

Abstract
Structural characterization of proteins in the gas phase is becoming increasingly popular, highlighting the need for a greater understanding of how proteins behave in the absence of solvent. It is clear that charged residues exert significant influence over structures in the gas phase due to strong Coulombic and hydrogen-bonding interactions. The net charge for a gaseous ion is easily identified by mass spectrometry, but the presence of zwitterionic pairs or salt bridges has previously been more difficult to detect. We show that these sites can be revealed by photoinduced electron transfer dissociation, which produces characteristic c and z ions only if zwitterionic species are present. Although previous work on small molecules has shown that zwitterionic pairs are rarely stable in the gas phase, we now demonstrate that charge-separated states are favored in larger molecules. Indeed, we have detected zwitterionic pairs in peptides and proteins where the net charge equals the number of basic sites, requiring additional protonation at nonbasic residues. For example, the small protein ubiquitin can sustain a zwitterionic conformer for all charge states up to 14+, despite having only 13 basic sites. Virtually all of the peptides/proteins examined herein contain zwitterionic sites if both acidic and basic residues are present and the overall charge density is low. This bias in favor of charge-separated states has important consequences for efforts to model gaseous proteins via computational analysis, which should consider not only charge state isomers that include salt bridges but also protonation at nonbasic residues.
Introduction
The existence of zwitterionic states in the gas phase has been a subject of intense interest for both theorists and experimentalists. Early calculations on individual amino acids, starting with glycine, revealed that zwitterionic states, known to be favored in aqueous solution, were not stable in the gas phase.1,2 Later experiments and calculations suggested that zwitterionic configurations for individual amino acids could be stabilized by the presence of an additional charge, though protons did not suffice.3 These and other fundamental studies led to an understanding of critical factors that influence zwitterion stability, including the basicity and acidity of the relevant functional groups, hydrogen bonding, solvation, and the magnitude of Coulomb stabilization gained by clustering oppositely charged ions.4−6 Small-molecule zwitterions can be favored, even in the absence of net charge,7 by increasing the basicity of the basic functional group or by clustering several amino acids together.
Importantly, zwitterion formation in biomolecules is not limited to the canonical amino acid functional group. Indeed, for arginine, it is the highly basic guanidinyl side chain that is crucial for zwitterion formation. When multiple amino acids are combined together into peptides, the relative importance of the N- and C- termini diminishes, and evaluation of zwitterionic states switches emphasis to consideration of the side chains. Zwitterionic pairings between side chains are often referred to as salt bridges.8 In the gas phase, arginine is clearly the most basic residue and is most likely to form salt bridges.4 In peptides and proteins, larger molecular size also leads to increased structural complexity, which obfuscates significantly the questions of zwitterion existence and stability. In order to form a salt bridge, side chains capable of holding charge must first be colocalized, and, second, the energetics must favor charge separation. Evaluation of the first criterion requires structure determination, or at least the generation of potential structures. Due to molecular size, this task is most efficiently carried out by molecular-mechanics-based approaches, which are not typically suitable for evaluating salt bridge stability.9 For most large molecules, searching will lead to a large number of potential structures. Evaluation of the second criterion (i.e., zwitterion stability) requires a higher level of theory.10 Unfortunately, many peptides and certainly all proteins are too large to examine thoroughly with high-level ab initio calculations. This problem is exacerbated by the fact that peptides can adopt a large number of energetically similar conformational states, meaning that high-level calculations would need to be repeated many times.
Experimental identification of salt bridge pairs by mass spectrometry (MS) would simplify calculations, but charge separation does not alter m/z, requiring the combination of MS with other methods. For example, it has been reported that salt bridges can be identified by CO2 loss following excitation with 157 nm light.11 In these experiments, CO2 loss was proposed to occur following electron migration away from a deprotonated acidic group, which would create an easily lost carboxyl radical. This promising approach is complicated by the need for monoisotopic isolation of the precursor ion, which becomes increasingly difficult at higher molecular weights, and by numerous competing fragmentation channels facilitated by high-energy 157 nm photons. Another approach utilizing infrared spectroscopy has been able to identify zwitterionic pairs in dipeptides,12 but the implementation of this approach in larger molecules is not straightforward. Theory has also been utilized to examine salt-bridge stability for a limited set of peptides,10 and although their existence has not typically been demonstrated, salt bridges have been invoked many times in discussions of protein structure in the gas phase.13−17
All of the issues described above are particularly relevant to the emerging area of gas-phase structural biology, which aims to leverage the speed and sensitivity of MS analysis for elucidation of three-dimensional protein structure.18,19 A number of experimental methods are currently being developed with this goal in mind, including ion mobility,20−22 spectroscopy,23,24 energy transfer,25−27 and fragmentation centered approaches.13,28 Although these methods vary widely in many respects, they are all unified by reliance on theory to fill in structural details. In other words, all current MS-based experimental methods fail to directly provide comprehensive atomic coordinates, for which they rely on some type of theoretical calculation. In order for theory to provide structures for comparison with the experimental data, relevant charge state isomers must first be assigned.29,30 This task can already be complicated if the number of basic sites exceeds the number of protons. If acidic residues are present, then consideration of zwitterionic pairs or salt bridges further complicates matters. An experimental method that can reliably identify the presence and location (in terms of sequence) of salt bridges in peptides and proteins is therefore critically needed in order to facilitate reliable structure generation by theoretical methods.
