

Summary
Immune homeostasis requires the tight, tissue‐specific control of the different CD4+ Foxp3+ regulatory T (Treg) cell populations. The cadherin‐binding inhibitory receptor killer cell lectin‐like receptor G1 (KLRG1) is expressed by a subpopulation of Treg cells with GATA3+ effector phenotype. Although such Treg cells are important for the immune balance, especially in the gut, the role of KLRG1 in Treg cells has not been assessed. Using KLRG1 knockout mice, we found that KLRG1 deficiency does not affect Treg cell frequencies in spleen, mesenteric lymph nodes or intestine, or frequencies of GATA3+ Treg cells in the gut. KLRG1‐deficient Treg cells were also protective in a T‐cell transfer model of colitis. Hence, KLRG1 is not essential for the development or activity of the general Treg cell population. We then checked the effects of KLRG1 on Treg cell activation. In line with KLRG1's reported inhibitory activity, in vitro KLRG1 cross‐linking dampened the Treg cell T‐cell receptor response. Consistently, lack of KLRG1 on Treg cells conferred on them a competitive advantage in the gut, but not in lymphoid organs. Hence, although absence of KLRG1 is not enough to increase intestinal Treg cells in KLRG1 knockout mice, KLRG1 ligation reduces T‐cell receptor signals and the competitive fitness of individual Treg cells in the intestine.
Keywords: Foxp3, intestine, KLRG1, T‐cell receptor
Abbreviations
- KLRG1
killer cell lectin‐like receptor G1
- KO
knockout
- TCR
T‐cell receptor
- Treg
CD4+ Foxp3+ regulatory T cells
Introduction
Foxp3‐expressing regulatory T (Treg) cells are essential for immune homeostasis, especially in the gut. However, Treg cells can also contribute to pathogenesis, most notably by inhibiting protective immune responses to tumours in mice and humans.1 Therefore Treg cells must be kept under strict control. We and others have previously shown that intestinal Treg cells can be regulated differently to Treg cells in other organs.2, 3, 4, 5, 6 Gut Treg cells express high levels of CD103 and/or the killer cell lectin‐like receptor G1 (KLRG1), the two known heterotypic receptors for E‐cadherin, a key component of epithelial intercellular junctions.7, 8, 9 Both receptors have been associated with activated Treg cells, and KLRG1 has been specifically associated with a population of effector Treg cells expressing GATA3+ in the gut.10
KLRG1 is an inhibitory receptor found on several immune subsets, most notably on natural killer cells and activated CD8+ T cells, and it is used as a marker for exhausted CD8+ T cells.11, 12 It has been described to bind cadherin family members, including E‐, N‐ and R‐cadherins.13 Our knowledge about KLRG1 function comes mainly from its interactions with E‐cadherin, as most cellular analyses were performed with E‐cadherin‐expressing cells. Cross‐linking T‐cell receptor (TCR) and KLRG1 has been shown to have an inhibitory role on TCR‐mediated signals.14 Like many inhibitory receptors, KLRG1 is expressed by Treg cells but, in contrast to the inhibitory receptor CTLA‐4, it is not found on every Treg cell but only on a subpopulation of effector‐like Treg cells.8, 9, 10 Although KLRG1 is often used as a marker for effector Treg cells, it has not been investigated whether and how it affects Treg cells.
Here we analyse the Treg cell population from KLRG1‐deficient animals. In line with the lack of phenotype of KLRG1 knockout (KO) mice, we found that KLRG1‐deficient animals have normal frequencies of Treg cells, also in the gut. The frequencies of the effector GATA3+ gut Treg population, which exhibit high levels of KLRG1, are similarly unaltered in KLRG1 KO mice. We also show that KLRG1 engagement with an antibody reduces Treg cell accumulation in response to TCR signals in vitro, and that under competitive settings KLRG1 limits Treg cell accumulation in the gut in a cell‐autonomous manner. Hence, although KLRG1 is dispensable for the functioning of the total Treg population, it can affect the survival potential of individual Treg cells in the gut.
