

Supplemental Digital Content is available in the text.
Keywords: atherosclerosis, endothelial cells, hematopoiesis, inflammation, microRNAs
Abstract
Rationale:
Inflammation is a key contributor to atherosclerosis. MicroRNA-146a (miR-146a) has been identified as a critical brake on proinflammatory nuclear factor κ light chain enhancer of activated B cells signaling in several cell types, including endothelial cells and bone marrow (BM)–derived cells. Importantly, miR-146a expression is elevated in human atherosclerotic plaques, and polymorphisms in the miR-146a precursor have been associated with risk of coronary artery disease.
Objective:
To define the role of endogenous miR-146a during atherogenesis.
Methods and Results:
Paradoxically, Ldlr−/− (low-density lipoprotein receptor null) mice deficient in miR-146a develop less atherosclerosis, despite having highly elevated levels of circulating proinflammatory cytokines. In contrast, cytokine levels are normalized in Ldlr−/−;miR-146a−/− mice receiving wild-type BM transplantation, and these mice have enhanced endothelial cell activation and elevated atherosclerotic plaque burden compared with Ldlr−/− mice receiving wild-type BM, demonstrating the atheroprotective role of miR-146a in the endothelium. We find that deficiency of miR-146a in BM-derived cells precipitates defects in hematopoietic stem cell function, contributing to extramedullary hematopoiesis, splenomegaly, BM failure, and decreased levels of circulating proatherogenic cells in mice fed an atherogenic diet. These hematopoietic phenotypes seem to be driven by unrestrained inflammatory signaling that leads to the expansion and eventual exhaustion of hematopoietic cells, and this occurs in the face of lower levels of circulating low-density lipoprotein cholesterol in mice lacking miR-146a in BM-derived cells. Furthermore, we identify sortilin-1(Sort1), a known regulator of circulating low-density lipoprotein levels in humans, as a novel target of miR-146a.
Conclusions:
Our study reveals that miR-146a regulates cholesterol metabolism and tempers chronic inflammatory responses to atherogenic diet by restraining proinflammatory signaling in endothelial cells and BM-derived cells.
Atherosclerosis is a chronic inflammatory vascular disease characterized by the narrowing of blood vessels caused by the growth of lipid-rich plaques.1 The initiation of atherogenesis relies on the recruitment of circulating leukocytes by activated endothelial cells (ECs) to regions of deposited oxidized low-density lipoprotein (LDL).2 Activated ECs and leukocytes use the nuclear factor κ light chain enhancer of activated B cells (NF-κB) signaling pathway to propagate inflammatory gene expression, including induction of adhesion molecules, chemoattractants, and cytokines to drive inflammation in the vessel wall.3,4 NF-κB signaling is tightly controlled, and this includes regulation by a network of microRNAs, which titrate the expression of signaling components post-transcriptionally.5 In particular, microRNA-146a (miR-146a) has been well characterized in both ECs and leukocytes as a negative regulator of NF-κB activity through its ability to target upstream adaptor proteins, including TRAF6 (TNF receptor–associated factor 6) and IRAK1 (interleukin receptor–associated kinase 1).6,7
In This Issue, see p 311
Characterization of miR-146a–deficient mice has revealed defects in multiple aspects of immune cell biology.8,9 Older (>1 year) miR-146a−/− mice develop multiorgan inflammation, bone marrow (BM) failure, splenomegaly, and lymphoadenopathy.9,10 When challenged by proinflammatory stimuli (eg, lipopolysaccharide [LPS] or IL-1β [interleukin-1β]), these mice have exacerbated NF-κB–dependent inflammatory responses and demonstrate expansion of proinflammatory Ly6Chi monocytes.6,10,11 Interestingly, the hyperactivation of NF-κB caused by low-grade inflammation during normal aging or through repeated LPS challenge drives the proliferation and eventual exhaustion of hematopoietic and progenitor stem cells in these mice, resulting in eventual loss of circulating leukocytes and lymphocytes.10
The NF-κB pathway is activated in ECs, macrophages, and smooth muscle cells within human atherosclerotic lesions.12 However, defining the role of NF-κB signaling in atherogenesis has been complicated, as ablation of NF-κB activity in ECs reduces atherogenesis,3 whereas inhibition within macrophages enhances atherogenesis.13 Of interest, recent studies have shown that injection of miR-146a mimic into atheroprone mice reduces atherogenesis, and it has been suggested that this is because of suppression of macrophage NF-κB signaling.14 The role of endogenous miR-146a in atherogenesis remains undefined. Here, we show that genetic ablation of miR-146a in BM-derived cells reduces atherogenesis and that this is paradoxically accompanied by enhanced circulating levels of proinflammatory cytokines despite reduced levels of circulating LDL cholesterol. Lack of miR-146a in BM-derived cells leads to monocytosis in response to high cholesterol diet (HCD), followed by BM exhaustion, depleting circulating levels of proatherogenic cells. Conversely, deletion of miR-146a in the vasculature promotes atherogenesis by increasing endothelial activation. Thus, unrestrained inflammatory signaling in miR-146a–deficient tissues has diverse consequences during atherogenesis, and our studies emphasize the importance of tight control of inflammatory pathways in the setting of hypercholesterolemia.
Methods
A complete description of Methods is included in the Online Data Supplement.
Results
miR-146a Expression Is Increased in ECs and Intimal Cells During Murine Atherogenesis
Ldlr−/− (low-density lipoprotein receptor null) mice were placed on a HCD for 18 weeks to visualize the expression of miR-146a in atherosclerotic plaque (Figure 1A). In situ polymerase chain reaction on aortic root cross-sections revealed that miR-146a was expressed in intimal cells, including Mac-2+ macrophages, and was robustly expressed in CD31+ ECs. The in situ signal was specific for miR-146a, as staining was not detected in miR-146a−/− mice (Figure 1A). Expression of miR-146a in the aortic root seemed to progressively increase in the intima during the progression of atherosclerosis (Online Figure I). The absence of signal in the media implies that contractile smooth muscle cells in the aortic root do not express miR-146a at sufficient levels to be detected by this technique. In addition, using quantitative reverse transcriptase-polymerase chain reaction at an early stage of atherogenesis (ie, Ldlr−/− mice, 4-week HCD), we found a significant elevation of miR-146a expression in the lesser curvature of the aortic arch, a region of the aorta where atherosclerotic plaque forms, compared with regions that are protected from atherosclerosis, namely the greater curvature of the aortic arch and the descending thoracic aorta (Figure 1B and 1C). However, miR-146a expression was at appreciable levels in all regions examined (not shown), which may reflect the known expression of miR-146a in the vascular endothelium.6
Figure 1.

MicroRNA-146a (miR-146a) is expressed in murine atherosclerotic plaques. A, Cross-sections of Ldlr−/− or Ldlr−/−;miR-146a−/− mouse aortic roots after 18 wk of high cholesterol diet (HCD). Expression of miR-146a, assessed by in situ polymerase chain reaction (red) overlaps with Mac-2–positive macrophages (purple) and CD31-positive endothelial cells (ECs; green) in the intima, and signal is absent in miR-146a−/− mice. miR-146a expression during the progression of atherosclerosis is shown in Online Figure I. B, Schematic of the aorta, indicating the aortic root (examined in A), the greater curvature (GC, atheroprotective) and lesser curvature (LC, atherosusceptible) of the aortic arch and the descending thoracic aorta (DTA, atheroprotective). C, Expression of miR-146a (normalized to U6 levels) in the specified regions of the aorta in Ldlr−/− mice after 4 wk of HCD (n=5). Ldlr indicates low-density lipoprotein receptor.
Global Deletion of miR-146a Activates Proinflammatory Pathways Yet Suppresses Atherogenesis and Is Accompanied by Reduced Circulating LDL Cholesterol Levels in Mice on HCD
To elucidate the role of miR-146a during atherogenesis, we generated global double knockout (DKO; miR-146a−/−; Ldlr−/−) mice by crossing miR-146a−/− mice with Ldlr−/− mice. Two time points (12 and 18 weeks of HCD) were assessed to determine the effect of miR-146a on the progression of atherosclerotic phenotypes (Figure 2A). Analyses of male and female mice were grouped together as we found no significant differences between sexes for the parameters measured, except for body weight (not shown). At the 12-week time point, no differences in body weight (Figure 2B) or aortic arch plaque burden (Figure 2D) were observed between Ldlr−/− and DKO mice. However, the circulating inflammatory marker, IL-6, was elevated in the majority of DKOs at this stage, although the difference did not reach statistical significance (Figure 2E). By 18 weeks, despite no differences in body weight (Figure 2B) or food intake (Figure 2C), DKO mice surprisingly had less lipid plaque in the aortic arch (Figure 2D). This decrease in atherosclerosis occurred despite signs of elevated systemic inflammatory signaling, including enhanced circulating levels of soluble intercellular adhesion molecule-1 (sICAM-1) and IL-6 (Figure 2E). Atherosclerotic plaque formation was unaltered in the descending thoracic aorta of DKO mice: this region is typically protected from atherosclerosis (Online Figure IIA). Plaque burden in the aortic root was also comparable between groups (Online Figure IIB and IIC).