Herein we report that a new intramolecular process we term photoelectron transfer dissociation (PETD) can be used to detect zwitterionic pairs or salt bridges in both peptides and proteins. In some cases, the site of deprotonation can be identified from characteristic fragment ions. In these experiments, electron transfer to a protonated site is initiated by ultraviolet excitation of the electron housed in the anionic portion of the salt bridge. PETD is closely related to electron capture dissociation (ECD) and electron transfer dissociation (ETD),31 and the same characteristic c- and z-type ions are produced. PETD was used to examine several peptides and ubiquitin. In general, most low charge state ions adopt zwitterionic charge configurations, although the highest charge states may not. However, interesting exceptions exist, including the 5+ charge state of beta-insulin, which is zwitterionic despite having only five basic residues. Similarly, high charge states of ubiquitin (>12+) also accommodate salt bridges, though not to the extent observed for lower charge states. These exceptions imply protonation at nonbasic residues is possible even in the presence of a nearby salt bridge and that zwitterionic states are more prevalent than previously imagined.
Experimental Section
The peptides RRLIEDNEYTARG (substrate for tyrosine protein kinase), RPPGFSPFR (bradykinin), YRVRFLAKENVTQDAEDNC (CD36 p93-110), DRVYIHPF (angiotensin II), and KKRAARATS-NH2 (myosin protein kinase) were purchased from American Peptide Company (Sunnyvale, CA, USA). Ac-DRVYIHPFHLLVYS was purchased from Bachem. Bovine ubiquitin, human insulin, and GIGAVLKVLTTGLPALISWIKRKRQQ-NH2 (melittin) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Before use, the disulfides of insulin were reduced and alkylated in the following manner; 5 μL of 1 mM insulin, 5 μL of 50 mM tris(2-carboxyethyl)phosphine (TCEP), and 20 μL of H2O were added to a vial and incubated overnight at room temperature. A manual microtrap from MICHROM Bioresources (Auburn, CA, USA) was used to remove excess salts; the sample was then lyophilized and incubated in an aqueous solution of ammonium acetate with excess iodoacetamide at pH 7.8 in the dark. The resulting reduced and alkylated beta chain was again purified by microtrap, lyophilized, and resuspended for electrospray. RRLIEDDEYTARG was synthesized using standard Fmoc chemistry. For our control experiment, RRLIEDDEYTARG was then methyl esterified by placing it in a vial with methanol and a catalytic amount of HCl while heating to 80 °C for about 1 h. Millipore (18.2 MΩ) water from a Synergy UV water purification system (Billerica, MA, USA) was used for all experiments. HPLC-grade acetonitrile was purchased from Fisher Scientific (Pittsburgh, PA, USA).
Mass Spectrometry and Photodissociation
PETD experiments were performed on a Thermo Fisher Scientific LTQ linear quadrupole ion trap and Thermo Fisher Scientific LTQ-Orbitrap Velos Pro mass spectrometer. The LTQ was modified to accept laser light from a 266 nm Nd:YAG laser (Continuum, Santa Clara, CA, USA), which is triggered by an externally connected delay generator (Berkley Nucleonics Corporation), directly into the ion trap. The LTQ-Orbitrap was also modified to directly accept laser light from a 266 nm Nd:YAG (Continuum), but through a quartz window placed in the back of the HCD vacuum housing (directly into the HCD cell) rather than the ion trap. All peptides were sprayed in 50:50 ACN/H2O at either 5 or 10 μM concentrations at 3 μL/min with electrospray voltages set between 3 and 4 kV and the capillary inlet temperature set to 200 °C. Multishot experiments were performed on the LTQ with a repetition rate of 10 Hz. Ions were given 200 ms postirradiation to cool back to the center of the ion trap before scan out.
Ab Initio Calculations
Theoretical calculations were carried out using density functional theory as implemented in Gaussian09.32 The B3LYP functional was used with the 6-31+G(d) basis set. Transition states were found using the QST3 approach and verified by calculating vibrational frequencies, for which a single imaginary value was obtained.
Molecular Dynamics
Molecular dynamics were performed using MacroModel (Schrodinger Inc., Portland, OR, USA) with the OPLS atomic force field. Simulations were initiated from the ubiquitin crystal structure (1UBQ) and run without solvent for 5 ns in 1.5 fs time steps with 1 ps equilibration time at slightly elevated temperatures (450 K) to simulate the mild heating that occurs during ionization. Charge state isomers were selected by starting with the 1UBQ crystal structure, followed by a simple minimization step, which leads primarily to modest repositioning of surface side-chain groups. Next, acidic functional groups were protonated, starting with those not found in salt bridges and then proceeding with acidic groups in less favorable salt bridges. Acidic residues in potential salt bridges with lysine were protonated first because arginine-based salt bridges have been shown to be more stable.4 However, the Lys11/Glu34 interaction is stabilized by two hydrogen bonds with an optimal distance of 2.89 Å between charge centers, so this zwitterionic pair was retained in all isomers. Additionally, the Asp39/Arg74 interaction exhibits a single hydrogen bond, and the charge centers are extended by ∼0.5 Å beyond the optimal value. Therefore, charge state isomers with deprotonation of the acidic residues were examined for the following pairs (one at a time): Asp39/Arg74, Glu16/Lys33, Glu18/Met1, Glu64/Lys63. In each case, the remaining acidic sites were protonated, yielding a net 6+ charge state in each case. For examination of the 12+ structure, Glu51 and Asp58 were alternately deprotonated due to the nearest sequence proximity to an arginine salt bridge (with Arg54). All basic sites were protonated to yield a net 12+ charge state.
