefore, we cotransfected myc-p300 and Flag-Stat3 into HEK293T (human embryonic kidney 293T) cells to recapitulate the process through which p300 acetylates Stat3. We then immunoprecipitated the resulting Flag-Stat3 complexes using antibodies against Flag-Stat3 and found that p300 increased acetylated Flag-tagged Stat3 levels in a dose-dependent manner (Figure 4F). To confirm the role of p300 in Stat3/RelA interactions, we cotransfected HEK293T cells with myc-p300, Flag-Stat3 and HA-RelA. Protein–protein interactions between Flag-Stat3 and HA-RelA were minimally detectable in HEK293T cells in which p300 was absent but were significantly enhanced in HEK293T cells in which exogenous myc-p300 was coexpressed (Figure 4G). By contrast, when NMCMs-TNFR1-KD were transfected with p300-specific siRNA (p300-siRNA), whose knockdown efficiency was tested previously (Online Figure IH), endogenous p300 downregulation attenuated the above enhancements of the interactions between Stat3 and RelA that were induced by TNFR2 activation in NMCMs (Figure 4H). Data collected by previous studies indicate that p300/CBP-associated factor (PCAF), another acetyltransferase, may also acetylate Stat3.60,61 To test the hypothesis that PCAF regulates Stat3/RelA interactions in response to TNFR2 activation, we transfected NMCMs-TNFR1-KD with mouse PCAF-specific siRNA (PCAF-siRNA), whose knockdown efficiency was tested previously (Online Figure II) and performed a co-IP assay using the methods described above. The results showed that Stat3 and RelA could still form a complex when PCAF was knocked down, suggesting that TNFR2 promotes Stat3/RelA interactions in a PCAF-independent manner (Online Figure III). The results also showed that transfecting NMCMs-TNFR1-KD with p300-siRNA eventually attenuated the enhancements of OPA1 expression induced by TNFR2 stimulation (Figure 4I). Taken together, the results of these experiments strongly supported our hypothesis that p300 modulates Stat3 acetylation to enhance Stat3/RelA interactions, leading to enhanced OPA1 expression in TNFR2-stimulated NMCMs.
Stat3 DNA-Binding Domain Acetylation by p300 Facilitated Stat3/RelA Interactions
As shown in Figure 4F through H, p300-induced Stat3 acetylation is essential for Stat3/RelA interactions. We subsequently aimed to elucidate the molecular mechanism responsible for the interactions. Previous studies have demonstrated that Stat3 contains several functional domains, including an N-terminal domain, a coiled-coil domain, a DNA-binding domain, a linker domain, an Src homology 2 domain, and a transactivation domain (TAD).62 We first conducted ZDOCK analysis to identify the potential sites at which Stat3 is acetylated, as ZDOCK studies can simulate Stat3-p300 protein–protein docking and thus identify multiple acetylation sites on full-length Stat3. Forty-eight lysine sites in Stat3, as well as their neighboring residues, were set as receptor-binding site residues,34 and 1 of the 3 residues in p300 carrying an acetyl group to acetylate substrates (such as Stat3), namely, ILE1456, ARG1461, and TRP1465, was set as a donor binding site. The most likely potential acetylation sites were listed according to their ZRNAK score. Of the 48 lysine sites in Stat3, 12 candidate residues (685, 531, 87, 409, 615, 383, 517, 49, 140, 631, 244, 370) were selected according to their ZRANK score (listed in Online Figure IV) and were assessed in subsequent experiments to quantify their ability to undergo acetylation.35 We also performed a parallel experiment, in which we generated a series of Flag-tagged truncated Stat3 mutants (Figure 5A and 5B) and transfected them into HEK293T cells alone or with HA-tagged RelA or myc-tagged p300. We then immunoprecipitated the cell extracts with Flag-specific antibodies before immunoblotting them using antibodies targeting either HA or Myc. The full-length Stat3 can bind to RelA in p300-over-expressing HEK293T cells. However, the truncated Stat3 mutants, which were devoid of their DBD (Figure 5A), lost the ability to interact with RelA, as demonstrated by the co-IP assay (Figure 5B). This observation suggested that the DBD (from amino acids 330 to 465) of Stat3 (Figure 5A) served as a RelA-affiliated binding domain. Importantly, this interaction was p300 dependent (lane 5 in Figure 5B). Notably, we observed that the depletion of more Stat3 domains (Figure 5A) gradually decreased the p300-binding capacity of Stat3 (lanes 9–11 on IB for myc-p300, Figure 5B), indicating that multiple acetylation sites in Stat3 may participate in its interaction with p300. Thus, our data indicate that the Stat3 DBD was the domain responsible for mediating Stat3/RelA interactions. Interestingly, the ZDOCK study results pertaining to the predicted Stat3 acetylation sites (listed in Online Figure IV) showed that lysine (LYS) 409, LYS383, and LYS370 were the most likely acetylation sites in the Stat3 DBD. Thus, we selected these 3 sites as receptor-binding residues; whereas residues of ILE1456, ARG1461, and TRP1465 within p300 that carry acetyl groups were set as donor-binding residues. We then analyzed the binding interface between Stat3 and p300 (Figure 5C). The distance between LYS370 and its closest donor site, ILE1456, was ≈3.457 Å, whereas the minimum distances between LYS383 and LYS409 (as the receptor residues) and their closest donor sites both exceeded 10 Å (Figure 5C). We also evaluated the binding interfaces between the remaining nine putative acetylated lysine residues in Stat3 and p300 (Online Figure Va–i).
Figure 5.
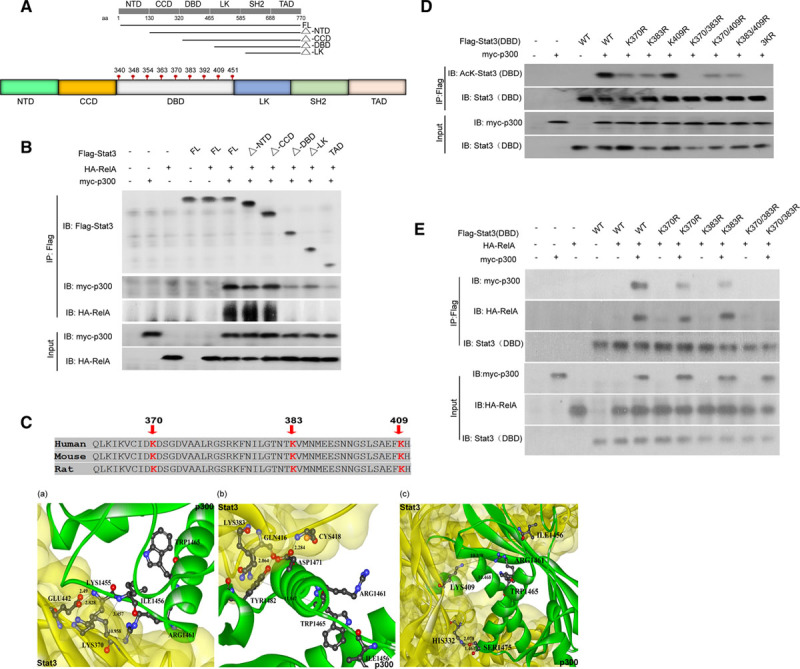
Acetylation of Stat3 at LYS(K)370, LYS(K)383, or both enhanced its interactions with RelA. A, Schematic diagram for various truncated mutant of Stat3 that span different domains of Stat3 as denoted by straight black line. The truncated mutant Stat3 lacking N-terminal domain (NTD) is denoted by ΔNTD. The same scheme was used for the other truncated mutants, which are noted by Δ coiled-coil domain (CCD), Δ DNA-binding domain (DBD), and Δ linker domain (LK). Different amino acid (aa) sites were also marked for each domain. All the lysine residues located within the DBD, which is labeled in gray, are outlined. B, HEK293T (human embryonic kidney 293T) cells were transfected with HA-RelA or myc-p300, as well as different vectors containing various Flag-tagged Stat3 mutants (as indicated in A). The Flag-tagged Stat3 mutants were immunoprecipitated and then immunoblotted using specific antibodies against Flag, myc, and HA. C, The Stat3–p300 interface was simulated by an in silico analysis focusing on the lysine residues K370 (a), K383 (b), and K409 (c), which served as binding sites. The distance between the residues on Stat3 and p300 is shown in the figure. D, Various vectors containing a Stat3 DBD in which the putative lysine residues found in Flag-Stat3(DBD), namely, LYS(K)409, K383, and K370, were mutated to arginine (R) either alone or in combination, as indicated, were generated and transfected into HEK293T cells along with myc-p300. Whole-cell proteins were then harvested for immunoprecipitation, which was performed using antibodies against Flag-tag, followed by immunoblotting using antibodies against acetyl-lysine to assess the acetylation of the mutant Stat3 DBD. E, The same HEK293T cells described in D were also transfected with HA-RelA before undergoing the same tests to determine whether p300-mediated Stat3 acetylation also affects Stat3/RelA interactions. ARG indicates arginine; ASP, aspartic acid; CYS, cysteine; FL, full-length Stat3; GLN, glutamine; GLU glutamic acid; HIS, histidine; ILE, isoleucine; SER, serine; SH2, Src homology 2 domain; TAD, that only the transactivation domain was present in the corresponding truncated mutant; TRP, tryptophan; and TYR, tyrosine.