Figure 2.

Reduced atherosclerosis in mice with global deletion of microRNA-146a (miR-146a). A, Schematic of high cholesterol diet (HCD) regimen for Ldlr−/− and Ldlr−/−;miR-146a−/− (double knockout [DKO]) mice. B, Weights of male mice after 12- or 18-wk HCD (n=3–5). Weights of female mice were also unchanged between genotypes (not shown). C, Food consumption in mice (n=4 mice per cage). T0 is 18 wk of HCD. D, Percentage of Oil Red-O (ORO) regions quantified from aortic arches of Ldlr−/− and DKO mice after HCD for 12 or 18 wk. Representative images are shown to the right. The descending side of the aorta is to the right. Aortic root and descending thoracic aorta analyses are shown in Online Figure II. n=18 to 22 for 12-wk time point and n=4 for 18-wk time point. E, Circulating levels of proinflammatory markers, IL-6 (interleukin-6) and soluble intercellular adhesion molecule-1 (sICAM-1) in wild-type and DKO mice (n=5–8). F, Time course of plasma cholesterol measurements (n=3–5; 1 group of mice were used for weeks 1, 6, and 9, and a separate group was used for weeks 12 and 18). Mice were fasted overnight before sample collection. G, FPLC (fast protein liquid chromatography) trace of cholesterol content in lipoprotein fractions in plasma after 18 wk of HCD (pooled analysis of 5 samples). H, Intrahepatic cholesterol and triglyceride levels in mice after 12-wk HCD (n=13–14). Eighteen-week HCD is shown in Online Figure IID. Bile cholesterol (n=3) and fecal cholesterol (n=4) in mice after 18-wk HCD. I, Assessment of very-low-density lipoprotein (VLDL) secretion by measurement of triglycerides and cholesterol in plasma after injection of Poloxamer 407 (12-wk HCD; n=4, 2). Ldlr indicates low-density lipoprotein receptor.
Unexpectedly, DKO mice displayed progressive lipid metabolism defects, resulting in lower circulating total cholesterol and LDL cholesterol. High-density lipoprotein levels and triglyceride levels were modestly affected (Figure 2F). Assessment of lipoprotein profiles by fast protein liquid chromatography (FPLC) revealed a striking decrease in cholesterol content in very-low-density lipoprotein (VLDL) fractions (Figure 2G). Measurement of total cholesterol and triglyceride levels in the liver revealed no significant differences at 12 weeks (Figure 2H) or 18 weeks of HCD (Online Figure IID). Likewise, cholesterol levels in bile and feces were unchanged at 12 weeks of HCD (Figure 2H). Assessment of VLDL secretion from the liver suggested a decrease in cholesterol and triglyceride secretion in DKO mice (Figure 2I). Taken together, these data demonstrate that lack of miR-146a decreases circulating VLDL/LDL cholesterol yet paradoxically enhances inflammatory signaling in mice on a proatherogenic diet.
Deletion of miR-146a in BM-Derived Cells Enhances Inflammatory Signaling, Yet Paradoxically Suppresses Atherogenesis and Alters Cholesterol Metabolism
Next, we performed BM transplantation (BMT) experiments to elucidate the role of miR-146a in BM-derived cells during atherogenesis. Ldlr−/− mice were lethally irradiated and reconstituted with either miR-146a+/+ (wild-type [WT]) or miR-146a−/− (knockout [KO]) BM (Figure 3A). Reconstitution of hematopoiesis after transplantation of WT or KO BM cells seemed to be normal, as circulating levels of leukocytes and lymphocytes were similar 8 weeks after BMT, before the administration of HCD (Online Figure IIIA). Body weight was similar between the 2 groups after 12 weeks of HCD (Figure 3B), as was food intake (Figure 3C). Although lipid plaque burden was not significantly altered at early stages (ie, 4-week HCD), mice receiving KO BM developed less lipid plaque in the aorta after 12 weeks of HCD (Figure 3D; Online Figure IIIB), and markers of macrophage content in the aortic arch were reduced (Online Figure IIIC). Plaque burden in the descending thoracic aorta (Online Figure IIIB) and aortic root (Online Figure IIID) appeared to be unchanged. The decrease in plaque burden in the aortic arch was paradoxically accompanied by signs of systemic inflammatory signaling, with higher levels of circulating sICAM-1, IL-6, and TNF-α (tumor necrosis factor-α) detected in the plasma of mice receiving KO BM after 12 weeks of HCD (Figure 3E), with a trend toward elevated IL-6 levels being observed after 4 weeks of HCD (Figure 3E). These findings suggest that loss of miR-146a expression in BM-derived cells surprisingly results in reduced atherosclerosis, despite the ability of miR-146a to restrain inflammatory signaling. The similarity in phenotypes observed in DKO mice and mice receiving KO BM suggests that loss of miR-146a function in BM-derived cells is the predominant contributor to the observed phenotypes.
Figure 3.

MicroRNA-146a (miR-146a) in bone marrow (BM)–derived cells contributes to atherogenesis. A, Ldlr−/− mice lethally irradiated and given BM transplantation (BMT) from wild-type (WT BM) or miR-146a−/− (knockout [KO] BM) donors followed by high cholesterol diet (HCD) for 4 or 12 wk. B, Body weights of female mice after 12-wk HCD (n=5–7). Weights of male mice were also unchanged (not shown). C, Food consumption in mice (n=4 mice per cage). T0 is 12 wk of HCD. D, Percentage of Oil Red-O (ORO) regions per aortic arch measured by en face imaging after 4- or 12-wk HCD (n=9–11). Representative images of plaque burden in aortas of Ldlr−/− mice with WT BM (top) and KO BM (bottom) after 12-wk HCD are shown to the right. Aortic root and descending thoracic aorta analyses are shown in Online Figure IIIB and IIID. E, Circulating proinflammatory markers, soluble intercellular adhesion molecule-1 (sICAM-1), IL-6 (interleukin-6), and TNF-α (tumor necrosis factor-α), measured by ELISA of plasma samples (n=4–7). F, Time course of plasma cholesterol measurement in Ldlr−/− mice receiving WT or KO BM (n=4–7; 1 group of mice was used for weeks 0 and 4, and a separate group was used for week 12). G, FPLC (fast protein liquid chromatography) trace of cholesterol content of lipoprotein fractions from plasma after 12 wk of HCD (pooled analysis of 4 samples). H, Intrahepatic total cholesterol (TC), free cholesterol (FC), and triglycerides (TG) after 12-wk HCD (n=4). Fecal cholesterol levels after 12 wk of HCD (n=8). I, Expression of inflammatory genes, Sort1, and a macrophage marker (F4/80) from liver tissues. Shown is a heat map of quantitative reverse transcriptase-polymerase chain reaction data (n=4–8). Values are relative to the controls for each group, as indicated. *Significant difference in expression. J, TG measurements in the media of cultured primary mouse hepatocytes treated with recombinant mouse IL-6 for 6 h (n=4). K, The predicted miR-146a binding in the human and mouse SORT1 3′ UTR (untranslated region; above) and luciferase analyses in bovine aortic endothelial cells (BAECs; n=5). HDL indicates high-density lipoprotein; and Ldlr, low-density lipoprotein receptor.