Collision Cross Section Modeling
Cross sections for ubiquitin 12+ were obtained using Collidoscope.33 Briefly, ubiquitin 12+ charge isomer outputs from dynamics simulations were exported as PDB files and run using the CCS algorithm with He buffer gas at 298 K.
Results and Discussion
The original concept for this work began with an investigation of photoinduced electron transfer from noncovalently adducted anions to protonated sites in peptides, and interesting data were obtained for a few systems. Figure 1a shows the results acquired following photoactivation of [RRLIEDNEYTARG+2H+Cl]+ with 266 nm photons. Several interesting features are present in this spectrum. First, backbone fragmentation is dominated by a string of c-type ions accompanied by a single a-type ion. Second, a small amount of charge-augmented doubly protonated peptide is generated. Importantly, this ion is observed only in conjunction with the loss of Cl. These results are best rationalized by a molecular ion composed of a charge-separated system where the doubly protonated peptide forms a salt complex with the chloride anion, yielding a net charge of 1+. Photoinduced electron detachment34 from the chloride anion creates a weakly bound Cl atom that is easily lost, yielding the naked, charge-increased doubly protonated peptide. Alternatively, if the electron is not detached, but instead transferred to a protonated site, fragmentation analogous to traditional ECD/ETD would be expected, explaining the observed sequence of c-type ions. The simultaneous observation of products from electron detachment and electron transfer following excitation at a single wavelength could be rationalized by the presence of multiple structural isomers with differing photodetachment energies. Importantly, loss of Cl from all assignable fragments strongly suggests that it is the source of the electron initiating the fragmentation; that is, the resulting chlorine radical is weakly bound and easily lost.
Figure 1.

PETD spectra for (a) [RRLIEDNEYTARG+2H+Cl]+ and (b) CD36 2+ and CID spectrum for (c) CD36 2+.
To investigate the possibility for detecting internal anionic groups in peptides, we examined YRVRFLAKE-NVTQDAEDNC (CD36 p93-110) in the 2+ charge state. Previous experiments suggested that CD36 exists in a zwitterionic charge configuration with salt bridges between Arg4/Asp15 and Arg2/C-terminus.35 Photoactivation of CD36 is shown in Figure 1b. Notably, a clear series of c ions are observed, as would be expected if an electron were ejected from an anionic carboxyl group and subsequently captured at a protonated residue. Similar types of ions are seen in traditional ETD of CD36 (see Figure S1). Several c-CO2 losses are also noteworthy, and a detailed discussion of these ions is provided further below. In contrast, collision-induced dissociation (CID) of CD36 2+ does not favor backbone cleavage yielding c/z ions; see Figure 1c. Instead, b/y-type ions are produced, consistent with fragmentation via the mobile proton model.36
If observation of c/z ions following photoactivation at 266 nm is a reliable paradigm for identifying zwitterionic pairs, then protonated peptides incapable of sustaining anionic sites should not yield c/z ions upon photoactivation. In order to test this hypothesis, we examined a series of control peptides. Figure 2a shows the results for KKRAARATS-NH2, which lacks any acidic side chains and is amidated at the C-terminus, making the existence of an anionic site on this peptide extremely unlikely. Photoactivation at 266 nm does not yield any c/z ions, consistent with a lack of PETD and absence of zwitterionic charge separation. Data obtained for the larger nonacidic protein, melittin, in the 4+ charge state is shown in Figure S2 (see Supporting Information). A distinct lack of c/z ions is again noted, but fragmentation is observed in the form of a series of b/y ions. In Figure 2b, results are shown for the 3+ charge state of RRLIE*D*D*E*YTARG* (* indicates methyl-esterification), where all acidic groups have been converted into methyl esters. No significant c/z ions are generated. In contrast, if the same peptide is examined with native acidic groups, abundant c/z ions are obtained as shown in Figure 2c. Interestingly, many of the c ions have additionally lost CO2, as was also observed in Figure 1b. Although the loss of CO2 is observed in traditional ECD/ETD as well, typically from the precursor ion,37 this degree of CO2 loss from fragment ions is unusual. A simple explanation can rationalize this observation. Loss of an electron from an acidic side chain or the C-terminus generates a neutral radical predisposed to lose CO2. DFT calculations indicate that barriers to this type of CO2 loss are minimal (see Scheme 1). For example, the transition state for CO2 loss from aspartic acid lies just ∼5.9 kJ/mol above reactants and leads to products −28.1 kJ/mol downhill in energy. Similar results are obtained for the side chain of glutamic acid and the C-terminus (Scheme S1). It should also be noted that we do not observe competing losses of 45 Da from our c-CO2 ions (Figure S3).
Figure 2.

PETD spectra for (a) KKRAARATS-NH2 2+, (b) RRLIE*D*D*E*YATRG* 3+ (* indicates methyl ester), (c) unmodified RRLIEDDEYTARG 3+, and (d) RRLIEDDEYTARG 4+.
Scheme 1. Optimized Structures for Radical and Transition State Preceding Loss of CO2 from Asp.
