

Abstract
The function and clinical implication of ArfGAP with SH3 domain, ankyrin repeat, and PH domain 3 (ASAP3) in colorectal cancer (CRC) remains undefined. In the present study, we showed that the expression level of ASAP3 was dramatically increased in CRC and its upregulation was associated with American Joint Committee on Cancer stage (P < 0.001) and poor prognosis (P = 0.0022). The combination of stage and ASAP3 expression improved the prediction of survival in CRC patients. Suppression of ASAP3 inhibited cell proliferation by inducing G1 phase arrest without influencing apoptosis. ASAP3 promoted growth of colon tumors in mice with colitis, and accelerated cell invasion and migration in vitro. Increased ASAP3 was associated with activation of the nuclear factor‐κB (NF‐κB) canonical pathway in CRC. Upregulation of ASAP3 increased the phosphorylation and nuclear translocation of the p65 NF‐κB subunit. Mechanistically, ASAP3 interacts with NF‐κB essential modulator (NEMO) and could reduce the polyubiquitinylation of NEMO. Overall, ASAP3 might regulate NF‐κB via binding to NEMO. ASAP3 acts as an oncogene in colonic cancer and could be a potential biomarker of colon carcinogenesis.
Keywords: ASAP3, colorectal cancer, NEMO, NFκB, oncogene
Colorectal cancer (CRC) is a leading cause of morbidity and mortality worldwide.1 Unfortunately, some newly discovered CRC patients are diagnosed at an advanced stage with poor prognosis.2 Metastasis also contributes to the high mortality of CRC patients. Despite great efforts being made to improve treatment strategies, there is an urgent need to identify targets that would be beneficial for CRC diagnosis and treatment. Accumulated evidence indicates that a diversity of genetic/epigenetic pathways facilitate cancer incidence,3 and there is a growing awareness that nuclear factor‐κB (NF‐κB) is constitutively activated in CRC,4, 5 playing a tumor‐promoting role by increasing cell proliferation and inducing metastasis.6, 7
The NF‐κB family comprises five members: RelA, RelB, c‐Rel, p50/p105, and p52/p100.8 Nuclear factor‐κB can be activated in two distinct ways, named the canonical and non‐canonical pathways. The canonical pathway demands activation by the RelA/p50 dimer, and various stimuli can activate the IκB kinase (IKK) complex, which comprises the catalytic subunits IKKα and IKKβ, and the regulatory subunit NF‐κB essential modulator (ΙΚΚγ/ΝΕΜΟ). Then IKK phosphorylates IκB to induce ubiquitin‐dependent degradation of IκBs, allowing translocation of NF‐κB into the nucleus, resulting in the altered expressions of NF‐κB downstream genes.9, 10 The non‐canonical pathway causes activation of RelB/p52 dimers, which is independent of ΙΚΚβ and ΙΚΚγ. Given the crucial role of NF‐κB in CRC development and progression, many studies have sought NF‐κB inhibitors to block cancer development.11 However, few NF‐κB inhibitors have been applied to CRC, and more research is needed to discover an inhibitor that can prevent NF‐κB activation without unanticipated adverse effects.
ArfGAP with SH3 domain, ankyrin repeat, and PH domain 3 (ASAP3) is a specific ARF6 GTPase‐activating protein (GAP), also termed DDEFL1 and ACAP4, which comprises ArfGAP and ankyrin repeat (ANK) domains in the C‐terminus and BAR and PH domains in the N‐terminus.12, 13 Several studies have reported that ASAP3 promotes metastasis of multiple cancers, 14, 15, 16, 17 however, the expression pattern and biological function of ASAP3 in CRC remain poorly understood. The present study aimed to identify the potential biological role of ASAP3 in CRC. First, ASAP3 expression profiles were examined in colorectal tumors and matched adjacent tissues. Second, we observed that upregulation of ASAP3 was related to poor survival of patients with CRC. Bioinformatic analyses identified that the NF‐κB canonical pathway had the strongest association with ASAP3 expression. ASAP3 could promote the activation of NF‐κB, thereby playing a functional role in CRC by altering the expressions of NF‐κB downstream genes. An Asap3 knockout mouse model was applied to verify the role of ASAP3 in vivo.
Materials and Methods
Patient specimens
Human CRC tissue samples and corresponding adjacent specimens were obtained from patients who underwent surgery at the Shanghai Renji Hospital (Shanghai, China) from January 2009 to December 2009. A total of 90 pathologically confirmed CRC patients were enrolled. The study was approved by the ethics committee of Shanghai Jiao Tong University, School of Medicine (Shanghai, China), and written informed consent was obtained from all patients at study entry. The Cancer Genome Atlas (TCGA) RNA‐Seq data was downloaded from the TCGA website (https://cancergenome.nih.gov/), which was collected and used following strict human subject protection guidelines, and with informed consent.
RNA extraction, RT‐PCR, and quantitative real‐time PCR
Total RNA was extracted from cultured cells using TRIzol reagent (Takara, Shiga, Japan) according to the manufacturer's instructions. Total RNA (1 μg) was reverse‐transcribed using a PrimeScript RT‐PCR Kit (Takara). The transcript level of the gene was quantified by real‐time PCR using a SYBR Premix ExTaq kit and was normalized to the expression of GAPDH. The change in cycle threshold method (2−∆∆Ct) was used to calculate relative changes in expression. The sequences of the primers are provided in Table S1.
Mice
C57BL/6 mice were obtained from the Experimental Animal Centre of Shanghai Institutes for Biological Sciences (Shanghai, China). Asap3 WT and KO littermates were required on a C57BL/6 × 129SV mixed background. The resultant Asap3 loxP/loxP mice were then crossed with UBC‐cre/ERT2 mice, and Asap3 −/− mice were generated by sequential peritoneal injection of tamoxifen for 7 days into adult offspring. Colon carcinoma was induced by treating the mice with azoxymethane (AOM) and dextran sulfate sodium (DSS). Six‐ to 8‐week‐old mice (Asap3 +/+ mice and Asap3 −/− mice) were injected i.p. with AOM. The DSS solution was replaced with autoclaved water after 5 days. The DSS solution was given again at day 24 and day 53. Tumor development was evaluated at day 85. Animal procedures were approved by the Institutional Animal Care and Use Committees at the Shanghai Research Center for Model Organisms and the Renji Hospital of Shanghai Jiao Tong University Medical School.
