

Abstract
Robust nanostructures have been obtained from otherwise non-assembling polymers using a novel ad hoc electrostatic self-assembly approach. The essence of this strategy involves the use of divalent counterions to temporarily perturb the packing features of ionic groups in a homopolymer, which results in a vesicle-like structure that is captured in situ through a simple crosslinking reaction. The fidelity of the assembly has been tested for molecular transport across the nanomembrane, both for the molecules encapsulated in the lumen and for those trapped in the membrane itself. The membranes are addressable for robust multifunctionalization of their surfaces and for tunable transmembrane molecular transport.
Keywords: Self-assembly, Electrostatic interaction, Programmable assemblies, Homopolymer
COMMUNICATION
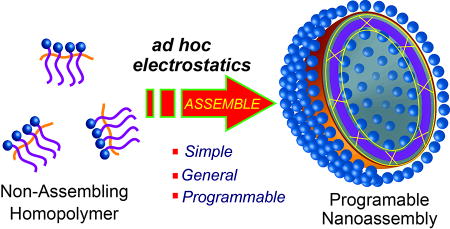
Multivalent counterions can be utilized to change a non-assembling polymer into a programmable nanoassembly with tunable interfacial and transport properties.
Self-assembling nanoscale systems have great potential in a variety of applications including in vitro diagnostics, delivery, nanoreactors and even artificial organelles.[1] While these artificial assemblies have had a significant impact, theories and strategies developed to predictably access such nanostructures are predominantly based on amphiphilic block copolymers.2,3 Here the block copolymers are designed with rationally designed block ratios as the final structure,[2] or more recently, from a propagating block copolymer chain in situ through polymerization induced self-assembly mechanism.[3] Current focus in nanoassembly research has shifted from structure to function, where there is a need for strategies that afford assemblies that are not only structurally robust, but also are functionally programmable.[4] We have been interested in designing nanostructructures with conveniently addressable surfaces, dilution-independent morphological stability, and tunable cross-membrane molecular transport. We stipulated here that the transport barriers that control the cross-membrane molecular transport should be based on covalent sterics, rather than hydrophobicity. We envisioned that such a system would obviate the issues that are typical for assemblies based solely on non-covalent forces, as they are prone to significant morphological changes or even complete disassembly upon dilution, exposure to different surroundings or surface functionalization.[5] The current work describes a simple, robust method to access programmable nanoassemblies, by temporarily perturbing the self-assembly of non-assembling polymers through ad hoc electrostatic interactions.
The basic molecular design premise here is that geometry alterations caused by electrostatic complexation between ionic groups of a polymer and common inorganic counterions, present during self-assembly, can greatly alter the assembly (Scheme 1). This assertion was inspired by the observations that the packing of small molecule surfactants can be significantly altered due to the presence of multivalent counterions.[6] This also should not be confused with polyionic complexes that concerns the co-assembly of two oppositely charged block polyelectrolytes.[7] Although self-assembly of amphiphilic homopolymers itself has been a subject of recent interest and thus reviews,[8] the possibility of directing a non-assembling homopolymer to self-assemble using electrostatic driving forces has not been studied to our knowledge.
Scheme 1.

Schematic illustration of the self-assembly of amphiphilic homopolymers by ad hoc electrostatic interactions using multivalent counterions. Structures of the cationic polymer P1 and anionic polymer P2, used in this study, are also shown here.