Materials and methods
Mouse strains
Wild‐type C57BL/6, congenic B6.SJL‐Cd45, C57BL/6 Klrg1−/− 15 and C57BL/6 Rag2 −/− mice were kept and bred under specific pathogen‐free conditions at the animal facility of the Max‐Planck Institute of Immunobiology and Epigenetics. All experiments were approved by the institutional review board of the Max Planck Institute of Immunobiology and Epigenetics and the local government in Freiburg.
Isolation of leucocytes from the lamina propria
Leucocytes from the lamina propria were isolated as described previously.4, 16 Briefly, small intestine and colon were removed and cleaned. After washing with ice‐cold PBS, intestines were washed twice in Hanks’ balanced salt solution containing 5 mm EDTA and 10 mm HEPES at 37° to remove the epithelial cell layer. The tissue was then minced finely and digested three times in Hanks’ balanced salt solution containing dispase (5 units/ml; BD Biosciences, Franklin Lakes, NJ, USA), collagenase IV (0·5 mg/ml; Worthington, Lakewood, NJ) and DNaseA (0·5 mg/ml; AppliChem, Darmstadt, Germany), at 37° with constant shaking. Supernatants were collected and lymphocytes were enriched after a gradient centrifugation using buffered Percoll (GE Healthcare, Freiburg, Germany).
Antibodies and flow cytometry
Single‐cell suspensions were stained in 96‐well plates (106 cells/well). The following conjugated antibodies were purchased from eBioscience (Affymetrix, Inc., Santa Clara, CA, USA): TCR‐β (H57‐597), CD3 (145‐2C11), CD4 (GK 1.5), KLRG1 (2F1), CD103 (2E7), CD45.1 (A20), CD45.2 (104), CD25 (PC61.5), Foxp3 (FJK‐16s) and Nur77 (12.14). Intracellular staining was performed with the eBioscience permeabilization and fixation kit. Anti‐Bcl‐2 (3F11) was purchased from BD Biosciences. Dead cells were excluded by staining with Fixable Viability Dye (eBioscience). All flow cytometry experiments were acquired using a BD LSR II cytometer or LSR Fortessa (BD Biosciences). flow jo Version 8.8.7 (Treestar Inc., Ashland, OR) was used for data analysis.
Bone marrow chimeras
Recipient CD45.1+ B6.SJL‐Cd45 mice were irradiated (2 × 300 Rad) and reconstituted 12 hr later using intravenous injection of CD45.1+ B6.SJL‐Cd45 bone marrow cells together with bone marrow cells from KLRG1 KO CD45.2+ or wild‐type C57BL/6 CD45.2+ mice (a total of 107 bone marrow cells were injected in a ratio of roughly 20 : 80 CD45.2+ : CD45.1+ cells). Two groups of mice were generated (KLRG1 KO + congenic bone marrow, control C57BL/6 + congenic bone marrow). Mice were analysed 6–8 months after reconstitution.
Prevention of colitis
Naive and Treg cells from C57BL/6 or KLRG1 KO mice were sorted as described.4 Briefly, CD4+ T cells from spleens of C57BL/6 and KLRG1 KO mice were enriched with a CD4+ isolation kit (Dynabeads® Untouched™ Mouse CD4 Cells, Thermo Fisher Scientific, Waltham, MA, USA), followed by FACS sorting of naive CD4+ CD45RBhi CD25− cells and Treg cell‐enriched CD4+ CD25+ T cells. Sort was performed with a cell sorter BD Aria (BD Biosciences).