Bioinformatics analysis
Gene Set Enrichment Analysis (GSEA) is a method to analyze and interpret microarray and similar data using biological knowledge.18 In our study, the GEO dataset of CRC was analyzed using GSEA2‐2.2.2 (Broad Institute, Harvard, MA, USA). The gene sets showing a false discovery rate of 0.25, a well‐established cut‐off for the identification of biologically relevant genes, were considered enriched between the classes under comparison. The “c2.all.v5.0.symbols.gmt” from the Molecular Signatures Database (http://www.broad.mit.edu/gsea/msigdb/index.jsp) were used to run GSEA and 1000 permutations were used to calculate the P‐value; the “permutation type” was set to “gene set”. All other parameters were set to default, except that the gene set should represent at least 15 genes.
Statistical analysis
Data are presented as the mean ± SD. All statistical analyses were carried out using spss 17 for Windows (SPSS, Chicago, IL, USA). Graphical representations were generated using GraphPad Prism 5 software (GraphPad, San Diego, CA, USA). Comparisons were undertaken using Student's t‐test, and χ2‐tests were used to assess the correlation between clinicopathologic parameters and Asap3 expression for CRC patients. The survival curves were assessed using the Kaplan–Meier method and a significant difference between two groups was evaluated using a log–rank test. Receiver operating characteristic analysis was used to calculate the sensitivity and specificity of ASAP3 as a diagnostic biomarker. P‐values < 0.05 were considered significant.
Additional materials and methods are detailed in Data S1.
Results
Upregulation of ASAP3 is associated with CRC and poor outcome
To test whether ASAP3 expression correlates with CRC prognosis, multiple microarray datasets of CRC from TCGA were analyzed, and we found that the ASAP3 expression level was significantly higher in CRC tissues than in normal tissues (Fig. 1a). Furthermore, we compared ASAP3 expression between paired colorectal cancer and normal tissues experimentally. Real‐time PCR data and immunohistochemical (IHC) staining (n = 90) showed that ASAP3 mRNA and protein expression were significantly increased in CRC tissues compared with adjacent normal tissues (Fig. 1b–d). We further investigated the association between ASAP3 expression levels and different clinicopathologic parameters of CRC. When the patients were stratified according to ASAP3 expression levels, significantly larger tumor size was found in the high ASAP3 group (P < 0.001), and expression of ASAP3 was correlated with lymph node metastasis (P = 0.002) and American Joint Committee on Cancer (AJCC) stage (P < 0.001, Table 1). In the univariate Cox regression analysis, high expression of ASAP3 in CRC was associated with cancer‐related death (hazard ratio, 3.089; 95% confidence interval, 1.439–6.632; P = 0.004) (Table S2). Multivariate Cox regression analysis indicated that tumor size, but not high expression of ASAP3, was considered an independent prognostic factor for CRC patients (Table S3). Our analysis showed that the high ASAP3 group had an unfavorable prognosis compared with the ASAP3 low group (P = 0.0022; Fig. 1e). Both the stratification by ASAP3 level and the widely used AJCC staging system (P < 0.0001; Fig. S1) showed high prognostic significance. Receiver operating characteristic curves were used to evaluate the sensitivity and specificity of the survival prediction based on the ASAP3 IHC intensity and AJCC stages (Fig. 1f). The area under the curve for the ASAP3‐based prediction was 0.653, whereas the AJCC‐based prediction was 0.702. However, combining both indexes further improved the survival prediction (area under the curve = 0.747). Taken together, these results suggested that ASAP3 might be involved in CRC.
Figure 1.
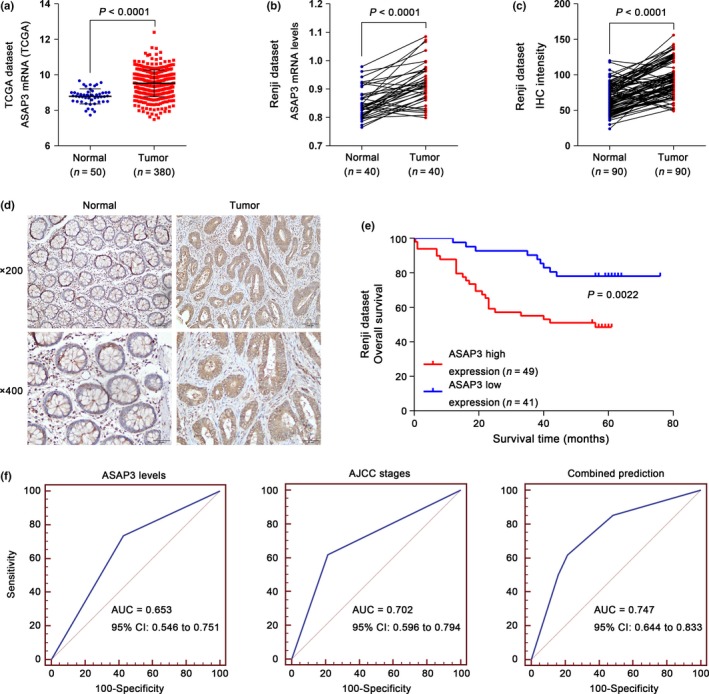
Overexpression of ArfGAP with SH3 domain, ankyrin repeat, and PH domain 3 (ASAP3) was correlated with poor survival in patients with colorectal cancer (CRC). (a, b) ASAP3 expression was significantly increased in CRC tumor tissues when compared with normal tissues from The Cancer Genome Atlas (TCGA) and Renji datasets (P < 0.0001). (c) Statistical analysis of ASAP3 protein expression in normal and CRC tissues as determined by immunohistochemistry (IHC) (n = 90; P < 0.0001). (d) Representative images of ASAP3 expression in 90 paired normal and cancerous colon tissues by IHC analysis from Renji Hospital (Shanghai, China). (e) Kaplan–Meier survival analysis of CRC patients stratified by ASAP3 expression level. (f) Receiver operating characteristic curves for survival prediction using ASAP3 level (left panel), American Joint Committee on Cancer (AJCC) stage (middle panel), or a combination of the two factors (right panel). The area under the curve (AUC) and the corresponding 95% confidence intervals (CI) are shown in the plots.
Table 1.