p300-Mediated Stat3 Acetylation at LYS370 and LYS383 Facilitated Its Interaction With RelA in HEK293T Cells
Based on the data obtained from the ZDOCK-based bioinformatic analysis, we determined the role of acetylation of the indicated lysine residues in Stat3/RelA interactions. We generated Stat3 mutants in which lysine (K) was replaced by arginine (R) to mimic the constitutively nonacetylated form of Stat3. We then constructed 8 different plasmids containing Flag-tagged WT Stat3 DBDs or Flag-tagged mutant Stat3 DBDs, which featured mutations at LYS370, LYS383, or LYS409 or combinations of these mutations (Figure 5D). We then cotransfected Flag-tagged WT Stat3 DBDs and different mutant Stat3 DBDs and myc-tagged p300 into HEK 293T cells. We first evaluated the roles of these 3 lysine residues in Stat3 DBD acetylation by co-IP assay. We observed WT Stat3 DBD acetylation in p300-overexpressing HEK 293T cells. Surprisingly, the K-to-R mutation on LYS409 (K409R) had no effect on Stat3 DBD acetylation, and the single K-to-R mutation on LYS370 (K370R) or LYS383 (K383R) and the combination of one of those mutations with the mutation on LYS409 (ie, K409/370R or K409/383R) had limited effects on Stat3 DBD acetylation (Figure 5D). Interestingly, K-to-R mutations on both LYS370 and LYS383 (K370/380R) or all 3 lysine residues, that is, LYS370, LYS383, and LYS409 (3K/R), abolished the capacity of Stat3 to undergo acetylation (Figure 5D), suggesting that p300 was capable of acetylating the Stat3 DBD at LYS370 and LYS383.
We then evaluated how the acetylation status of the Stat3 DBD affects its interactions with RelA. We cotransfected HEK 293T cells with different Flag-tagged mutant Stat3 DBDs and myc-p300, as well as HA-tagged RelA. Consistent with our previous results (refer to Figure 4G), the interaction between the WT Stat3 DBD and WT-RelA was significantly enhanced by p300 overexpression (Figure 5E). Single K-to-R mutations (either K370R or K383R) partially reduced the ability of the Stat3 DBD to bind WT-RelA. By contrast, double K-to-R mutations (K370/383R) abolished the interaction between Stat3 and RelA (Figure 5E).
MDS Analysis of the Contribution of Stat3 Acetylation to Stat3/RelA Interactions
As co-IP assay enables only semiquantitative assessments of protein-protein interactions, we conducted MDS to simulate the binding of RelA to an unmodified Stat3 (S1), a LYS(K)383-acetylated Stat3 (S2), a K370-acetylated Stat3 (S3), or both acetylated Stat3 species (Online Figure IV; Figure 6A). After establishing the 4 binding models, that is, S1-RelA, S2-RelA, S3-RelA, and S4-RelA, we calculated the binding free energy (ΔGtotal) value for each model using the molecular mechanics/Poisson–Boltzmann surface area equation (Figure 6B). S3-RelA had the lowest binding free energy of −77.37 kcal/mol and the binding free energy of the other models gradually increased from S4-RelA to S2-RelA and then to S1-RelA (Figure 6B). In addition, the electrostatic interaction energy (ΔEele) is a greater contributor to the above protein–protein interactions than van der Waals interaction energy (ΔEvdw). Moreover, in silico analysis showed that Stat3 acetylation at K370 alone led to the formation of the most stable Stat3–RelA binding interface.
Figure 6.
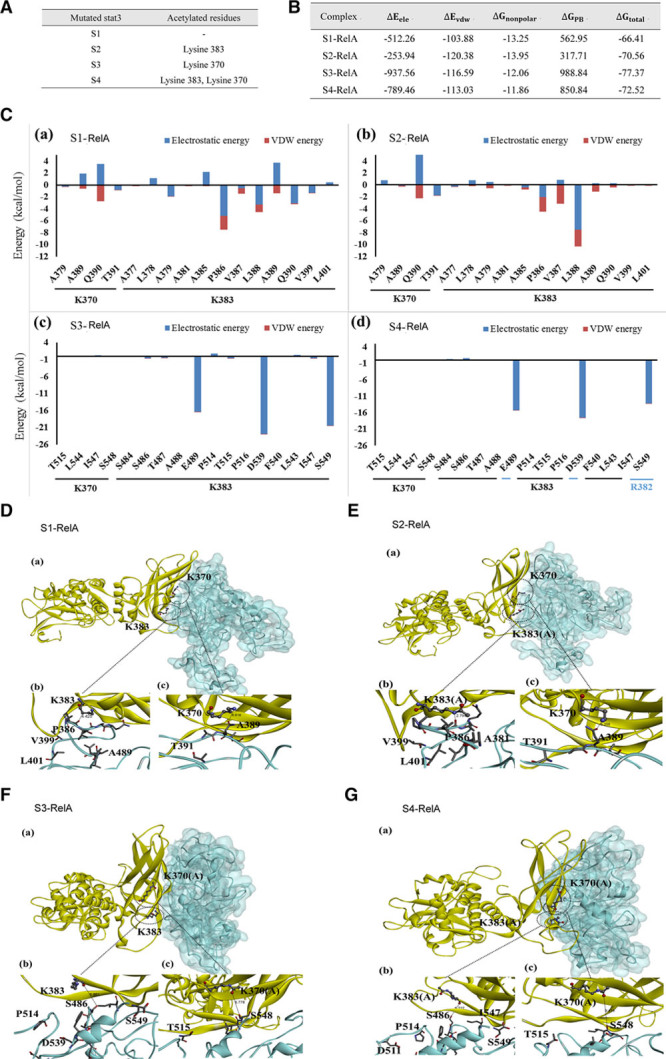
Molecular dynamics simulation (MDS) analysis of the contribution of Stat3 acetylation to Stat3/RelA interactions. A, Four Stat3 mutants with different acetylated residues were constructed for MDS. S1, S2, S3, and S4 denote nonacetylated Stat3, K383-acetylated Stat3, K370-acetylated Stat3, and K370/K383-acetylated Stat3, respectively. B, Free binding energy calculation for the Stat3–RelA binding complexes, namely, S1-RelA, S2-RelA, S3-RelA, and S4-RelA. The energy for each interaction was expressed in kcal/mol. The definition of each equation term was provided in text. C, The energies for the interactions between each Stat3 mutant (ie, (a) S1, (b) S2, (c) S3, and (d) S4) and the residues located on RelA. The electrostatic and van der Waals contributions to the interactions are colored in blue and red, respectively. D–G, The interfaces for the 4 binding complexes described in B and C are illustrated. (a) The RelA (with cyan surface)–Stat3 (in yellow) interactions involving residues K370 and K383 are represented by balls and sticks; (b) Close-up view of K383 in Stat3 and the residues on RelA. (c) Close-up view of K370 in Stat3 and the residues on RelA. K383(A) and K370(A) denote K383 acetylation and K370 acetylation, respectively. A indicates alanine; D, aspartic acid; E, glutamic acid; F, phenylalanine; I, isoleucine; L, leucine; P, proline; Q, glutamine; S, serine; T, threonine; and V, valine.
We also investigated how the acetylation statuses of K370 and K383 affect the binding free energy that drives their interactions with their respective RelA binding residues using information provided by the docking model. We calculated the electrostatic and van der Waals interaction energies from 20 snapshots of the last 1 ns of the corresponding trajectories.
When K383 was acetylated, the repulsive interactions between this site and A385 and A389 of RelA decreased (Figure 6Ca and b). As shown in Figure 6Db and 6Eb, the distance between S2 and RelA was smaller than that between S1 and RelA. The shorter distance between S2 and RelA resulted in stronger electrostatic interactions between L388 and K383. These results indicated that acetylation at K383 may facilitate stronger binding between Stat3 and RelA.
When K370 was acetylated, the dynamics of the binding interface fluctuated and the binding spectrum of the amino acids of RelA changed from A377-L401 (Figure 6Cb) to S484-S549 (Figure 6Cc). Careful quantification revealed that the energy levels of the interactions between K370 and the residues on RelA (namely, T515, L544, I547, and S548) were <1 kcal/mol (Figure 6Cc), which indicated that the acetylation of K370 resulted in a weaker interaction between Stat3 and RelA and that the K370 residue on Stat3 was not spatially adjacent to the corresponding residues on RelA (Figure 6Fc). Intriguingly, K370 acetylation contributes to enhanced electrostatic interactions between K383 and residues on RelA, enhancements that are partially attributable to strong electrostatic interactions between the positively charged lysine and the indicated negatively charged amino acids (aspartate and glutamate; Figure 6Cc and d).
We also assessed the major interactions that occurred following the acetylation of both K370 and K383 and found that both residues were neutral (Figure 6Cd). The interaction between S4 and RelA (Figure 6G) was weaker than that between S3 and RelA (Figure 6F), as the acetylated K383 was neutral, and the electrostatic interactions between K383 on Stat3 and the E489, D539, and S549 residues on RelA in the indicated model were weaker than the corresponding interactions in the S3-RelA model. Acetylation at both K370 and K383 also enhanced the electrostatic interactions between R382, a residue in close proximity to K383 on Stat3, and S549 on RelA (Figure 6Cd). These findings indicated that the variations in electrostatic energy that characterized the indicated binding interface arose from the acetylated residues and their adjacent residues. The backbone root mean square deviation (RMSD) changes that occurred during 40 ns of MDS (overall, 5000 snapshots were taken during the 50-ns trajectory; the first 10 ns represented the equilibrium run, and the last snapshot was set as the RMSD reference point) are shown in Online FigureVIa through d. All RMSDs were converged during the simulation, and the last 1-ns trajectory was used to calculate the binding free energy.