C57BL/6 RAG2−/− were injected intraperitoneally with either 4 × 105 naive CD4+ CD45RBhi CD25− T cells or 4 × 105 naive CD4+ CD45RBhi CD25− plus 105 regulatory CD4+ CD25+ T cells. Mice were killed for colitis assessment when symptoms of clinical disease (significant weight loss or diarrhoea) became apparent, or after 4 months. Intestinal samples were fixed in paraformaldehyde and stained with haematoxylin & eosin, and intestinal inflammation was assessed. Inflammation was assessed as described previously.4 Briefly, each sample was graded semi‐quantitatively from 0 to 3 for the four following criteria: degree of epithelial hyperplasia and goblet cell depletion; leucocyte infiltration in the lamina propria; area of tissue affected; and the presence of markers of severe inflammation such as crypt abscesses, submucosal inflammation and ulcers. Scores for each criterion were added to give an overall inflammation score for each sample of 0–12. The total colonic score was calculated as the average of the individual scores from the sections of proximal colon, mid‐colon and distal colon. Images were obtained using Plan‐Neofluar 5 × 0·15 or 10 × 0·30 objectives on a Zeiss Axioplan II with axiovs40 V4.8.2.0 software connected to a camera AxioCam MR5 (Carl Zeiss, Jena, Germany).
Regulatory T cell culture
Total CD4+ CD25+ or CD4+ CD25+ KLRG1+ and CD4+ CD25+ KLRG1− cells were sorted from wild‐type C57BL/6 (CD45.2+) or B6.SJL‐Cd45 (CD45.1+) spleens. Flat‐bottom 96‐well plates were pre‐coated with 5 μg/ml purified anti‐ mouse CD3ε (145‐2C11, Biolegend, San Diego, CA, USA) with or without 5 μg/ml grade purified anti‐mouse KLRG1 (2F1, eBioscience). After 8 hr or 3 days of incubation at 37° in RPMI‐1640 containing 10% fetal calf serum and 1 U/ml interleukin‐2 (PeproTech, Hamburg, Germany), cells were harvested and analysed by flow cytometry.
To test in vitro suppression, 105 CD4+ CD25− T sorted T cells were labelled with CFSE (eBioscience) according to the manufacturer's instructions and cultured in U‐shaped 96‐well plates with 105, 5 × 104 or 104 CD4+ CD25+ KLRG1− or CD4+ CD25+ KLRG1+ cells with anti‐CD3 anti‐CD28 coated beads (ThermoFisher) for 3 days without exogenous interleukin‐2. The frequency of divided cells was calculated using flowjo software.
Immunofluorescence
For immunofluorescence, cryosections were fixed in ice‐cold acetone/methanol (75%/25%) and then blocked with 10% normal rat serum and a Streptavidin and Biotin Blocking Kit (Invitrogen Life Technologies, Darmstadt, Germany) for biotinylated antibodies. Sections were then incubated with Alexa Fluor® 647‐conjugated rat anti‐mouse Foxp3 antibody (clone FJK‐16s; Affymetrix eBioscience, Santa Clara, CA), phycoerythrin‐conjugated or biotinylated hamster anti‐mouse KLRG1 antibody (clone 2F1, Biolegend) followed by streptavidin‐Cy3 conjugate (Jackson Immuno Research, Suffolk, UK) and mouse anti‐mouse E‐cadherin (clone 36, BD Biosciences, Heidelberg, Germany) followed by goat anti‐mouse antibody conjugated to Alexa Fluor® 488‐conjugated anti‐rat E‐cadherin antibody (clone DECMA‐1, eBioscience). Stained sections were mounted in Fluoromount‐DAPI (Invitrogen, Carlsbad, CA) and analysed with a fluorescence microscope (Axio Imager.Z1 with axiovision 4·8 software, Zeiss) using Plan‐Apochromat 20 × 0·8 or Plan‐Neofluar 40 × 0·75 objectives connected to a camera AxioCam MRm (Carl Zeiss).
Statistical analysis
Statistical analysis was performed using graphpad prism using a two‐tailed unpaired Student's t‐test when only two groups were tested, or analysis of variance with Bonferroni Post‐test where more than two groups were tested. Differences were considered statistically significant when P < 0·05.