Clinicopathological characteristics of ArfGAP with SH3 domain, ankyrin repeat and PH domain 3 (ASAP3) expression in patients with colorectal cancer
| Variable | No. of patients | ASAP3 expression | P‐value† | |
|---|---|---|---|---|
| High | Low | |||
| Age | 90 | 67.67 ± 11.11 | 63.29 ± 14.61 | 0.110 |
| Gender | ||||
| Male | 47 | 23 | 24 | 0.273 |
| Female | 43 | 26 | 17 | |
| Location | ||||
| Proximal colon | 47 | 22 | 25 | 0.128 |
| Distal colon | 43 | 27 | 16 | |
| Tumor size, cm3 | ||||
| <30 | 45 | 16 | 29 | <0.001*** |
| ≥30 | 45 | 33 | 12 | |
| T stage | ||||
| T1 + T2 | 13 | 8 | 5 | 0.579 |
| T3 + T4 | 77 | 41 | 36 | |
| Lymph node metastasis | ||||
| Absent | 59 | 25 | 34 | 0.002** |
| Present | 31 | 24 | 7 | |
| Distant metastasis | ||||
| M0 | 87 | 46 | 41 | 0.248 |
| M1a | 3 | 3 | 0 | |
| Histological grade | ||||
| I–II | 76 | 39 | 37 | 0.165 |
| II–III | 14 | 10 | 4 | |
| AJCC stage | ||||
| I–II | 57 | 23 | 34 | <0.001*** |
| III–IV | 33 | 26 | 7 | |
†χ2‐test, significant at <0.05. AJCC, American Joint Committee on Cancer. **P < 0.01, ***P < 0.001.
ASAP3 promotes the proliferation of CRC cells
To explore the effect of ASAP3 on CRC tumorigenesis, we first investigated ASAP3 expression in CRC cell lines by real‐time PCR (Fig. 2a) and Western blot analysis (Fig. 2b,c). Both RKO and SW480 cell lines showed a higher expression of ASAP3, whereas SW1116 showed a lower level. Second, specific siRNAs were used to knockdown ASAP3 expression in RKO and SW480 cell lines. The cell counting kit‐based viability assay detected a significant decrease in the proliferation of RKO and SW480 cells after inhibition of ASAP3 (Fig. 2d,e). However, cDNA‐mediated ectopic expression of ASAP3 increased the proliferation rate of SW1116 cells dramatically (Fig. 2f).
Figure 2.
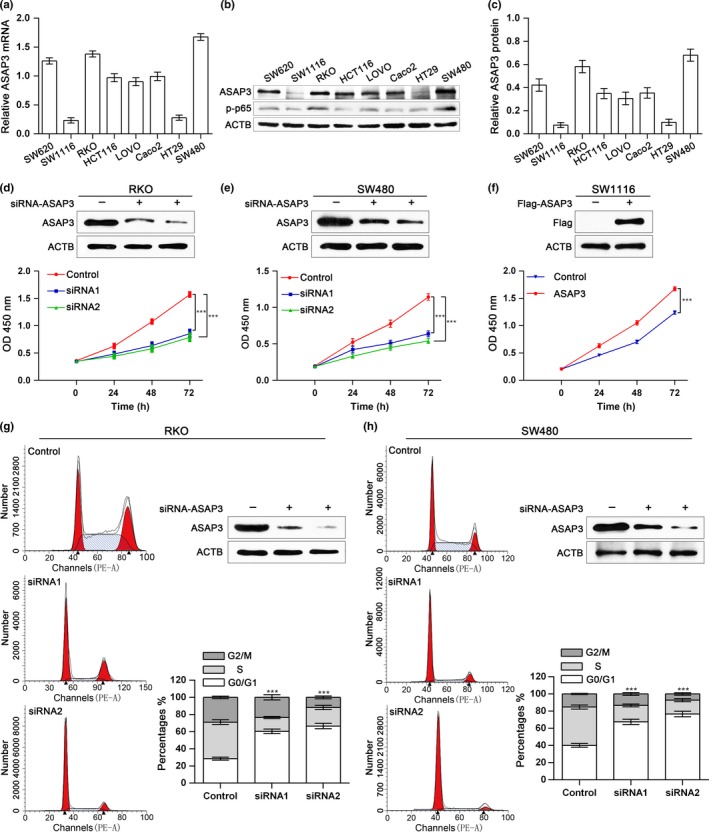
Depletion of ArfGAP with SH3 domain, ankyrin repeat, and PH domain 3 (ASAP3) inhibited proliferation and induced cell cycle arrest of colorectal cancer (CRC) cells. (a) ASAP3 mRNA levels in different CRC cell lines. (b) ASAP3 and phosphorylated (p‐)p65 protein levels in various colon cancer cell lines. (c) Statistical analysis of ASAP3 protein levels in CRC cell lines. (d–f) Knockdown of ASAP3 inhibited CRC cells proliferation in vitro, whereas ectopic expression of ASAP3 accelerated the growth of CRC cells. RKO (d) and SW480 (e) cells were transfected with ASAP3 or control siRNA1/2. SW1116 (f) cells were transfected with ASAP3 cDNA or control cDNA. (g, h) Cell cycle arrest after ASAP3 knockdown in RKO (g) and SW480 (h) CRC cells, as assessed by flow cytometry based on phycoerythrin‐conjugated annexin V (PE‐A). Statistical results and representative images for cell cycle phases are shown (n = 3). ***P < 0.001.
Furthermore, to probe how ASAP3 affects proliferation, a flow cytometry assay was applied to examine the effects of ASAP3 on CRC cell‐cycle progression and apoptosis. As illustrated in Figure 2(g,h), suppression of ASAP3 significantly blocked the cell cycle at the G1/S phase transition, concomitant with an increase in G1 phase. However, phycoerythrin‐conjugated annexin V staining revealed no alteration in cell apoptosis in RKO or SW480 cells after changes in ASAP3 levels (data not shown). These results show that ASAP3 inhibits cell cycle progression, which is consistent with the observation that ASAP3 expression correlates with tumor size.
ASAP3 promotes migration and invasion of colon cancer cells
To determine whether ASAP3 plays a role on CRC cell invasion and migration, wound‐healing and Transwell assays were carried out. The wound‐healing assay, reflecting the migration ability of cells, showed significantly decreased migration after suppression of ASAP3 in RKO and SW480 cells (Fig. 3). Consistently, downregulation of ASAP3 in RKO (Fig. 3b) and SW480 cells (Fig. 3d) revealed a substantial decrease in the number of invasive cells, which was assessed by Transwell migration and invasion assays, suggesting impaired migration and invasion ability for both cell lines. In addition, cDNA‐mediated overexpression of ASAP3 increased migration, as assessed by the wound‐healing assay (Fig. 3e). The Transwell migration and invasion assays showed that upregulation of ASAP3 increased the migration and invasion of SW1116 cells (Fig. 3f). These findings suggested that ASAP3 might play a pivotal role in CRC development.
