

Abstract
During development, neural crest cells are induced by signaling events at the neural plate border of all vertebrate embryos. Initially arising within the central nervous system, neural crest cells subsequently undergo an epithelial to mesenchymal transition to migrate into the periphery, where they differentiate into diverse cell types. Here we provide evidence that postnatal human epidermal keratinocytes, in response to FGF2 and IGF1 signals, can be reprogrammed toward a neural crest fate. Genome-wide transcriptome analyses show that keratinocyte-derived neural crest cells are similar to those derived from human embryonic stem cells. Moreover, they give rise in vitro and in vivo to neural crest derivatives such as peripheral neurons, melanocytes, Schwann cells and mesenchymal cells (osteocytes, chondrocytes, adipocytes and smooth muscle). By demonstrating that human KRT14+ keratinocytes can form neural crest cells, even from clones of single cells, our results have important implications in stem cell biology and regenerative medicine.
Keywords: Neural crest, Epidermal keratinocytes, Neural plate border, Neural crest induction, Reprogramming
Graphical abstract
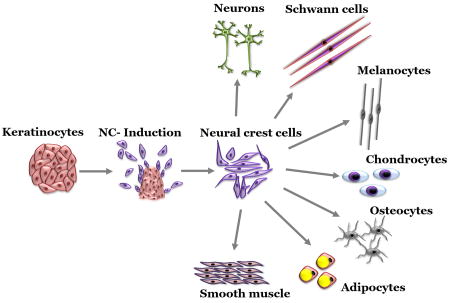
In this study we demonstrate that human epidermal keratinocytes (KC) can be efficiently reprogrammed to acquire neural crest (NC) fate in response to FGF2 and IGF1 signals. Clonal KC gave rise to NC cells suggesting KC possess NC competence at the single cell level. KC derived NC (KC-NC) differentiated into multiple NC derivatives both in vitro and in vivo and were clonally multipotent. This study has significant implications for stem cell biology and regenerative medicine.
INTRODUCTION
In vertebrate embryos, neural crest (NC) cells form important components of the peripheral nervous system, craniofacial skeleton and pigmentation of the skin. Because of their diverse developmental potential, there has been great interest in producing NC cells from human embryonic stem cells (hESC)[1] and induced pluripotent stem cells (iPSC)[2]. In fact, recently it has been shown that introduction of a single transcription factor, SOX10, plus environmental factors was sufficient to reprogram fibroblasts toward a NC fate[3]. However, cell reprogramming typically involves introduction of genes via lentiviral infection[4] with variable transduction efficiency, thereby often requiring selection or sorting of the minority transduced subpopulation. Moreover, virally-mediated genomic integration runs the risk of mutations that may lead to tumorigenesis.
An alternative approach is to use growth factors to reprogram cells in order to recapitulate the embryonic process of NC formation under conditions reflecting the endogenous state. In the embryo, NC cells are induced at the neural plate border by neighboring tissues that provide signals like WNT, FGF, BMP, and NOTCH[5–11]. These imbue the border region with the ability to form NC via an evolutionarily conserved gene regulatory network (GRN)[12]. Newly formed neural crest cells express a characteristic set of transcription factors including FOXD3, PAX3/7, SNAI2, TFAP2A, MSX1/2, c-MYC, and SOX9, which in turn activate the transition to a migratory and multipotent population[5]. For example, the combination of FGF2 and WNT activation or BMP inhibition is sufficient to commit naïve embryonic ectoderm into NC cells [6, 13].
Here we report that human inter-follicular epidermal keratinocytes (KC) - derived from non-neural ectoderm - express several neural plate border genes. Notably, in response to FGF2 and IGF1, KC activate a NC program at the clonal level. NC cells derived from KC (KC-NC) display a global gene expression profile similar to that of human embryonic stem cell derived NC. Under differentiation conditions, clonal KC-NC give rise to functional NC lineages including peripheral neurons, Schwann cells, melanocytes and mesenchymal stem cell derivatives. Moreover, human KC-NC migrate and differentiate into NC derivatives upon implantation into chicken embryos. Our results show that postnatal human epidermal KC can be rapidly and easily reprogrammed into neural crest cells, demonstrating their utility for regenerative medicine and stem cell biology.
MATERIALS & METHODS
Isolation of human inter-follicular epidermal keratinocytes
Human studies were performed as per institutional guidelines. Glabrous (lacking hair follicles) foreskin from 1-3 day old neonates was procured from Women and Children Hospital, Buffalo. Adult (45, 64 and 96 years old) skin (abdomen or thigh) tissues were procured from the School of Medicine, University at Buffalo, SUNY. Keratinocytes (KC) were isolated as described previously[14]. Briefly, skin samples were washed 3 times with PBS, cut into pieces (~5mm×5mm), enzymatically digested with dispase I (Zen-Bio) for 15-20 hours at 4°C. Afterwards, epidermis was separated from dermis manually using fine forceps. The epidermis was further treated with trypsin-EDTA (Life Technologies) for 10-15 min at 37°C, filtered through 70 µm cell strainer (BD Biosciences), centrifuged and plated on a confluent monolayer of growth-arrested 3T3-J2 mouse fibroblast feeder cells in keratinocyte growth medium consisting of a 3:1 mixture of DMEM (high glucose) and Ham's F-12 medium (Life Technologies) supplemented with 10%(v/v) fetal bovine serum (FBS, Gibco), 100 nM cholera toxin (Vibrio Cholerae, Type Inaba 569 B, Calbiochem), 5 µg/ml transferrin (Life Technologies), 0.4 µg/ml hydrocortisone (Sigma), 0.13 U/ml insulin (Sigma), 1.4x10−4 M adenine (Sigma), 2x10−9 M triiodo-L-thyronine thyronine (Sigma), 1x antibiotic-antimycotic (Life Technologies) and 10 ng/ml epidermal growth factor (EGF, BD Biosciences). After 8–10 days of culture, 3T3-J2 feeder layer was detached using versene leaving behind KC colonies. KC colonies were treated with Trypsin-EDTA and then rinsed with serum-free and EGF-free keratinocyte growth medium. KC were further cultured in EpiLife medium (Life Technologies) before using them for NC induction. Passage 1-3 KC were used in all experiments.
Induction of KC into neural crest stem cell fate
For induction into the NC fate, KC were cultured at a density of 10-15×103 cells/ cm2 in collagen type I coated dishes (10µg collagen type I per cm2; BD Biosciences) in the presence of neural crest induction medium (NCIM) comprising of basal medium (EBM-2 medium; Lonza) plus 2% (v/v) FBS, 10 µg per ml heparin, 100 µg per ml ascorbic acid and 0.5 µg per ml hydrocortisone, 1x Gentamicin/Amphotericin-B supplemented with 10 ng/ml fibroblast growth factor 2 (FGF2, BD Biosciences), 10 ng/ml Insulin like growth factor 1 (IGF1, BD Biosciences). Other induction factors and inhibitors tested were EGF, WNT1 (Life Technologies), Chir99021 (Tocris), NRG1 (Sigma), BMP4 (Gibco), and PD173074 (Cayman).
Neural crest induction of KC clones
To ensure each KC colony was derived from a single cell, we plated 0.5 cells per 100 µl KC growth medium per well of a 96 well plate on 3T3-J2 feeder cells. Wells with more than one colonies were excluded. Total 174 single cells KC clones from three different donors grew and all the clones were used for NC induction treatment irrespective of their colony size.
Differentiation of KC-NC into NC derivatives
For peripheral neuronal differentiation, after 10 day of induction KC-NC were cultured on poly-L ornithine (Sigma) (2 µg/cm2)/ laminin (Life Technology) (5 µg/cm2) coated dishes in presence of NCIM plus 1 ng/ml of transforming growth factor-beta 1 (TGF-β1, Biolegend) for 24 hours followed by one week culture in neuronal differentiation media (EBM2 basal medium, 1% (v/v) FBS, 50 ng/ml brain derived neurotrophic factor (BDNF, Peprotech), 50 ng/ml glial derived nerve factor (GDNF, Peprotech), 200 ng/ml nerve growth factor (NGF, Peprotech), 20 ng/ml neurotrophin 3 (NT3, Peprotech), 0.5µM N(6),2'-O-dibutyryladenosine 3':5' cyclic monophosphate (dbcAMP) (Sigma), 0.5x Glutamax, (Life Technology)).
For melanocyte differentiation, after 7 days of NC induction treatment, KC-NC were cultured in melanocyte differentiation medium (EBM2 basal medium, 5% (v/v) FBS, 100 ng per ml SCF (Life Technology), 200 nM Endothelin 3 (EDN3, Sigma), 50 ng/ml WNT1, 10 ng/ml FGF2, 5µg/ml Insulin, 1 pM cholera toxin, 10 nM 12-O-tetra-decanoylphorbol-13-acetate (TPA, Sigma) and 10µM SB431542 (Cayman) for 5 weeks. For L-DOPA assay, KC-NC derived melanocytes were fixed with 4% (w/v) paraformaldehyde for 20 min at room temperature, washed three times with PBS and incubated with freshly prepared 5 mM L-DOPA (Sigma) overnight at 37°C. After incubation, cells were post-fixed with 4% paraformaldehyde for 20 min at room temperature, washed with PBS and visualized for melanin pigment under bright field microscopy. Primary human melanocytes used as control were isolated from foreskin tissues using previously described protocols [14] and cultured in medium M254 (Thermo Fisher Scientific) supplemented with human melanocyte growth supplement (HMGS) (Thermo Fisher Scientific).