To test our design hypothesis, we designed polymers P1 and P2, where each repeat unit contains an ionic hydrophilic group and a crosslinkable functionality. Since the hydrophobicity of the crosslinkable unit is very small, we conceived that these polymers would not self-assemble in the aqueous phase. The polymers were synthesized using an amine-induced ring opening of a g-thiolactone side chain followed by capture of the free thiol using Aldrithiol (see supporting information for details). Indeed, while these polymers do not exhibit any discernible assembly in the aqueous phase, their self-assembly features of both P1 and P2 are drastically affected in the presence of divalent counterions (Na2HPO4 and MgCl2 respectively) (Figure 1a,d). These assemblies were furthered covalently stabilized through a self-crosslinking protocol using a reductive thiol in the form of dithiothreitol (DTT).[9] The fact that the size of the assembly remains the same before and after crosslinking suggests that the crosslinking is predominantly intra-assembly (Figure S1). Interestingly, a morphological evolution was observed in both P1 and P2, along with increasing concentration of Na2HPO4 and MgCl2. Beyond a specific salt concentration, vesicular assemblies are observed, as shown by transmission electron microscopy (TEM) (Figure 1B,E). The the vesicular morphology is further supported by the hollow nature visualized in partially broken particles. The concentration for Na2HPO4 and MgCl2 to induce the vesicle formation from P1 and P2 are 2.0 mM and 3.6 mM (Figure S2). The size of vesicle increases upon increasing salt concentration, which is likely due to salt induced fusion of vesicles indicated by the observation of fused vesicle from TEM (Figure 1E). The wall thickness of vesicles is about 2 nm and no significant change in this thickness was observed at different salt concentrations. Solution size of the assemblies, measured by dynamic light scattering (DLS), correlate very closely with those measured by TEM and by atomic force microscopy (AFM). In addition, the vesicular morphology was also confirmed through scattering experiments. The radius of gyration (Rg) of these assemblies was analyzed using static light scattering (SLS), while their hydrodynamic radius (Rh) was obtained from DLS. Rg/Rh of non-vesicular P2 assembly with hydrodynamic diameter of 50 nm was found to be 1.35 (Figure S3). While Rg/Rh value of P2 assembly with hydrodynamic diameter of 110 nm were found to be 0.99 (Figure 1G), further supporting the vesicular morphology of the assemblies in solution.[10]
Figure 1.

A) Na2HPO4 concentration dependent size of P1 assembly; B) TEM and C) AFM image of P1 assembly in 2.5 mM Na2HPO4; D) MgCl2 concentration dependent size of P2 assembly. e) TEM and f) AFM image of P2 assembly in 4.2 mM MgCl2; g) Rg of P2 assembly in 4.8 mM MgCl2 by SLS. (TEM scale bars are 100 nm).
To further test the basis for the assembly formation and the generality of this phenomenon, P1 was tested in the presence of other divalent anions. The vesicular morphologies of nanoassemblies can be obtained from all divalent salts tested (Figure S4,S5). Similarly, the influence of CaCl2 and BaCl2 on the self-assembly of P2 was also found to be similar to that of MgCl2 (Figure S4,S5). Sizes of ~6 nm were consistently obtained in the presence of monovalent salts and neither the morphology nor the size changed with the salt concentration (Fig S4,S6). It is thus clear that the vesicular morphology formation requires the counterions to be at least divalent, in order for these non-assembling polymers to morph into the observed nanoassemblies.
Next, fluorescent counterions were used to investigate the role of counterions in self-assembly. If fluorescent counterions were bound to the polymer assembly, then the local concentration of the fluorophore around the polymer assembly would be higher than the global solution concentration (Figure S7). The counterions were chosen to report on this feature.[11] For example, the tetra-anionic calcein can be a counterion for P1 and is known to exhibit concentration-dependent self-quenching. Similarly, the excimer fluorescence from pyrene, is used to study P2 by using a pyrene-functionalized lysine molecule as the di-cationic counterion. If these counterions were electrostatically bound to the polymer, the resultant proximity among the probes would be manifested as increased local concentration of the probe molecule to exhibit significant self-quenching for calcein and excimer emission for pyrene. This is indeed the case, as shown in Figure 2a. This complexation-induced probe aggregation was also confirmed by the observation of red-shift in the absorbance spectrum of the probes (Figure 2B). When these fluorescent probes were mixed with the like-charged polymer, neither quenching/excimer formation nor absorbance red-shift was observed (Figure S7).
Figure 2.

A) Effect of complexation of polymers with oppositely charged probes on probe fluorescence; B) absorbance-matching of the fluorescent probes in the presence and absence of the complementary polymers; evolution of the C) absorption and D) fluorescence spectra of calcein upon titrating the calcein-P1 complex with Na2SO4; E) Molecular dynamics (MD) simulations on Mg2+ stabilization of a P2 bilayer; F), MD simulation: Mg2+ gluing action on an intrinsically instable P2 reverse bilayer. Followed by self-assembly energy (ΔEass) in kcal mol−1 as a function of simulation time (values per-monomer).