Acetylation of K370 and K383 Was Essential for OPA1 Expression and, ultimately, Mitochondrial Fusion in NMCMs After TNFR2 Activation
As stated above, we confirmed that p300-induced acetylation of the K370 and K383 residues of Stat3 was indispensable for the interactions between Stat3 and RelA in HEK293T cells. We also showed that knocking down p300 can abolish TNFR2-induced enhancements of OPA1 expression in NMCMs (Figure 4I). Thus, we aimed to confirm that acetylation of Stat3 by p300 at residues K370 and K383 played key roles in its binding to RelA and the subsequent formation of a transcriptional complex (Stat3/RelA complex) that promoted OPA1 expression at the transcriptional level after TNFR2 activation.
We knocked down endogenous Stat3 in freshly isolated NMCMs and cotransfected the cells with various vectors, including WT Stat3, acetylation-mimetic Stat3 (K370Q-Stat3) and acetylation-resistant Stat3 (K370/383R-Stat3), to re-express exogenous shRNA-resistant Stat3. We then identified the resulting Stat3-knockdown and put-back cells (Online Figure VII). The co-IP assay showed that the interactions between Flag-tagged WT-Stat3 and endogenous RelA increased significantly after TNFR2 stimulation (Figure 7A). Moreover, expressing acetylation-mimetic K370Q-Stat3 in endogenous Stat3-deprived NMCMs-TNFR1-KD substantially enhanced the interactions between Stat3 and endogenous RelA even in the absence of TNFR2 stimulation (Figure 7A). By contrast, expressing deacetylation-resistant K370/383R-Stat3 in these cells resulted in a failure of Stat3 to bind endogenous RelA even after TNFα stimulation (Figure 7A). Re-expressing WT Stat3 in the indicated cells resulted in increased OPA1 expression after TNFα therapy, whereas expressing K370Q-Stat3 increased OPA1 protein expression even in the absence of TNFR2 stimulation (Figure 7B). However, as expected, no increase in OPA1 expression was observed in cells transfected with K370/383R-Stat3 (Figure 7B). Thus, our data supported the idea that K370- and K383-dependent Stat3 acetylation plays an essential role in the Stat3/RelA interactions that lead to enhanced OPA1 expression.
Figure 7.
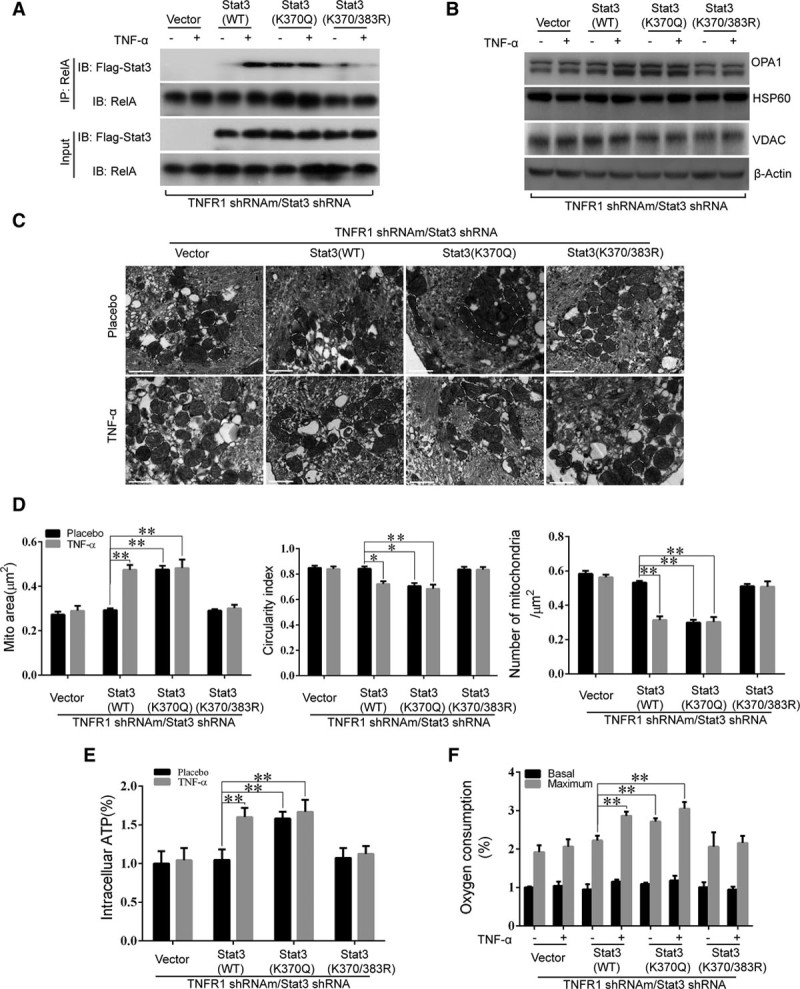
Acetylation of K370 and K383 was essential for OPA1 (optic atrophy 1) expression and, consequently, mitochondria fusion in TNFR2 (TNFα receptor 2)–treated cardiac myocytes. A and B, Neonatal mouse cardiac myocytes (NMCMs)-TNFR1-KD were first transfected with Stat3-shRNA to knock down Stat3. Adenoviruses containing wild-type (WT) Stat3, Stat3 with a K370Q or K370/380R mutation, and empty vectors (which served as controls) were simultaneously transfected into the cells, which were then treated with either TNFα (tumor necrosis factor α; 0.5 ng/mL) or placebo for 3 h. Co-immunoprecipitation (Co-IP) was performed using antibodies against RelA precipitation followed by immunoblotting (IB) using antibodies against Flag or RelA. OPA1 protein expression levels were detected by Western blotting with HSP60 (heat shock protein 60), VDAC (voltage-dependent anion-selective channel), and β-actin as loading controls. C and D, transmission electron microscopic images of the mitochondria were obtained using cells treated as described in A and B, and the average mitochondrial area (μm2), circularity index and number of mitochondria per micrometer square were quantified in a bar graph (n=3, 100 mitochondria per group). E and F, Intracellular ATP levels and basal and maximal oxygen consumption capacity were evaluated (F, n=3 per group). **P<0.01.
When we assessed mitochondrial morphology (Figure 7C and 7D) (mitochondrial area, the circularity index, and mitochondrial density) and function (Figure 7E and 7F) (ATP production and both basal and maximal oxygen consumption), we found that the patterns of the changes in mitochondrial morphology induced by the above treatments were consistent with the corresponding changes in Stat3/RelA interactions and OPA1 expression (as shown in Figure 7A and 7B). We observed no changes in the above parameters in response to TNFR2 activation when NMCMs-TNFR1-KD were deprived of endogenous Stat3; however, re-expressing WT Stat3 re-established the abovementioned response to TNFR2 activation. Interestingly, spontaneous increases (ie, increases not induced by TNFα stimulation) in mitochondrial fusion (Figure 7C and 7D) and function (Figure 7E and 7F) were observed when K370Q-Stat3 was put back. By contrast, no increases in mitochondrial fusion (Figure 7C and 7D) and function (Figure 7E and 7F) occurred when NMCMs-TNFR1-KD were transfected with K370/383R-Stat3.
Effects of TNFR2 on Mitochondrial Fusion in TAC-Induced Heart Failure
To determine whether TNFR2 activation-induced OPA1 expression protects the heart against stress, we generated TAC mice model to create pressure-overload–induced heart failure. A total of 60, 8-week-old mice, including 30 TNFR1-knockout male mice and 30 TNFR1/TNFR2 double (TNFR1/2)-knockout male mice, were evaluated in the in vivo study. Mice were randomly assigned to either sham (n=12 of each mouse genotype) or TAC surgery (n=18 of each mouse genotype) groups and followed up for 8 weeks after TAC surgery. All sham-operated TNFR1-knockout and TNFR1/2-knockout mice survived the 8-week postoperative period, whereas 7 TAC-treated TNFR1/2-knockout mice died during the indicated period. By contrast, only 3 TAC-treated TNFR1-knockout mice died; thus, survival in this group was superior to that in the double-knockout group (Online Figure VIII). Autopsy revealed that the mice in the 2 groups that underwent sham surgery did not differ with respect to their heart weight-to-body weight or wet lung weight-to-body weight ratios. However, TNFR1/2-knockout mice subjected to TAC surgery had significantly increased heart weight-to-body weight and lung weight-to-body weight ratios compared with sham-operated mice. These changes were significantly attenuated in TNFR1-knockout mice subjected to TAC surgery (Figure 8A and 8B; Online Table IV).
Figure 8.