For Schwann cell differentiation, after 7 days of induction treatment, KC-NC were plated on poly-L ornithine/laminin coated dishes and cultured in Schwann cell differentiation medium (EBM2 basal medium, 2% (v/v) FBS, 100ng/ml ciliary neurotrophic factor (Life Technologies), 100 ng/ml NRG1, 4 ng/ ml FGF2, 200µg/ ml ascorbic acid, 0.5x Glutamax, 10µM SB431542 for 5 weeks.
For mesenchymal differentiation, after 10 days of induction KC-NC were transferred to mesenchymal growth medium (MGM) comprising of DMEM plus 10% (v/v) mesenchymal stem cell qualified-FBS (MSC-FBS, Life Technologies) for 3 weeks. At this point, KC-NC were analyzed for MSC immunophenotype and induced into mesenchymal lineages by lineage specific differentiation medium[15]. Bone marrow derived mesenchymal stem cells (Stem Cell technologies) were cultured in DMEM plus 10% (v/v) MSC-FBS and used as positive control. Osteogenic, chondrogenic and adipogenic differentiation was induced and functionally analyzed as described previously[15]. Smooth muscle cell (SMC) differentiation was induced by MGM supplemented with 10 ng/ml TGF-β1 for one week.
Clonal multipotency of KC-NC
Single cell KC-NC were plated in 96 well plate by limiting dilution. Each well was examined microscopically to ensure that only one cell gave rise to the clone. We derived 6 KC-NC clones per donor from 4 different neonatal donors (total 24 KC-NC clones). Each clone was expanded in NCIM and split into 24 wells of a 96 well plate for differentiation experiments. Six wells were used for differentiation into each of the following lineages: smooth muscle cells (SMC), melanocytes (Mel), Schwann cells (SC) and neurons (N). Each lineage was evaluated by immunostaining using lineage specific marker antibodies.
Gel compaction assay and vasoreactivity assay for testing KC-NC derived smooth muscle cell function
Gel compaction assay measures the ability of SMC to generate force and compact fibrin hydrogels. To this end, 1×106 KC-NC derived SMC or control human aortic smooth muscle cells (ASMC, Life Technologies) were mixed with 0.8 ml of fibrinogen (Enzyme Research Laboratories) that was polymerized by addition of 0.2 ml thrombin (Sigma) in a 24-well plate at 37°C for 1 hr. The final concentration of fibrinogen and thrombin in the fibrin hydrogel was 2.5mg/ml and 2.5 U/ml, respectively. The resulting KC-NC derived SMC constructs were incubated in MGM supplemented with 2ng/ml TGF-β1. After 1 hr incubation, the fibrin gels were released from the surface of the plate to enable gel compaction, an indicator of SMC contractile function. At indicated times (0, 24, 48, 72, and 96 hr) images were acquired using a EC3 imaging system (UVP) and the area of each hydrogel (A) was measured using Image J software and normalized to the initial hydrogel area (A0). The results were plotted as the ratio A/A0 and therefore, the smaller the area the higher the contractile function of KC-NC derived SMC.
Vasoreactivity assay was performed as described previously[15]. Towards this end, fibrin hydrogels (final concentration of fibrinogen and thrombin at 2.5 mg/ml and 2.5 U/ml, respectively) containing 1×106 KC-NC derived SMC or control ASMC were polymerized around a 6.0 mm diameter mandrel of poly(di-methyl siloxane). After 1 hr, hydrogels were detached from the walls and incubated in a medium comprising of MGM plus 2 µg/ml insulin, 2 ng/ml TGF-β1, 300 µM ascorbic acid phosphate (Alfa Aesar) and 2 mg/ml ε-amino-n-caproic acid (Sigma) for 2 weeks. After two weeks, compacted hydrogels were released from the mandrels and mounted on an isolated tissue bath containing Krebs–Ringer solution at 37°C. Each hydrogel was mounted on two hooks through the lumen; one hook was fixed and the other was connected to a force transducer. Hydrogels were equilibrated at a basal tension of 1.0 g and constant length for approximately 1 hr. After equilibration, vasocontractile agonists (endothelin-1 (20 nM) (Sigma), the thromboxane A2 mimetic U46619 (1 µM, Sigma) or potassium chloride (KCl; 118 mM, Sigma) were added to the tissue bath and isometric contractions were recorded using a PowerLab data acquisition unit and analyzed with Chart5 software (ADInstruments). To determine the mechanical properties (Young’s modulus (YM) and ultimate tensile stress (UTS)) of the hydrogels, samples were incrementally stretched until they broke using the Instron 3343 (Instron). Contractility and UTS/YM were normalized to the cross-sectional area of the hydrogels and expressed in Pa and kPa, respectively.
Electrophysiological measurements
KC-NC derived neurons were differentiated on coverslips and transferred into a perfusion chamber with a solution containing the following components (in mM): 140 NaCl, 4 KCl, 2 CaCl22 MgCl210 HEPES, 10 D-glucose, 10 sucrose (pH 7.4 with NaOH)) for electrophysiological recordings. To measure action potentials, pipettes were fire-polished and filled with an intracellular solution containing (in mM): 135 K-gluconate, 7.3 KCl, 10 phosphocreatine, 10 HEPES, 2 MgATP, 0.3 Na2GTP (pH 7.3 with KOH; 300 mOsm) with a final resistance of 1-2 MΩ. Following the formation of a GΩ seal and establishment of electrical access to the cell, action potentials were measured either by applying a very brief (1-2 ms) but large (0.8 nA) current injection or by injecting current for a more prolonged amount of time (200 ms) with each subsequent recording increasing by 50 pA until an action potential was fired. To verify the involvement of voltage-gated Na+ channels which are required for action potential formation, tetrodotoxin (TTX, 1 µM) was applied and allowed to diffuse before recording. To measure current through Nav and Kv channels, voltage-clamp was employed. Pipettes and recording solutions were identical to those used to measure action potentials. Following formation of a GΩ seal, cells were electrically accessed through the application of negative suction pressure. Pipette capacitance and series resistance compensation were performed according to standard procedure. Cells were held at hyperpolarized potentials (−90 mV) and stepped to various voltages (Δ10 mV; 200 ms) following a step back to the hyperpolarized potential for a sufficient amount of time (5 s) to allow for recovery from inactivation. Nav and Kv channel currents were blocked using 1µm TTX and 40mM TEA, respectively. Following acquisition of data from steps to −100 to +50 mV, traces were analyzed using pClamp (Molecular Devices) and displayed as current/voltage plot.
Quantitative real time PCR
At the indicated times total RNA was isolated using RNeasy kit (Qiagen) as per manufacturer’s instructions. First strand cDNA was synthesized from 1 µg of total RNA using QuantiTect kit (Qiagen). To determine the kinetics of gene expression during differentiation, quantitative real time PCR was performed on the Bio-Rad CFX96 Real-Time PCR detection system with the SYBR Green mix (Bio-Rad) according to manufacturer’s instructions. Primers information is given in supplementary table 1. KC (day 0, before start of induction treatment) were included as a calibrator in our qRT-PCR analysis. The level of each mRNA was quantified using the ΔΔCT method and normalized with the expression level of the housekeeping gene, 18sRNA. The specificity of each product was verified by melt curve analysis and gel electrophoresis.
RNA sequencing
KC and KC-NC were characterized in terms of their global expression profile by next generation RNA sequencing using Illumina platform. To this end, total RNA was isolated from both KC (n=3) and the respective KC-NC (n=3) and quality control analysis was performed by RNA gel and Agilent Bio-analyzer. Sequencing libraries were prepared as per standard Illumina protocols and sequenced in pair-end (2x50 bp) rapid run mode. After quality control of raw data using Illumina pipeline, RNA-Seq reads were mapped to the human genome (hg19) using Tophat (version 2.0.11) with default settings. The mapping quality was examined by RSeQC 2.3.9. Then the read counts for each gene in the accepted hits are counted by the R package qRNASeq. For each gene, total reads within the exon regions were counted and overlapped regions from different transcripts were united. The differentially expressed genes between KC and KC-NC were detected by the R package DESeq2, using a design formula considering patient variability. Hierarchical clustering and 3D multi-dimensional plot (3D MDS) were generated in R using hclust() and cmdscale functions, respectively. Heat maps were generated using the ‘heatmap.plus’ function in R. The KC-NC gene expression profile was compared to that of human embryonic stem cell derived neural crest cell signatures reported in the literature [1, 16] as well as KC, using ROCR and verification libraries in R Bioconductor (v3.1.3). RNA sequencing data is available on the NCBI Gene Expression Omnibus (GEO) and accessible through GEO Series accession number GSE72268.
SOX10 promoter methylation assay
SOX10 CpG Island (Chr22: 38379093-38379964 (hg19), containing 82 CpG) whose methylation status inversely correlates with SOX10 transcription [17] was examined by MethylScreen technology[18], using the Epitect methyl II PCR assay (catalog# EPHS109833-1A, Qiagen) as per manufacturer's instructions (EpiTect Methyl II PCR Assay Handbook – Qiagen). Genomic DNA was isolated from KC and the corresponding KC-NC (n=5 donors) using PureLink genomic DNA isolation kit (Life Technologies). One µg of genomic DNA from both KC and the respective KC-NC was mock-digested (no enzyme) or digested with methylation-dependent (digests methylated DNA) and methylation-sensitive (digests unmethylated and partially methylated DNA) restriction enzymes (provided with EpiTect Methyl II DNA Restriction Kit (Qiagen)) individually or together. Methylation status of the target sequence was measured using quantitative real time PCR with target sequence specific probes. The raw ∆CT values from all 4 restriction digestion groups were plugged in the data analysis spreadsheet (http://www.sabiosciences.com/dna_methylation_data_analysis.php), which automatically calculates the relative amount of methylated and unmethylated DNA fractions.