Interestingly, we found a progressive decrease in total cholesterol, LDL, triglycerides, and high-density lipoprotein levels in the plasma of mice receiving KO BM (Figure 3F). FPLC revealed a marked reduction in cholesterol content in VLDL fractions (Figure 3G). However, levels of total and free cholesterol and triglycerides in the liver were not significantly different (Figure 3H), neither were cholesterol esters (not shown), and fecal cholesterol levels were also unchanged (Figure 3H). To determine potential mechanisms for the altered lipid metabolism, we assessed gene expression in livers of Ldlr−/− and DKO mice (18-week HCD), and Ldlr−/− mice receiving WT or KO BM (12-week HCD). We observed an elevation of a macrophage marker (F4/80), as well as several proinflammatory cytokines, such as IL-1β and IL-6, and an increase in IL-10, in DKO livers and in the livers of Ldlr−/− mice receiving KO BM, compared with their respective controls (Figure 3I). Importantly, dysregulation of IL-6 and IL-10 has previously been implicated in altered lipid metabolism.15–17 Indeed, we found that exposing primary hepatocytes to IL-6 decreased triglyceride secretion (Figure 3J). Acute phase response genes were elevated in DKO livers, but not in recipients of KO BM (not shown). We also assessed the expression of a panel of 84 lipid signaling and cholesterol metabolism genes by quantitative reverse transcriptase-polymerase chain reaction arrays. A small number of genes were significantly dysregulated in either experimental group (9 genes in DKOs compared with Ldlr−/− mice, and 19 genes in KO BMT recipients compared with WT BMT recipients; Online Figure IV; Online Table I). However, the only genes that were significantly decreased in both models were ApoB (apolipoprotein B; 1.25-fold decrease in DKOs versus Ldlr−/− and 1.61-fold decrease in KO BMT versus WT BMT recipients) and Cnbp (CCHC-type zinc finger nucleic acid binding protein; 1.43-fold decrease in DKOs versus Ldlr−/− and 1.69-fold decrease in KO BMT versus WT BMT recipients), but these changes were modest. Although not on the quantitative reverse transcriptase-polymerase chain reaction array, we also assessed the expression of sortilin-1(Sort1) because it is a known regulator of circulating LDL levels that was identified by genome-wide association studies in humans,18 and it has been shown to promote IL-6 signaling and secretion in macrophages in mouse models.19 Interestingly, Sort1 is also predicted to be an miR-146a target gene (Figure 3K). We found that Sort1 expression in the liver was elevated in both models (2.21-fold increase in DKOs versus Ldlr−/− and 1.68-fold increase in KO BMT versus WT BMT recipients; Figure 3I). Furthermore, we confirmed that Sort1 is a bone fide miR-146a target gene by luciferase assay (Figure 3K). Thus, loss of miR-146a from BM-derived cells perturbs cholesterol metabolism, potentially through dysregulated NF-κB–dependent inflammatory pathways in the liver, including macrophage accumulation and IL-6 secretion, and perhaps through regulation of Sort1. Of note, despite the lower levels of VLDL/LDL cholesterol, miR-146a−/− mice display an exaggerated inflammatory response to HCD.
Diet- and Age-Dependent Hematopoiesis Defects in miR-146a−/− Mice
Strikingly, the spleens of DKO mice fed HCD for 18 weeks (6–7 months of age) were ≈2.5× larger than Ldlr−/− mice on the same atherogenic diet (Figure 4A). At earlier stages (ie, 12 weeks of HCD; 5–6 months of age) spleen weight was not significantly changed (Figure 4A). Of note, aged miR-146a−/− mice (>8 months of age) have previously been shown to spontaneously develop splenomegaly, which is accompanied by BM hematopoiesis defects.10 Because we observed a splenomegaly phenotype in young mice on HCD, this suggests that atherogenic diet may accelerate the development of splenomegaly. Similar to global KOs, mice receiving KO BMT and fed HCD developed larger spleens and had pale femurs, suggestive of BM dysfunction (Figure 4B and 4C). We previously showed that prolonged hypercholesterolemia results in the outsourcing of hematopoiesis from the BM to the spleen.20 It seems that this phenotype may be accelerated and exaggerated in miR-146a−/− mice, even in the face of lower circulating VLDL/LDL cholesterol levels.
Figure 4.
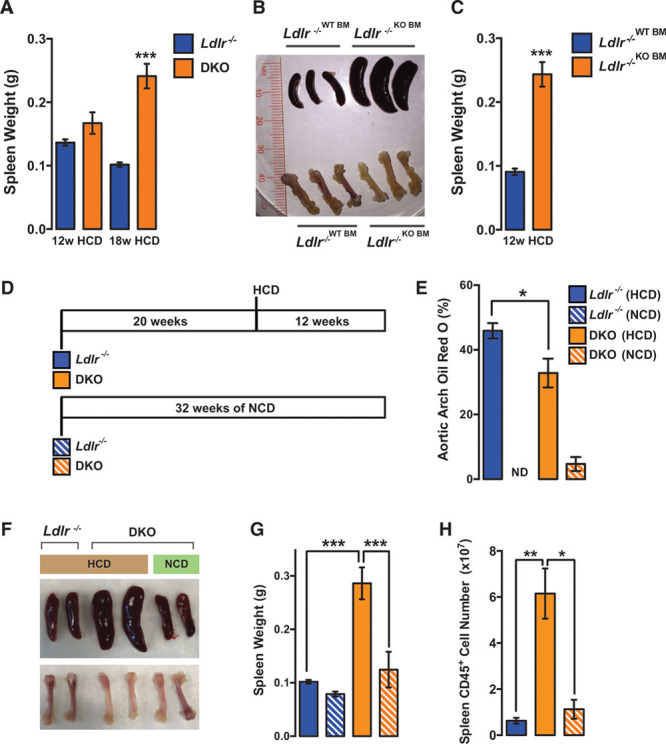
Diet- and age-dependent splenomegaly in double knockout (DKO) mice. A, Spleen weight in Ldlr−/− or DKO mice after 12 or 18 wk of high cholesterol diet (HCD; n=12–16 for 12-wk HCD; n=5 for 18-wk HCD). B, Representative images of spleens and femurs in wild-type (WT) and KO bone marrow transplant (BMT) mice on HCD for 12 wk. C, Quantification of spleen weight in WT and KO BMT mice on HCD for 12 wk (n=5–7). D, Mice were placed on HCD or normal chow diet (NCD) at 20 wk of age for 12 wk. E, Percentage of Oil Red-O (ORO) region per aortic arch measured en face (n=3–4). Ldlr−/− mice on NCD were from a separate experiment and are included for comparison purposes (n=5). F, Representative images of spleens and femurs. G, Quantification of spleen weights (n=3–4). Ldlr−/− mice on NCD were from a separate experiment and are included for comparison purposes (n=5). H, Quantification of total CD45+ cells in spleens by fluorescence-activated cell sorting (FACS) analysis (n=3–4). Ldlr indicates low-density lipoprotein receptor.
To further investigate the effects of aging on splenomegaly and atherogenesis, DKO mice were fed a 12-week HCD regime starting at 20 weeks of age (rather than the typical 10 weeks of age; Figure 4D). In contrast to younger DKO mice, which had unaltered plaque burden in the aorta after 12 weeks of HCD (Figure 2D), older mice had reduced atherosclerosis in the aortic arch after the same duration of diet (Figure 4E). No differences in plaque formation were observed in the descending thoracic aorta (not shown). This reduction in aortic arch atherosclerosis was accompanied by splenomegaly (Figure 4F and 4G). Importantly, the splenomegaly phenotype at this age was dependent on exposure to HCD, as this was not observed in DKO mice on a regular chow diet (Figure 4F and 4G). The pale femur phenotype in older DKO mice also seemed to be dependent on exposure to HCD (Figure 4F). The increased spleen size in older DKO mice on HCD corresponded with an increase in splenic CD45+ leukocytes (Figure 4H). Intriguingly, these findings highlight a potential relationship between the reduced atherogenesis observed in DKO mice on HCD and development of splenomegaly and pale femurs, suggesting that defective hematopoiesis may contribute to the phenotype. Although previous studies have linked splenomegaly with reduced circulating cholesterol,21 the contribution of splenomegaly to reduced LDL cholesterol in miR-146a−/− mice remains unclear. Although leukocyte content in the spleen at 12 weeks of HCD was not significantly different in DKOs (Online Figure VA) and spleens were not significantly larger (Figure 4A)—despite reduced levels of plasma cholesterol at this stage (Figure 2F)—mice receiving miR-146a−/− BMT had greatly enlarged spleens at 12 weeks of HCD (Figure 4C), which coincided with reduced circulating LDL (Figure 3F). Furthermore, oxidized LDL uptake and cholesterol efflux were similar in WT and miR-146a−/− macrophages (Online Figure VB and VC), suggesting that miR-146a–deficient macrophages seem not to be more avid at sequestering cholesterol. However, expansion of macrophages in the liver and spleen may contribute to the sequestering of cholesterol from circulation. The contribution of splenomegaly to cholesterol lowering in miR-146a−/− mice will require further exploration.