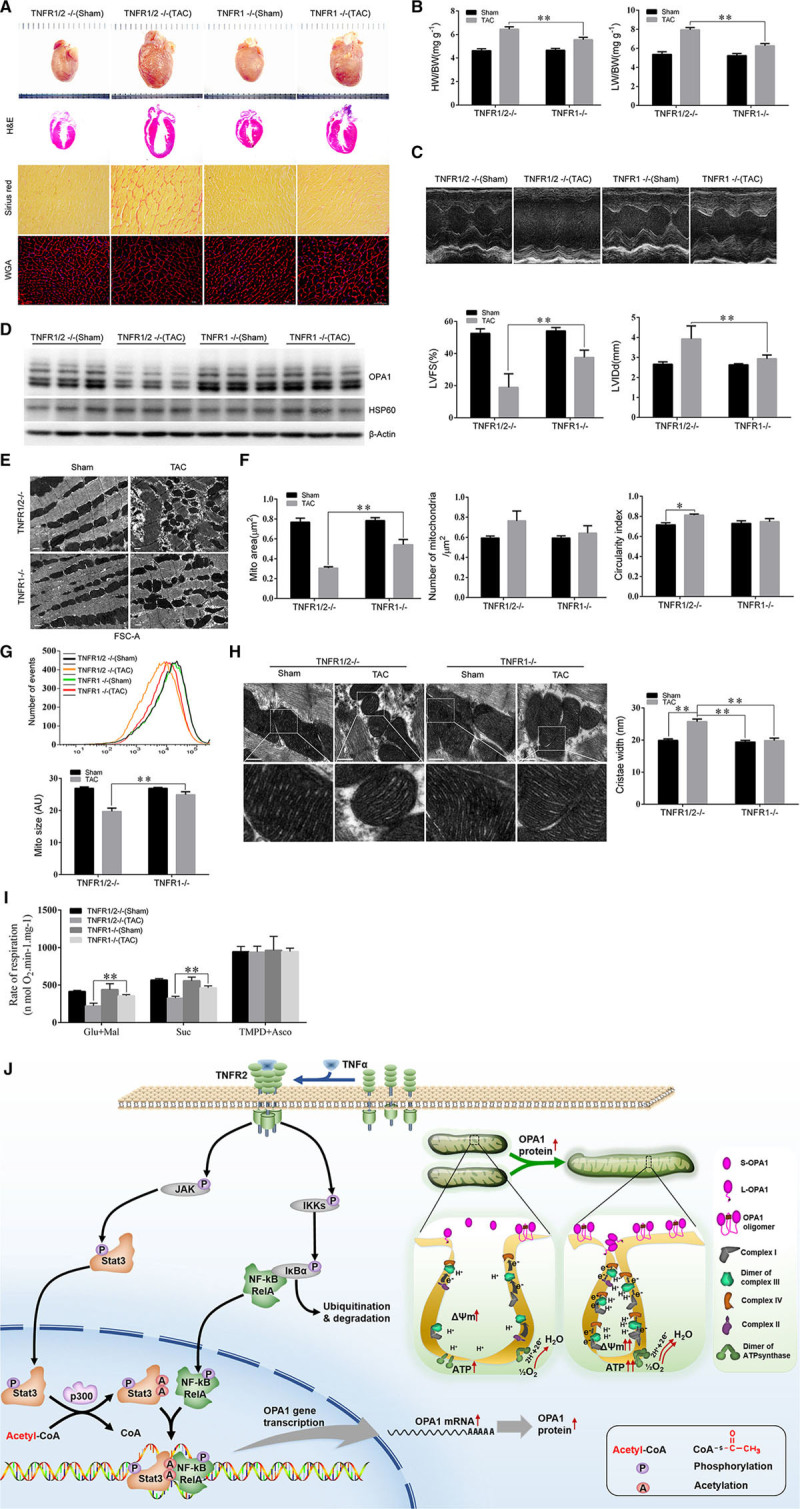
TNFR2 (TNFα receptor 2) activation prevented pressure-overload–induced mitochondrial fragmentation and improved cardiac function in vivo. A, Cardiac autopsy was performed 8 wk after transverse aortic constriction (TAC) induction in each group of mice, and hematoxylin and eosin (H&E), picrosirius red and wheat germ agglutinin (WGA) staining were performed. B, The heart weight-to-body weight (HW/BW) and lung weight (LW)-to-BW ratios for each group of mice are also shown (n=7–8). C, Echocardiographic examinations were performed, and representative M-mode images are shown. Left ventricular fractional shortening (LVFS) and left ventricular end-diastolic diameter (LVEDD) were measured to evaluate cardiac morphology and function (n=7–8/group). D, OPA1 (optic atrophy 1) and HSP60 (heat shock protein 60) protein expression levels were detected by western blotting. E, Transmission electron microscopic (TEM) examination was done to show the myofibrillary array and mitochondrial morphology. F, The mitochondrial area, mitochondrial circularity index, and number of mitochondria per micrometer square in the indicated groups were also quantified (n=3, 200 mitochondria per group). G, Mitochondria were isolated from the heart and then stained with Mito-tracker Red before being subjected to flow cytometry analysis. Mitochondrial sizes were quantified using the FSC-A (forward scatter) index (n=4–6). H, Representative TEM images of the mitochondrial cristae (scale bar, 500 nm) are shown, and mitochondrial cristae widths were analyzed morphometrically (n=3, 80 mitochondria per group). I, The specific substrate-driven oxygen consumption rate of each mitochondrial complex (complex I, complex II, and complex IV) was determined using freshly isolated mitochondria from each group of mice. *P<0.05; **P<0.01. J, Schematic figure demonstrating the role of p300-mediated acetylation in the Stat3-NFκB(RelA) cross talk responsible for TNFR2-induced OPA1 upregulation and, consequently, enhancements of mitochondrial fusion and function.
Echocardiography showed that sham-operated TNFR1-knockout and TNFR1/2-knockout mice did not differ with respect to their left ventricular end-diastolic diameters or left ventricular fractional shortening measurements at 8 weeks after the procedure (P>0.05, respectively; Figure 8C; Online Table IV). However, TNFR1/2 knockout mice subjected to TAC surgery exhibited signs of severe cardiac remodeling, as well as significant increases in left ventricular end-diastolic diameter and decreases in left ventricular fractional shortening compared with sham-operated mice (P<0.05, respectively; Online Table IV); however, these changes were significantly attenuated in TNFR1-knockout mice subjected to TAC surgery (P<0.05, compared with TNFR1/2-knockout TAC mice; Online Table IV; Figure 8C).
We then quantified OPA1 protein expression levels in each group. Sham-operated TNFR1-knockout and TNFR1/2-knockout mice did not differ with respect to OPA1 expression at 8 weeks post-surgery; by contrast, TNFR1/2 knockout mice subjected to TAC displayed significantly decreased OPA1 expression levels compared with sham-operated mice. These changes were significantly attenuated in TNFR1-knockout mice, whose OPA1 expression levels were similar to those of sham-operated mice (Figure 8D).
To evaluate the beneficial effects of preserving of OPA1 expression, we analyzed mitochondrial morphology and function. Transmission electron microscope showed that sham-operated mice displayed round or rectangular mitochondria with normal morphological features (Figure 8E and 8F). By contrast, TAC-treated TNFR1/2-knockout mice displayed fragmented mitochondria exhibiting significantly decreased areas and increased circularity (P<0.05). These changes were significantly attenuated in TAC-treated TNFR1-knockout mice (P<0.05; Figure 8E and 8F). Flow cytometric assay of isolated mitochondria from freshly harvested hearts showed that mitochondrial size decreased in TNFR1/2-knockout mice subjected to TAC surgery compared with sham-operated mice. This change was partially attenuated in TNFR1-knockout mice subjected to TAC surgery (Figure 8G).
OPA1 is critical for cristae junction tightening. As expected, cristae widths were similar between TNFR1-knockout and TNFR1/2-knockout mice subjected to sham surgery. By contrast, cristae were significantly wider (26%) in TAC-treated TNFR1/2-knockout mice compared with sham-operated mice and were unaltered in TAC-treated TNFR1-knockout mice compared with sham-operated mice (Figure 8H). We also quantified mitochondrial respiratory capacity in each group mice. Specifically, we measured the substrate-driven OCRs for ETC complexes I, II, and IV and found that complex I and II activity levels were significantly decreased in TAC-treated TNFR1-knockout mice compared with sham-operated mice (P<0.01) and were even more significantly decreased in TAC-treated TNFR1/2-knockout mice compared with sham-operated mice (Figure 8I).
Finally, histological examination demonstrated that TAC-treated TNFR1-knockout mice displayed significantly less severe fibrosis than TAC-treated TNFR1/2-knockout mice (Figure 8A), indicating that that the former group of mice experienced better outcomes than the latter group of mice after TAC (Online Figure VIII)
Discussion
The protective effects of TNFR2 against cardiac injury have been recognized5,6,63; however, the mechanisms underlying these effects remain poorly understood. In this study, we showed for the first time that TNFR2 activation confers mitochondria-dependent cardioprotective effects. As a mitochondrial profusion protein, OPA1 has received much attention because it plays significant roles in modulating mitochondrial morphology and function.15,23,25,64 Our data demonstrated that TNFR2 activation mediates interactions between Stat3 and RelA to synergistically upregulate OPA1 expression, an effect that can be abolished by knocking down either Stat3 or RelA. Furthermore, using in vitro and in silico (MDS) studies, we verified that the acetylation statuses of the 2 lysine residues within the Stat3 DBD, that is, K370 and K383, can be modulated by p300, which facilitates Stat3/RelA interactions, and that K370 acetylation resulted in a more stable Stat3/RelA complex than K383 acetylation. We confirmed that Stat3 acetylation plays an essential role in TNFR2 activation-induced increases in OPA1 expression via the following 2 approaches: first, we knocked down endogenous Stat3 in NMCMs and ectopically expressed the acetylation-mimetic K370Q mutant (a knockdown-put-back approach) to recapitulate its pro-OPA1 expression effects even in the absence of TNFα stimulation. Second, we transfected NMCMs deprived of endogenous Stat3 with an acetylation-resistant K370/383R mutant. We found that no changes in OPA1 expression occurred in these cells despite TNFR2 stimulation. Taken together, our data indicate that TNFR2 can activate p300/Stat3/RelA/OPA1 signaling to improve mitochondrial fusion and function and thus ameliorate heart failure.