Immunocytochemistry and flow cytometry
Cells were fixed (4% (w/v) paraformaldehyde), permeabilized (PBS with 0.1% (v/v) triton X-100, Sigma), blocked (5% (v/v) normal goat serum in PBS, Life Tech or BlokHen solution (Aves labs)) and incubated with primary antibodies (Supplementary Table 2) followed by appropriate secondary antibodies conjugated with Alexa 488 or Alexa 594. Hoechst 33342 (Thermo Fisher Scientific) was used for nuclear staining. Cells that were incubated with only secondary antibody served as controls. For flow cytometry detection of KRT14, SOX10, NES and PMEL, cells were fixed with 4% (w/v) paraformaldehyde, permeabilized with 0.1% (v/v) triton X-100, and incubated with the respective primary antibodies (Supplementary Table 2). Alexa 488 or Alexa 647-R conjugated secondary antibodies were used. For all surface proteins, cells were detached with 5mM EDTA and stained with appropriate primary and secondary antibodies. Cells were run in a FACS Calibur (BD) or CytoFLEX (Beckman Coulter) flow cytometer and the data were analyzed using CellQuest software (BD) or FlowJo (FlowJo LLC), respectively.
Imaging and Image analysis
Images were acquired using a Zeiss Axio Observer Z1 inverted microscope with an ORCA-ER CCD camera (Hamamatsu, Japan). Fluorescence intensity quantification was performed using NIH ImageJ as described previously[19]. Briefly, cells from all time points were stained in one batch and images were acquired using fixed exposure time for each fluorescent secondary antibody. Corrected total cell fluorescence intensity (CTCF) was calculated as per following formula:
In ovo transplantations
To this end, KC and the respective KC-NC were transduced with lentivirus containing minimal CMV promoter driven EGFP reporter. Approximately 50-60% cells were transduced by the lentivirus as assessed by examining EGFP+ cells under fluorescence. KC-NC (n=8) or KC (n=3) were trypsinized and ≈ 30-60 cells per embryo were transplanted in ovo onto the head mesenchyme to join the migrating neural crest cells of 10-13 somite stage chick embryos (Fig. 5B). The eggs were sealed, kept humidified by adding Ringer’s balanced salt solution once a day, and fixed 2-3 days later in 4% (v/v) paraformaldehyde overnight at 4°C and washed 2x with PBS. Finally, the embryos were embedded in 15% (w/v) sucrose / 30% (w/v) gelatin in PBS and cryosectioned (12µm sections), and de-gelatinized by incubation in 42°C PBS for 1.5h prior to incubation in blocking buffer (5% (v/v) donkey, 5% (v/v) goat serum, 1% (v/v) DMSO in PBS-0.1% (v/v) tween 20) and mounting. First, the slides were screened under the microscope to look for EGFP+ cells and the appropriate slides were marked. The EGFP+ cells were distinguished from the highly autofluorescent blood cells found abundantly in the capillaries in the mesenchyme by checking their fluorescence additionally on the red and blue channels, which makes the blood cells to look white in the images (see Fig. 5C). Depending on the location of the EGFP+ cells, the marked sections were decoverslipped (PBST treatment for 1-2 days) and stained with antibodies accordingly. The following primary antibodies were used (overnight, 4°C): BLBP (ABN14 Millipore, 1:200), SMA (A5228 Sigma, 1:1000), Tuj-1 (Covance MMS-435P, 1:400), GFAP (SMI22 Sternberger Monoclonals, Covance 1:800) and human nuclear antibody (MAB 1281 Millipore, 1:100). Subsequently the following Alexa Fluor conjugated secondary antibodies (overnight, 4°C were used: 488 donkey anti-rabbit, 647 donkey anti-rabbit, 568 goat anti-mouse IGg2a, 633 goat anti-mouse IGg2a (Molecular Probes).
Figure 5. KC-NC migrate and differentiate into neural crest lineages in ovo.

KC-NC migrate and differentiate into neural crest lineages in ovo as shown in 12µm transverse sections of 2-3 days old chicken embryos. Percentages of transplanted cells detected in each target structure in the developing chick embryos (n=8 embryos; total number of detected EGFP+ cells = 151 out of ~ 400 transplanted cells) (A). The EGFP+ KC-NC or KC were transplanted to the head mesenchyme (hm) of 10-13 somite host chick embryos on Day 0 and the cells were traced 2-3 days post transplantation (B). A Tuj/EGFP double positive neuron in the trigeminal ganglion (TGG) (day 3). The transplanted EGFP+ cells are not visible on other channels and thus are not to be mixed with highly autofluorescent blood cells (BC) that are found throughout the mesenchyme in vessels and small capillaries; merged image with red, blue and green fluorescence makes the blood cells look white (C). BLBP+/EGFP+ double positive glial cells in the TGG (day 3) (D). EGFP+ putative Schwann cells localized around a nerve bundle at the outer edge of a cranial ganglion (day 3) (E). A cranial blood vessel surrounded by αSMA+ cells with one of them originating from the transplanted cells as indicated by co-expression of the human specific nuclear marker (day 2) (F). Differentiating EGFP+ putative melanocytes were detected under the cranial ectoderm (day 3). Blood cells are highly autofluorescent on both green and blue channels (light blue) (G). Scale bar 50 µm. hm= head mesenchyme, nt=neural tube, som= somites, LV=lateral ventricle, TGG=trigeminal ganglion, BC=blood cells.
Clonogenic assay and population doubling
Clonogenic assay was performed as described previously[15]. Briefly, KC-NC were seeded (10 cells/cm2) in a 100 mm culture dish and cultured for one week in NCIM. Afterwards, plates were fixed with a solution of methanol and acetic acid (3:1 v/v), stained with trypan blue and photographed. Images were analyzed using ImageJ to determine the area and effective diameter of each clone. For proliferation studies, 40,000 KC-NC were seeded in culture plates in triplicates or quadruplicates and grown in NCIM. Every 3 days, the cells were counted and the doubling time and cumulative cell numbers were calculated assuming geometric growth.
Immunoblotting
Immunoblotting on KC and the respective KC-NC protein lysate for CDH1 (E-cadherin) and CDH2 (N-cadherin) was done as described previously[15].
Statistical analysis
Experiments were performed with KC from at least 3 human donors (range: 3–30 human donors) and each experiment was repeated at least three times. Data were expressed as mean ± standard deviation except electrophysiological data, which is mean ± SEM. Comparison among groups were performed using one-way ANOVA followed by post-hoc analysis using Tukey’s HSD test or two-tailed t-test. Wilcoxon rank-sum tests were performed for paired comparisons. Statistical significance was defined as p < 0.05.
RESULTS
Human Keratinocytes (KC) display neural plate border characteristics
An adult neural crest population has been reported to be present in hair follicle’s bulge region[20]. In addition, skin derived precursors (SKPs) having similar characteristics as neural crest cells have been derived from mouse[21] and human dermis[22]. To avoid possible contamination from bulge neural crest, we isolated human interfollicular[23] epidermal KC from glabrous (lacking hair follicles) foreskins (1-3 day old neonates). Furthermore, we manually separated epidermis from dermis after enzymatic tissue digestion to avoid the presence of dermal SKPs in our cultures. KC on 3T3-J2 feeder layer grow as distinct compact colonies of cuboidal cells. Versene treatment and multiple subsequent PBS washes removed the 3T3-J2 cells and any melanocytes that might be present on top of the feeder cells (Fig.S1A). Indeed, we confirmed the purity of our KC population by performing immunocytochemistry and flow cytometry analysis for melanocyte markers (n=3 donors). As shown by flow cytometry and immunostaining, primary human foreskin derived melanocytes express the melanocyte marker PMEL (also known as SILV or HMB45) and lack KRT14 (a KC marker; Fig.S1B,C). In contrast, KC lack PMEL (and MITF) and express KRT14 (Fig.S1B,C). Finally, single cell derived clonal cultures of KC were established to ensure that the starting cell population in our experiments is bona fide KC.
All experiments were done with KC from at least three human donors (range: 3-30 donors) and each experiment was repeated at least three times. Passage 1 to 3 KC were used for all experiments. Human foreskin KC expressed type I intermediate filament KRT14 (99.6% cells), TP63 (97.9%), ITGB1 (99.7%) with variable immunofluorescence intensity (n=3 donors; Fig.S2A), indicative of basal/transit amplifying KC. When examined for the presence of genes at the embryonic neural plate border, we found that KC expressed several neural crest and neural plate border markers (MYC, SOX9, SNAI2 MSX2, IRX2, DLX3, TFAP2A , and KLF4) when compared with human dermal fibroblasts that were of mesodermal origin (n=3 donors) from the same donor and used as a control (Fig.S2B). Immunostaining revealed that 81.8% of KRT14+ KC were positive for SNAI2, 60% were MYC+ and 96.7% were TFAP2A+ (n=3 donors; Fig.S2A). We further examined adult human KC for the expression of neural plate (NP) border markers. To this end, we isolated abdominal or thigh skin KC from three human donors (45, 64 and 96 years old). qRT-PCR for MYC, SOX9, SNAI2 MSX2, IRX2, DLX3, TFAP2A and KLF4 (Fig.S3A) and immunostaining for TFAP2A, SNAI2 and KLF4 (Fig.S3B) showed that adult KC express NP border markers, albeit at lower level compared to neonatal KC. The enrichment of several neural plate border markers in KC led to the intriguing possibility that they might be reprogrammed to a NC fate under appropriate environmental conditions.