Loss of miR-146a Leads to Reduced BM Hematopoiesis While Promoting Extramedullary Hematopoiesis in the Spleen in Mice Fed a HCD
The enlarged spleens in DKO mice on 18 weeks of HCD contained more CD45+ leukocytes and lymphocytes (Figure 5A; Online Figure VI for flow cytometry gating strategies). This was in contrast to the depletion of these cells from the BM of DKO mice (Figure 5B). Previous studies have observed defects in BM hematopoietic stem cell (HSC) longevity in aged (>8 months) miR-146a−/− mice, or in younger mice after repeated challenge with LPS.10 Assessing the spectrum of hematopoietic cells in the BM revealed normal levels of hematopoietic progenitors cells 1 and 2 and HSCs, but levels of multipotent progenitor cells were significantly decreased in DKOs (Figure 5C; Online Figure VIIA). The consequences of decreased multipotent progenitor cell levels were further evident in the decreased numbers of downstream progenitor cells (eg, Sca-1 [stem cells antigen-1]–negative progenitors, common myeloid progenitors, granulocyte–macrophage progenitors, megakaryocyte–erythroid progenitors; Figure 5C). Taken together, these data are suggestive of an HSC functional defect in the BM. Interestingly, despite this defect in HSC function in the BM, HSCs and downstream progenitor cells appeared in the spleens of DKO mice on HCD for 18 weeks but not in Ldlr−/− mice (Figure 5D), demonstrating that loss of miR-146a accelerates extramedullary hematopoiesis. The dysregulation of hematopoiesis in the spleen and BM was accompanied by modest effects on circulating leukocytes, such as neutrophils and B cells, although anti-inflammatory Ly6Clo monocytes were significantly increased (Online Figure VIIB).
Figure 5.
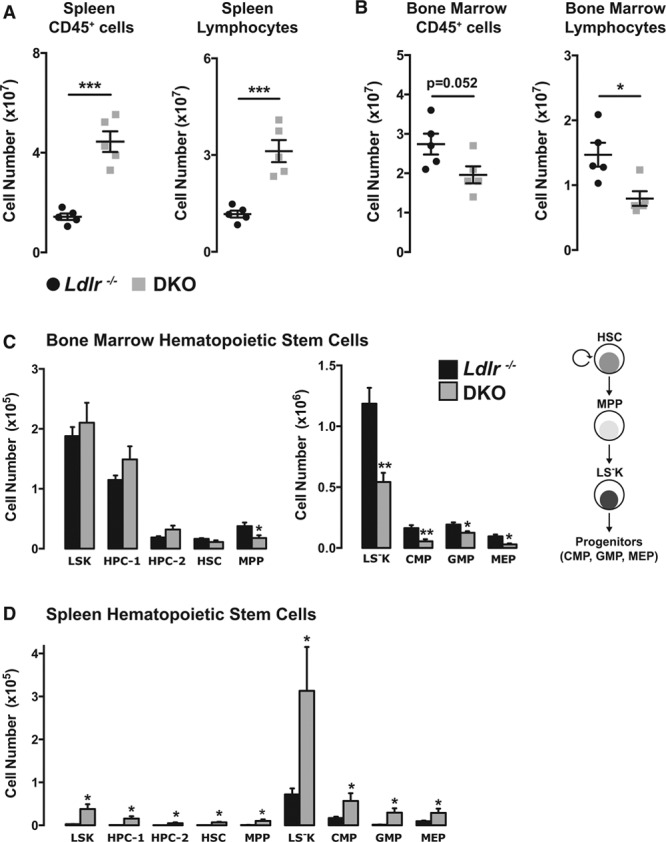
Global loss of microRNA-146a (miR-146a) inhibits bone marrow (BM) hematopoiesis and promotes extramedullary hematopoiesis in the spleen. Increase of splenic (A) and decrease in BM (B) CD45+ leukocytes and Ly6G−/CD115− lymphocytes in DKO mice on diet for 18 wk, determined by fluorescence-activated cell sorting (FACS) analysis (n=5). C, Decrease of multipotent progenitor cells (MPPs) and downstream progenitor cells (eg, Sca-1 [stem cells antigen-1]–negative progenitor [LS−K], megakaryocyte–erythroid progenitor [MEP], common myeloid progenitor [CMP], and granulocyte–macrophage progenitor [GMP]) in BM of double knockout (DKO) mice after 18 wk of high cholesterol diet (HCD; n=5). D, Increase of splenic hematopoietic and multipotent stem cells in DKO mice after 18 wk of HCD (n=5; Online Figure VII). HPC indicates hematopoietic progenitors cell; HSC, hematopoietic stem cell; Ldlr, low-density lipoprotein receptor; and LSK, lineage- Sca-1+ Kit+.
miR-146a in BM-Derived Cells Regulates BM and Extramedullary Hematopoiesis and Levels of Circulating Leukocytes and Lymphocytes
Similar to the nontransplanted DKO mice on 18-week HCD, mice receiving KO BM accumulated more CD45+ leukocytes and lymphocytes in their spleens; however, this occurred after just 12-week HCD (Figure 6A). The BM in these mice was depleted of these cells by 12-week HCD (Figure 6B). This was accompanied by elevated NF-κB signaling in the BM of KO mice, as well as enhanced expression of TRAF6, an miR-146a target gene (Figure 6C). Progenitors downstream of HSCs, namely multipotent progenitor cells, Sca-1–negative progenitors, common myeloid progenitors, granulocyte–macrophage progenitors and megakaryocyte–erythroid progenitors, were diminished in the BM (Figure 6D), whereas extramedullary hematopoiesis was evident in mice receiving KO BM (Figure 6E). Correspondingly, mice receiving KO BM had decreased levels of circulating atherogenic leukocytes, including neutrophils, B cells, and Ly6Chi monocytes, but levels of atheroprotective Ly6Clo monocytes were increased after 12 weeks of HCD (Figure 6F). Assessing circulating levels of leukocytes and lymphocytes at earlier stages (ie, 4 weeks of HCD) revealed monocytosis in mice receiving KO BM (Figure 6F), suggesting that the reduction of hematopoiesis at later stages of atherosclerosis is preceded by enhanced hematopoiesis at earlier stages, similar to previous studies that revealed HSC exhaustion in KO mice in the context of repeated LPS stimulation.10
Figure 6.

MicroRNA-146a (miR-146a) in bone marrow (BM)–derived cells regulates BM and extramedullary hematopoiesis and levels of circulating leukocytes and lymphocytes. Lethally irradiated Ldlr−/− mice (mix of males and females) were reconstituted with BM from wild-type (WT BM) or miR-146a−/− (knockout [KO] BM) donors, followed by high cholesterol diet (HCD) for 12 wk. Increase of splenic (A) and a decrease of BM (B) leukocytes and lymphocytes, as assessed by fluorescence-activated cell sorting (FACS) analysis (n=5–7). C, Western blot of TRAF6 (TNF receptor–associated factor 6) and phospho-p65 (normalized to β-actin and total p65, respectively), in mice receiving WT or KO BM after 12 wk of HCD. Phospho-p65/p65 blots from WT/KO animals are from the same membrane with identical imaging parameters. D, Decrease of a subset of multipotent stem cells, but no changes to the long-term HSCs in the BM of mice receiving KO BM transplants (BMT; n=5–7). E, Increase of splenic hematopoietic and multipotent stem cells in mice receiving KO BM (n=5–7). F, Early monocytosis (4-wk HCD), followed by a decrease in peripheral blood (PB) proatherogenic cells (neutrophils, B Cells, and Ly6Chi monocytes), and an increase of antiatherogenic Ly6Clo monocytes after 12 wk of HCD (n=3 for 4-wk HCD; n=9–11 for 12-wk HCD). CMP indicates common myeloid progenitor; GMP, granulocyte–macrophage progenitor; HPC, hematopoietic progenitors cell; HSC, hematopoietic stem cell; Ldlr, low-density lipoprotein receptor; LS−K, Sca-1 (stem cells antigen-1)–negative progenitor; MEP, megakaryocyte–erythroid progenitor; and MPP, multipotent progenitor cell.