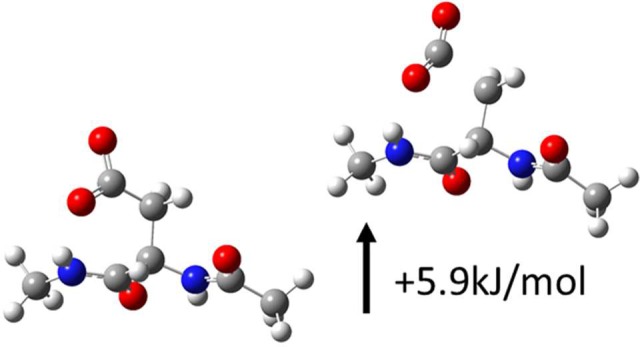
C atoms represented in gray, N in blue, O in red, and H in white.
Taken together, these results suggest that acidic functional groups are required for PETD to occur and that the structure adopted by the 3+ charge state of unmodified RRLIEDNEYTARG contains salt bridges (Figure 2c). A final control is illustrated in Figure 2d, where another proton is added to RRLIEDDEYTARG, leading to complete protonation of all basic sites. Photoactivation of the 4+ ion does lead to fragmentation, though not of the c/z type. Instead, b/y ions are the primary products, consistent with internal conversion of photon energy into vibrational excitation.
The results in Figure 2b and d also serve as controls for the possibility of PETD from a chromophore (rather than an anion). In principle, an excited electron could be transferred from tyrosine to a protonated site, yielding the observed c/z ions. The results in Figure 2b and d illustrate that this does not occur for RRLIEDDEYTARG, where tyrosine is present. The electron affinity of isolated acetate anion is ∼3.2 eV, while the ionization energies for phenol and indole are ∼8.5 and 8.3 eV, respectively.38 Although both the electron affinity and ionization energies could be modified considerably in the context of a protonated peptide, it is likely that the trend will still hold. In other words, it will be easier to transfer an electron from an anion than from a chromophore to initiate PETD. Photons at 266 nm, or 4.6 eV, may not be capable of electron transfer from tyrosine or tryptophan, although it is conceivable that absorption at these chromophores could be followed by energy transfer to anionic sites and help facilitate electron transfer.
Thanks to pioneering work by Williams and co-workers, singly protonated bradykinin is the prototypical gas-phase zwitterion.39 In Figure 3a, photoactivation of bradykinin in the 1+ charge state is shown. Loss of CO2 and phenylalanine side chain are abundant, but a distinct series of c/z ions is absent. Instead, a5–a8, c4, c5, y7, and y8 are observed. Although y ions are typical of CID, the series of a ions and c ions are not. For comparison, the radical-directed dissociation (RDD) spectrum for bradykinin 1+ is shown in Figure 3b. These ions were generated by photolytically cleaving an appended carbon–iodine bond to create a hydrogen-deficient radical, which was subjected to collisional activation.40 RDD typically produces abundant a-type ions with c/z ions and side-chain losses.41 The results in Figure 3a and b are very similar, and both are consistent with RDD. It has been recognized previously that there is an inherent connection between ETD and RDD and that many ETD fragmentation pathways actually occur via hydrogen-deficient chemistry.42 These pathways should be more favorable in PETD because the peptide is never formally hydrogen abundant, as is the case in traditional ETD/ECD. The RDD-like fragments observed in Figure 3a are likely generated by radical chemistry. Fragmentation via hydrogen-deficient pathways following electron transfer is consistent with the results and indicative of the existence of a zwitterionic state.
Figure 3.

PETD of (a) Bradykinin 1+ compared to (b) RDD of 4-iodobenzoic acid-modified bradykinin 1+.
Results for the beta-chain of insulin are shown in Figure 4. Abundant c/z ions are observed in Figure 4a and b for the 3+ and 4+ charge states, suggesting these ions are zwitterionic. Once again, accompanying CO2 loss is abundant; however, in this case, the loss of CO2 from fragment ions can be used to localize the site of deprotonation. For the 3+ ion, a series of c-CO2 ions are observed near the C-terminal end of the peptide, as indicated by the red cleavage marks above each sequence. In contrast, the c ions near the N-terminus do not exhibit loss of CO2. From this information, it can be concluded that the site of deprotonation is one of the two internal glutamic acid residues. For the 4+ charge state, a pair of c-CO2 ions in the center of the peptide are only consistent with deprotonation at Glu13, which is clearly the source of the electron for at least some fraction of the ions. Additional c-CO2 ions are also observed near the C-terminus, which is consistent with deprotonation at either internal glutamic acid residue.
Figure 4.