Reprogramming of KC into NC cells
To identify factors that may induce NC fate in human KC, we tested signaling pathways previously implicated in neural crest formation[24–26] using FGF2, WNT1, WNT activator Chir99021, EGF, IGF1, BMP4, NRG1 and combinations thereof[5, 6]. We found that FGF2 significantly promoted NC fate as evidenced by SOX10 and NES (Nestin) dual positive KC-NC cells (1,313±394 cells out of 3,000 plated cells; p=1.08x10−8, n=3 donors) after 6 days of induction treatment (Fig.1B). NES is an intermediate filament protein that is expressed both in CNS progenitors and neural crest stem cells [20, 27]. This induction was blocked by FGFR chemical inhibitor PD173074, yielding a drastic decrease in SOX10+NES+ dual positive cells (p=0.0013, n=3 donors; Fig.1C). The combination of FGF2 and IGF1 further potentiated the induction process as shown by significant increase in SOX10+NES+ cells compared to FGF2 alone (p=2.39x10−6, n=3 donors; Fig.1D). It also led to significant increase in expression of neural crest genes (SOX10, NES, PAX3, FOXD3, NGFR and B3GAT1), EMT genes (TWIST1) and the cell proliferation marker, KI67 (n=3 donors; Fig.S2C). As NC migration is influenced by the extracellular matrix environment[28], we tested the effects of different ECM coatings (fibronectin, laminin, collagen type 1) to promote NC fate. All three ECM molecules supported expression of NC genes (NGFR, PAX3, SOX10, and NES) to a similar extent (Fig.S2D). However, both fibronectin and collagen type I showed similar levels of SOX+NES+ cells (p=0.68, n=3 donors; Fig.S2E), which were significantly higher than that observed on laminin-coated or non-coated surfaces (fibronectin: 2.9±0.42 fold, collagen type I: 2.8±0.13 fold, p=0.004, n=3 donors; Fig.S2E).
Figure 1. Keratinocytes induction into neural crest fate.

Scheme of NC induction of KC (A). FGF2 significantly promotes NC induction of KC as shown by increase in SOX10 and NES dual positive (SOX10+NES+) cells (n=3 donors, *p < 0.001) (B). KC to NC induction is blocked by FGFR inhibitor PD173074 (n=3 donors, #p < 0.01) (C). IGF1 potentiates KC to NC induction as shown by increase in SOX10+NES+ cells (n=3 donors, *p < 0.001 and $p < 0.05) (D). Phase images of KC to NC induction process; after 3 days of induction treatment, small spindle shaped cells migrated outwards from KC clusters and proliferated, while newer cells continued to delaminate from the KC clusters (n= 30 donors) (E). Emigrated KC-NC cells reduced KRT14 expression and upregulated NES as determined by immunofluorescence (F) and flow cytometry (G). KC-NC expressed SOX10, NGFR, FOXD3 and PAX3 as confirmed by immunofluorescence (H, J) and by flow cytometry for SOX10 and NGFR (I). KC possess NC competence at the clonal level as single KC derived clones gave rise to KRT14 negative and NES positive NC cells. A representative KC clone is shown (KLM). Percentage of KC clones (from 3 donors) that gave rise to KC-NC (N). Scale bars, 50µm (except for Ki, which indicate 400µm). All values are mean±SD. Experiments were repeated three times independently.
Cultured KC assumed typical cuboidal morphology in low calcium (0.06mM) serum free medium (Fig.1E). Upon induction with FGF2 and IGF1 in high calcium (1.8mM) medium, KC formed compact cell clusters (n=30 donors; Fig.1E). As early as day 3, small spindle shaped cells started to delaminate from KC clusters, migrated outwards and started to proliferate, while newer cells continued to delaminate from the KC clusters. Delaminated cells detached from the surface by trypsin/EDTA treatment much faster (~1 min) than KC clusters. Thus using short-term (~1 min) trypsin/EDTA treatment, we were able to separate and collect delaminating cells from KC clusters, which remained intact. When analyzed for the presence of NC markers, the cells that delaminated from KC clusters displayed molecular characteristics of NC cells and thus termed KC derived NC cells (KC-NC). KC-NC were highly proliferative as evidenced by a short population doubling time of ~19.6 hr (n=2 donors; Fig.S2F) and high clonogenic ability (n=3 donors; Fig.S2G). KC-NC could be maintained for 20 days without loss of proliferation potential. KC-NC downregulated KRT14 and upregulated NES protein expression as determined by immunofluorescence (n=6 donors; Fig.1F) and flow cytometry (KC, 99.4±0.26% KRT14+, 0% NES+; KC-NC, 0% KRT14+, 99.6±0.2% NES+;n=3 donors; Fig.1G). Notably, KC-NC were uniformly positive for SOX10 (98.3±0.4%), while 75.4±1.18% were positive for NGFR (p75NTR) as determined by immunofluorescence (n=3 donors; Fig.1H) and flow cytometry (n=4 donors, Fig.1I). In addition, KC-NC expressed other key NC genes such as PAX3 (93.5±3.46%), FOXD3 (90.1±3.58%) and KIT (96.8±1.04%) (n=3-6 donors; Fig.1H,J).
To determine relative efficiency of KC reprogramming towards NC fate, we plated cells at clonal density i.e. one cell per well of a 96 well plate containing 3T3-J2 feeder cells and grew a total 174 single cell clones from 3 different donors (Fig.1N). The 3T3-J2 feeder cells were removed by Versene treatment leaving behind KC clones that were induced into NC phenotype using NCIM (Fig.1K, L, M). Prior to NC induction cells from each clone were stained for KRT14 and NES to ascertain KC identity. After induction, the NC forming efficiency ranged from 6.06% to 7.9% among different KC donors as evidenced by loss of KRT14 and acquisition of NES and SOX10 by immunostaining (Fig.1 L, M, S2H). These results confirm the clonal ability of KC to convert to NC phenotype.
Detailed Molecular Profile of KC-NC
We followed the kinetics of gene expression as KC turned into KC-NC using qRT-PCR. NC specific genes were expressed for a window of time between day 3 and 12 post-induction, with some NC specific genes (SOX10, FOXD3, ID2, B3GAT1 (HNK1)) exhibiting peak expression by day 6 and reduced expression thereafter, and others (PAX3, NES, KIT, NGFR, NR2F1) exhibiting sustained expression up to 12-18 days (n=3 donors; Fig.2A,B). We also examined the protein levels of SOX10, PAX3, FOXD3, NES and NGFR by immunostaining and quantified relative changes in protein expression using ImageJ software. Similar to RNA expression, KC-NC expressed NC specific proteins between day 3 and 12 (n=3 donors; Fig.S4A,B). Concomitantly, KC specific genes TP63, KRT14, KRT5 and KRT8 were downregulated, suggesting loss of epidermal phenotype in KC-NC (n=5 donors; Fig.2C). Taken together, the data suggest that the NC phenotype of KC-NC is transient, similar to what was previously reported for embryonic NC cells[24].
Figure 2. KC-NC display global molecular characteristics of NC cells.

KC-NC express NC specific genes (SOX10, NES, PAX3, FOXD3, NGFR, KIT, ID2, B3GAT1, NR2F1) transiently during induction (n=3 donors) (A, B) and downregulate KC specific genes (KRT14, KRT5, KRT8, and TP63) (n=3-5 donors) (C). KC-NC induction is marked by upregulation of EMT genes (ZEB1, ZEB2, SNAI1, FOXC2 and TWIST1) including classical cadherin switch (CDH1 to CDH2) (n=3-5 donors) (DE). Transcriptome sequencing (RNA-Seq) analysis of KC-NC and their respective parental KC (n=3 donors) shows basal epidermis and epidermal stratification genes were downregulated in KC-NC (F) while NC specific genes (G) and EMT genes (H) were upregulated. The genomic region known to regulate SOX10 transcription was severely hypomethylated in KC-NC, while heavily methylated in KC (methylation: 97±1.2% of input genomic DNA for KC and 0.54±0.3% of input genomic DNA for KC-NC, p = 5.45x10−15, n=5 donors) (I). Hierarchical clustering performed on all pairwise distances between the global transcriptome-wide RNA-seq profiles of KC and respective KC-NC shows distinct clustering between the two groups (J). Three-dimensional multi-dimensional scaling (3D-MDS) of all differentially expressed genes demonstrates that KC and KC-NC populations clustered separately (K). KC-NC displayed molecular signatures similar to human embryonic stem cell derived NC cells as confirmed by AUC (area under curve) of ROC plots (L). Statistical significance of ROC tests was confirmed using Wilcoxon rank-sum test. ([16], AUC: 0.597; p=4.304x10−5[1] AUC: 0.644; p= 5.454 x10−5 and parental KC AUC: 0.0003461405; p= 1). Scale bars, 50µm. All values are mean±SD. Experiments were repeated three times independently.
NC cells delaminate from the neural tube via an epithelial to mesenchymal transition (EMT) to acquire migratory phenotype[24]. Since KC-NC appear to undergo a similar EMT program, we examined changes in ZEB1 and ZEB2 (SIP1)[29], which mediate an E-Cadherin (CDH1) to N-Cadherin (CDH2) switch during EMT. Both were upregulated by day 3 of KC-NC reprogramming (n=3 donors; Fig.2D). Subsequently, CDH1 was downregulated while CDH2 was upregulated in KC-NC both at the mRNA (n=3 donors; Fig.2D) and protein level (n=5 donors; Fig.2E, S4C). Interestingly, we observed β-catenin, normally at adherens junctions of epithelial cells, translocated into the nucleus of KC-NC after undergoing EMT possibly reflecting WNT activation (n=3 donors; Fig.S4D,E). In addition, other key mediators of EMT such as SNAI1, TWIST1 and FOXC2 were significantly upregulated during KC-NC reprogramming (n=3 donors; Fig.2D). During EMT, epithelial cells shut off the expression of keratins (class I, II intermediate filaments) and induce expression of VIM (class III intermediate filament protein), which reportedly mediates cell shape and motility changes[30]. Indeed, we observed that KC-NC abolished KRT14 and expressed VIM (n=3 donors; Fig.S4F). Taken together, these results suggest that the KC to NC conversion invokes a coordinated gene regulatory machinery to induce EMT.