In the setting of ischemia-reperfusion injury, Stat3 signaling, which is also a key component of the survival activating factor enhancement pathway, plays an essential role in the protection conferred by ischemic pre- or postconditioning.5,6 Given the short time frame associated with the indicated setting, it is difficult to believe that Stat3 would exert its effects as a transcription factor and/or cofactor. It is more likely that phosphorylation at serine 727 in Stat3 mediates its mitochondrial translocation65 and that this noncanonical Stat3 signaling pathway may ameliorate ischemia-reperfusion injury by reducing ROS production and inhibiting mitochondrial permeability transition pore opening at the onset of reperfusion.66 Whether and how mitochondria are involved in the cardioprotective effects of Stat3 under chronic stress conditions has seldom been reported. We showed for the first time that Stat3 signaling plays an essential protective role in settings characterized by chronic stress, such as chronic pressure-overload–induced cardiac remodeling, during which Stat3 activation enhances mitochondrial integrity and function by regulating OPA1 transcriptional activity.
NF-kB has been shown to have multiple physiological effects in cardiac disease, effects that can be either protective or detrimental.67,68 Intriguingly, both TNFR1 and TNFR2 have been shown to be able to activate RelA, a central component of NF-kB signaling.69 Therefore, it is reasonable to speculate that TNFR1 and TNFR2 may recruit different cofactors for RelA, thereby enhancing either prosurvival or proapoptotic gene expression. Based on data obtained from previous studies, as well as data obtained by our group, we surmised that p300 served as a cotranscriptional factor to regulate RelA to produce different gene repertoires. It is known that p300-dependent RelA acetylation increases RelA nuclear retention and transcriptional activity.48 Moreover, p300-mediated Stat3 acetylation can guide NF-kB transcriptional activity to regulate gene expression and induce shifts in transcriptional patterns resulting in the downregulation of proinflammatory or apoptotic genes and the upregulation of prosurvival genes. These shifts in transcriptional patterns, which are driven by different upstream regulators, such as TNFR1/RelA and TNFR2/p300/RelA, were validated in our study, which showed that TNFR1/RelA activation had no effect on OPA1 expression (Online Figure IX).
Some studies have also demonstrated that p300-dependent Stat3 acetylation plays an essential role in regulating mitochondrial function. It has been shown that p300-mediated Stat3 acetylation increases its dimerization and nuclear retention and also enhances its transcriptional activity.70 Heart failure is characterized by and may be ascribed to increased glucose and reduced fatty acid metabolism in cardiac myocytes.71 p300-induced Stat3 acetylation inhibits gluconeogenesis,51 which may counteract the conversion of mitochondrial metabolic substrates from fatty acid to glucose to attenuate heart failure progression. We showed that in addition to inhibiting gluconeogenesis, p300-induced Stat3 acetylation also enhanced mitochondrial respiratory chain supercomplex assembly and stability to increase mitochondrial respiration efficiency. These findings indicated that p300/Stat3 signaling is important for mitochondrial function in the heart.
Lysine acetylation is an evolutionarily conserved post-translational modification that regulates a variety of cellular processes, particularly nuclear transcription and cytoplasmic metabolism.72–74 Protein acetylation is regulated by enzymes with contrasting effects, histone acetyltransferases,75 and histone deacetylases.73 A series of nonhistone proteins, which serve as signal transducers and transcription factors, are also substrates of histone acetyltransferases and histone deacetylases.50 Our results highlight that p300, a histone acetyltransferase family member, plays an important role in regulating mitochondrial dynamics by upregulating OPA1 expression. The data obtained in the present study support the notion that p300 plays a critical role in maintaining cardiac mitochondrial function and sustaining cell survival in postnatal hearts.76,77 Unsurprisingly, mitochondrial fragmentation was observed in p300C/H3 transgenic mice in which p300 lacked a functional domain.76 Histone deacetylase inhibitors have previously been reported to promote mitochondrial fusion, which can be abolished by either OPA1 or Mfn1 downregulation.78,79 Thus, we could not totally exclude the possibility that histone deacetylase inhibitors played a role in modulating Stat3 acetylation levels in our study; nevertheless, our data strongly supported the idea that Stat3 acetylation and RelA play key roles in upregulating OPA1 expression and thus improving mitochondrial dynamics.
It should be noted that Stat3 phosphorylation at Y705 is a prerequisite for Stat3 translocation to the nucleus. The role of p300-mediated Stat3 acetylation in Stat3/RelA interactions and, consequently, OPA1 expression modulation has been thoroughly investigated; however, the p300-dependent Stat3 acetylation at K370 and K383 that occurred in the nucleus was originally facilitated by Stat3 phosphorylation. We observed that TNFR2 activation increased both Stat3 (at tyrosine 705 and serine 727) and RelA phosphorylation levels (Online Figure XA). In addition, ruxolitinib, a Stat3 phosphorylation inhibitor, could prevent Stat3 accumulation in the nucleus and p300-mediated Stat3 acetylation at K370 and K383, which eventually blocked the interaction between Stat3 and RelA (Online Figure XB). Moreover, in silico analysis of the docking interface between Stat3 and RelA enabled us to determine whether Stat3-Y705 and Stat3-S727 phosphorylation was associated with Stat3/RelA interactions. As shown in Online Figure XI, the indicated phosphorylation sites, Y705 and S727, are located far away from the docking interface (the Stat3 DBD), a finding that supports the idea that Stat3 acetylation, but not Stat3 phosphorylation, contributes to Stat3/RelA interactions.
In summary, the present study has provided novel evidence indicating that TNFR2 activation can modulate mitochondrial dynamics by upregulating OPA1 expression, a process in which p300-mediated Stat3 acetylation at K370 and K383 played an essential role, as the indicated events enabled Stat3 to interact with RelA to activate OPA1 at the transcriptional level. Thus, TNFR2 activation, which facilitates enhancements of mitochondrial integrity and function, can serve as another novel therapeutic target for the treatment or prevention of cardiac dysfunction and remodeling (Figure 8J).
Sources of Funding
This study was supported by the National Basic Research Program of China (973 Program, No. 2014CB965100, 2014CB965103), the National High-tech R&D 863 Program (No. 2015AA020922 for XBL) and grants from the National Natural Science Foundation of China (No. 81320108003 and 31371498 for J. Wang; No. 81370346 for W. Zhu; No 81370247, 81622006, and 81670261 for X. Hu; and No. 81573641 for L. Zhang), the Science and Technology Department of Zhejiang Province Public Welfare Projects (No. 2013C37054 for J. Wang), the Science and Technology Department of Zhejiang Province Major Development Projects and Public Welfare Projects (No. 2013C37054 for J. Wang), the Fundamental Research Funds for the Central Universities (No.2016XZZX002-03 for X. Hu, No.BSF-001-00* for J. Wang, and No. 2017FZA7011 for L. Zhang), the National Key Research and Development Program of China (No.2016YFC1301204 for J. Wang), the Major Scientific and Technological Project of the Zhejiang Province Science and Technology Department (No. 2013C03043-4 for Y. Sun), the Education Department of Zhejiang Province (No. Y201329862 for W. Zhu), and the Zhejiang Provincial Natural Science Foundation (No. LY16H280003 for L. Zhang).
Disclosures
None.
Supplementary Material
Nonstandard Abbreviations and Acronyms
- ChIP
- chromatin immunoprecipitation
- co-IP
- co-immunoprecipitation
- Drp1
- dynamin-related protein 1
- Mfn1
- mitofusin 1
- Mfn2
- mitofusin 2
- NMCMs
- neonatal mouse cardiac myocytes
- NMCMs-TNFR1-KD
- NMCMs with TNFR1 knocked down
- NMCMs-TNFR2-KD
- NMCMs with TNFR2 knocked down
- OPA1
- optic atrophy 1
- PCAF
- p300/CBP-associated factor
- PGC-1α
- peroxisome proliferator–activated receptor gamma coactivator 1-alpha
- TAD
- transactivation domain
- TAC
- transverse aortic constriction
- TNFα
- tumor necrosis factor α
- TNFR1
- tumor necrosis factor α receptor 1
- TNFR2
- tumor necrosis factor α receptor 2
- TNFR1-shRNAm
- shRNA targeting mouse TNFR1
- TNFR1-shRNAr
- shRNA targeting rat TNFR1
- TNFR2-shRNAm
- shRNA targeting mouse TNFR2
- TNFR2-shRNAr
- shRNA targeting rat TNFR2
- Wort
- wortmannin
- WT
- wild-type
In May 2017, the average time from submission to first decision for all original research papers submitted to Circulation Research was 12.28 days.
These authors contributed equally to this article.
The online-only Data Supplement is available with this article at http://circres.ahajournals.org/lookup/suppl/doi:10.1161/CIRCRESAHA.117.311143/-/DC1.