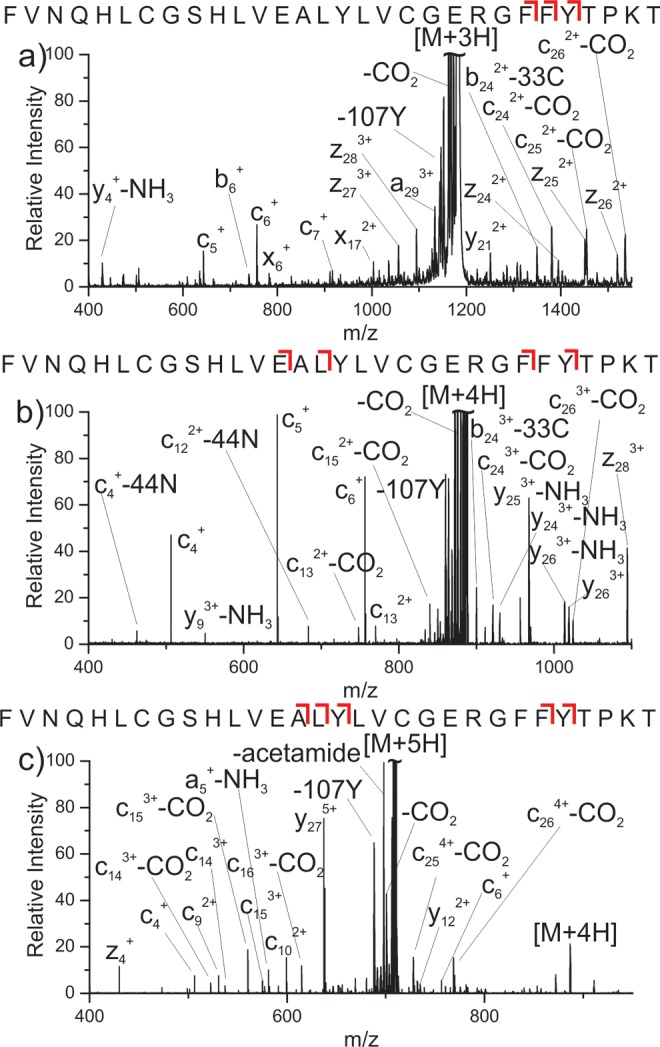
Mass spectra resulting from PETD of insulin (a) 3+, (b) 4+, and (c) 5+. The red brackets indicate c-CO2 fragmentation points relative to the peptide sequence.
Inspection of the 5+ charge state (Figure 4c) reveals an important result. Numerous c/z ions are observed, indicating the 5+ charge state is zwitterionic despite having only five basic sites. This suggests protonation of the peptide backbone is stable despite the presence of a zwitterion. When compared with RRLIEDDEYTARG, the charge density for beta-insulin is much lower (0.31 versus 0.17 net charge/residue, respectively). It is possible that the reduced relative charge density in beta-insulin allows for protonation of the backbone, yet it is very unlikely that the zwitterionic and protonated backbone sites are colocalized. Rather, we hypothesize a stable salt bridge composed of basic and acidic residues in one portion of the structure, with protonation of the backbone accommodated elsewhere. There is ample precedent for protonation of the peptide backbone, under appropriate circumstances, in the supercharging literature.43,44 Inspection of the sequence reveals the most likely site for the persistent salt bridge. Given that the 5+ charge state will experience significant Coulombic repulsion, the proximal Glu21/Arg22 and Glu13/Arg22 pairs are the most likely sites to support a zwitterion.
We expanded the scope of our experiments further by examining ubiquitin, an ∼8.6 kDa protein. The results are shown in Figure 5. Photoexcitation of lower charge states, 5+, 6+, and 7+ (Figure S4), yields numerous c/z ions, consistent with zwitterionic pairs. Several of these ions additionally lose CO2. Ion mobility results have previously revealed that these charge states are compact45 and should be able to accommodate numerous intramolecular interactions, including salt bridges.13,46 Inspection of the crystal structure (PDB 1UBQ)47 reveals ubiquitin has the potential to form several salt bridges among residues on the protein surface. In order to explore whether these interactions might be retained in the gas phase, molecular dynamics calculations were carried out on the 6+ charge state (as detailed in the Experimental Section).
Figure 5.

Summary of PETD results for ubiquitin. An illustrative mass spectrum is shown for the 5+ along with its corresponding c/z fragmentation map directly below. Also shown are the c/z fragmentation maps for the 6+, 12+, and 13+ charge states, with CO2 losses marked by an *.
Backbone structures obtained for several charge state isomers after 5 ns of annealing at 450 K are overlaid with the original crystal structure in Figure 6a. Although the backbone shifts upon desolvation, the primary features, including all beta sheets and alpha helices, are preserved in all conformers. The native-like structures are stabilized by several +-+ salt-bridge arrangements. Such structures are ideal for stabilizing charge separation due to favorable Coulombic terms derived from charge clustering (see Introduction). In fact, an extended +-+-+ salt-bridge cluster between Lys27-Asp52-Arg42-Glu51-Arg72 (see Figure S5) is observed in several isomers, which provides both a net charge to the molecule and extensive structural stabilization. Numerous hydrogen bonds further support this salt-bridge cluster, which serves as an anchor for the native-like state. Extended salt-bridge clusters have also been observed to be consistent with experimental results in previous experiments.13
Figure 6.

(a) Superposition of five ubiquitin 6+ charge isomers obtained with molecular dynamics and the 1UBQ crystal structure. (b) Structures obtained by molecular dynamics for the 12+ ion. All basic sites are protonated, and the lone deprotonated site is indicated.