The global transcription profile of KC-NC is similar to hESC derived NC
Next we performed transcriptome analysis of KC-NC and KC from three donors. Between the two cell types, there were 3,894 differentially expressed genes; some of the quintessential KC-NC genes were verified by qRT-PCR (Fig.S4G). The data show that several NP border genes were expressed in KC but decreased as KC acquired NC fate (Fig.S4H). Concomitantly, genes that were expressed in the basal epidermal layer or during epidermal stratification were downregulated (Fig.2F). Instead, KC acquired a NC signature as suggested by upregulation of several NC (Fig.2G), EMT (Fig.2H) and ECM (Fig.S4I) genes. RTKs and WNTs were upregulated, while NOTCH and TGFβ pathways were downregulated (as indicated by increased expression of TGFβ pathway inhibitors) (Fig.S4J)[31]. In addition, histone demethylase (KDM6A, KDM5D), histone methyltransferase (EZH1), de novo DNA methyltransferases (DNMT3A/B) and NAD-dependent deacetylase SIRT1 were upregulated during KC-NC reprogramming (Fig.S4K), consistent with epigenetic modifications occurring during NC induction[5, 32]. Change in expression of a subset of those genes (EZH1, EZH2, DNMT3A, DNMT3B) was also confirmed with qRT-PCR (n=3 donors; Fig.S4L).
Promoter demethylation of the critical neural crest gene, SOX10, is essential for acquisition of NC fate[32, 33]. Therefore, we examined SOX10 promoter methylation in both KC and the corresponding KC-NC populations using the Epitect Methyl II PCR Array (QIAGEN) by examining 871 bp long region of Chr22 (38379093-38379964 (hg19), containing 82 CpG) for CpG island methylation, whose status inversely correlates with SOX10 expression[17]. SOX10 promoter was heavily methylated in KC but severely hypomethyled in KC-NC (methylation KC: 97±1.2% of input genomic DNA; methylation KC-NC: 0.54±0.3% of input genomic DNA, p=5.45x10−15, n=5 donors), consistent with high expression of SOX10 in KC-NC (Fig.2I).
We next compared KC and KC-NC transcriptomes using average linkage hierarchical clustering on all pairwise distances between their global transcriptome-wide RNA-seq profiles (Fig.2J) and 3D multidimensional scaling analysis (Fig.2K). KC and KC-NC transcriptome were significantly different as shown by distinct clustering (Fig.2J,K). Similarities with published neural crest signatures were analyzed using ROCR and verification libraries in R Bioconductor package. The results show that KC-NC are very similar to the human embryonic stem cell derived NC cells (Mica et al. 2013[16], AUC: 0.597; p=4.304x10−5 and Lee et al.[1], AUC: 0.644; p= 5.454 x10−5; Fig.2L), as opposed to parental KC (AUC: 0.0003461405; p=1; Fig.2L).
KC-NC give rise to functional neural crest derivatives in vitro
Peripheral neurons
KC-NC differentiated into neurons (n=5 donors) with typical neuronal morphology (Fig.3Ai), expressing TUBB3 gene (a.k.a. TUJ1) (Fig.3Aii), as well as proteins TUBB3, PRPH (Peripherin), NeuN (neuronal nuclei) and MAP2, indicative of their maturity and peripheral identity (Fig.3Aiii,iv,v). Using whole-cell current-clamp method (Fig.3Bi), the resting membrane potential (RMP) of KC-NC derived neurons was −47±5mV (Mean±SEM; n=25 cells) and membrane capacitance was 48±8 pF (Mean±SEM; n=25cells). On current-clamp recordings, they displayed evoked action potentials upon injecting 50pA step currents with duration of 6.4±0.9ms (Mean±SEM; n=15 cells; Fig.3Bii). KC-NC neurons expressed sustained outward current ranging from few hundred pA to ~4 nA (n=10 cells; Fig.3Biii,iv), which displayed voltage dependence and kinetics typical of tetraethylammonium (TEA) sensitive delayed rectifier potassium channels (Fig.3Biv,v). We also detected KC-NC derived neurons displaying inward currents (ranging from few hundreds of pA to ~2 nA (n=4cells)) with kinetics typical of TTX sensitive voltage activated sodium channels (Fig.3Biii,iv,vi). Taken together these results suggest functional maturity of KC-NC derived neurons.
Figure 3. KC-NC in vitro differentiate into functional neural crest derivatives.

KC-NC differentiate into functional peripheral neurons (A). Phase image of KC-NC derived neurons (A,i). KC-NC neurons expressed the TUBB3 gene (A,ii) and positively immunostained for PRPH (Peripherin) and TUBB3 (A,iii) suggestive of their peripheral origin. KC-NC neurons expressed MAP2 (A,iv) and NEUN (A,v). KC were negative for neuronal markers (A,vi; IgG control is shown in inset). KC-NC neurons are electrophysiologically mature (B). KC-NC neurons displayed evoked action potentials upon 50pA step current injections (n=15 cells) (B,i,ii). KC-NC neurons expressed tetrodotoxin (TTX) sensitive inward Na+ currents (n=4 cells) (B,iii) and tetraethylammonium (TEA) sensitive outward K+ currents (n=10 cells) (B,iv) . Red lines under traces in (B,iii) and (B,iv) represent portions of traces expanded for visual ease. Current-voltage relationship for K+ currents (B,v) and Na+ currents (B,vi) are shown (error bars, SEM). KC-NC differentiate into functional melanocytes (C). MITF and PMEL (HMB45) dual positive melanocytes (C,i), melanin containing melanocytes (C,ii) and centrifuged melanocyte pellet (C,iii). KC-NC derived melanocytes upregulated key melanocyte markers (TYR, PMEL, DCT, MITF, GPNMB, S100B) (C,iv) and produced melanin in response to L-DOPA (C,v). KC lacked melanocyte markers. (Cvi,vii; IgG control is shown in inset). KC-NC differentiate into Schwann cells (D) as shown by increased expression of Schwann cell specific genes (MPZ, PMP22, NFATC4, EGR2, S100B) (D,i) and positive immunofluorescence for MPZ (D,ii), CNPase and PLP1 (D,iii), GFAP (D,iv), S100B (D,v), . KC were negative for Schwann cell markers (Dvi,vii; IgG control is shown in inset). KC-NC differentiate into functional mesenchymal lineages i.e. osteocytes (E), chondrocytes (F) and adipocytes (G) as shown by RT-PCR and functional stains for each lineage. KC-NC give rise to functional smooth muscle cells as confirmed by gene and protein expression for SMC markers (H). Compaction of fibrin hydrogels embedded with KC-NC-SMC. The area of the hydrogels was imaged and quantified using image J. Aortic smooth muscle cells were used as positive control (I). Fibrin-based tissue constructs fabricated with KC-NC-SMC displayed isometric contractions in response to vasoagonists (U46619, Endothelin-1 and KCl). ASMC were used as positive control (J). Scale bars, 50µm. All values are mean±SD. $p < 0.05. Experiments were repeated three times independently. Inset images are uninduced controls. N.I. = non-induced; I. = induced
Melanocytes
KC-NC efficiently differentiated into melanocytes (n=3 donors; Fig.3C). After 3 weeks of differentiation, ~20% cells expressed MITF a key transcription factor for melanocyte development and ~33% of MITF+ cells also expressed the marker of mature melanocytes, PMEL (a.k.a. HMB45; Fig.3Ci), and synthesized melanin after 5 weeks (Fig.3Cii,iii). They abundantly expressed melanocyte genes (TYR, PMEL, DCT, MITF, GPNMB, and S100B) as determined by qRT-PCR (Fig.3Civ) and displayed functional tyrosinase activity as evidenced by the L-DOPA assay[34] (Fig.3Cv).
Schwann cells
KC-NC differentiated into Schwann cells as determined by qRT-PCR analysis of key Schwann cell genes and immunostaining (n=3 donors; Fig.3D). They upregulated MPZ, EGR2 (a.k.a. KROX20), PMP22, S100B and NFATC4 (Fig.3Di) genes and were MPZ+ (98±1.5%), S100B+ (94±1.3%), GFAP+ (92.6±1.6%) and dual positive for CNP (CNPase)+/ PLP1 (97.5±1.8%) (Fig.3Dii,iii,iv,v).
Mesenchymal Stem Cells
Upon differentiation KC-NC formed mesenchymal lineage as evidenced by differentiation towards osteogenic, chondrogenic, adipogenic and smooth muscle lineages and expression of mesenchymal markers CD73, CD90, CD44, CD105, CD49b (n=3 donors; Fig.S5A). Upon osteogenic differentiation (n=3 donors), cells expressed RUNX2 and ALP (alkaline phosphatase) and deposited calcium phosphate as shown by Von Kossa staining (Fig.3E). Chondrogenic differentiation (n=3 donors) was assessed by expression of COL2A1 and mature marker ACAN (Aggrecan) as well as by Alcian Blue staining for glycosaminoglycans (Fig.3F). Upon adipocyte differentiation (n=3 donors), they expressed PPAR-γ and LPL and deposited oil droplets, as shown by Red-O staining (Fig.3G).