Figure 3.
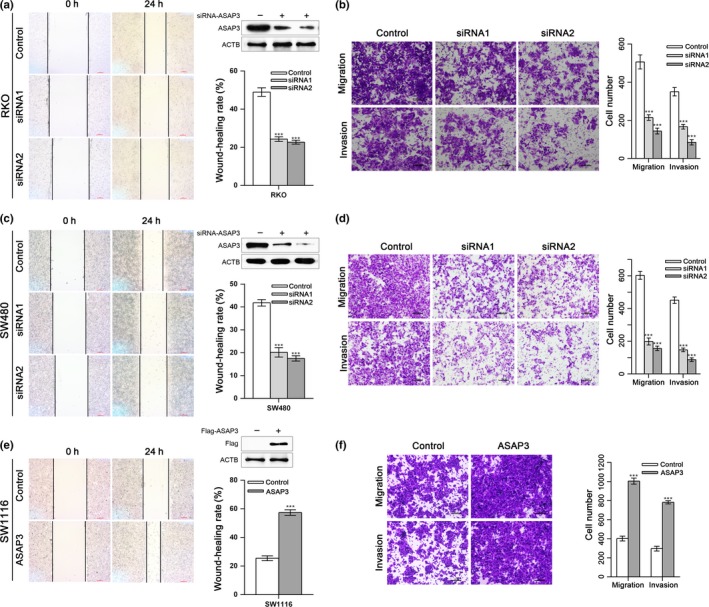
ArfGAP with SH3 domain, ankyrin repeat, and PH domain 3 (ASAP3) regulates migration and invasion of colorectal cancer cells. The expression of ASAP3 was suppressed by specific siRNAs or upregulated by vector‐mediated cDNA overexpression in human colon cancer RKO (a, b), SW480 (c, d), and SW1116 (e, f) cells. (a, c, e) Representative images of wound‐healing assays for RKO, SW480, and SW1116 cells 24 h after treatment. Scale bar = 200 μm. Statistical results of wound‐healing rates are also shown. (b, d, f) Transwell assays were used to quantify cell migration and invasion ability. The images show the number of cells that penetrated the Boyden chambers; top panels show migration ability, and lower panels show invasion assays. Statistical results are based on three independent experiments. Scale bar = 50 μm. ***P < 0.001.
ASAP3 is critical for colon tumorigenesis in vivo
To check whether ASAP3 plays a similar function in colon carcinogenesis in vivo to that determined in vitro, we used a well‐established inflammation‐driven colon carcinoma model based on AOM plus DSS. In contrast to WT mice, Asap3 KO mice showed markedly decreased tumor size and number (Fig. 4). Tumors were located mainly in the middle to distal colon. During the experiment, we discovered that Asap3 +/+ mice suffered frequently from colitis, with bloody stools. Consistent with this, the Asap3 +/+ mice showed more bodyweight loss and colon shortening compared with the Asap3 KO mice (Fig. 4b,d). The colonic tumors in the Asap3 +/+ mice had a higher degree of inflammatory cell infiltration (Fig. 4f), which agreed with a report that the severity of colitis correlates with increased tumor incidence.19 Thus, Asap3 KO decreased tumor incidence. Survival analysis revealed that Asap3 KO mice had a longer overall survival time than WT mice (Fig. 4e). Immunohistochemical staining of colon tissue indicated that there was almost no expression of ASAP3 in the KO mice (Fig. 4g). Immunohistochemical staining showed a significant decrease of cell proliferation markers, including Ki67, in the KO mice (Fig. 4h). Thus, ASAP3 promotes growth of inflammation‐driven cancer in mice.
Figure 4.
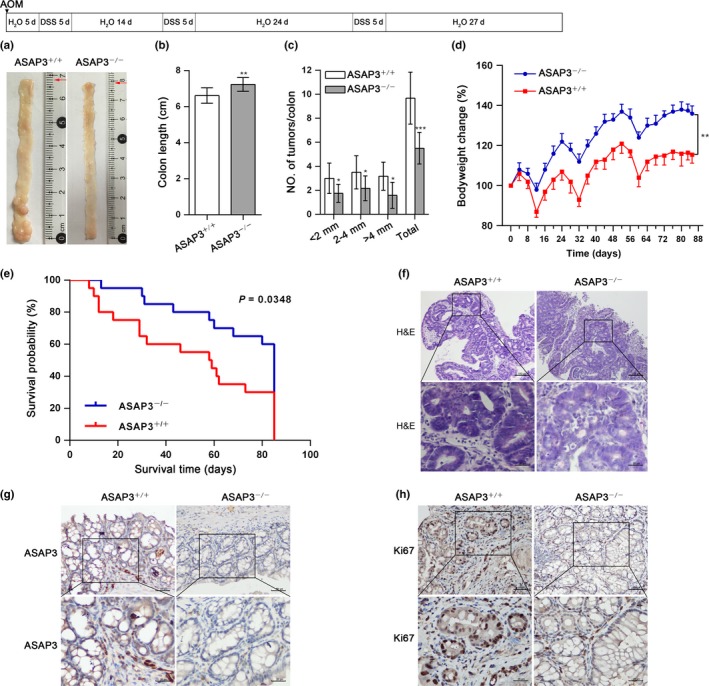
ArfGAP with SH3 domain, ankyrin repeat, and PH domain 3 (ASAP3) is critical for colon tumorigenesis in vivo. (a) Scheme for the procedure of azoxymethane (AOM)‐induced colon carcinogenesis with inflammation‐driven tumor progression. Deletion of Asap3 decreased tumor incidence. Representative images are shown. d, days; DSS, dextran sulfate sodium. Colon length (b) and the number of tumors in the colon and diameters of the tumors (c) are summarized. (d) Alteration in bodyweight was detected throughout the experiment. (e) Survival analysis was executed in Asap3 +/+ and Asap3 −/− mice (n = 20; P < 0.05). (f) Representative H&E staining shows colonic tumor tissue from Asap3 +/+ and Asap3 −/− mice. Scale bar = 100 μm; zoomed sections in the upper images (boxed) are shown in the lower images. (g) Immunohistochemical staining validated the KO efficiency of Asap3 in mice colonic tissues. Scale bar = 50 μm; zoomed sections are shown at the bottom. (h) Representative immunohistochemical staining for Ki67 of colonic tissue from Asap3 +/+ and Asap3 −/− mice. Scale bar = 50 μm; zoomed sections are shown at the bottom. **P < 0.01; ***P < 0.001.