We then tested the possibility of restoring the fluorescence of these probes in the presence of the divalent ions that induce self-assembly. The hypothesis here is that if the inorganic counterions were to be bound to the polymer, then these should be able to competitively displace the fluorophores into solution, where its normal solution fluorescence would be restored. Indeed, when a P1/calcein mixture was titrated with sulfate ions, a concentration-dependent evolution of calcein’s spectroscopic features (both absorbance and the fluorescence) towards that in bulk solution was observed (Figure 2C, D). To test whether the fluorescent probe is indeed electrostatically bound to the polymer, we investigated the effect of increase in ionic strength of the solution upon the probe fluorescence. Indeed, probe displacement was observed at high ionic strengths, supporting the electrostatic nature of the interaction. The extent of displacement by NaCl is understandably lesser than that by Na2SO4, underscoring the strength of multivalent electrostatic complexation (Figure S8).
To further gain insights into the role of multivalency in self-assembly, molecular dynamics (MD) simulations were used to test the preservation of pre-organized lamellar structures in the presence of divalent and monovalent counterions.[12] MD run on a pre-organized bilayer structure from P2 suggests that it is stabilized by divalent Mg2+, by ~10 kcal mol−1 per-monomer, compared to the monovalent Na+ ions (Figure 2E). Simulations were also carried out on a reverse bilayer configuration with the ions initially fitted in the interspace between the anionic groups of P2. Surprisingly, Mg2+ strongly stabilizes this intrinsically unstable, but necessary, arrangement for mutilamellar structures by gluing the two anionic layers. The stabilization energy from Mg2+ is about 30–40 kcal mol−1 compared to Na+ (Figure 2F). These simulations show that divalent counterions strongly interact with the ionic moieties and that these interactions can substantially modify the polymer conformational flexibility and promote interchain interactions.
Next, we tested the possibility of addressing the outer surface of these nanoassemblies in terms of their functionalization. We envisaged that the crosslinked nature of the assembly should allow for robust surface modification (Figure 3A), which was tested by PEGylation. Successful surface modification was initially ascertained through the change from a positively or negatively charged surface to a charge-neutral surface (Figure 3B). Interestingly, we found that only ~40% of the amines were accessible for the PEGylation in P1 assembly, as quantified through a fluorescamine assay (Figure 3C). This is understandable, since these assemblies are likely multilamellar and thus many amine moieties must be present in the inner leaflets of the assembly, which are likely to be inaccessible. This assertion is supported by the fact the surface of the assembly is neutral, from the zeta potential measurements (Figure 3B).
Figure 3.

A) Schematic representation of addressable surface; B) zeta potential of vesicles upon PEGylation; C) quantification of PEGylation by fluorescamine assay; D) absorbance evolution after DTT treatment.
Next, we examined the possibility of concurrently introducing multiple functional groups on their surfaces using the two independent functionalization handles, one of which is reversible. To examine this, the assembly based on P1 was functionalized with tetramethylrhodamine isothiocyanate (TRITC) and a thiol-terminated fluorescein, using the amine and the PDS moieties in the polymer, respectively. Since fluorescein is attached through a reversible disulfide bond and rhodamine is linked through a thiourea bond, the former fluorophore can be released from the assembly with a thiol-based reducing agent. This was directly demonstrated through the DTT-induced decrease of fluorescein absorbance with no concomittant change in rhodamine absorbance (Figure 3D) The ability to multifunctionalize the membrane endows these assemblies with reversible and irreversible surface modulation features, which should allow for tuning their interfacial properties.
We were interested in assessing whether the assemblies can be programmed to be responsive to a change in the external environment. The crosslinked nature of the membrane provides a physical barrier to limit cross-membrane molecular transport. Since these crosslinks are based on cleavable disuflide bonds, we hypothesized that this barrier can be compromised with a redox trigger. To test this, self-quenching fluorophores (rhodamine-6G and calcein) were non-covalently encapsulated within the vesicles. Leakage of these molecules across the membrane tests its fidelity as a steric barrier for the molecular transport. There was no apparent change in the self-quenched fluorescence over a long time, indicating the membrane provides a strong barrier for molecular diffusion (Figure 4A,B). To test whether this barrier can be compromised using a specific stimulus, we disrupted this barrier using a high concentration of a reducing agent (5 mM DTT) that uncrosslinks the membrane, in both cationic and anionic nanoassemblies. The systematic temporal increase in the fluorescence intensity shows that the membrane barrier is indeed be compromised by the redox trigger. Moreover, the membrane itself can also act as a reservoir to stably encapsulate hydrophobic guests, such as DiI. Release of DiI caused by membrane disruption using reducing agent further confirms that these crosslinked structures provides a steric barrier for molecular release (Figure 4C). The incomplete release of DiI is attributed to the possible association of hydrophobic DiI with the degraded polymer.