KC-NC derived smooth muscle cells (KC-NC-SMC) (n=3 donors) showed transcriptional upregulation of SMC specific genes (ACTA2, TAGLN, SMTN, CALD1, MYH11) and displayed fibrillar organization of contractile proteins (ACTA2, CNN1, MYH11) (Fig.3H). KC-NC-SMC compacted fibrin hydrogels as a function of time to a similar extent as human aortic smooth muscle cells (ASMC) (p=0.095, n=3 donors; Fig.3I), indicating development of ability to generate force [35]. In addition, KC-NC-SMC remodeled fibrin hydrogels possibly by synthesizing ECM (collagen and elastin) as evidenced by increase in ultimate tensile stress (Fig.S5B). Most notably, KC-NC-SMC displayed receptor and non-receptor mediated isometric contractions upon treatment with vasoconstrictors[15] (U46619, Endothelin-1, KCl; n=3 donors; Fig.3J). These results suggest functional SMC differentiation of KC-NC.
KC-NC are clonally multipotent
To ascertain the multipotency of KC-NC, we plated KC-NC at clonal density and picked 6 clones per donor from 4 different KC donors (total 24 KC-NC clones). KC from each donor gave rise to KC-NC that differentiated into all four lineages (Table 1). Each clone was induced to differentiate into smooth muscle (SMC), melanocytes (Mel), Schwann cells (SC) and neurons (N) as evidenced by immunostaining for lineage specific markers.
Table 1.
Multilineage differentiation potential of KC-NC clones
| DONOR 1 | SMC | Mel | Schwann cells | Neurons |
|---|---|---|---|---|
| Clone 1 | Yes | Yes | Yes | Yes |
| Clone 2 | No | No | No | Yes |
| Clone 3 | Yes | No | Yes | Yes |
| Clone 4 | Yes | No | No | Yes |
| Clone 5 | Yes | No | Yes | No |
| Clone 6 | Yes | No | Yes | No |
| DONOR 2 | SMC | Mel | Schwann cells | Neurons |
| Clone 1 | Yes | No | No | No |
| Clone 2 | Yes | Yes | Yes | Yes |
| Clone 3 | Yes | Yes | Yes | No |
| Clone 4 | Yes | No | No | Yes |
| Clone 5 | Yes | No | No | No |
| Clone 6 | Yes | No | Yes | No |
| DONOR 3 | SMC | Mel | Schwann cells | Neurons |
| Clone 1 | Yes | No | No | No |
| Clone 2 | Yes | No | No | Yes |
| Clone 3 | Yes | Yes | Yes | Yes |
| Clone 4 | Yes | No | Yes | No |
| Clone 5 | Yes | No | Yes | No |
| Clone 6 | No | Yes | Yes | No |
| DONOR 4 | SMC | Mel | Schwann cells | Neurons |
| Clone 1 | Yes | No | Yes | No |
| Clone 2 | Yes | Yes | Yes | Yes |
| Clone 3 | Yes | Yes | Yes | Yes |
| Clone 4 | Yes | No | No | Yes |
| Clone 5 | Yes | Yes | No | No |
| Clone 6 | Yes | No | Yes | Yes |
We found that 21% of the clones (5 out of 24 clones) differentiated into all four lineages; 12% (3 out of 24 clones) were tripotent; 50% (12 out of 24 clones) were bipotent; and 17% (4 out of 24 clones) were unipotent (Fig. 4A). Immunostaining for MPZ (P0), PLP1 and S100B proteins showed that 62.5 ± 7.2% clones acquired Schwann cell fate (Fig. 4B,D); while 33.3 ± 11.7% clones gave rise to melanocytes as evidenced by PMEL (SILV) immunostaining and L-DOPA reaction positivity (Fig. 4B,E). The majority of clones (91.6 ± 8.3%) differentiated into SMC as determined by ACTA2 and CNN1 positive cells (Fig. 4B,F). Finally, immunostaining for neuronal markers TUBB3, PRPH and MAP2 showed that 50 ± 16.6% of clones differentiated into neuronal phenotype (Fig. 4B, G). Interestingly, KC-NC derived neurons in each TUBB3+ clone were uniformly positive for 5-HT (serotonin), a marker of enteric autonomic neurons and 32 ± 2.5% were found to be positive for BRN3A (POU4F1), indicative of their sensory identity (Fig.4G). These results suggest that KC-NC give rise to both enteric autonomic and sensory neurons. The differentiation potential of KC-NC derived from each donor was summarized in Fig. 4C. Despite variations in the multi-lineage potential at the clonal level, KC-NC derived from all donors were capable of differentiation towards all the stereotypical lineages of NC cells.
Figure 4. KC-NC are clonally multipotent.

KC-NC single cell clones (24 clones from 4 donors) were grown and differentiated into SMC, melanocytes (Mel), Schwann cells (SC) or neurons (N) and examined by immunostaining with lineage specific antibodies. Distribution of multipotency of KC-NC clones: 21% KC-NC clones were multipotent, 12% tripotent, 50% bipotent and 17% unipotent (A). KC-NC clones gave rise to Schwann cells (~62.5%) as determined by S100B, MPZ and PLP1 immunostaining (B,D). Fewer clones (~33%) gave rise to melanocytes as shown by PMEL immunostaining and L-DOPA assay (B,E). The majority of clones (~92%) differentiated into SMC, as shown by ACTA2 and CNN1 immunostaining (B,F). About half (~50%) of KC-NC clones differentiated into neurons as identified by TUBB3, MAP2 and PRPH protein expression. KC-NC derived neurons were positive for 5-HT (Serotonin) as well as BRN3A, indicating differentiation towards the enteric and sensory phenotype (B,G). Percentage of clones from each donor that differentiated into the indicated NC lineages (C). Scale bars, 50µm. All values are mean±SD.
KC derived NC migrate and differentiate into NC lineages in ovo
We transplanted KC or KC-NC labeled with EGFP into the head mesenchyme of 10-13 somite host chick embryos (Fig.5B) that were analyzed either 2 days (n=4 embryos) or 3 days (n=4 embryos) after the transplantation. The results show that normal KC did not contribute to neural crest derivatives, but were found in the mesenchyme (a total of 7 cells detected in 3 embryos). In contrast, a total of 151 transplanted human KC-NC EGFP+ cells (from total 400 transplanted KC-NC) were found in sections from 8 embryos in locations typical for neural crest. They contributed to the full repertoire of neural crest derivatives, from neural and glial cells to mesenchymal and pigment cells (Fig.5A). They gave rise to TUBB3 (TUJ1)+ neurons (Fig.5C), BLBP+ glial cells (Fig.5D) in the trigeminal ganglia as well as putative Schwann cells around axon bundles (Fig.5E). We also detected transplanted cells that were localized in blood vessel walls and expressed α-SMA (ACTA2), indicating differentiation into smooth muscle cells (Fig.5F). KC-NC gave rise to presumptive melanoblasts below the ectoderm (Fig.5G). Additionally, transplanted cells were detected in the branchial arches, the gut wall, the heart or the mesenchyme (Fig.5A). These results confirm that KC-NC behave similar to embryonic NC in ovo and can contribute to multiple NC derivatives.
DISCUSSION
We report for the first time that KRT14+ postnatal human epidermal KC can be reprogrammed to a NC fate upon FGF2 stimulation, further potentiated by IGF1. KC derived NC cells displayed a global transcription profile similar to human embryonic stem cell (hESC) derived NC cells[1, 16] and differentiated into functional NC derivatives including peripheral neurons, melanocytes, Schwann cells and mesenchymal stem cell derivatives (osteocytes, chondrocytes, adipocytes and smooth muscle cells). Upon transplantation into chicken embryos, KC-NC migrated along stereotypical pathways and gave rise to multiple NC derivatives.
Previous work showed that WNT1 expressing cells in the bulge of the murine hair follicle represent a NC cell population[20]. Notably, WNT1 expression was limited to the bulge and dermal papilla, while inter-follicular epidermis was devoid of WNT1 expressing cells, suggesting absence of NC cells in the inter-follicular epidermis[20]. Pioneering work by the Sieber-Blum laboratory suggested the presence of melanocyte precursors in the murine bulge region as a potential source of melanocytes responsible for hair pigmentation. Human inter-follicular epidermis contains unipotent melanocyte precursor/melanocyte stem cells that could potentially contaminate our KC-NC population. However, we have accumulated evidence that strongly suggests that this is highly unlikely and that KC-NC originate from KC. Specifically, we isolated inter-follicular KC from glabrous (lacking hair follicles) foreskin[23], ruling out contamination from hair follicle bulge cells. We further ensured that our starting KC population does not contain melanocytes (Fig. S1). Foreskin derived KC expressed KRT14, TP63 and ITGB1 but lacked PMEL and MITF. Importantly, each KC clone was examined by KRT14 immunostaining prior to NC induction to confirm the purity of KC and lack of melanocytes. Upon induction, clonally derived KRT14+ KC gave rise to NC cells, confirming their reprogramming potential. Finally, in our KC-NC clonal multipotency experiments, we employed 24 single cell clones from 4 donors and did not find a single KC-NC clone that gave rise only to melanocytes and no other lineage, further suggesting the lack of unipotent melanocyte precursor in our starting KC populations.
We speculate that expression of various neural plate border genes (SNAI2, MYC, TFAP2A, SOX9, DLX3/5, MSX1/2, IRX2 etc.) may predispose KRT14+ KC with NC potential. Indeed, the higher propensity of KC to reprogram to the pluripotent state - 100 times more efficiently than fibroblasts – was attributed to native expression of pluripotency factors MYC and KLF4[36]. On the other hand, SOX10 overexpression was required to reprogram human fibroblast into NC cells[3] and neural progenitors into oligodendrocyte progenitors[37], suggesting that the molecular nature of the starting cell population may be critical to the reprogramming process.