ASAP3 promotes the activation of NF‐κB
To investigate the ASAP3‐associated pathways that influence tumor progression in CRC, we used GSEA analysis. The results showed that the NF‐κB canonical pathway was significantly relevant to ASAP3 (Fig. 5a). Considering that ASAP3 dramatically influenced tumor incidence, based on the inflammation‐driven colon carcinoma mice model, we assessed the effects of changing ASAP3 levels on NF‐κB canonical pathway members. We discovered that overexpression of Flag‐tagged ASAP3 in SW1116 cells resulted in substantially increased phosphorylation of the NF‐κB subunit p65 compared with control cells. Accordingly, phosphorylation of the kinases IKKα and IKKβ, and the inhibitory cytoplasmic NF‐κB chaperone IκBα were improved. Upregulation of ASAP3 also increased the level of NEMO and degradation of IκBα; however, the total levels of IKKα, IKKβ and p65 showed no changes (Fig. 5b). After ASAP3 was reduced in RKO and SW480, the opposite results were obtained. Knockdown of ASAP3 potently inhibited the phosphorylation of p65, IKKα/IKKβ, and IκBα. Reduction of ASAP3 decreased NEMO expression and the degradation of IκBα. There was still no alteration in the total levels of IKKα, IKKβ, and p65 (Fig. 5c). Consistent with these findings, the translocation of p65 into the nucleus was enhanced in ASAP3‐overexpressing cells compared with controls (Fig. 5d). The immunofluorescence also revealed that upregulation of ASAP3 increased the nuclear translocation of p65 (Fig. 5e). Furthermore, we investigated whether ASAP3 also activated the NF‐κB canonical pathway in vivo. Immunohistochemical staining for p‐p65 in the colonic tissue of Asap3 −/− mice revealed a significantly higher decrease of phosphorylated p65 than in Asap3 +/+ mice (Fig. 5f); however, there was no difference in total p65 among them (Fig. 5g). The IHC staining also showed a reduced expression of NEMO in the KO mice (Fig. 5h).
Figure 5.

ArfGAP with SH3 domain, ankyrin repeat, and PH domain 3 (ASAP3) promotes the activation of nuclear factor‐κB (NF‐κB). (a) Gene Set Enrichment Analysis confirmation of significant associations between ASAP3 and the NF‐κB canonical pathway. (b, c) Proteins involved in the NF‐κB canonical pathway were determined by Western blot analysis. Flag‐tagged ASAP3 was transfected into SW1116 cells (b); siRNAs targeting ASAP3 were transfected into RKO and SW480 cells (c). (d) Levels of p65 in nuclear and cytoplasmic extracts were measured by Western blotting in SW1116 cells. (e) Immunofluorescent images show the nuclear accumulation of p65 (red) in SW1116 cells 48 h after transfection with Flag‐tagged ASAP3 (green) plasmids. Cell nucleus was stained with DAPI (blue). Scale bar = 5 μm. (f–h) Representative immunohistochemical staining for phosphorylated (p‐)p65 (f), p65 (g), and NF‐κB essential modulator (NEMO) (h) in colonic tissues from Asap3 +/+ and Asap3 −/− mice. Scale bar = 50 μm; zoomed sections are shown at the bottom. ACTB, β‐actin; IKK, IκB kinase.
ASAP3 interacts with NEMO and could reduce the polyubiquitinylation of NEMO
ASAP3 was observed to alter the expression of NEMO, therefore, we further investigated the interaction between ASAP3 and NEMO. According to the information provided by the UniProt website (http://www.uniprot.org), we constructed mutant NEMO with truncations of amino acids (a.a.) 44–111, a.a. 150–257, a.a. 242–350, and a.a. 251–419. SW1116 cells were transfected with Flag‐tagged ASAP3 and WT HA‐tagged NEMO or mutant NEMO. Co‐immunoprecipitation assays revealed that the NEMO interacted with ASAP3 through the region of a.a. 44–111 (Fig. 6a). We also constructed ASAP3 mutants with deletion of the BAR, PH, or ArfGAP domains. Unfortunately, we failed to construct an ASAP3 mutant with deleted ANK domain. The ASAP3 mutants lacking BAR, PH, or ArfGAP domain did not affect the interaction between ASAP3 and NEMO. Interestingly, we found that ASAP3 could regulate NEMO protein expression, whereas the mRNA expression level of NEMO remained essentially unchanged after suppression or enhancement of ASAP3 expression. Enhanced ASAP3 decreased NEMO polyubiquitination in SW1116 cells (Fig. 6b), whereas ASAP3 KO cells increased the polyubiquitination of NEMO (Fig. 6c), indicating that the ubiquitinated form of NEMO might be required for ASAP3‐mediated NEMO alteration. In addition, treatment with MG132, a proteasome inhibitor, partly restored the NEMO protein levels that were reduced by treatment with the ASAP3 siRNA (Fig. 6d), suggesting that ASAP3 might affect the proteasome‐dependent degradation of polyubiquitinated NEMO.
Figure 6.

ArfGAP with SH3 domain, ankyrin repeat, and PH domain 3 (ASAP3) interact with NF‐κB essential modulator (NEMO) and could reduce the polyubiquitinylation of NEMO. (a) Immunoblot analysis of immunoprecipitates (IP) and whole‐cell lysates (WCL) of SW1116 cells transfected with vectors encoding Flag‐tagged ASAP3 (Flag‐ASAP3) and HA‐tagged WT NEMO (HA‐NEMO(WT)) or mutant NEMO with various truncations. (b) Co‐immunoprecipitation revealed the polyubiquitination (Ub) of NEMO in SW1116 cells transfected with ASAP3 cDNA or control cDNA. (c) Co‐immunoprecipitation showed the polyubiquitination of NEMO in SW480 cells transfected with ASAP3 siRNA or control siRNA. (d) SW480 cells were transfected with ASAP3 siRNA or control siRNA. The cells were treated with DMSO or MG132 (5 μm/L) for 6 h. ACTB, β‐actin.