Results
KLRG1 is not essential for Treg cell generation or function
We checked the expression of the two E‐cadherin receptors on Treg cells. As described previously,9 KLRG1 and CD103 can be co‐expressed on Treg cells (Fig. 1a) and are found mostly in the gut (Fig. 1b,c); however, the frequency of KLRG1+ Treg cells was lower than the frequency of CD103+ Treg cells in all organs tested (Fig. 1b), and the fraction of KLRG1+ among Treg cells declined with age (Fig. 1d). On the other hand, the per cell levels of KLRG1 on intestinal Treg, as measured by flow cytometry, were higher than for other intestinal KLRG1‐expressing populations, such as CD8+ and ILC2 cells (Fig. 1e). The high KLRG1 expression suggests that KLRG1 can play a role in Treg cells.
Figure 1.
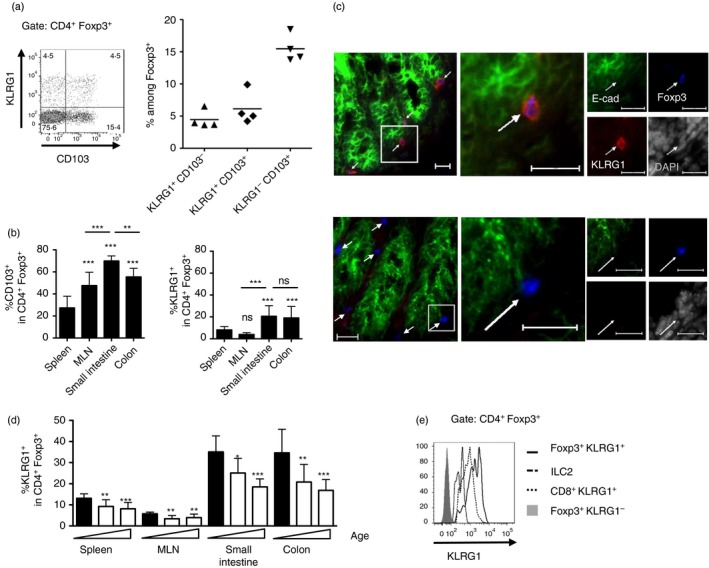
Killer cell lectin‐like receptor G1 (KLRG1) expression on gut regulatory T (Treg) cells. (a) KLRG1 and CD103 on spleen CD4+ Foxp3+ cells. Data show a representative FACS plot (left) and frequencies among Foxp3+ cells (right); each dot represents an individual 6‐week‐old mouse. Horizontal bars represent arithmetical mean. (b) KLRG1 expression in tissue Treg cells. Graph shows frequency of CD103+ and KLRG1+ cells among CD4+ Foxp3+ in spleen, mesenteric lymph nodes (MLN), and intestinal lamina propria from colon and small intestine from 24‐week‐old wild‐type mice. Significance is compared with the spleen (no bars) or between indicated subsets, data pooled from at least nine mice per group from four different experiments. Bar graphs represent arithmetical mean + SD. (c) Top : KLRG1+ Foxp3+ cells in the colonic lamina propria of a 3‐week‐old mouse (top) and control KLRG1 KO mouse (bottom). E‐cadherin marks epithelial cells. Bar, 20 μm. (d) Frequency of KLRG1+ cells among Foxp3+ in spleen, MLN, and intestinal lamina propria from colon and small intestine from or 2‐, 6‐ and 24‐week‐old mice. Significance is compared with 2‐week‐old mice (left, filled bar) for the same organ. Data pooled from at least four mice per group from four different experiments. Bar graphs represent arithmetical mean + SD. (e) Comparison of KLRG1 levels on different KLRG1+ small intestinal lymphoid populations. Solid thick line: CD4+ Foxp3+ KLRG1+ cells, thin dotted line: CD8+ KLRG1+ cells, dashed line: TCR‐β − GATA3hi KLRG1+ ILC2 cells. Grey histogram, control CD4+ Foxp3+ KLRG1− cells. (b) and (d) show mean + SD.