Novelty and Significance
What Is Known?
Previous clinical trials evaluating anti-TNFα (tumor necrosis factor α) therapies failed to show beneficial effects against heart failure, possibly because of activation of the existence of TNFR2 (TNFα receptor 2).
Mitochondrial fusion and fission process (mitochondrial dynamics) orchestrates the metabolic performance of the cardiac myocytes.
Cardiac diseases are closely associated with dysregulation of mitochondrial dynamics.
What New Information Does This Article Contribute?
We report that TNFR2 activation upregulates OPA1 (optic atrophy 1) expression, enhances mitochondrial fusion, promotes respiratory activity, and increases ATP content.
Acetylation of Stat3 at lysine 370 or 383 by p300 is essential for the interaction of Stat3 and RelA and binding to the promoter region of OPA1 and enhancing transcription.
We show in an in vivo transverse aortic constriction–induced heart failure mouse model that activation of TNFR2 in TNFR1 knockout mice improved mitochondrial morphology and respiratory activity, leading to improved cardiac function and survival rate, as compared with TNFR1/2 double knockout mice.
Our data demonstrated that TNFR2 activation enhances mitochondria function via an OPA1-mediated mitochondrial fusion process. Thus, the TNFR2 signaling pathway might be a therapeutic target in heart failure.
References
- 1.Levine B, Kalman J, Mayer L, Fillit HM, Packer M. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N Engl J Med. 1990;323:236–241. doi: 10.1056/NEJM199007263230405. doi: 10.1056/NEJM199007263230405. [DOI] [PubMed] [Google Scholar]
- 2.Duerrschmid C, Crawford JR, Reineke E, Taffet GE, Trial J, Entman ML, Haudek SB. TNF receptor 1 signaling is critically involved in mediating angiotensin-II-induced cardiac fibrosis. J Mol Cell Cardiol. 2013;57:59–67. doi: 10.1016/j.yjmcc.2013.01.006. doi: 10.1016/j.yjmcc.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Higuchi Y, McTiernan CF, Frye CB, McGowan BS, Chan TO, Feldman AM. Tumor necrosis factor receptors 1 and 2 differentially regulate survival, cardiac dysfunction, and remodeling in transgenic mice with tumor necrosis factor-alpha-induced cardiomyopathy. Circulation. 2004;109:1892–1897. doi: 10.1161/01.CIR.0000124227.00670.AB. doi: 10.1161/01.CIR.0000124227.00670.AB. [DOI] [PubMed] [Google Scholar]
- 4.Garlie JB, Hamid T, Gu Y, Ismahil MA, Chandrasekar B, Prabhu SD. Tumor necrosis factor receptor 2 signaling limits β-adrenergic receptor-mediated cardiac hypertrophy in vivo. Basic Res Cardiol. 2011;106:1193–1205. doi: 10.1007/s00395-011-0196-6. doi: 10.1007/s00395-011-0196-6. [DOI] [PubMed] [Google Scholar]
- 5.Wang M, Crisostomo PR, Markel TA, Wang Y, Meldrum DR. Mechanisms of sex differences in TNFR2-mediated cardioprotection. Circulation. 2008;118:S38–S45. doi: 10.1161/CIRCULATIONAHA.107.756890. doi: 10.1161/CIRCULATIONAHA.107.756890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lacerda L, Somers S, Opie LH, Lecour S. Ischaemic postconditioning protects against reperfusion injury via the SAFE pathway. Cardiovasc Res. 2009;84:201–208. doi: 10.1093/cvr/cvp274. doi: 10.1093/cvr/cvp274. [DOI] [PubMed] [Google Scholar]
- 7.Lecour S, Smith RM, Woodward B, Opie LH, Rochette L, Sack MN. Identification of a novel role for sphingolipid signaling in TNF alpha and ischemic preconditioning mediated cardioprotection. J Mol Cell Cardiol. 2002;34:509–518. doi: 10.1006/jmcc.2002.1533. doi: 10.1006/jmcc.2002.1533. [DOI] [PubMed] [Google Scholar]
- 8.Deuchar GA, Opie LH, Lecour S. TNFalpha is required to confer protection in an in vivo model of classical ischaemic preconditioning. Life Sci. 2007;80:1686–1691. doi: 10.1016/j.lfs.2007.01.040. doi: 10.1016/j.lfs.2007.01.040. [DOI] [PubMed] [Google Scholar]
- 9.Lacerda L, McCarthy J, Mungly SF, Lynn EG, Sack MN, Opie LH, Lecour S. TNFα protects cardiac mitochondria independently of its cell surface receptors. Basic Res Cardiol. 2010;105:751–762. doi: 10.1007/s00395-010-0113-4. doi: 10.1007/s00395-010-0113-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tartaglia LA, Goeddel DV. Two TNF receptors. Immunol Today. 1992;13:151–153. doi: 10.1016/0167-5699(92)90116-O. doi: 10.1016/0167-5699(92)90116-O. [DOI] [PubMed] [Google Scholar]
- 11.Dopp JM, Mackenzie-Graham A, Otero GC, Merrill JE. Differential expression, cytokine modulation, and specific functions of type-1 and type-2 tumor necrosis factor receptors in rat glia. J Neuroimmunol. 1997;75:104–112. doi: 10.1016/s0165-5728(97)00009-x. [DOI] [PubMed] [Google Scholar]
- 12.Anker SD, Coats AJ. How to RECOVER from RENAISSANCE? The significance of the results of RECOVER, RENAISSANCE, RENEWAL and ATTACH. Int J Cardiol. 2002;86:123–130. doi: 10.1016/s0167-5273(02)00470-9. [DOI] [PubMed] [Google Scholar]
- 13.Coletta AP, Clark AL, Banarjee P, Cleland JG. Clinical trials update: RENEWAL (RENAISSANCE and RECOVER) and ATTACH. Eur J Heart Fail. 2002;4:559–561. doi: 10.1016/s1388-9842(02)00121-6. [DOI] [PubMed] [Google Scholar]
- 14.Chung ES, Packer M, Lo KH, Fasanmade AA, Willerson JT Anti-TNF Therapy Against Congestive Heart Failure Investigators. Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure: results of the anti-TNF Therapy Against Congestive Heart Failure (ATTACH) trial. Circulation. 2003;107:3133–3140. doi: 10.1161/01.CIR.0000077913.60364.D2. doi: 10.1161/01.CIR.0000077913.60364.D2. [DOI] [PubMed] [Google Scholar]
- 15.Sanchis-Gomar F, García-Giménez JL, Gómez-Cabrera MC, Pallardó FV. Mitochondrial biogenesis in health and disease. Molecular and therapeutic approaches. Curr Pharm Des. 2014;20:5619–5633. doi: 10.2174/1381612820666140306095106. [DOI] [PubMed] [Google Scholar]
- 16.Valero T. Mitochondrial biogenesis: pharmacological approaches. Curr Pharm Des. 2014;20:5507–5509. doi: 10.2174/138161282035140911142118. [DOI] [PubMed] [Google Scholar]
- 17.Song M, Dorn GW., 2nd Mitoconfusion: noncanonical functioning of dynamism factors in static mitochondria of the heart. Cell Metab. 2015;21:195–205. doi: 10.1016/j.cmet.2014.12.019. doi: 10.1016/j.cmet.2014.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Losón OC, Song Z, Chen H, Chan DC. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol Biol Cell. 2013;24:659–667. doi: 10.1091/mbc.E12-10-0721. doi: 10.1091/mbc.E12-10-0721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Formosa LE, Ryan MT. Mitochondrial fusion: reaching the end of mitofusin’s tether. J Cell Biol. 2016;215:597–598. doi: 10.1083/jcb.201611048. doi: 10.1083/jcb.201611048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Song Z, Chen H, Fiket M, Alexander C, Chan DC. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J Cell Biol. 2007;178:749–755. doi: 10.1083/jcb.200704110. doi: 10.1083/jcb.200704110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zick M, Rabl R, Reichert AS. Cristae formation-linking ultrastructure and function of mitochondria. Biochim Biophys Acta. 2009;1793:5–19. doi: 10.1016/j.bbamcr.2008.06.013. doi: 10.1016/j.bbamcr.2008.06.013. [DOI] [PubMed] [Google Scholar]
- 22.Mannella CA. Structure and dynamics of the mitochondrial inner membrane cristae. Biochim Biophys Acta. 2006;1763:542–548. doi: 10.1016/j.bbamcr.2006.04.006. doi: 10.1016/j.bbamcr.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 23.Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, Bartoli D, Polishuck RS, Danial NN, De Strooper B, Scorrano L. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. 2006;126:177–189. doi: 10.1016/j.cell.2006.06.025. doi: 10.1016/j.cell.2006.06.025. [DOI] [PubMed] [Google Scholar]
- 24.Cipolat S, Rudka T, Hartmann D, et al. Mitochondrial rhomboid PARL regulates cytochrome c release during apoptosis via OPA1-dependent cristae remodeling. Cell. 2006;126:163–175. doi: 10.1016/j.cell.2006.06.021. doi: 10.1016/j.cell.2006.06.021. [DOI] [PubMed] [Google Scholar]