ASAP3 regulates the NF‐κB downstream genes related to cell cycle and metastasis
To elucidate how ASAP3 affects CRC tumorigenesis and metastasis, bioinformatics analyses were used to identify gene sets related to the cell cycle and metastasis that were positively correlated with ASAP3 upregulation in CRC tissues (Fig. 7). Real‐time PCR and Western blot analysis results showed the levels of cyclin D1, CDK4, and CDK6 decreased after knockdown ASAP3 in RKO and SW480 cells (Fig. 7b,c), which was consistent with G1 phase arrest after knockdown of ASAP3. We then checked the expression of genes that are critical to promote metastasis in the NF‐κB pathway. Consistently, knockdown of ASAP3 contributed to increase the expression of E‐cadherin and depress the expression of mesenchymal markers (N‐cadherin, SNAIL, and TWIST) in RKO and SW480 cells (Fig. 7e,f). Upregulated ASAP3 expression had the opposite effects (Fig. 7g). These results revealed that ASAP3 could increase NF‐κB downstream genes related to cell cycle and metastasis.
Figure 7.

ArfGAP with SH3 domain, ankyrin repeat, and PH domain 3 (ASAP3) regulates the nuclear factor‐κB (NF‐κB) downstream genes related to cell cycle and metastasis in colorectal cancer. (a, d) Gene Set Enrichment Analysis showing a set of activated genes related to cell cycle and metastasis between colorectal cancer tissues and normal tissues. (b, c) Modulating ASAP3 expression significantly affected genes related to the cell cycle. The mRNA and protein levels of target genes of NF‐κB were measured by real‐time PCR and Western blotting after KO of ASAP3 in RKO (b) and SW480 (c) cells. (e–g) Suppression of ASAP3 inhibited NF‐κB pathway downstream genes, which contributed to invasion and migration in RKO (e) and SW480 cells (f). Enhanced ASAP3 had the opposite effect in SW1116 cells (g). **P < 0.01; ***P < 0.001. ACTB, β‐actin; FDR, false discovery rate; NES, normalized enrichment score.
ASAP3 might act as an oncogene in CRC through the NF‐κB pathway
In order to investigate whether ASAP3 regulates cell proliferation and metastasis through the NF‐κB pathway, SW1116 cells were treated with andrographolide after transfection with expression plasmids for ASAP3. Andrographolide, an NF‐κB inhibitor, can reduce the phosphorylation of p65.20 Treatment with andrographolide inhibited the increases of cyclin D1, CDK4, and CDK6 that were induced by overexpression of ASAP3 in SW1116 cells (Fig. 8a). Similarly, andrographolide also decreased the expression of genes that promoted cell metastasis (Fig. 8b). The activation of NF‐κB contributed to ASAP3‐associated cell proliferation and metastasis in CRC. ASAP3 might act as an oncogene in CRC through the NF‐κB pathway (Fig. 8c).
Figure 8.

ArfGAP with SH3 domain, ankyrin repeat, and PH domain 3 (ASAP3) might act as an oncogene in colorectal cancer through the nuclear factor‐κB (NF‐κB) pathway. (a, b) Western blot analyses of the genes related to cell cycle (a) and cell metastasis (b) in SW1116 cells transfected with control or Flag‐ASAP3 expression plasmids for 48 h, followed by treatment with DMSO or andrographolide (120 mg/L) for 24 h. (c) Schematic illustration of ASAP3‐mediated NF‐κB activation in colorectal cancer. ACTB, β‐actin; IKK, IκB kinase; NEMO, NF‐κB essential modulator; P, phosphorylation; Ub, ubiquitination.
Discussion
Previous research has indicated the expression of ASAP3 in multiple tumors.16, 21 However, the biological function and clinical implication of ASAP3 in CRC remain poorly understood. Our present study revealed a possible functional role for ASAP3 in colon tumorigenesis.
We discovered that ASAP3 was amplified in CRC tissues compared with adjacent normal tissues, and upregulation of ASAP3 was associated with poor prognosis of patients with CRC. The results were consistent with a previous study that found that increased ASAP3 contributes to poor clinical outcomes in patients with non‐small‐cell lung cancer and hepatocellular cancer.16 Moreover, higher ASAP3 significantly enhanced tumor volume and lymph node metastasis, which suggested that ASAP3 might be a promising marker for the diagnosis and prognosis of patients with CRC.
Bioinformatics analysis revealed that the canonical NF‐κB, cell cycle, and metastasis pathways were positively associated with alterations of ASAP3 in CRC. We confirmed this analysis using in vivo and in vitro experiments. ASAP3 increased the phosphorylation of p65 and facilitated p65 translocation into the nucleus through the canonical pathway, without altering the total levels of p65. Previous studies noted that ASAP3 maintained the amount of GTP‐bound “active” ARF6 through GTP hydrolysis.13, 22 ARF6 functions effectively in the ERK and P38MAPK pathways; 23, 24 however, we detected no significant change in the MAPK pathway (Fig. S2). The IHC staining of colon tissue showed decreased phosphorylation of NF‐κB in Asap3 KO mice but no difference in total NF‐κB (Fig. 5g). Nuclear factor‐κB was shown to be constitutively active in both colon cancer cell lines and colorectal tissues.25, 26 The canonical IKK/NF‐κB activation pathway is indispensable for colonic tumor development.27, 28 The mechanism of NF‐κB involvement in oncogenesis is the promotion of cell proliferation and suppression of apoptosis, in which NF‐κB promotes cyclin D1 transcription.29, 30 Depletion of ASAP3 markedly inhibited growth in cultured cells through decreases in cyclin D1 and CDK4/6, which induced G1 phase arrest. However, ASAP3 had no effect on apoptosis in CRC. The AOM/DSS colon tumor model was used to verify the function of ASAP3, and we found that ASAP3 KO dramatically decreased tumorigenesis. A previous study also showed that ASAP3 contributed to cell proliferation.31
Nuclear factor‐κB also plays a pivotal role in tumor metastasis and induces epithelial–mesenchymal transition through activation of transcription factors, such as TWIST and SNAIL.32, 33 We also detected that ASAP3 promoted invasion and migration in vitro. Upregulation of ASAP3 decreased E‐cadherin and increased mesenchymal markers (N‐cadherin, SNAIL, and TWIST), whereas ASAP3 knockdown had the reverse effect. However, no distinct difference in invasion or migration was found between the WT and Asap3 −/−, as the AOM‐DSS model is seldom invasive.19 Previous research showed that ASAP3 mediates invasion through control of molecular plasticity and plasma membrane dynamics, and that the ARF6‐GTPase cycle is also required for the invasive potential.14, 15, 34 However, we showed that ASAP3 could promote invasion and migration through modulation of the expressions of NF‐κB downstream genes. The treatment with andrographolide suggested that the activation of NF‐κB contributed to ASAP3‐associated cell proliferation and metastasis in colorectal cancer.