Figure 4.

Fluorescence recovery in A) R6G loaded cationic vesicle; B) calcein loaded anionic vesicle; C) DiI release profile; D) Illustration of the need of membrane integrity of vesicle to prevent trypsinization of lysozyme; E) Mass spectra of trypsinized lysozyme fragment (top: lysozyme loaded vesicle middle: lysozyme loaded vesicle incubated with DTT, bottom: free lysozyme).
Finally, formulating biomacromolecules into nanostructures for therapeutic or catalysis application is of great interest, because of their potential utility as a delivery vehicle or a nanoreactor. Lysozyme (MW: 14 kDa, pI 11.4) was chosen as a model protein to be encapsulated in to the cationic vesicle. By forming the assembly in the presence of the protein, it was captured within the assemblies. To test whether the protein is encapsulated in the assembly and thus protected from environmental influences, we subjected the assembly to proteolytic digestion with trypsin. The rationale for this assay is that membrane of vesicle can serve as a barrier preventing trypsin from accessing the encapsulated lysozyme and therefore protect it from being digested (Figure 4D). Once vesicle membrane is disrupted, if the encapsulated lysozyme were to be released to the bulk solution, it will be prone to trypsinization. We monitored these possibilities using matrix-assisted laser desorption ionization mass spectrometry (MALDI-MS). Indeed, the characteristic peaks attributed to peptide fragment from lysozyme were observed, only when the lysozyme-containing vesicles were disrupted by DTT treatment before tryptic digest (Figure 4E). This shows that lysozyme can be successfully encapsulated, protected from its surroundings, and be reductively triggered to release. Similarly, the anionic assemblies was successfully used to encapsulate 3 wt% of myoglobin (Mw: 17 kDa, pI: 7.2) supported by absorbance at 408 nm, which was attributed to the porphyrin moiety in myoglobin (Figure S9).
In summary, we show here that ad hoc electrostatic interactions can be utilized to change a non-assembling polyelectrolyte into a well-defined assembly, which can be readily converted into a programmable nanoassembly. Once constructed, the assembly does not require continued multivalent electrostatic interactions to maintain its fidelity. The nanoassembly can be conveniently reversibly and irreversibly functionalized to produce multi-functional surfaces, which lends itself for robust control over the interfacial properties. Molecular transport across and from the membrane of the nanoassembly is controlled by the steric barriers, which can be compromised on-command in response to a specific trigger. The simplicity and potential generality of this approach facilitate the generation of programmable nanoassemblies with tunable interfacial and transport properties, which could find use in applications such as drug delivery, sensing, nanoreactor design, and self-healing materials. Such utility is exemplified by the encapsulation of enzymes and their triggered release.
Supplementary Material
Acknowledgments
We thank NIGMS of the NIH (GM-065255) and Amy Research Office (W911NF-15-1-0568) for funding. G.M.P. acknowledges the support from the Swiss National Science Foundation (SNSF grant 200021_162827) We thank Michael Leaf for the static light scattering measurements and Meizhe Wang for MALDI measurements.
References
- 1.a) Tanner P, Baumann P, Enea R, Onaca O, Palivan C, Meier W. Acc. Chem. Res. 2011;44:1039–1049. doi: 10.1021/ar200036k. [DOI] [PubMed] [Google Scholar]; b) Marguet M, Bonduelle C, Lecommandoux S. Chem. Soc. Rev. 2013;42:512–529. doi: 10.1039/c2cs35312a. [DOI] [PubMed] [Google Scholar]; c) Discher DE, Ahmed F. Annu. Rev. Biomed. Eng. 2006;8:323–341. doi: 10.1146/annurev.bioeng.8.061505.095838. [DOI] [PubMed] [Google Scholar]; d) Vriezema DM, Garcia PML, Oltra NS, Natzakis NS, Kuiper SM, Nolte RJM, Rowan AE, van Hest JCM. Angew. Chem. Int. Ed. 2007;46:7378–7382. doi: 10.1002/anie.200701125. [DOI] [PubMed] [Google Scholar]; e) Choi H-J, Montemagno CD. Nano Lett. 2005;5:2538–2542. doi: 10.1021/nl051896e. [DOI] [PubMed] [Google Scholar]; f) Ben-Haim N, Broz P, Marsch S, Meier W, Hunziker P. Nano Lett. 2008;8:1368–1373. doi: 10.1021/nl080105g. [DOI] [PubMed] [Google Scholar]
- 2.a) Yu G, Yu W, Shao L, Zhang Z, Chi X, Mao Z, Cao C, Huang F. Adv. Funct. Mater. 2016 doi: 10.1002/ADFM.20161770. [DOI] [Google Scholar]; b) Sun G, Fang H, Cheng C, Lu P, Zhang K, Walker AV, Taylor JA, Wooley KL. ACS Nano. 2009;3:673–681. doi: 10.1021/nn8007977. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Napoli A, Valentini M, Tirelli N, Muller M, Hubbell JA. Nat. Mater. 2004;3:183–189. doi: 10.1038/nmat1081. [DOI] [PubMed] [Google Scholar]; d) Mai Y, Eisenberg A. Chem. Soc. Rev. 2012;41:5969–5985. doi: 10.1039/c2cs35115c. [DOI] [PubMed] [Google Scholar]; e) Zhu Y, Yang B, Chen S, Du J. Prog. Polym. Sci. 2016 http://dx.doi.org/10.1016/j.progpolymsci.2015.05.001.