For higher charge states (see Figure 5 and Supporting Information), c/z ions are increasingly replaced with b/y ions, suggesting increased abundance of nonzwitterionic structure. Nevertheless, a few small c/z-type ions are still observed for the 14+ charge state. Ion mobility indicates the structure of this ion is largely extended,48 but some pairs of basic and acidic residues share very close sequence proximity, for example, 32DKE34 and 51EDGRTLSD58. It is possible that salt bridges in these regions could remain intact even for significantly unfolded structures. Given the greater stability of arginine-based salt bridges, we examined charge isomers for the 12+ ion where all basic sites were protonated and either Glu51, Asp52, or Asp58 was deprotonated. Starting again from the native structure, molecular dynamics were carried out for 15 ns at 450 K (to allow time for more significant structural rearrangements). The protein rapidly unfolds due to Coulombic repulsion, and the lowest energy structures from each run are shown in Figure 6b. The Asp58 charge isomer is nearly completely unfolded, with only a small knot retained at the site of the zwitterion and retention of the N-terminal beta-strand due to solvation of the protonated N-terminus. Our calculated cross section for this ion (2035 Å2)33 does not agree well with experiment (1890 Å2)49 and suggests that the structure for this charge isomer has unfolded too much. Both the Glu51 and Asp52 isomers retain more substantial folded structure, including part of the native α helix. The calculated cross sections (1947 and 1897 Å2, respectively) are also in better agreement with experiment. Interestingly, the cleavage site yielding the z20 ion (i.e., at Leu56) is in close proximity to the zwitterionic site for all three isomers, consistent with the persistent observation of this ion.
The structures in Figure 6b also suggest additional charges can be accommodated without disrupting the salt bridges. The charge density for ubiquitin is low, at 0.18 charge/residue for the 14+ charge state, which is close to the 0.17 charge/residue for the 5+ charge state of beta-insulin. For ubiquitin, it is likely the extra proton is located in the region between Thr12 and Val36, which lacks basic residues and contains four acidic residues and a proline. If the charge state of ubiquitin is driven higher via the addition of supercharging reagents,50,51 no c/z-type ions are observed above 14+ (see Figure S6).
The full list of peptides and charge states examined herein is shown in Table 1. Most of the peptides capable of supporting zwitterionic pairs do so. In three instances, salt bridges are detected when the charge state equals or exceeds the number of basic sites. These results reveal that the propensity to form salt bridges in the gas phase is not restricted to a handful of peptides, but, rather, is likely to be a pervasive characteristic.
Table 1. Summary of Results.
| sequence/protein | zwitterion theoretically possible? | charge states examined | zwitterionic charge states observed | zwitterion at max charge? | charge density of fully protonated sequence |
|---|---|---|---|---|---|
| DRVYIHPF | yes | 1+, 2+ | 1+, 2+ | no | 0.38 |
| RPPGFSPFR | yes | 1+, 2+, 3+ | 1+, 2+ | no | 0.33 |
| KKRAARATS-NH2 | no | 2+ | none | no | 0.56 |
| RRLIEDNEYTARG | yes | 2+, 3+, 4+ | 2+, 3+ | no | 0.31 |
| RRLIEDDEYTARG Methyl ester | no | 3+ | none | no | 0.31 |
| Ac-DRVYIHPFHLLVYS | yes | 2+, 3+ | 2+, 3+ | yes | 0.21 |
| YRVRFLAKENVTQDAEDNC CD36 (93-110) | yes | 2+, 3+, 4+ | 2+, 3+ | no | 0.21 |
| GIGAVLKVLTTGLPALISWIKRKRQQ-NH2 | no | 3+, 4+ | none | no | 0.23 |
| insulin β-chain | yes | 3+, 4+, 5+ | 3+, 4+, 5+ | yes | 0.17 |
| bovine ubiquitin | yes | 5–17+ | 5–14+ | yes | 0.17 |
Conclusions
Our results demonstrate that zwitterionic pairs are common in large gaseous peptides and proteins. Although charge separation is difficult to stabilize in the gas phase at the amino acid level, it appears that the abundance of hydrogen bond partners and opportunities for charge clustering increase the probability for zwitterionic pairs in larger peptides and proteins. Strikingly, strong evidence suggests that salt bridges can be stabilized when the charge state is equal to the number of basic residues if the overall charge density is low and at least one acidic and one basic residue are in close sequence proximity. In these cases, additional protonation at a nonbasic residue must occur to yield the correct net charge for the system. For proteins, it is likely that salt bridges will significantly influence the structure adopted in the gas phase and salt bridges may facilitate kinetic trapping of native-like states. Cumulatively, these findings imply that selection of charge state isomers for theoretical calculations should include not only consideration of zwitterionic pairs but also protonation at nonbasic residues. In summary, zwitterions offer both promise, in the form of stabilizing native-like structures, and complication, due to charge state isomer identification. It is clear that both will impact future progress in gas-phase structural biology.
Acknowledgments
The authors thank the NSF (CHE 1401737) and NIGMS (R01GM107099) for funding.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b02428.