We next assessed the functionality of WT (CD45.1) and KO (CD45.2) BM-derived cells in a competitive 1:1 BMT. After reconstitution of the BM compartment of lethally irradiated Ldlr−/− mice for 8 weeks, mice were placed on either normal chow diet (NCD) or HCD diet. Assessing circulating levels of leukocytes in mice fed an NCD revealed that KO BM cells preferentially contributed to neutrophil and Ly6Chi monocyte populations compared with WT BM cells (Figure 7A). A short duration on HCD (4 weeks) expanded the leukocyte populations examined, and KO cells were predominant compared with WT cells (Figure 7A). This was especially the case for neutrophils and Ly6Chi monocytes. However, in mice that received HCD for 12 or 32 weeks, the abundance of KO BM-derived cells was decreased. WT BM-derived cells were less affected (Figure 7A). This suggests that long-term HCD impairs the ability of KO BM-derived cells to contribute to circulating leukocyte populations. Assessing the abundance of WT versus KO leukocytes in the aorta at advanced stages of atherosclerosis revealed that neutrophils, macrophages, and monocytes (Ly6Chi and Ly6Clo) seemed to be primarily WT BM derived, whereas B- and T-cell populations had similar contributions from WT and KO cells (Figure 7B). This was in contrast to the aorta in mice fed an NCD, where the majority of the cells seemed to be derived from KO cells (Figure 7B). Consistent with the reduced abundance of KO BM-derived cells in the circulation and atherosclerotic plaques in mice fed HCD, hematopoietic cells in the BM seemed to be primarily of WT origin under conditions of HCD feeding (Figure 7C). However, the opposite was observed in mice fed an NCD (Figure 7C).
Figure 7.

MicroRNA-146a (miR-146a)–deficient cells seem to be outcompeted by wild-type (WT) cells in the bone marrow (BM), circulation, and in atherosclerotic plaques in high cholesterol diet (HCD)–treated animals, but not in normal chow diet (NCD)–treated animals. Competitive BM transplantation (BMT) was performed into Ldlr−/− recipients. A 1:1 mix of BM from WT (CD45.1) and knockout (KO; CD45.2) was used. A, Peripheral blood was analyzed by fluorescence-activated cell sorting (FACS) after NCD or HCD for 4, 12, or 32 wk (n=8, 5, 4, 2, respectively). Comparison was made between WT and KO within each time point. B, Cells in the aorta (B) and the BM (C) were analyzed by FACS in animals receiving NCD or HCD for 32 wk (n=2 per group). CMP indicates common myeloid progenitor; GMP, granulocyte–macrophage progenitor; HPC, hematopoietic progenitors cell; HSC, hematopoietic stem cell; Ldlr, low-density lipoprotein receptor; LS−K, Sca-1 (stem cells antigen-1)–negative progenitor; MEP, megakaryocyte–erythroid progenitor; and MPP, multipotent progenitor cell.
miR-146a in the Vasculature Restrains EC Activation and Atherosclerosis
Deletion of miR-146a has a major effect on BM-derived cell function, promoting systemic inflammatory signaling, extramedullary hematopoiesis, BM failure, and lipid dysregulation. To further distinguish the role of miR-146a in BM-derived cells versus the rest of the body, we transplanted lethally irradiated Ldlr−/− and DKO mice with miR-146a+/+ (WT) BM (Figure 8A). Transplanted mice were placed on HCD for 12 weeks. Interestingly, we found no differences in circulating IL-6, sICAM-1, or TNF-α levels (Figure 8B) or circulating cholesterol or lipoproteins (Figure 8C). This suggests that the dysregulation of inflammation and circulating lipoprotein levels are dependent on deletion of miR-146a from BM-derived cells, rather than in other cell types, such as hepatocytes. In addition, no changes were observed in spleen size (Figure 8D). Levels of leukocytes in the spleen and in the circulation were also normalized, and only a modest decrease in leukocyte levels in the BM was seen (Figure 8E). Interestingly, NF-κB–dependent cytokines known to accelerate HSC proliferation (ie, IL-6, TNF-α, and IL-10)10,22,23 were highly expressed in the BM of Ldlr−/− mice reconstituted with KO BM, but this was not observed in DKO mice reconstituted with WT BM (Figure 8F). Finally, with the normalization of these parameters after transplantation of WT BM in DKO mice, lipid plaque burden in the aorta was elevated compared with Ldlr−/− mice receiving WT BMT (Figure 8G).
Figure 8.
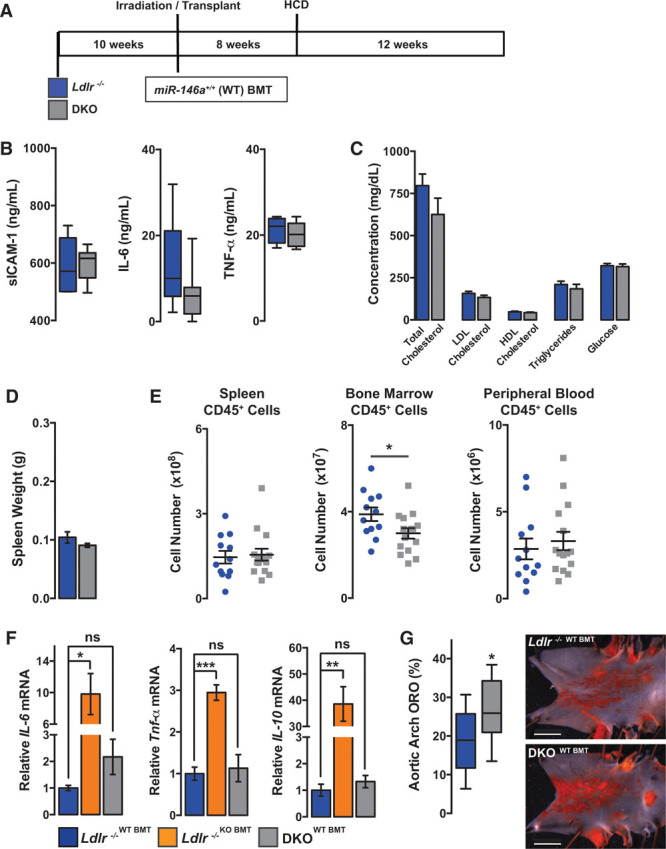
MicroRNA-146a (miR-146a) in the vasculature restrains endothelial cell (EC) activation and atherosclerosis. A, Schematic of lethally irradiated Ldlr−/− or double knockout (DKO) mice given bone marrow transplantation (BMT) from wild-type (WT BM) donors followed by high cholesterol diet (HCD) for 12 wk. B, Circulating proinflammatory markers, soluble intercellular adhesion molecule-1 (sICAM-1), IL-6 (interleukin-6), and TNF-α (tumor necrosis factor-α), measured by ELISA (n=5–8). C, Circulating cholesterol, high-density lipoprotein (HDL), triglycerides (TG), low-density lipoprotein (LDL) and glucose levels after 12 wk of HCD (n=5). D, Quantification of spleen weight after 12 wk of HCD (n=12–15). E, Fluorescence-activated cell sorting (FACS) analysis of myeloid cells from spleen, BM, and peripheral blood (n=12–15). F, Gene expression in BM cells from BM transplanted animals (n=3–7). G, Percentage of Oil Red-O (ORO) region per aortic arch measured by en face staining (n=11–14). Representative images are shown to the right. Data on EC activation in the aorta are given in Online Figure VIII.
To determine whether miR-146a in the vasculature affects EC activation in the aorta, we stimulated WT or KO mice with the proinflammatory cytokine, IL-1β. We found that miR-146a target genes (eg, HuR [human antigen R] and TRAF6) were elevated in the aortic arch of KO mice, and that levels of VCAM-1 (vascular cell adhesion molecule-1), E-Selectin (SELE), and ICAM-1 (intercellular adhesion molecule-1) were induced to a greater extent in KO compared with WT mice (Online Figure VIIIA and VIIIB). In the setting of atherosclerosis, we found that expression of adhesion and chemokine genes seemed to be elevated in intimal cells of the aorta from DKO mice receiving WT BM compared with Ldlr−/− mice receiving WT BM (Online Figure VIIIC). These observations are consistent with our previous study that demonstrated that miR-146a restrains EC activation.6
Discussion
miR-146a has been identified as a vital brake in inflammatory signaling pathways,6,8,10,24 and levels are elevated in human atherosclerotic plaques.25 Recent studies have also uncovered a single-nucleotide polymorphism in the miR-146a gene that influences miR-146a expression and susceptibility to coronary artery disease.26–29 However, no studies have directly assessed the function of endogenous miR-146a during atherogenesis. Here, we report that deletion of miR-146a within BM-derived cells surprisingly reduces atherosclerotic plaque formation, whereas deletion of miR-146a in the vasculature enhances endothelial activation and atherogenesis. These diverse phenotypes arise from a common defect in distinct cellular compartments, namely unrestrained NF-κB–dependent inflammatory signaling.