- 3.a) Derry MJ, Fielding LA, Armes SP. Prog. Polym. Sci. 2016;52:1–18. [Google Scholar]; d) Warren NJ, Mykhaylyk OO, Ryan AJ, Williams M, Doussineau T, Dugourd P, Antoine R, Portale G, Armes SP. J. Am. Chem. Soc. 2015;137:1929–1937. doi: 10.1021/ja511423m. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Azzam T, Eisenberg A. Angew. Chem. Int. Ed. 2006;45:7443–7447. doi: 10.1002/anie.200602897. [DOI] [PubMed] [Google Scholar]; d) Wang W, Tetley L, Uchengbu IF. J. Colloid. Interf. Sci. 2001;237:200–207. doi: 10.1006/jcis.2001.7463. [DOI] [PubMed] [Google Scholar]
- 4.a) Li B, Martin AL, Gillies ER. Chem. Commun. 2007;30:5217–5219. doi: 10.1039/b713569f. [DOI] [PubMed] [Google Scholar]; b) Egli S, Nussbaumer MG, Balasubramanian V, Chami M, Bruns N, Palivan C, Meier W. J. Am. Chem. Soc. 2011;133:4476–4483. doi: 10.1021/ja110275f. [DOI] [PubMed] [Google Scholar]; c) Opsteen JA, Brinkhuis RP, Teeuwen RLM, Lowik DWPM, van Hes JCM. Chem. Commun. 2007;30:3136–3138. doi: 10.1039/b704568a. [DOI] [PubMed] [Google Scholar]; Xu H, Meng F, Zhong Z. J. Mater. Chem. 2009;19:4183–4190. [Google Scholar]; d) Gaitzsch J, Appelhans D, Wang L, Battaglis G, Voit B. Angew. Chem. Int. Ed. 2012;51:4448–4451. doi: 10.1002/anie.201108814. [DOI] [PubMed] [Google Scholar]; e) Wang X, Hu J, Liu G, Tian J, Wang H, Gong M, Liu S. J. Am. Chem. Soc. 2015;137:15262–15275. doi: 10.1021/jacs.5b10127. [DOI] [PubMed] [Google Scholar]; f) Yan Q, Wang J, Yin Y, Yuang J. Angew. Chem. Int. Ed. 2013;52:5070–5073. doi: 10.1002/anie.201300397. [DOI] [PubMed] [Google Scholar]; g) Chi X, Ji X, Xia D, Huang F. J. Am. Chem. Soc. 2015;137:1440–1443. doi: 10.1021/ja512978n. [DOI] [PubMed] [Google Scholar]; h) Chi X, Yu G, Shao L, Chen J, Huang F. J. Am. Chem. Soc. 2016;138:3168–3174. doi: 10.1021/jacs.5b13173. [DOI] [PubMed] [Google Scholar]
- 5.a) Du J, O’Reilly RK. Soft Matter. 2009;5:3544. [Google Scholar]; b) Su L, Wang C, Polzer F, Lu Y, Chen G, Jiang M. ACS Macro Lett. 2014;3:534–539. doi: 10.1021/mz500211v. [DOI] [PubMed] [Google Scholar]; c) Blanazs A, Ryan A, Armes SP. Macromolecules. 2012;45:5099–5107. [Google Scholar]; d) Azagarsamy M, Yesilyurt V, Thayumanavan S. J. Am. Chem. Soc. 2010;132:4550–4551. doi: 10.1021/ja100746d. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Molla MR, Prasad P, Thayumanavan S. J. Am. Chem. Soc. 2015;137:7286. doi: 10.1021/jacs.5b04285. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Sakai R, Nagai A, Tago Y, Sato S, Nishimura Y, Arai T, Satoh T, Kakuchi T. Macromolecules. 2012;45:4122–4127. [Google Scholar]; g) Maiti C, Banerjee R, Maiti S, Dhara D. Langmuir. 