Additional mass spectra, molecular dynamics structures, and DFT calculations (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Ding Y.; Krogh-Jespersen K. Chem. Phys. Lett. 1992, 199, 261. 10.1016/0009-2614(92)80116-S. [DOI] [Google Scholar]
- Jensen J. H.; Gordon M. S. J. Am. Chem. Soc. 1995, 117, 8159. 10.1021/ja00136a013. [DOI] [Google Scholar]
- Jockusch R. A.; Price W. D.; Williams E. R. J. Phys. Chem. A 1999, 103, 9266. 10.1021/jp9931307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julian R. R.; Hodyss R.; Beauchamp J. L. J. Am. Chem. Soc. 2001, 123, 3577. 10.1021/ja003105a. [DOI] [PubMed] [Google Scholar]
- Wyttenbach T.; Witt M.; Bowers M. T. J. Am. Chem. Soc. 2000, 122, 3458. 10.1021/ja992546v. [DOI] [Google Scholar]
- Kamariotis A.; Boyarkin O. V.; Mercier S. R.; Beck R. D.; Bush M. F.; Williams E. R.; Rizzo T. R. J. Am. Chem. Soc. 2006, 128, 905. 10.1021/ja056079v. [DOI] [PubMed] [Google Scholar]
- Julian R. R.; Jarrold M. F. J. Phys. Chem. A 2004, 108, 10861. 10.1021/jp047369l. [DOI] [Google Scholar]
- Kumar S.; Nussinov R. J. Mol. Biol. 1999, 293, 1241. 10.1006/jmbi.1999.3218. [DOI] [PubMed] [Google Scholar]
- Rogalewicz F.; Ohanessian G.; Gresh N. J. Comput. Chem. 2000, 21, 963.. [DOI] [Google Scholar]
- Marchese R.; Grandori R.; Carloni P.; Raugei S. PLoS Comput. Biol. 2010, 6, 1. 10.1371/journal.pcbi.1000775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjeldsen F.; Silivra O. A.; Zubarev R. A. Chem. - Eur. J. 2006, 12, 7920. 10.1002/chem.200600248. [DOI] [PubMed] [Google Scholar]
- Prell J. S.; O’Brien J. T.; Steill J. D.; Oomens J.; Williams E. R. J. Am. Chem. Soc. 2009, 131, 11442. 10.1021/ja901870d. [DOI] [PubMed] [Google Scholar]
- Skinner O. S.; McLafferty F. W.; Breuker K. J. Am. Soc. Mass Spectrom. 2012, 23, 1011. 10.1007/s13361-012-0370-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Julian R. R. Int. J. Mass Spectrom. 2011, 308, 225. 10.1016/j.ijms.2011.07.010. [DOI] [Google Scholar]
- Zhang Z.; Browne S. J.; Vachet R. W. J. Am. Soc. Mass Spectrom. 2014, 25 (4), 604–613. 10.1007/s13361-013-0821-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goth M.; Lermyte F.; Schmitt X. J.; Warnke S.; von Helden G.; Sobott F.; Pagel K. Analyst 2016, 141 (19), 5502–5510. 10.1039/C6AN01377E. [DOI] [PubMed] [Google Scholar]
- Liu Z. J.; Cheng S. J.; Gailie D. R.; Julian R. R. Anal. Chem. 2008, 80 (10), 3846–3852. 10.1021/ac800176u. [DOI] [PubMed] [Google Scholar]
- Benesch J. L.; Ruotolo B. T. Curr. Opin. Struct. Biol. 2011, 21, 641. 10.1016/j.sbi.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heck A. J. R. Nat. Methods 2008, 5, 927. 10.1038/nmeth.1265. [DOI] [PubMed] [Google Scholar]
- Bush M. F.; Hall Z.; Giles K.; Hoyes J.; Robinson C. V.; Ruotolo B. T. Anal. Chem. 2010, 82, 9557. 10.1021/ac1022953. [DOI] [PubMed] [Google Scholar]
- Silveira J. A.; Fort K. L.; Kim D.; Servage K. A.; Pierson N. A.; Clemmer D. E.; Russell D. H. J. Am. Chem. Soc. 2013, 135, 19147. 10.1021/ja4114193. [DOI] [PubMed] [Google Scholar]
- Pierson N. A.; Chen L.; Russell D. H.; Clemmer D. E. J. Am. Chem. Soc. 2013, 135, 3186. 10.1021/ja3114505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagornova N. S.; Rizzo T. R.; Boyarkln O. V. J. Am. Chem. Soc. 2010, 132 (12), 4040. 10.1021/ja910118j. [DOI] [PubMed] [Google Scholar]
- Burke N. L.; DeBlase A. F.; Redwine J. G.; Hopkins J. R.; McLuckey S. A.; Zwier T. S. J. Am. Chem. Soc. 2016, 138, 2849. 10.1021/jacs.6b00093. [DOI] [PubMed] [Google Scholar]
- Hendricks N. G.; Lareau N. M.; Stow S. M.; Mclean J. A.; Julian R. R. J. Am. Chem. Soc. 2015, 136, 13363. 10.1021/ja507215q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly S.; Poussigue F.; Simon A. L.; MacAleese L.; Bertorelle F.; Chirot F.; Antoine R.; Dugourd P. Anal. Chem. 2014, 86, 8798. 10.1021/ac502027y. [DOI] [PubMed] [Google Scholar]
- Czar M. F.; Zosel F.; Konig I.; Nettels D.; Wunderlich B.; Schuler B.; Zarrine-Afsar A.; Jockusch R. A. Anal. Chem. 2015, 87, 7559. 10.1021/acs.analchem.5b01591. [DOI] [PubMed] [Google Scholar]
- Ly T.; Julian R. R. J. Am. Chem. Soc. 2010, 132, 8602. 10.1021/ja910665d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison L. J.; Brodbelt J. S. Analyst 2016, 141, 166. 10.1039/C5AN01819F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnier P. D.; Gross D. S.; Williams E. R. J. Am. Soc. Mass Spectrom. 1995, 6, 1086. 10.1016/1044-0305(95)00532-3. [DOI] [PubMed] [Google Scholar]
- Tureček F.; Julian R. R. Chem. Rev. 2013, 113 (8), 6691. 10.1021/cr400043s. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Petersson G. A.; Nakatsuji H.; Li X.; Caricato M.; Marenich A.; Bloino J.; Janesko B. G.; Gomperts R.; Mennucci B.; Hratchian H. P.; Ortiz J. V.; Izmaylov A. F.; Sonnenberg J. L.; Williams-Young D. F.; Lipparini F.; Egidi F.; Goings J.; Peng B.; Petrone A.; Henderson T.; Ranasinghe D.; Zakrzewski V. G.; Gao J.; Rega N.; Zheng G.; Liang W.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Throssell K.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Keith T.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Millam J. M.; Klene M.; Adamo C.; Cammi R.; Ochterski J. W.; Martin R. L.; Morokuma K.; Farkas O.; Foresman J. B.; Fox D. J.. Gaussian 09; Gaussian, Inc.: Wallingford, CT, 2016.