NC induction during embryonic development involves activation of a gene regulatory network (GRN)[5],[24]. Similar to the transcriptional dynamics during NC formation in vivo[12] , expression of neural plate border genes (TFAP2A, MYC, SNAI2, MSX1/2, DLX3/5, SOX9), in KC was followed by sequential upregulation of EMT (ZEB1, ZEB2, SNAI1, FOXC2, TWIST) and NC specification (SOX10, FOXD3, PAX3, TFAP2A, NR2F1, ID2) gene module during KC to NC reprogramming ,. Consistent with upregulation of NC specific genes the SOX10 promoter region[17] became hypomethylated. The drastic change in methylation status was accompanied by transcriptional changes in several epigenetic modifiers (DNMT3A/B, EZH1/2, SIRT1).
The ability of KC-NC to migrate along the routes of endogenous NC cells within chicken embryos and differentiate into multiple NC derivatives provides very strong support of their NC character [11]. Similar to endogenous and hESC derived NC, KC-NC differentiated into functional peripheral neurons, Schwann cells, melanocytes and mesenchymal stem cell derivatives (osteocytes, chondrocytes, adipocytes and smooth muscle cells) in vitro, further supporting the NC phenotype of these cells.
Although dermal fibroblasts can be reprogrammed into specific cell types (neurons, glial cells, cardiomyocytes and hepatocytes etc.)[4], the virally-mediated genetic modification of these reprogrammed cells restricts their potential clinical applicability. Moreover, poor proliferation of terminally reprogrammed cells[4] further limits their clinical utility as cell based regenerative therapies require large number (~107) of cells.
Given the accessibility, higher proliferative capacity and ease of reprogramming epidermal KC without the need for genetic manipulation, KC-NC represent a potentially useful source of functional therapeutic cells for regenerative medicine and tissue engineering applications as well as a model for analysis of human neurocristopathies[38, 39], similar to human iPSC. Taken together, our study presents human epidermal KC as a novel inducible source of functional NC cells without genetic modification. This readily accessible source of abundant, highly proliferative, autologous NC cells has significant implications for stem cell biology, drug discovery and regenerative medicine.
Supplementary Material
A human epidermal KC colony morphology after removal of the fibroblast feeder layer (A). Flow cytometry (B) and immunofluorescence (C) of KC shows lack of melanocyte specific marker, PMEL (SILV) and uniform expression of KC marker, KRT14. Primary human melanocytes lack KRT14 and express PMEL and served as positive control (Representative data from 2 of 3 donors are shown). Scale bars, 400µm.
Human epidermal KC express basal KC proteins (KRT14, ITGB1, TP63) (A). Neonatal KC express neural plate border specific genes (MYC, SOX9, SNAI2, MSX2, IRX2, DLX3, TFAP2A, and KLF4) (A) and proteins (SNAI2, TFAP2A, MYC, KLF4) (B). KC to NC induction by FGF2 is potentiated by IGF1 as shown by qRT-PCR for key NC genes (NES, PAX3, FOXD3, TWIST1, NGFR, SOX10, B3GAT1) and proliferation marker KI67 (C). Collagen type I, fibronectin and laminin ECM coatings supported expression of NC genes to a similar extent (D). Collagen type I and fibronectin increase KC to NC induction as shown by increased number of SOX+NES+ cells ((E). Proliferation kinetics and clonogenic ability of KC-NC (F,G). KC clones give rise to SOX10 and NES positive NC cells (H). Scale bars, 50µm. All values are mean±SD (*p < 0.001, #p < 0.01).
Adult KC express neural plate border specific genes (MYC, SOX9, SNAI2, MSX2, IRX2, DLX3, TFAP2A, and KLF4) (A). Adult KC express the basal KC marker KRT14 as well as neural plate border specific proteins (SNAI2, KLF4, and TFAP2A) (B). Scale bars, 50µm.
KC-NC express NC lineage proteins (SOX10, FOXD3, PAX3, NGFR and NES) as determined by measurements of immunofluorescence intensity using imageJ (A,B). KC-NC undergo EMT as seen by upregulation of CDH2, downregulation of CDH1 (C,D,E), expression of VIM (Vimentin) (F) and several ECM genes (I). As CDH1 is repressed during EMT, β-catenin (CTNNB1) translocates to the nucleus of KC-NC (E) as compared to the cell border in KC (D). Transcriptome (RNA-Seq) analysis was performed on KC and respective KC-NC and was in good agreement with qRT-PCR results as examined for NC markers (n=3 donors) (G). KC expressed several neural plate border genes (H) and their induction into NC fate was marked by changes in the expression of several signal transduction (J), epigenetic and cell cycle related genes (K,L) (n=3donors). Scale bars, 50µm. All values are mean±SD.
KC-NC derived mesenchymal cells expressed CD73, CD90, CD44, CD105 and CD49b and lacked pan-leukocyte marker, CD45 as determined by flow cytometry (n=3 donors) (A). Compared to ASMC, KC-NC derived SMC significantly remodeled fibrin hydrogels as evidenced by increased ultimate tensile stress of KC-NC-SMC based tissue constructs (n=3 donors) (B). $p < 0.05.
Acknowledgments
This work was supported by a grant (IMPACT Award) from the University at Buffalo to S.T.A. K.A.C. was supported by grant F31 NS 084668. G.P. was supported by grants F31 NS 084668 and AHA 12EIA9100012. M.E.B. was supported by R01DE024157. Authors thank Deepika Verma for her help in creating schematic diagram.
Footnotes
AUTHOR CONTRIBUTIONS V.K.B. conceptualized and designed the study and performed the experiments. V.K.B., S.T.A. designed experiments and performed data analysis and interpretation. L.K., M.E.B. performed chicken embryo experiments. K.A.C., G.P. did electrophysiological analysis. G.T., X.W., P.L. performed experiments. B.L., S.L., V.K.B. did RNA sequencing analysis. All authors read the manuscript and provided their inputs. V.K.B., S.T.A., M.E.B. wrote the manuscript.
CONFLICT OF INTERESTS
None
ACCESSION NUMBERS
GEO: GSE72268
SUPPLEMENTARY INFORMATION
Supplemental figures and list of antibodies and primer pairs used in this study are provided as supplementary information.
References
- 1.Lee G, Kim H, Elkabetz Y, et al. Isolation and directed differentiation of neural crest stem cells derived from human embryonic stem cells. Nat Biotechnol. 2007;25:1468–1475. doi: 10.1038/nbt1365. [DOI] [PubMed] [Google Scholar]
- 2.Lee G, Chambers SM, Tomishima MJ, et al. Derivation of neural crest cells from human pluripotent stem cells. Nature protocols. 2010;5:688–701. doi: 10.1038/nprot.2010.35. [DOI] [PubMed] [Google Scholar]
- 3.Kim YJ, Lim H, Li Z, et al. Generation of multipotent induced neural crest by direct reprogramming of human postnatal fibroblasts with a single transcription factor. Cell stem cell. 2014;15:497–506. doi: 10.1016/j.stem.2014.07.013. [DOI] [PubMed] [Google Scholar]
- 4.Xu J, Du Y, Deng H. Direct lineage reprogramming: strategies, mechanisms, and applications. Cell stem cell. 2015;16:119–134. doi: 10.1016/j.stem.2015.01.013. [DOI] [PubMed] [Google Scholar]
- 5.Simoes-Costa M, Bronner ME. Establishing neural crest identity: a gene regulatory recipe. Development. 2015;142:242–257. doi: 10.1242/dev.105445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mayor R, Morgan R, Sargent MG. Induction of the prospective neural crest of Xenopus. Development. 1995;121:767–777. doi: 10.1242/dev.121.3.767. [DOI] [PubMed] [Google Scholar]
- 7.Garcia-Castro MI, Marcelle C, Bronner-Fraser M. Ectodermal Wnt function as a neural crest inducer. Science. 2002;297:848–851. doi: 10.1126/science.1070824. [DOI] [PubMed] [Google Scholar]
- 8.Endo Y, Osumi N, Wakamatsu Y. Bimodal functions of Notch-mediated signaling are involved in neural crest formation during avian ectoderm development. Development. 2002;129:863–873. doi: 10.1242/dev.129.4.863. [DOI] [PubMed] [Google Scholar]
- 9.Monsoro-Burq AH, Fletcher RB, Harland RM. Neural crest induction by paraxial mesoderm in Xenopus embryos requires FGF signals. Development. 2003;130:3111–3124. doi: 10.1242/dev.00531. [DOI] [PubMed] [Google Scholar]
- 10.Liem KF, Jr, Tremml G, Roelink H, et al. Dorsal differentiation of neural plate cells induced by BMP-mediated signals from epidermal ectoderm. Cell. 1995;82:969–979. doi: 10.1016/0092-8674(95)90276-7. [DOI] [PubMed] [Google Scholar]
- 11.Kerosuo L, Nie S, Bajpai R, et al. Crestospheres: Long-Term Maintenance of Multipotent, Premigratory Neural Crest Stem Cells. Stem cell reports. 2015;5:499–507. doi: 10.1016/j.stemcr.2015.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Green SA, Simoes-Costa M, Bronner ME. Evolution of vertebrates as viewed from the crest. Nature. 2015;520:474–482. doi: 10.1038/nature14436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.LaBonne C, Bronner-Fraser M. Neural crest induction in Xenopus: evidence for a two-signal model. Development. 1998;125:2403–2414. doi: 10.1242/dev.125.13.2403. [DOI] [PubMed] [Google Scholar]
- 14.Rheinwald JG, Green H. Serial cultivation of strains of human epidermal keratinocytes: the formation of keratinizing colonies from single cells. Cell. 1975;6:331–343. doi: 10.1016/s0092-8674(75)80001-8. [DOI] [PubMed] [Google Scholar]