To our surprise, ablation of miR-146a from BM-derived cells reduced atherosclerosis, while paradoxically elevating indices of systemic inflammatory signaling (ie, proinflammatory cytokines and sICAM-1; an overview of miR-146a–deficient phenotypes is given in Online Figure IX). This increase in circulating cytokines would typically be accompanied by abundant inflammatory immune cells in circulation. However, we observed a decrease in proatherogenic cells, including Ly6Chi monocytes, T cells and neutrophils, and an increase in atheroprotective Ly6Clo monocytes. This implies that miR-146a–deficient leukocytes present in circulation are likely to be especially proinflammatory, demonstrating that miR-146a is important in quelling their activation. The paucity of circulating immune cells is the consequence of defective BM hematopoiesis, which likely arises because of hematopoietic cell exhaustion. Hypercholesterolemia stimulates hematopoiesis in the BM and spleen to produce proinflammatory cells, such as Ly6Chi monocytes that contribute to plaque growth.20,30–33 We find evidence of precocious monocytosis at early stages of atherogenesis in mice that received miR-146a−/− BM. In addition, transplanted miR-146a–deficient cells outcompete transplanted WT cells in the BM and in circulation during early atherogenesis. However, prolonged exposure to hypercholesterolemia seems to lead to a defect in the contribution of miR-146a−/− cells to hematopoietic cell populations in the BM, circulation, and in atherosclerotic plaques, implying that the activation of BM hematopoiesis by HCD cannot be sustained in the absence of miR-146a. Consequently, these mice initiate extramedullary hematopoiesis in the spleen. Although spleen-derived Ly6Chi monocytes can contribute to atherogenesis,20 the circulating cells generated in the spleen of mice receiving miR-146a–deficient BM seem to be insufficient to compensate for the reduction in leukocyte output from the BM.
Defects in hematopoiesis have previously been observed in aged miR-146a−/− mice.10 In this case, older mice (>8 months of age) developed a progressive loss of hematopoietic stem and progenitor cells as a result of increased NF-κB–dependent IL-6 production in the BM. Enhanced IL-6 production in miR-146a−/− mice promoted hematopoietic cell proliferation leading to eventual exhaustion. This phenotype could be accelerated by repeated challenge with LPS, which drives IL-6 production.10 Similarly, in our model of atherosclerosis, we observed an increase in TRAF6 expression, NF-κB activity, and IL-6 expression (an NF-κB–regulated cytokine) in the BM, as well as IL-6 protein levels in circulation. Because hypercholesterolemia drives stress-induced hematopoiesis33–35 and premature HSC aging and senescence,36 it is likely that increased cycling of hematopoietic cells leads to stem cell exhaustion in the absence of miR-146a.
A notable phenotype observed in both the global and BM-restricted miR-146a loss-of-function models is the reduction in VLDL/LDL cholesterol in circulation. Defects in cholesterol homeostasis in miR-146a global KOs could be rescued by transplantation of WT BM, demonstrating that miR-146a in BM-derived cells, rather than hepatocytes, regulates lipid metabolism. It is important to note that the defects that we observe in miR-146a−/− mice (ie, enhanced levels of circulating cytokines, monocytosis, outsourcing of hematopoiesis to extramedulary sites, splenomegaly, and eventual BM failure) are known to be driven by hypercholesterolemia.20,32,33 That we observe these phenotypes even in the face of lower VLDL/LDL cholesterol suggests that it is not lower cholesterol per se that is solely responsible for the decrease in atherosclerosis in these mice, although it likely contributes. miR-146a−/− mice are especially sensitive to the inflammatory effects of hypercholesterolemia (even though VLDL/LDL cholesterol levels are decreased), and the hematopoietic phenotypes that we observe are not apparent in mice fed an NCD. The reduced atherosclerosis seems to be at least partly attributable to an unchecked chronic inflammatory response to hypercholesterolemia that drives BM hematopoiesis defects and a decrease in circulating proatherogenic inflammatory cells. The role of miR-146a in monocyte recruitment and macrophage biology in the plaque has not been explored here, but should be assessed in future studies.
We demonstrate that a lack of miR-146a seems to impair VLDL secretion from the liver. This is accompanied by macrophage accumulation, inflammatory gene expression (including IL-6), and an increase in Sort1 expression in the liver; each of which may contribute to the phenotype. Although miR-146a–deficient BM-derived macrophages have unaltered oxidized LDL uptake and cholesterol efflux, the expansion of macrophages in the liver or spleen may influence circulating cholesterol levels through cholesterol sequestration. Altered VLDL secretion also occurs in response to proinflammatory cytokines such as IL-6,37–39 and we confirm that exposure to IL-6 reduces triglyceride secretion from cultured primary mouse hepatocytes. Finally, we identify Sort1 as a novel miR-146a target gene. SORT1 has been shown to play an important, though controversial role in VLDL secretion.40 Genome-wide association studies in humans identified SORT1 as a causative gene in the regulation of circulating LDL levels and risk of atherosclerotic disease.18 Overexpression of SORT1 in the liver of mice enhances VLDL destruction in the liver and inhibits secretion.18 SORT1 can also bind extracellular LDL and direct its catabolism.41 However, other studies have found that deletion of Sort1 in mice can also result in reduced LDL levels,19,42 suggesting that the contribution of SORT1 to cholesterol metabolism remains to be fully resolved. Finally, additional studies found that lack of SORT1 in macrophages can inhibit secretion and signaling of cytokines, such as IL-6,19 whereas overexpression can enhance LDL uptake.43 This is of interest considering that Sort1 is likely dysregulated in BM-derived cells in our miR-146a loss-of-function models. Further studies will be required to delineate the contribution of miR-146a–dependent Sort1 regulation within BM-derived cells to the VLDL/LDL phenotype. Furthermore, additional miR-146a target genes in BM-derived cells may contribute.
Our study emphasizes the important role that miR-146a plays in controlling the output of inflammatory signaling pathways in ECs and hematopoietic cells in the setting of an atherogenic diet and further confirms the critical role of hypercholesterolemia in hematopoietic cell stress. Our findings are intriguing in light of polymorphisms in miR-146a in human patients that alters susceptibility to coronary artery disease. Furthermore, our studies suggest that elevating the expression of miR-146a in BM-derived cells or ECs is likely to suppress human atherogenesis by restraining NF-κB signaling, which is in agreement with recent studies in mouse models.14,44
Sources of Funding
This research was supported by a group grant to J.E. Fish, A. Schober, and C.M. Boulanger (Canadian Institutes of Health Research [CIHR] MTA-118968), an operating grant from the Heart & Stroke Foundation of Canada (NA-7282), a bridge grant from CIHR (OCN-126570), a seed grant from the CIHR-funded Canadian Vascular Network and a Leaders Opportunity Fund/Canada Foundation for Innovation equipment grant (to J.E. Fish, No. 26422). J.E. Fish was supported by an Early Researcher Award from the Ontario Ministry of Research and Innovation and a Canada Research Chair from CIHR. H.S. Cheng received funding from the Heart & Stroke Richard Lewar Centre of Excellence in Cardiovascular Research, the Ontario Graduate Studentship program, and the Canadian Vascular Network.
Disclosures
None.
Supplementary Material
Nonstandard Abbreviations and Acronyms
- BM
- bone marrow
- BMT
- bone marrow transplant
- DKO
- double knockout
- EC
- endothelial cell
- HCD
- high cholesterol diet
- HSC
- hematopoietic stem cell
- HuR
- human antigen R
- ICAM-1
- intercellular adhesion molecule-1
- IL
- interleukin
- IRAK1
- interleukin receptor–associated kinase 1
- KO
- knockout
- LDL
- low-density lipoprotein
- LDLR
- low-density lipoprotein receptor
- LPS
- lipopolysaccharide
- miR-146a
- microRNA-146a
- NCD
- normal chow diet
- NF-κB
- nuclear factor κ light chain enhancer of activated B cells
- SELE
- E-selectin
- sICAM-1
- soluble intercellular adhesion molecule-1
- SORT1
- sortilin-1
- TNF-α
- tumor necrosis factor-α
- TRAF6
- TNF receptor–associated factor 6
- VCAM-1
- vascular cell adhesion molecule-1
- VLDL
- very-low-density lipoprotein
- WT
- wild type
In May 2017, the average time from submission to first decision for all original research papers submitted to Circulation Research was 12.28 days.
The online-only Data Supplement is available with this article at http://circres.ahajournals.org/lookup/suppl/doi:10.1161/CIRCRESAHA.116.310529/-/DC1.
Novelty and Significance
What Is Known?
MicroRNA-146a (miR-146a) suppresses inflammatory responses in endothelial cells and bone marrow (BM)–derived cells by targeting adaptor proteins in the nuclear factor κ light chain enhancer of activated B cells signaling pathway.