- Ewing S. A.; Donor M. T.; Wilson J. W.; Prell J. S. J. Am. Soc. Mass Spectrom. 2017, 28 (4), 587–596. 10.1007/s13361-017-1594-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larraillet V.; Vorobyev A.; Brunet C.; Lemoine J.; Tsybin Y. O.; Antoine R.; Dugourd P. J. Am. Soc. Mass Spectrom. 2010, 21, 670. 10.1016/j.jasms.2010.01.015. [DOI] [PubMed] [Google Scholar]
- Hendricks N. G.; Julian R. R. Phys. Chem. Chem. Phys. 2015, 17, 25822. 10.1039/C5CP01617G. [DOI] [PubMed] [Google Scholar]
- Dongre A. R.; Jones J. L.; Somogyi A.; Wysocki V. H. J. Am. Chem. Soc. 1996, 118, 8365. 10.1021/ja9542193. [DOI] [Google Scholar]
- Savitski M. M.; Nielsen M. L.; Zubarev R. A. Anal. Chem. 2007, 79, 2296. 10.1021/ac0619332. [DOI] [PubMed] [Google Scholar]
- Chemistry WebBook, NIST Standard Reference Database Number 69; Linstrom P. J.; Mallard W. G., Eds.; National Institute of Standards and Technology: Gaithersburg, MD, 20899, doi: 10.18434/T4D303. [DOI] [Google Scholar]
- Schnier P. D.; Price W. D.; Jockusch R. A.; Williams E. R. J. Am. Chem. Soc. 1996, 118, 7178. 10.1021/ja9609157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ly T.; Zhang X.; Sun Q.; Moore B.; Tao Y.; Julian R. R. Chem. Commun. 2011, 47, 2835. 10.1039/c0cc03363d. [DOI] [PubMed] [Google Scholar]
- Sun Q.; Nelson H.; Ly T.; Stoltz B. M.; Julian R. R. J. Proteome Res. 2009, 8, 958. 10.1021/pr800592t. [DOI] [PubMed] [Google Scholar]
- Moore B. N.; Ly T.; Julian R. R. J. Am. Chem. Soc. 2011, 133, 6997–7006. 10.1021/ja1096804. [DOI] [PubMed] [Google Scholar]
- Sterling H. J.; Daly M. P.; Feld G. K.; Thoren K. L.; Kintzer A. F.; Krantz B. A.; Williams E. R. J. Am. Soc. Mass Spectrom. 2010, 21, 1762. 10.1016/j.jasms.2010.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomeli S. H.; Yin S.; Ogorzalek Loo R. R.; Loo J. A. J. Am. Soc. Mass Spectrom. 2009, 20, 593. 10.1016/j.jasms.2008.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentine S. J.; Counterman A. E.; Clemmer D. E. J. Am. Soc. Mass Spectrom. 1997, 8 (9), 954–961. 10.1016/S1044-0305(97)00085-8. [DOI] [Google Scholar]
- Zhang Z.; Browne S. J.; Vachet R. W. J. Am. Soc. Mass Spectrom. 2014, 25 (4), 604–613. 10.1007/s13361-013-0821-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijaykumar S.; Bugg C. E.; Cook W. J. J. Mol. Biol. 1987, 194 (3), 531–544. 10.1016/0022-2836(87)90679-6. [DOI] [PubMed] [Google Scholar]
- Purves R. W.; Barnett D. A.; Ells B.; Guevremont R. J. Am. Soc. Mass Spectrom. 2001, 12, 894–901. 10.1016/S1044-0305(01)00272-0. [DOI] [PubMed] [Google Scholar]
- Bush M. F.; Hall Z.; Giles K.; Hoyes J.; Robinson C. V.; Ruotolo B. T. Anal. Chem. 2010, 82 (22), 9557–9565. 10.1021/ac1022953. [DOI] [PubMed] [Google Scholar]
- Sterling H. J.; Daly M. P.; Feld G. K.; Thoren K. L.; Kintzer A. F.; Krantz B. A.; Williams E. R. J. Am. Soc. Mass Spectrom. 2010, 21 (10), 1762–1774. 10.1016/j.jasms.2010.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomeli S. H.; Peng I. X.; Yin S.; Ogorzalek Loo R. R.; Loo J. A. J. Am. Soc. Mass Spectrom. 2010, 21 (1), 127–131. 10.1016/j.jasms.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.