A diffuse cytoplasmic distribution of ASAP3 was evident in cultured cells evaluated by immunofluorescence. In vitro and in vivo experiments showed that ASAP3 could change the expression of NEMO. Co‐immunoprecipitation analysis also revealed that NEMO bound to ASAP3 through the region of a.a 44–111; however, we did not identify the region by which ASAP3 interact with NEMO. ASAP3 reduced the polyubiquitylation of NEMO and might participate in the proteasome‐dependent degradation of polyubiquitinated NEMO. It was reported that NEMO can regulate NF‐κB activation by binding to Lys63‐linked polyubiquitin chains or linear polyubiquitin chains.35, 36, 37 Moreover, Lys27‐mediated polyubiquitylation of NEMO is critical for activation of NF‐κB.38, 39, 40 Conjugated to K27‐linked ubiquitin, NEMO can promote degradation of NEMO and interfere with the activation of NF‐κB. However, further study is needed to illustrate how ASAP3 binds to NEMO and how ASAP3 affects the polyubiquitylation of NEMO.
In conclusion, our results showed that ASAP3 plays a pivotal role in colonic carcinogenesis, and it might be a potential prognostic marker and therapeutic target in CRC.
Disclosure Statement
The authors have no conflict of interest.
Supporting information
Fig. S1. Kaplan–Meier survival analysis of colorectal cancer patients with different American Joint Committee on Cancer (AJCC) tumor stages (I–II versus III–IV) and ArfGAP with SH3 domain, ankyrin repeat, and PH domain 3 (ASAP3) expression level (high versus low).
Fig. S2. ArfGAP with SH3 domain, ankyrin repeat, and PH domain 3 (ASAP3) has no effect on the MAPK pathway.
Table S1. Primers for real‐time RT‐PCR.
Table S2. Univariate Cox regression analysis of potential prognostic factors for colorectal cancer patients.
Table S3. Multivariate Cox regression analysis of potential prognostic factors for colorectal cancer patients.
Data S1. Additional materials and methods.
Acknowledgments
This study was supported by grants from the National Natural Science Foundation of China (Grant Nos. 81572326, 81322036, 30971330, 81272383, 81421001, 31371420, 81320108024, 81000861, and 81302090), the Shanghai “Oriental Scholars” project (Grant No. 2013XJ), Shanghai Municipal Education Commission – Gaofeng Clinical Medicine Grant Support (Grant No. 20152514), National Key Technology Support Program (Grant No. 2015BAI13B07), the Doctoral Innovation Fund Projects from Shanghai Jiao Tong University School of Medicine (Grant No. BXJ201419 to J.Q.), and the Natural Science Foundation of Shang Hai (SWY13ZR1457200).
Cancer Sci 108 (2017) 1544–1555
Funding Information
National Natural Science Foundation of China (81572326, 81322036, 30971330, 81272383, 81421001, 31371420, 81320108024, 81000861, 81402390, and 81302090); Shanghai “Oriental Scholars” project (2013XJ); Shanghai Municipal Education Commission – Gaofeng Clinical Medicine Grant Support (20152514); National Key Technology Support Program (2015BAI13B07); the Doctoral Innovation Fund Projects from Shanghai Jiao Tong University School of Medicine (BXJ201419); the Natural Science Foundation of Shang Hai (SWY13ZR1457200); Shanghai Youth Science and technology talents sailing program(14YF1402500).
Contributor Information
Jie Xu, Email: xujieletter@gmail.com.
Jing‐Yuan Fang, Email: fangjingyuan_new@163.com.
References
- 1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin 2011; 61: 69–90. [DOI] [PubMed] [Google Scholar]
- 2. Yamashita K, Watanabe M. Clinical significance of tumor markers and an emerging perspective on colorectal cancer. Cancer Sci 2009; 100: 195–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vaiopoulos AG, Kostakis ID, Koutsilieris M, Papavassiliou AG. Colorectal cancer stem cells. Stem Cells 2012; 30: 363–71. [DOI] [PubMed] [Google Scholar]
- 4. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell 2010; 140: 883–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yu LL, Yu HG, Yu JP, Luo HS, Xu XM, Li JH. Nuclear factor‐kappaB p65 (RelA) transcription factor is constitutively activated in human colorectal carcinoma tissue. World J Gastroenterol 2004; 10: 3255–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Karin M. Nuclear factor‐kappaB in cancer development and progression. Nature 2006; 441: 431–6. [DOI] [PubMed] [Google Scholar]
- 7. Kim HJ, Hawke N, Baldwin AS. NF‐kappaB and IKK as therapeutic targets in cancer. Cell Death Differ 2006; 13: 738–47. [DOI] [PubMed] [Google Scholar]
- 8. Perkins ND. The diverse and complex roles of NF‐kappaB subunits in cancer. Nat Rev Cancer 2012; 12: 121–32. [DOI] [PubMed] [Google Scholar]
- 9. Smale ST. Hierarchies of NF‐kappaB target‐gene regulation. Nat Immunol 2011; 12: 689–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Perkins ND. Integrating cell‐signalling pathways with NF‐kappaB and IKK function. Nat Rev Mol Cell Biol 2007; 8: 49–62. [DOI] [PubMed] [Google Scholar]
- 11. Li F, Zhang J, Arfuso F et al NF‐kappaB in cancer therapy. Arch Toxicol 2015; 89: 711–31. [DOI] [PubMed] [Google Scholar]
- 12. Zhao X, Wang D, Liu X et al Phosphorylation of the Bin, Amphiphysin, and RSV161/167 (BAR) domain of ACAP4 regulates membrane tubulation. Proc Natl Acad Sci USA 2013; 110: 11023–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fang Z, Miao Y, Ding X et al Proteomic identification and functional characterization of a novel ARF6 GTPase‐activating protein, ACAP4. Mol Cell Proteomics 2006; 5: 1437–49. [DOI] [PubMed] [Google Scholar]
- 14. Yu X, Wang F, Liu H et al ACAP4 protein cooperates with Grb2 protein to orchestrate epidermal growth factor‐stimulated integrin beta1 recycling in cell migration. J Biol Chem 2011; 286: 43735–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ha VL, Bharti S, Inoue H et al ASAP3 is a focal adhesion‐associated Arf GAP that functions in cell migration and invasion. J Biol Chem 2008; 283: 14915–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fan C, Tian Y, Miao Y et al ASAP3 expression in non‐small cell lung cancer: association with cancer development and patients’ clinical outcome. Tumour Biol 2014; 35: 1489–94. [DOI] [PubMed] [Google Scholar]