- 25.Cogliati S, Frezza C, Soriano ME, Varanita T, Quintana-Cabrera R, Corrado M, Cipolat S, Costa V, Casarin A, Gomes LC, Perales-Clemente E, Salviati L, Fernandez-Silva P, Enriquez JA, Scorrano L. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell. 2013;155:160–171. doi: 10.1016/j.cell.2013.08.032. doi: 10.1016/j.cell.2013.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hahn WS, Kuzmicic J, Burrill JS, Donoghue MA, Foncea R, Jensen MD, Lavandero S, Arriaga EA, Bernlohr DA. Proinflammatory cytokines differentially regulate adipocyte mitochondrial metabolism, oxidative stress, and dynamics. Am J Physiol Endocrinol Metab. 2014;306:E1033–E1045. doi: 10.1152/ajpendo.00422.2013. doi: 10.1152/ajpendo.00422.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sente T, Van Berendoncks AM, Fransen E, Vrints CJ, Hoymans VY. Tumor necrosis factor-α impairs adiponectin signalling, mitochondrial biogenesis, and myogenesis in primary human myotubes cultures. Am J Physiol Heart Circ Physiol. 2016;310:H1164–H1175. doi: 10.1152/ajpheart.00831.2015. doi: 10.1152/ajpheart.00831.2015. [DOI] [PubMed] [Google Scholar]
- 28.Moe GW, Marin-Garcia J, Konig A, Goldenthal M, Lu X, Feng Q. In vivo TNF-alpha inhibition ameliorates cardiac mitochondrial dysfunction, oxidative stress, and apoptosis in experimental heart failure. Am J Physiol Heart Circ Physiol. 2004;287:H1813–H1820. doi: 10.1152/ajpheart.00036.2004. doi: 10.1152/ajpheart.00036.2004. [DOI] [PubMed] [Google Scholar]
- 29.Mariappan N, Elks CM, Fink B, Francis J. TNF-induced mitochondrial damage: a link between mitochondrial complex I activity and left ventricular dysfunction. Free Radic Biol Med. 2009;46:462–470. doi: 10.1016/j.freeradbiomed.2008.10.049. doi: 10.1016/j.freeradbiomed.2008.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Müller-Rischart AK, Pilsl A, Beaudette P, et al. The E3 ligase parkin maintains mitochondrial integrity by increasing linear ubiquitination of NEMO. Mol Cell. 2013;49:908–921. doi: 10.1016/j.molcel.2013.01.036. doi: 10.1016/j.molcel.2013.01.036. [DOI] [PubMed] [Google Scholar]
- 31.Drabarek B, Dymkowska D, Szczepanowska J, Zabłocki K. TNFα affects energy metabolism and stimulates biogenesis of mitochondria in EA.hy926 endothelial cells. Int J Biochem Cell Biol. 2012;44:1390–1397. doi: 10.1016/j.biocel.2012.05.022. doi: 10.1016/j.biocel.2012.05.022. [DOI] [PubMed] [Google Scholar]
- 32.Makrecka-Kuka M, Krumschnabel G, Gnaiger E. High-resolution respirometry for simultaneous measurement of oxygen and hydrogen peroxide fluxes in permeabilized cells, tissue homogenate and isolated mitochondria. Biomolecules. 2015;5:1319–1338. doi: 10.3390/biom5031319. doi: 10.3390/biom5031319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stadlmann S, Renner K, Pollheimer J, Moser PL, Zeimet AG, Offner FA, Gnaiger E. Preserved coupling of oxidative phosphorylation but decreased mitochondrial respiratory capacity in IL-1beta-treated human peritoneal mesothelial cells. Cell Biochem Biophys. 2006;44:179–186. doi: 10.1385/CBB:44:2:179. doi: 10.1385/CBB:44:2:179. [DOI] [PubMed] [Google Scholar]
- 34.Chen R, Li L, Weng Z. ZDOCK: an initial-stage protein-docking algorithm. Proteins. 2003;52:80–87. doi: 10.1002/prot.10389. doi: 10.1002/prot.10389. [DOI] [PubMed] [Google Scholar]
- 35.Pierce B, Weng Z. ZRANK: reranking protein docking predictions with an optimized energy function. Proteins. 2007;67:1078–1086. doi: 10.1002/prot.21373. doi: 10.1002/prot.21373. [DOI] [PubMed] [Google Scholar]
- 36.Ryckaert J-P, Ciccotti G, Berendsen HJ. Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys. 1977;23:327–341. [Google Scholar]
- 37.Darden T, York D, Pedersen L. Particle mesh Ewald: An nlog (n) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092.. [Google Scholar]
- 38.Weiser J, Shenkin PS, Still WC. Approximate atomic surfaces from linear combinations of pairwise overlaps (lcpo). J Comput Chem. 1999;20:217–230.. [Google Scholar]
- 39.Parra V, Verdejo HE, Iglewski M, Del Campo A, Troncoso R, Jones D, Zhu Y, Kuzmicic J, Pennanen C, Lopez-Crisosto C, Jana F, Ferreira J, Noguera E, Chiong M, Bernlohr DA, Klip A, Hill JA, Rothermel BA, Abel ED, Zorzano A, Lavandero S. Insulin stimulates mitochondrial fusion and function in cardiac myocytes via the akt-mtor-nfkappab-opa-1 signaling pathway. Diabetes. 2014;63:75–88.. doi: 10.2337/db13-0340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Song M, Mihara K, Chen Y, Scorrano L, Dorn GW., 2nd Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab. 2015;21:273–285. doi: 10.1016/j.cmet.2014.12.011. doi: 10.1016/j.cmet.2014.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Piquereau J, Caffin F, Novotova M, Prola A, Garnier A, Mateo P, Fortin D, Huynh le H, Nicolas V, Alavi MV, Brenner C, Ventura-Clapier R, Veksler V, Joubert F. Down-regulation of OPA1 alters mouse mitochondrial morphology, PTP function, and cardiac adaptation to pressure overload. Cardiovasc Res. 2012;94:408–417. doi: 10.1093/cvr/cvs117. doi: 10.1093/cvr/cvs117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Varanita T, Soriano ME, Romanello V, et al. The OPA1-dependent mitochondrial cristae remodeling pathway controls atrophic, apoptotic, and ischemic tissue damage. Cell Metab. 2015;21:834–844. doi: 10.1016/j.cmet.2015.05.007. doi: 10.1016/j.cmet.2015.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goukassian DA, Qin G, Dolan C, et al. Tumor necrosis factor-alpha receptor p75 is required in ischemia-induced neovascularization. Circulation. 2007;115:752–762. doi: 10.1161/CIRCULATIONAHA.106.647255. doi: 10.1161/CIRCULATIONAHA.106.647255. [DOI] [PubMed] [Google Scholar]
- 44.Constantin CE, Mair N, Sailer CA, Andratsch M, Xu ZZ, Blumer MJ, Scherbakov N, Davis JB, Bluethmann H, Ji RR, Kress M. Endogenous tumor necrosis factor alpha (TNFalpha) requires TNF receptor type 2 to generate heat hyperalgesia in a mouse cancer model. J Neurosci. 2008;28:5072–5081. doi: 10.1523/JNEUROSCI.4476-07.2008. doi: 10.1523/JNEUROSCI.4476-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fischer R, Maier O, Siegemund M, Wajant H, Scheurich P, Pfizenmaier K. A TNF receptor 2 selective agonist rescues human neurons from oxidative stress-induced cell death. PLoS One. 2011;6:e27621. doi: 10.1371/journal.pone.0027621. doi: 10.1371/journal.pone.0027621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Defer N, Azroyan A, Pecker F, Pavoine C. TNFR1 and TNFR2 signaling interplay in cardiac myocytes. J Biol Chem. 2007;282:35564–35573. doi: 10.1074/jbc.M704003200. doi: 10.1074/jbc.M704003200. [DOI] [PubMed] [Google Scholar]
- 47.Kesanakurti D, Chetty C, Rajasekhar Maddirela D, Gujrati M, Rao JS. Essential role of cooperative NF-κB and Stat3 recruitment to ICAM-1 intronic consensus elements in the regulation of radiation-induced invasion and migration in glioma. Oncogene. 2013;32:5144–5155. doi: 10.1038/onc.2012.546. doi: 10.1038/onc.2012.546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee H, Herrmann A, Deng JH, Kujawski M, Niu G, Li Z, Forman S, Jove R, Pardoll DM, Yu H. Persistently activated Stat3 maintains constitutive NF-kappaB activity in tumors. Cancer Cell. 2009;15:283–293. doi: 10.1016/j.ccr.2009.02.015. doi: 10.1016/j.ccr.2009.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang H, Zhou L, Shi Q, Zhao Y, Lin H, Zhang M, Zhao S, Yang Y, Ling ZQ, Guan KL, Xiong Y, Ye D. SIRT3-dependent GOT2 acetylation status affects the malate-aspartate NADH shuttle activity and pancreatic tumor growth. EMBO J. 2015;34:1110–1125. doi: 10.15252/embj.201591041. doi: 10.15252/embj.201591041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Glozak MA, Sengupta N, Zhang X, Seto E. Acetylation and deacetylation of non-histone proteins. Gene. 2005;363:15–23. doi: 10.1016/j.gene.2005.09.010. doi: 10.1016/j.gene.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 51.Nie Y, Erion DM, Yuan Z, Dietrich M, Shulman GI, Horvath TL, Gao Q. STAT3 inhibition of gluconeogenesis is downregulated by SirT1. Nat Cell Biol. 2009;11:492–500. doi: 10.1038/ncb1857. doi: 10.1038/ncb1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee JL, Wang MJ, Chen JY. Acetylation and activation of STAT3 mediated by nuclear translocation of CD44. J Cell Biol. 2009;185:949–957. doi: 10.1083/jcb.200812060. doi: 10.1083/jcb.200812060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ray S, Boldogh I, Brasier AR. STAT3 NH2-terminal acetylation is activated by the hepatic acute-phase response and required for IL-6 induction of angiotensinogen. Gastroenterology. 2005;129:1616–1632. doi: 10.1053/j.gastro.2005.07.055. doi: 10.1053/j.gastro.2005.07.055. [DOI] [PubMed] [Google Scholar]