2015;31:32–41. doi: 10.1021/la504165e. [DOI] [PubMed] [Google Scholar]; h) Savic R, Azzam T, Eisenberg A, Maysinger D. Langmuir. 2006;22:3570–3578. doi: 10.1021/la0531998. [DOI] [PubMed] [Google Scholar]; i) Allen C, Maysinger D, Eisenberg A. Colloids Surf. B. 1999;16:3–27. [Google Scholar]; k) Versluis F, Tomatsu I, Kehr S, Fregonesse C, Tepper AWJW, Stuart MCA, Ravoo BJ, Koning RI, Kros A. J. Am. Chem. Soc. 2009;131:13186–13187. doi: 10.1021/ja9026264. [DOI] [PubMed] [Google Scholar]
- 6.a) Tian H, Wang D, Xu W, Song A, Hao J. Langmuir. 2013;29:3538–3545. doi: 10.1021/la4003669. [DOI] [PubMed] [Google Scholar]; b) Liu Z, Cao M, Chen Y, Fan Y, Wang D, Xu H, Wang Y. J. Phys. Chem. B. 2016;120:4102–4113. doi: 10.1021/acs.jpcb.6b02897. [DOI] [PubMed] [Google Scholar]; c) Yu D, Huang X, Deng M, Lin Y, Jiang L, Huang J, Wang Y. J. Phys. Chem. B. 2010;114:14955–14964. doi: 10.1021/jp106031d. [DOI] [PubMed] [Google Scholar]; d) Oda R, Huc I, Schmutz M, Candau SJ, Mackintosh FC. Nature. 1999;399:566–569. doi: 10.1038/21154. [DOI] [PubMed] [Google Scholar]; e) Lee H-Y, Hashizaki K, Diehn K, Raghavan RS. Soft Matter. 2013;9:200–207. [Google Scholar]; f) Li G, Zhang S, Wu N, Cheng Y, You J. Adv. Funct. Mater. 2014;24:6204–6209. [Google Scholar]
- 7.a) Anraku Y, Kishimura A, Oba M, Yamasaki Y, Kataoka K. J. Am. Chem. Soc. 2010;132:1631–1636. doi: 10.1021/ja908350e. [DOI] [PubMed] [Google Scholar]; b) Koide A, Kishimura A, Osada K, Jang W-D, Yamasaki Y, Kataoka K. J. Am. Chem. Soc. 2006;128:5988–5989. doi: 10.1021/ja057993r. [DOI] [PubMed] [Google Scholar]
- 8.a) Zhang J, Liu K, Müllen K, Yin M. Chem. Commun. 2015;51:11541–11555. doi: 10.1039/c5cc03016a. [DOI] [PubMed] [Google Scholar]; b) Kale T, Klaikherd A, Popere B, Thayumanavan S. Langmuir. 2009;25:9660–9670. doi: 10.1021/la900734d. [DOI] [PubMed] [Google Scholar]
- 9.a) Ryu J-H, Bickerton S, Zhuang J, Thayumanavan S. Biomacromolecules. 2012;13:1515–1522. doi: 10.1021/bm300201x. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhuang J, Chacko R, Amado Torres DF, Wang H, Thayumanavan S. ACS Macro Lett. 2014;3:1–5. doi: 10.1021/mz400515s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Benoit H, Froehlich D. Light Scattering from Polymer Solutions. Academic Press; London: 1972. [Google Scholar]
- 11.a) Grohn F, Klein K, Brand S. Chem. Eur. J. 2008;14:6866–6869. doi: 10.1002/chem.200800650. [DOI] [PubMed] [Google Scholar]; b) Yildiz UH, Koynov K, Grohn F. Macromol. Chem. Phys. 2009;210:1678–1690. [Google Scholar]
- 12.Detailed information on molecular dynamics simulation is discussed in SI.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.