- 17. Luo Y, Kong F, Wang Z et al Loss of ASAP3 destabilizes cytoskeletal protein ACTG1 to suppress cancer cell migration. Mol Med Rep 2014; 9: 387–94. [DOI] [PubMed] [Google Scholar]
- 18. Subramanian A, Tamayo P, Mootha VK et al Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA 2005; 102: 15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Greten FR, Eckmann L, Greten TF et al IKKbeta links inflammation and tumorigenesis in a mouse model of colitis‐associated cancer. Cell 2004; 118: 285–96. [DOI] [PubMed] [Google Scholar]
- 20. Wang ZM, Kang YH, Yang X et al Andrographolide radiosensitizes human esophageal cancer cell line ECA109 to radiation in vitro. Dis Esophagus 2016; 29: 54–61. [DOI] [PubMed] [Google Scholar]
- 21. Okabe H, Furukawa Y, Kato T, Hasegawa S, Yamaoka Y, Nakamura Y. Isolation of development and differentiation enhancing factor‐like 1 (DDEFL1) as a drug target for hepatocellular carcinomas. Int J Oncol 2004; 24: 43–8. [PubMed] [Google Scholar]
- 22. Donaldson JG. Multiple roles for Arf6: sorting, structuring, and signaling at the plasma membrane. J Biol Chem 2003; 278: 41573–6. [DOI] [PubMed] [Google Scholar]
- 23. Knizhnik AV, Kovaleva OV, Komelkov AV et al Arf6 promotes cell proliferation via the PLD‐mTORC1 and p38MAPK pathways. J Cell Biochem 2012; 113: 360–71. [DOI] [PubMed] [Google Scholar]
- 24. Tague SE, Muralidharan V, D'Souza‐Schorey C. ADP‐ribosylation factor 6 regulates tumor cell invasion through the activation of the MEK/ERK signaling pathway. Proc Natl Acad Sci USA 2004; 101: 9671–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lind DS, Hochwald SN, Malaty J et al Nuclear factor‐kappa B is upregulated in colorectal cancer. Surgery 2001; 130: 363–9. [DOI] [PubMed] [Google Scholar]
- 26. Kojima M, Morisaki T, Sasaki N et al Increased nuclear factor‐kB activation in human colorectal carcinoma and its correlation with tumor progression. Anticancer Res 2004; 24: 675–81. [PubMed] [Google Scholar]
- 27. Karin M, Lin A. NF‐kappaB at the crossroads of life and death. Nat Immunol 2002; 3: 221–7. [DOI] [PubMed] [Google Scholar]
- 28. Maeda S, Chang L, Li ZW, Luo JL, Leffert H, Karin M. IKKbeta is required for prevention of apoptosis mediated by cell‐bound but not by circulating TNFalpha. Immunity 2003; 19: 725–37. [DOI] [PubMed] [Google Scholar]
- 29. Guttridge DC, Albanese C, Reuther JY, Pestell RG, Baldwin AS Jr. NF‐kappaB controls cell growth and differentiation through transcriptional regulation of cyclin D1. Mol Cell Biol 1999; 19: 5785–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gupta S, Afaq F, Mukhtar H. Involvement of nuclear factor‐kappa B, Bax and Bcl‐2 in induction of cell cycle arrest and apoptosis by apigenin in human prostate carcinoma cells. Oncogene 2002; 21: 3727–38. [DOI] [PubMed] [Google Scholar]
- 31. Randazzo PA, Weiss O, Kahn RA. Preparation of recombinant ADP‐ribosylation factor. Methods Enzymol 1992; 219: 362–9. [DOI] [PubMed] [Google Scholar]
- 32. Huber MA, Azoitei N, Baumann B et al NF‐kappaB is essential for epithelial‐mesenchymal transition and metastasis in a model of breast cancer progression. J Clin Invest 2004; 114: 569–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lopez‐Novoa JM, Nieto MA. Inflammation and EMT: an alliance towards organ fibrosis and cancer progression. EMBO Mol Med 2009; 1: 303–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ding X, Deng H, Wang D et al Phospho‐regulated ACAP4‐Ezrin interaction is essential for histamine‐stimulated parietal cell secretion. J Biol Chem 2010; 285: 18769–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wu CJ, Conze DB, Li T, Srinivasula SM, Ashwell JD. Sensing of Lys 63‐linked polyubiquitination by NEMO is a key event in NF‐kappaB activation. Nat Cell Biol 2006; 8: 398–406. [DOI] [PubMed] [Google Scholar]
- 36. Ni CY, Wu ZH, Florence WC et al Cutting edge: K63‐linked polyubiquitination of NEMO modulates TLR signaling and inflammation in vivo. J Immunol 2008; 180: 7107–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tokunaga F, Sakata S, Saeki Y et al Involvement of linear polyubiquitylation of NEMO in NF‐kappaB activation. Nat Cell Biol 2009; 11: 123–32. [DOI] [PubMed] [Google Scholar]
- 38. Liu J, Han C, Xie B et al Rhbdd3 controls autoimmunity by suppressing the production of IL‐6 by dendritic cells via K27‐linked ubiquitination of the regulator NEMO. Nat Immunol 2014; 15: 612–22. [DOI] [PubMed] [Google Scholar]
- 39. Ashida H, Kim M, Schmidt‐Supprian M, Ma A, Ogawa M, Sasakawa C. A bacterial E3 ubiquitin ligase IpaH9.8 targets NEMO/IKKgamma to dampen the host NF‐kappaB‐mediated inflammatory response. Nat Cell Biol 2010; 12: 66–73; sup pp 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Arimoto K, Funami K, Saeki Y et al Polyubiquitin conjugation to NEMO by triparite motif protein 23 (TRIM23) is critical in antiviral defense. Proc Natl Acad Sci USA 2010; 107: 15856–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Kaplan–Meier survival analysis of colorectal cancer patients with different American Joint Committee on Cancer (AJCC) tumor stages (I–II versus III–IV) and ArfGAP with SH3 domain, ankyrin repeat, and PH domain 3 (ASAP3) expression level (high versus low).
Fig. S2. ArfGAP with SH3 domain, ankyrin repeat, and PH domain 3 (ASAP3) has no effect on the MAPK pathway.
Table S1. Primers for real‐time RT‐PCR.
Table S2. Univariate Cox regression analysis of potential prognostic factors for colorectal cancer patients.
Table S3. Multivariate Cox regression analysis of potential prognostic factors for colorectal cancer patients.
Data S1. Additional materials and methods.