- 54.Lee H, Zhang P, Herrmann A, Yang C, Xin H, Wang Z, Hoon DS, Forman SJ, Jove R, Riggs AD, Yu H. Acetylated stat3 is crucial for methylation of tumor-suppressor gene promoters and inhibition by resveratrol results in demethylation. Proc Natl Acad Sci USA. 2012;109:7765–7769.. doi: 10.1073/pnas.1205132109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dhar K, Rakesh K, Pankajakshan D, Agrawal DK. SOCS3 promotor hypermethylation and STAT3-NF-κB interaction downregulate SOCS3 expression in human coronary artery smooth muscle cells. Am J Physiol Heart Circ Physiol. 2013;304:H776–H785. doi: 10.1152/ajpheart.00570.2012. doi: 10.1152/ajpheart.00570.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu Z, Kone BC. The STAT3 DNA-binding domain mediates interaction with NF-kappaB p65 and inducible nitric oxide synthase transrepression in mesangial cells. J Am Soc Nephrol. 2004;15:585–591. doi: 10.1097/01.asn.0000114556.19556.f9. [DOI] [PubMed] [Google Scholar]
- 57.Chen Y, Wu R, Chen HZ, Xiao Q, Wang WJ, He JP, Li XX, Yu XW, Li L, Wang P, Wan XC, Tian XH, Li SJ, Yu X, Wu Q. Enhancement of hypothalamic STAT3 acetylation by nuclear receptor Nur77 dictates leptin sensitivity. Diabetes. 2015;64:2069–2081. doi: 10.2337/db14-1206. doi: 10.2337/db14-1206. [DOI] [PubMed] [Google Scholar]
- 58.Yue H, Li W, Desnoyer R, Karnik SS. Role of nuclear unphosphorylated STAT3 in angiotensin II type 1 receptor-induced cardiac hypertrophy. Cardiovasc Res. 2010;85:90–99. doi: 10.1093/cvr/cvp285. doi: 10.1093/cvr/cvp285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hou T, Ray S, Lee C, Brasier AR. The STAT3 NH2-terminal domain stabilizes enhanceosome assembly by interacting with the p300 bromodomain. J Biol Chem. 2008;283:30725–30734. doi: 10.1074/jbc.M805941200. doi: 10.1074/jbc.M805941200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zheng X, Xu M, Yao B, Wang C, Jia Y, Liu Q. IL-6/STAT3 axis initiated CAFs via up-regulating TIMP-1 which was attenuated by acetylation of STAT3 induced by PCAF in HCC microenvironment. Cell Signal. 2016;28:1314–1324. doi: 10.1016/j.cellsig.2016.06.009. doi: 10.1016/j.cellsig.2016.06.009. [DOI] [PubMed] [Google Scholar]
- 61.Cai K, Wan Y, Wang Z, Wang Y, Zhao X, Bao X. C5a promotes the proliferation of human nasopharyngeal carcinoma cells through PCAF-mediated STAT3 acetylation. Oncol Rep. 2014;32:2260–2266. doi: 10.3892/or.2014.3420. doi: 10.3892/or.2014.3420. [DOI] [PubMed] [Google Scholar]
- 62.Zhang HF, Lai R. STAT3 in cancer-friend or foe? Cancers (Basel) 2014;6:1408–1440. doi: 10.3390/cancers6031408. doi: 10.3390/cancers6031408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lecour S, James RW. When are pro-inflammatory cytokines SAFE in heart failure? Eur Heart J. 2011;32:680–685. doi: 10.1093/eurheartj/ehq484. doi: 10.1093/eurheartj/ehq484. [DOI] [PubMed] [Google Scholar]
- 64.Dorn GW., 2nd Mitochondrial dynamism and heart disease: changing shape and shaping change. EMBO Mol Med. 2015;7:865–877. doi: 10.15252/emmm.201404575. doi: 10.15252/emmm.201404575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kramer AH, Edkins AL, Hoppe HC, Prinsloo E. Dynamic mitochondrial localisation of STAT3 in the cellular adipogenesis model 3T3-L1. J Cell Biochem. 2015;116:1232–1240. doi: 10.1002/jcb.25076. doi: 10.1002/jcb.25076. [DOI] [PubMed] [Google Scholar]
- 66.Szczepanek K, Xu A, Hu Y, Thompson J, He J, Larner AC, Salloum FN, Chen Q, Lesnefsky EJ. Cardioprotective function of mitochondrial-targeted and transcriptionally inactive STAT3 against ischemia and reperfusion injury. Basic Res Cardiol. 2015;110:53. doi: 10.1007/s00395-015-0509-2. doi: 10.1007/s00395-015-0509-2. [DOI] [PubMed] [Google Scholar]
- 67.Gutiérrez SH, Kuri MR, del Castillo ER. Cardiac role of the transcription factor NF-kappaB. Cardiovasc Hematol Disord Drug Targets. 2008;8:153–160. doi: 10.2174/187152908784533702. [DOI] [PubMed] [Google Scholar]
- 68.Gordon JW, Shaw JA, Kirshenbaum LA. Multiple facets of NF-κB in the heart: to be or not to NF-κB. Circ Res. 2011;108:1122–1132. doi: 10.1161/CIRCRESAHA.110.226928. doi: 10.1161/CIRCRESAHA.110.226928. [DOI] [PubMed] [Google Scholar]
- 69.Cabal-Hierro L, Lazo PS. Signal transduction by tumor necrosis factor receptors. Cell Signal. 2012;24:1297–1305. doi: 10.1016/j.cellsig.2012.02.006. doi: 10.1016/j.cellsig.2012.02.006. [DOI] [PubMed] [Google Scholar]
- 70.Yuan ZL, Guan YJ, Chatterjee D, Chin YE. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science. 2005;307:269–273. doi: 10.1126/science.1105166. doi: 10.1126/science.1105166. [DOI] [PubMed] [Google Scholar]
- 71.Fukushima A, Milner K, Gupta A, Lopaschuk GD. Myocardial energy substrate metabolism in heart failure: from pathways to therapeutic targets. Curr Pharm Des. 2015;21:3654–3664. doi: 10.2174/1381612821666150710150445. [DOI] [PubMed] [Google Scholar]
- 72.Kim SC, Sprung R, Chen Y, Xu Y, Ball H, Pei J, Cheng T, Kho Y, Xiao H, Xiao L, Grishin NV, White M, Yang XJ, Zhao Y. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol Cell. 2006;23:607–618. doi: 10.1016/j.molcel.2006.06.026. doi: 10.1016/j.molcel.2006.06.026. [DOI] [PubMed] [Google Scholar]
- 73.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 74.Zhao S, Xu W, Jiang W, et al. Regulation of cellular metabolism by protein lysine acetylation. Science. 2010;327:1000–1004. doi: 10.1126/science.1179689. doi: 10.1126/science.1179689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lee KK, Workman JL. Histone acetyltransferase complexes: one size doesn’t fit all. Nat Rev Mol Cell Biol. 2007;8:284–295. doi: 10.1038/nrm2145. doi: 10.1038/nrm2145. [DOI] [PubMed] [Google Scholar]
- 76.Nakagawa Y, Kuwahara K, Takemura G, et al. p300 plays a critical role in maintaining cardiac mitochondrial function and cell survival in postnatal hearts. Circ Res. 2009;105:746–754. doi: 10.1161/CIRCRESAHA.109.206037. doi: 10.1161/CIRCRESAHA.109.206037. [DOI] [PubMed] [Google Scholar]
- 77.Jain S, Wei J, Mitrani LR, Bishopric NH. Auto-acetylation stabilizes p300 in cardiac myocytes during acute oxidative stress, promoting STAT3 accumulation and cell survival. Breast Cancer Res Treat. 2012;135:103–114. doi: 10.1007/s10549-012-2069-6. doi: 10.1007/s10549-012-2069-6. [DOI] [PubMed] [Google Scholar]
- 78.Lee JS, Yoon YG, Yoo SH, Jeong NY, Jeong SH, Lee SY, Jung DI, Jeong SY, Yoo YH. Histone deacetylase inhibitors induce mitochondrial elongation. J Cell Physiol. 2012;227:2856–2869. doi: 10.1002/jcp.23027. doi: 10.1002/jcp.23027. [DOI] [PubMed] [Google Scholar]
- 79.Zhu M, Li WW, Lu CZ. Histone decacetylase inhibitors prevent mitochondrial fragmentation and elicit early neuroprotection against MPP+. CNS Neurosci Ther. 2014;20:308–316. doi: 10.1111/cns.12217. doi: 10.1111/cns.12217. [DOI] [PMC free article] [PubMed] [Google Scholar]