Increased levels of miR-146a have been detected in human atherosclerotic plaques, and polymorphisms in the miR-146a precursor are associated with risk of coronary artery disease.
Injection of exogenous miR-146a reduces atherogenesis in mouse models.
What New Information Does This Article Contribute?
Deletion of miR-146a in BM-derived cells enhances the production of proinflammatory cytokines, but paradoxically reduces circulating proatherogenic leukocytes, ultimately resulting in decreased atherosclerosis.
miR-146a in BM-derived cells protects against high cholesterol diet–induced hematopoietic progenitor cell exhaustion in the BM and prevents extramedullary hematopoiesis and splenomegaly.
Circulating very-low-density lipoprotein levels are progressively decreased in mice lacking miR-146a in the BM, and this is accompanied by enhanced inflammation in the liver and dysregulation of a newly identified miR-146a target gene, sortilin 1 (Sort1).
Elevation of miR-146a expression in atherosclerotic plaques in humans and polymorphisms in the miR-146a precursor that are associated with coronary artery disease are suggestive of a role for this microRNA in atherogenesis. Although numerous studies have placed miR-146a among the echelon of anti-inflammatory microRNAs, the role of endogenous miR-146a in atherosclerosis remains unknown. Surprisingly, despite the ability of this microRNA to restrain cytokine production in BM-derived cells, loss of this microRNA resulted in reduced atherosclerosis. This was accompanied by hematopoietic stem cell exhaustion and a corresponding reduction in levels of circulating proatherogenic cells. Enhanced inflammatory signaling occurred even though circulating levels of very-low-density lipoprotein cholesterol were diminished in these mice. Within the vasculature, miR-146a restrained endothelial activation, and loss of miR-146a in the vasculature enhanced atherosclerosis. This study reveals a critical function for a single microRNA in the control of the intensity of inflammatory responses to hypercholesterolemia and highlights the detrimental effects of unrestrained inflammatory signaling in multiple organs: BM (hematopoietic stem cell exhaustion), spleen (extramedullary hematopoiesis and splenomegaly), liver (cholesterol homeostasis defects), and the vasculature (enhanced endothelial cell activation and monocyte recruitment). Importantly, these findings provide a further impetus to therapeutically augment miR-146a expression/function in atherosclerosis.
References
- 1.Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685–1695. doi: 10.1056/NEJMra043430. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 2.Weber C, Noels H. Atherosclerosis: current pathogenesis and therapeutic options. Nat Med. 2011;17:1410–1422. doi: 10.1038/nm.2538. doi: 10.1038/nm.2538. [DOI] [PubMed] [Google Scholar]
- 3.Gareus R, Kotsaki E, Xanthoulea S, van der Made I, Gijbels MJ, Kardakaris R, Polykratis A, Kollias G, de Winther MP, Pasparakis M. Endothelial cell-specific NF-kappaB inhibition protects mice from atherosclerosis. Cell Metab. 2008;8:372–383. doi: 10.1016/j.cmet.2008.08.016. doi: 10.1016/j.cmet.2008.08.016. [DOI] [PubMed] [Google Scholar]
- 4.Kempe S, Kestler H, Lasar A, Wirth T. NF-kappaB controls the global pro-inflammatory response in endothelial cells: evidence for the regulation of a pro-atherogenic program. Nucleic Acids Res. 2005;33:5308–5319. doi: 10.1093/nar/gki836. doi: 10.1093/nar/gki836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheng HS, Njock MS, Khyzha N, Dang LT, Fish JE. Noncoding RNAs regulate NF-κB signaling to modulate blood vessel inflammation. Front Genet. 2014;5:422. doi: 10.3389/fgene.2014.00422. doi: 10.3389/fgene.2014.00422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheng HS, Sivachandran N, Lau A, Boudreau E, Zhao JL, Baltimore D, Delgado-Olguin P, Cybulsky MI, Fish JE. MicroRNA-146 represses endothelial activation by inhibiting pro-inflammatory pathways. EMBO Mol Med. 2013;5:1017–1034. doi: 10.1002/emmm.201202318. doi: 10.1002/emmm.201202318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci USA. 2006;103:12481–12486. doi: 10.1073/pnas.0605298103. doi: 10.1073/pnas.0605298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao JL, Rao DS, Boldin MP, Taganov KD, O’Connell RM, Baltimore D. NF-kappaB dysregulation in microRNA-146a-deficient mice drives the development of myeloid malignancies. Proc Natl Acad Sci USA. 2011;108:9184–9189. doi: 10.1073/pnas.1105398108. doi: 10.1073/pnas.1105398108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boldin MP, Taganov KD, Rao DS, Yang L, Zhao JL, Kalwani M, Garcia-Flores Y, Luong M, Devrekanli A, Xu J, Sun G, Tay J, Linsley PS, Baltimore D. miR-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. J Exp Med. 2011;208:1189–1201. doi: 10.1084/jem.20101823. doi: 10.1084/jem.20101823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao JL, Rao DS, O’Connell RM, Garcia-Flores Y, Baltimore D. MicroRNA-146a acts as a guardian of the quality and longevity of hematopoietic stem cells in mice. Elife. 2013;2:e00537. doi: 10.7554/eLife.00537. doi: 10.7554/eLife.00537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Etzrodt M, Cortez-Retamozo V, Newton A, et al. Regulation of monocyte functional heterogeneity by miR-146a and Relb. Cell Rep. 2012;1:317–324. doi: 10.1016/j.celrep.2012.02.009. doi: 10.1016/j.celrep.2012.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brand K, Page S, Rogler G, Bartsch A, Brandl R, Knuechel R, Page M, Kaltschmidt C, Baeuerle PA, Neumeier D. Activated transcription factor nuclear factor-kappa B is present in the atherosclerotic lesion. J Clin Invest. 1996;97:1715–1722. doi: 10.1172/JCI118598. doi: 10.1172/JCI118598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kanters E, Pasparakis M, Gijbels MJ, Vergouwe MN, Partouns-Hendriks I, Fijneman RJ, Clausen BE, Förster I, Kockx MM, Rajewsky K, Kraal G, Hofker MH, de Winther MP. Inhibition of NF-kappaB activation in macrophages increases atherosclerosis in LDL receptor-deficient mice. J Clin Invest. 2003;112:1176–1185. doi: 10.1172/JCI18580. doi: 10.1172/JCI18580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li K, Ching D, Luk FS, Raffai RL. Apolipoprotein E enhances microRNA-146a in monocytes and macrophages to suppress nuclear factor-κB-driven inflammation and atherosclerosis. Circ Res. 2015;117:e1–e11. doi: 10.1161/CIRCRESAHA.117.305844. doi: 10.1161/CIRCRESAHA.117.305844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Von Der Thüsen JH, Kuiper J, Fekkes ML, De Vos P, Van Berkel TJ, Biessen EA. Attenuation of atherogenesis by systemic and local adenovirus-mediated gene transfer of interleukin-10 in LDLr-/- mice. FASEB J. 2001;15:2730–2732. doi: 10.1096/fj.01-0483fje. doi: 10.1096/fj.01-0483fje. [DOI] [PubMed] [Google Scholar]
- 16.Hashizume M, Yoshida H, Koike N, Suzuki M, Mihara M. Overproduced interleukin 6 decreases blood lipid levels via upregulation of very-low-density lipoprotein receptor. Ann Rheum Dis. 2010;69:741–746. doi: 10.1136/ard.2008.104844. doi: 10.1136/ard.2008.104844. [DOI] [PubMed] [Google Scholar]
- 17.Lehtimäki T, Ojala P, Rontu R, Goebeler S, Karhunen PJ, Jylhä M, Mattila K, Metso S, Jokela H, Nikkilä M, Wuolijoki E, Hervonen A, Hurme M. Interleukin-6 modulates plasma cholesterol and C-reactive protein concentrations in nonagenarians. J Am Geriatr Soc. 2005;53:1552–1558. doi: 10.1111/j.1532-5415.2005.53484.x. doi: 10.1111/j.1532-5415.2005.53484.x. [DOI] [PubMed] [Google Scholar]