We therefore assessed the frequencies of Treg cells in KLRG1 KO mice, comparing them with KLRG1‐expressing KLRG1 heterozygote littermates. KLRG1 KO mice15 do not develop spontaneous inflammation, suggesting that KLRG1 is not essential for Treg cell generation. As KLRG1 inhibits TCR signals,14 and TCR is important for Treg cell accumulation,17, 18 we hypothesized that KLRG1 KO mice might have altered Treg cell frequencies. However, KLRG1 KO mice did not show an increase in intestinal Treg cells (Fig. 2a). When we assessed the frequencies of CD103+ Treg cells, we found a small increase in the colon of KLRG1 KO mice compared with control BL/6 (Fig. 2b). This increase did not take place in the spleen, mesenteric lymph nodes or small intestine.
Figure 2.
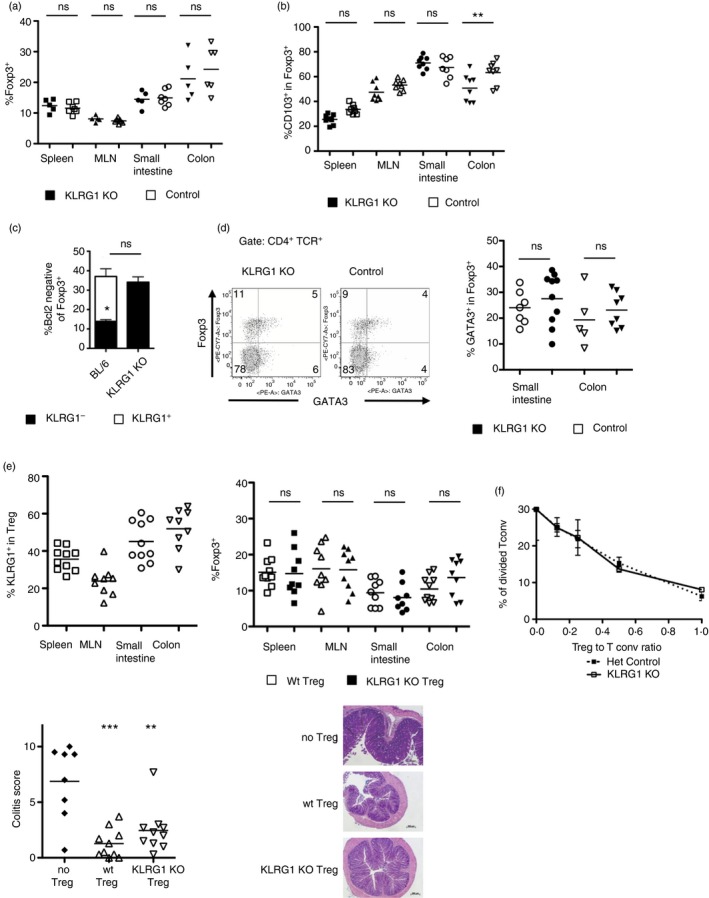
Killer cell lectin‐like receptor G1 (KLRG1) is not essential for regulatory T (Treg) cell accumulation or function. (a) Frequency of Foxp3+ cells among CD4+ T lymphocytes in 3 week‐old KLRG1 knockout (KO) and littermate KLRG1‐heterozygote mice. Each point corresponds to an individual mouse. Horizontal bars represent arithmetical mean. (b) Frequency of CD103+ cells among Foxp3+ CD4+ T lymphocytes in KLRG1 KO mice and wild‐type C57BL/6 controls. Each point corresponds to an individual mouse. Data are pooled from three independent analyses. Horizontal bars represent arithmetical mean. (c) Frequency of Bcl‐2low cells among small intestinal CD4+ Foxp3+ cells in 2‐week‐old KLRG1 KO mice and wild‐type C57BL/6 controls. For C57BL/6 mice, Bcl‐2low cells are divided in KLRG1+ (clear) and KLRG1− (black). ns, the frequency of total Bcl2low cells is not significantly different between KLRG1 KO and controls. *P < 0·05 for the frequency of Bcl2− KLRG1− in control mice compared with the frequency of Bcl2− cells in KLRG1 KO mice. Graph shows mean + SD of three independent experiments with at least two mice per experiment per group. (d) Representative plots (left) and individual values (right) of GATA3 versus Foxp3 expression among intestinal lamina propria CD4+ T cells from 3‐week‐old KLRG1 KO and heterozygote littermate controls. Each dot represents an individual mouse. FACS plot shows small intestinal cells. Horizontal bars represent arithmetical mean. (e) KLRG1 KO Treg cells can prevent colitis in a T‐cell transfer model. RAG2−/− mice were transferred with naive CD45.1+ CD4+ T cells on their own or together with CD45.2+ wild‐type or CD45.2+ KLRG1 KO CD4+ CD25+ cells. Top left, frequency of KLRG1+ among CD4+ CD45.1− Foxp3+ cells after transfer of wild‐type CD4+ CD25+. Top right, frequency of CD45.1− Foxp3+ cells among CD4+ cells