- 15.Bajpai VK, Mistriotis P, Loh YH, et al. Functional Vascular Smooth Muscle Cells Derived From Human Induced Pluripotent Stem Cells Via Mesenchymal Stem Cell Intermediates. Cardiovascular research. 2012 doi: 10.1093/cvr/cvs253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mica Y, Lee G, Chambers SM, et al. Modeling neural crest induction, melanocyte specification, and disease-related pigmentation defects in hESCs and patient-specific iPSCs. Cell reports. 2013;3:1140–1152. doi: 10.1016/j.celrep.2013.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iwamoto K, Bundo M, Yamada K, et al. DNA methylation status of SOX10 correlates with its downregulation and oligodendrocyte dysfunction in schizophrenia. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2005;25:5376–5381. doi: 10.1523/JNEUROSCI.0766-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ordway JM, Bedell JA, Citek RW, et al. Comprehensive DNA methylation profiling in a human cancer genome identifies novel epigenetic targets. Carcinogenesis. 2006;27:2409–2423. doi: 10.1093/carcin/bgl161. [DOI] [PubMed] [Google Scholar]
- 19.Burgess A, Vigneron S, Brioudes E, et al. Loss of human Greatwall results in G2 arrest and multiple mitotic defects due to deregulation of the cyclin B-Cdc2/PP2A balance. Proc Natl Acad Sci U S A. 2010;107:12564–12569. doi: 10.1073/pnas.0914191107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sieber-Blum M, Grim M, Hu YF, et al. Pluripotent neural crest stem cells in the adult hair follicle. Developmental dynamics : an official publication of the American Association of Anatomists. 2004;231:258–269. doi: 10.1002/dvdy.20129. [DOI] [PubMed] [Google Scholar]
- 21.Toma JG, Akhavan M, Fernandes KJ, et al. Isolation of multipotent adult stem cells from the dermis of mammalian skin. Nature cell biology. 2001;3:778–784. doi: 10.1038/ncb0901-778. [DOI] [PubMed] [Google Scholar]
- 22.Toma JG, McKenzie IA, Bagli D, et al. Isolation and characterization of multipotent skin-derived precursors from human skin. Stem cells. 2005;23:727–737. doi: 10.1634/stemcells.2004-0134. [DOI] [PubMed] [Google Scholar]
- 23.Kaur P. Interfollicular epidermal stem cells: identification, challenges, potential. J Invest Dermatol. 2006;126:1450–1458. doi: 10.1038/sj.jid.5700184. [DOI] [PubMed] [Google Scholar]
- 24.Sauka-Spengler T, Bronner-Fraser M. A gene regulatory network orchestrates neural crest formation. Nat Rev Mol Cell Biol. 2008;9:557–568. doi: 10.1038/nrm2428. [DOI] [PubMed] [Google Scholar]
- 25.Prasad MS, Sauka-Spengler T, LaBonne C. Induction of the neural crest state: control of stem cell attributes by gene regulatory, post-transcriptional and epigenetic interactions. Dev Biol. 2012;366:10–21. doi: 10.1016/j.ydbio.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Larue L, Bellacosa A. Epithelial-mesenchymal transition in development and cancer: role of phosphatidylinositol 3' kinase/AKT pathways. Oncogene. 2005;24:7443–7454. doi: 10.1038/sj.onc.1209091. [DOI] [PubMed] [Google Scholar]
- 27.Lothian C, Lendahl U. An evolutionarily conserved region in the second intron of the human nestin gene directs gene expression to CNS progenitor cells and to early neural crest cells. The European journal of neuroscience. 1997;9:452–462. doi: 10.1111/j.1460-9568.1997.tb01622.x. [DOI] [PubMed] [Google Scholar]
- 28.Perris R, Perissinotto D. Role of the extracellular matrix during neural crest cell migration. Mechanisms of development. 2000;95:3–21. doi: 10.1016/s0925-4773(00)00365-8. [DOI] [PubMed] [Google Scholar]
- 29.Rogers CD, Saxena A, Bronner ME. Sip1 mediates an E-cadherin-to-N-cadherin switch during cranial neural crest EMT. The Journal of cell biology. 2013;203:835–847. doi: 10.1083/jcb.201305050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mendez MG, Kojima S, Goldman RD. Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2010;24:1838–1851. doi: 10.1096/fj.09-151639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Betancur P, Bronner-Fraser M, Sauka-Spengler T. Assembling neural crest regulatory circuits into a gene regulatory network. Annual review of cell and developmental biology. 2010;26:581–603. doi: 10.1146/annurev.cellbio.042308.113245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu N, Strobl-Mazzulla P, Sauka-Spengler T, et al. DNA methyltransferase 3A as a molecular switch mediating the neural tube-to-neural crest fate transition. Genes & development. 2012;26:2380–2385. doi: 10.1101/gad.198747.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hu N, Strobl-Mazzulla PH, Simoes-Costa M, et al. DNA methyltransferase 3B regulates duration of neural crest production via repression of Sox10. Proc Natl Acad Sci U S A. 2014;111:17911–17916. doi: 10.1073/pnas.1318408111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Slominski A, Moellmann G, Kuklinska E, et al. Positive regulation of melanin pigmentation by two key substrates of the melanogenic pathway, L-tyrosine and L-dopa. J Cell Sci. 1988;89(Pt 3):287–296. doi: 10.1242/jcs.89.3.287. [DOI] [PubMed] [Google Scholar]
- 35.Bajpai VK, Mistriotis P, Andreadis ST. Clonal multipotency and effect of long-term in vitro expansion on differentiation potential of human hair follicle derived mesenchymal stem cells. Stem cell research. 2012;8:74–84. doi: 10.1016/j.scr.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aasen T, Raya A, Barrero MJ, et al. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nature biotechnology. 2008;26:1276–1284. doi: 10.1038/nbt.1503. [DOI] [PubMed] [Google Scholar]
- 37.Wang J, Pol SU, Haberman AK, et al. Transcription factor induction of human oligodendrocyte progenitor fate and differentiation. Proc Natl Acad Sci U S A. 2014;111:E2885–2894. doi: 10.1073/pnas.1408295111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Snider TN, Mishina Y. Cranial neural crest cell contribution to craniofacial formation, pathology, and future directions in tissue engineering. Birth defects research Part C, Embryo today : reviews. 2014;102:324–332. doi: 10.1002/bdrc.21075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Butler Tjaden NE, Trainor PA. The developmental etiology and pathogenesis of Hirschsprung disease. Translational research : the journal of laboratory and clinical medicine. 2013;162:1–15. doi: 10.1016/j.trsl.2013.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A human epidermal KC colony morphology after removal of the fibroblast feeder layer (A). Flow cytometry (B) and immunofluorescence (C) of KC shows lack of melanocyte specific marker, PMEL (SILV) and uniform expression of KC marker, KRT14. Primary human melanocytes lack KRT14 and express PMEL and served as positive control (Representative data from 2 of 3 donors are shown). Scale bars, 400µm.
Human epidermal KC express basal KC proteins (KRT14, ITGB1, TP63) (A). Neonatal KC express neural plate border specific genes (MYC, SOX9, SNAI2, MSX2, IRX2, DLX3, TFAP2A, and KLF4) (A) and proteins (SNAI2, TFAP2A, MYC, KLF4) (B). KC to NC induction by FGF2 is potentiated by IGF1 as shown by qRT-PCR for key NC genes (NES, PAX3, FOXD3, TWIST1, NGFR, SOX10, B3GAT1) and proliferation marker KI67 (C). Collagen type I, fibronectin and laminin ECM coatings supported expression of NC genes to a similar extent (D). Collagen type I and fibronectin increase KC to NC induction as shown by increased number of SOX+NES+ cells ((E). Proliferation kinetics and clonogenic ability of KC-NC (F,G). KC clones give rise to SOX10 and NES positive NC cells (H). Scale bars, 50µm. All values are mean±SD (*p < 0.001, #p < 0.01).
Adult KC express neural plate border specific genes (MYC, SOX9, SNAI2, MSX2, IRX2, DLX3, TFAP2A, and KLF4) (A). Adult KC express the basal KC marker KRT14 as well as neural plate border specific proteins (SNAI2, KLF4, and TFAP2A) (B). Scale bars, 50µm.
KC-NC express NC lineage proteins (SOX10, FOXD3, PAX3, NGFR and NES) as determined by measurements of immunofluorescence intensity using imageJ (A,B). KC-NC undergo EMT as seen by upregulation of CDH2, downregulation of CDH1 (C,D,E), expression of VIM (Vimentin) (F) and several ECM genes (I). As CDH1 is repressed during EMT, β-catenin (CTNNB1) translocates to the nucleus of KC-NC (E) as compared to the cell border in KC (D). Transcriptome (RNA-Seq) analysis was performed on KC and respective KC-NC and was in good agreement with qRT-PCR results as examined for NC markers (n=3 donors) (G). KC expressed several neural plate border genes (H) and their induction into NC fate was marked by changes in the expression of several signal transduction (J), epigenetic and cell cycle related genes (K,L) (n=3donors). Scale bars, 50µm. All values are mean±SD.
KC-NC derived mesenchymal cells expressed CD73, CD90, CD44, CD105 and CD49b and lacked pan-leukocyte marker, CD45 as determined by flow cytometry (n=3 donors) (A). Compared to ASMC, KC-NC derived SMC significantly remodeled fibrin hydrogels as evidenced by increased ultimate tensile stress of KC-NC-SMC based tissue constructs (n=3 donors) (B). $p < 0.05.