- 18.Musunuru K, Strong A, Frank-Kamenetsky M, et al. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature. 2010;466:714–719. doi: 10.1038/nature09266. doi: 10.1038/nature09266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mortensen MB, Kjolby M, Gunnersen S, Larsen JV, Palmfeldt J, Falk E, Nykjaer A, Bentzon JF. Targeting sortilin in immune cells reduces proinflammatory cytokines and atherosclerosis. J Clin Invest. 2014;124:5317–5322. doi: 10.1172/JCI76002. doi: 10.1172/JCI76002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Robbins CS, Chudnovskiy A, Rauch PJ, et al. Extramedullary hematopoiesis generates Ly-6C(high) monocytes that infiltrate atherosclerotic lesions. Circulation. 2012;125:364–374. doi: 10.1161/CIRCULATIONAHA.111.061986. doi: 10.1161/CIRCULATIONAHA.111.061986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aviram M, Brook JG, Tatarsky I, Levy Y, Carter A. Increased low-density lipoprotein levels after splenectomy: a role for the spleen in cholesterol metabolism in myeloproliferative disorders. Am J Med Sci. 1986;291:25–28. doi: 10.1097/00000441-198601000-00006. [DOI] [PubMed] [Google Scholar]
- 22.Rezzoug F, Huang Y, Tanner MK, Wysoczynski M, Schanie CL, Chilton PM, Ratajczak MZ, Fugier-Vivier IJ, Ildstad ST. TNF-alpha is critical to facilitate hemopoietic stem cell engraftment and function. J Immunol. 2008;180:49–57. doi: 10.4049/jimmunol.180.1.49. [DOI] [PubMed] [Google Scholar]
- 23.Baldridge MT, King KY, Goodell MA. Inflammatory signals regulate hematopoietic stem cells. Trends Immunol. 2011;32:57–65. doi: 10.1016/j.it.2010.12.003. doi: 10.1016/j.it.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang L, Boldin MP, Yu Y, Liu CS, Ea CK, Ramakrishnan P, Taganov KD, Zhao JL, Baltimore D. miR-146a controls the resolution of T cell responses in mice. J Exp Med. 2012;209:1655–1670. doi: 10.1084/jem.20112218. doi: 10.1084/jem.20112218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Raitoharju E, Lyytikäinen LP, Levula M, Oksala N, Mennander A, Tarkka M, Klopp N, Illig T, Kähönen M, Karhunen PJ, Laaksonen R, Lehtimäki T. miR-21, miR-210, miR-34a, and miR-146a/b are up-regulated in human atherosclerotic plaques in the Tampere Vascular Study. Atherosclerosis. 2011;219:211–217. doi: 10.1016/j.atherosclerosis.2011.07.020. doi: 10.1016/j.atherosclerosis.2011.07.020. [DOI] [PubMed] [Google Scholar]
- 26.Bao MH, Xiao Y, Zhang QS, Luo HQ, Luo J, Zhao J, Li GY, Zeng J, Li JM. Meta-analysis of miR-146a polymorphisms association with coronary artery diseases and ischemic stroke. Int J Mol Sci. 2015;16:14305–14317. doi: 10.3390/ijms160714305. doi: 10.3390/ijms160714305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.He Y, Yang J, Kong D, Lin J, Xu C, Ren H, Ouyang P, Ding Y, Wang K. Association of miR-146a rs2910164 polymorphism with cardio-cerebrovascular diseases: a systematic review and meta-analysis. Gene. 2015;565:171–179. doi: 10.1016/j.gene.2015.04.020. doi: 10.1016/j.gene.2015.04.020. [DOI] [PubMed] [Google Scholar]
- 28.Ramkaran P, Khan S, Phulukdaree A, Moodley D, Chuturgoon AA. miR-146a polymorphism influences levels of miR-146a, IRAK-1, and TRAF-6 in young patients with coronary artery disease. Cell Biochem Biophys. 2014;68:259–266. doi: 10.1007/s12013-013-9704-7. doi: 10.1007/s12013-013-9704-7. [DOI] [PubMed] [Google Scholar]
- 29.Xiong XD, Cho M, Cai XP, Cheng J, Jing X, Cen JM, Liu X, Yang XL, Suh Y. A common variant in pre-miR-146 is associated with coronary artery disease risk and its mature miRNA expression. Mutat Res. 2014;761:15–20. doi: 10.1016/j.mrfmmm.2014.01.001. doi: 10.1016/j.mrfmmm.2014.01.001. [DOI] [PubMed] [Google Scholar]
- 30.Ma X, Feng Y. Hypercholesterolemia tunes hematopoietic stem/progenitor cells for inflammation and atherosclerosis. Int J Mol Sci. 2016;17:E1162. doi: 10.3390/ijms17071162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murphy AJ, Tall AR. Disordered haematopoiesis and athero-thrombosis. Eur Heart J. 2016;37:1113–1121. doi: 10.1093/eurheartj/ehv718. doi: 10.1093/eurheartj/ehv718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Feng Y, Schouteden S, Geenens R, Van Duppen V, Herijgers P, Holvoet P, Van Veldhoven PP, Verfaillie CM. Hematopoietic stem/progenitor cell proliferation and differentiation is differentially regulated by high-density and low-density lipoproteins in mice. PLoS One. 2012;7:e47286. doi: 10.1371/journal.pone.0047286. doi: 10.1371/journal.pone.0047286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Soehnlein O, Swirski FK. Hypercholesterolemia links hematopoiesis with atherosclerosis. Trends Endocrinol Metab. 2013;24:129–136. doi: 10.1016/j.tem.2012.10.008. doi: 10.1016/j.tem.2012.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ye YX, Calcagno C, Binderup T, et al. Imaging macrophage and hematopoietic progenitor proliferation in atherosclerosis. Circ Res. 2015;117:835–845. doi: 10.1161/CIRCRESAHA.115.307024. doi: 10.1161/CIRCRESAHA.115.307024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Westerterp M, Gourion-Arsiquaud S, Murphy AJ, Shih A, Cremers S, Levine RL, Tall AR, Yvan-Charvet L. Regulation of hematopoietic stem and progenitor cell mobilization by cholesterol efflux pathways. Cell Stem Cell. 2012;11:195–206. doi: 10.1016/j.stem.2012.04.024. doi: 10.1016/j.stem.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tie G, Messina KE, Yan J, Messina JA, Messina LM. Hypercholesterolemia induces oxidant stress that accelerates the ageing of hematopoietic stem cells. J Am Heart Assoc. 2014;3:e000241. doi: 10.1161/JAHA.113.000241. doi: 10.1161/JAHA.113.000241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, Osterreicher CH, Takahashi H, Karin M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell. 2010;140:197–208. doi: 10.1016/j.cell.2009.12.052. doi: 10.1016/j.cell.2009.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Glund S, Krook A. Role of interleukin-6 signalling in glucose and lipid metabolism. Acta Physiol. 2008;192:37–48. doi: 10.1111/j.1748-1716.2007.01779.x. doi: 10.1111/j.1748-1716.2007.01779.x. [DOI] [PubMed] [Google Scholar]
- 39.Murthy S, Mathur S, Bishop WP, Field EJ. Inhibition of apolipoprotein B secretion by IL-6 is mediated by EGF or an EGF-like molecule in CaCo-2 cells. J Lipid Res. 1997;38:206–216. [PubMed] [Google Scholar]
- 40.Westerterp M, Tall AR. SORTILIN: many headed hydra. Circ Res. 2015;116:764–766. doi: 10.1161/CIRCRESAHA.115.306036. doi: 10.1161/CIRCRESAHA.115.306036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Strong A, Ding Q, Edmondson AC, et al. Hepatic sortilin regulates both apolipoprotein B secretion and LDL catabolism. J Clin Invest. 2012;122:2807–2816. doi: 10.1172/JCI63563. doi: 10.1172/JCI63563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kjolby M, Andersen OM, Breiderhoff T, Fjorback AW, Pedersen KM, Madsen P, Jansen P, Heeren J, Willnow TE, Nykjaer A. Sort1, encoded by the cardiovascular risk locus 1p13.3, is a regulator of hepatic lipoprotein export. Cell Metab. 2010;12:213–223. doi: 10.1016/j.cmet.2010.08.006. doi: 10.1016/j.cmet.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 43.Patel KM, Strong A, Tohyama J, Jin X, Morales CR, Billheimer J, Millar J, Kruth H, Rader DJ. Macrophage sortilin promotes LDL uptake, foam cell formation, and atherosclerosis. Circ Res. 2015;116:789–796. doi: 10.1161/CIRCRESAHA.116.305811. doi: 10.1161/CIRCRESAHA.116.305811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ma S, Tian XY, Zhang Y, Mu C, Shen H, Bismuth J, Pownall HJ, Huang Y, Wong WT. E-selectin-targeting delivery of microRNAs by microparticles ameliorates endothelial inflammation and atherosclerosis. Sci Rep. 2016;6:22910. doi: 10.1038/srep22910. doi: 10.1038/srep22910. [DOI] [PMC free article] [PubMed] [Google Scholar]