after transfer of naive CD45.1+ CD4+ T cells together with CD45.2+ wild‐type (empty symbols) or CD45.2+ KLRG1 KO (filled symbols) CD4+ CD25+ cells. Bottom left, colitis scores of mice transferred with naive CD45.1+ CD4+ T cells on their own (filled circles) or together with CD45.2+ wild‐type (empty upward triangles) or CD45.2+ KLRG1 KO (empty downward triangles) CD4+ CD25+ cells. Bottom right, representative pictures of histology sections from colon. Data are pooled from three independent experiments. Horizontal bars represent arithmetical mean. (f) Treg cells from KLRG1 KO and control heterozygous littermates suppress CD4+ CD25− T‐cell proliferation. CFSE‐labelled CD25− CD4+ (Tconv) T cells were cultured with KLRG1 KO or control CD25+ T cells at different ratios. The graph shows the frequency of divided CFSE+ cells after 3 days of culture. Data show mean ± SD of three replicates and they are representative of two independent experiments. **P < 0·01; ***P < 0·001; ns, not significant. Bars show mean.
We then checked if KLRG1 was necessary for the effector Treg subpopulation. Treg cells can be divided into ‘naive’ and ‘effector’ following different markers such as Bcl2. In young mice, KLRG1+ Treg cells account for the majority of intestinal Bcl2− effector Treg cells (Fig. 2c). We found that the frequency of intestinal Bcl2− Treg cells was not reduced in the gut of KLRG1 KO mice, indicating that KLRG1 is not essential for the generation of the KLRG1+ Bcl2− Treg cell subset (Fig. 2c). Similarly, the frequencies of GATA3+ Treg cells, a subset characterized by high KLRG1 expression,10 were comparable in KLRG1 KO mice and control littermates (Fig. 2d).
We then tested the ability of KLRG1 KO Treg cells to prevent inflammation in an established model of T‐cell transfer‐mediated colitis. In this system, where inflammation is absent, KLRG1 is expressed by Treg cells in the protected mice (see refs 10 and 19, and Fig. 2e); however, KLRG1 KO Treg cells accumulated and prevented colitis in a similar way to control Treg cells (Fig. 2e). KLRG1 KO Treg cells also suppressed T‐cell proliferation in vitro as efficiently as control Treg cells (Fig. 2f). Hence, KLRG1 is dispensable for the protective ability of Treg cells. Together, our data show that KLRG1 is dispensable for Treg cell generation and function.
KLRG1 ligation reduces TCR signals and Treg cell accumulation in vitro
KLRG1 ligation by 2F1 antibody or by E‐cadherin inhibits TCR signalling in a reporter cell line model.14 As KLRG1+ Treg cell lines are not available, we used ex vivo T cells to assess the effects of KLRG1 ligation by 2F1 antibody. We used Nur77 as a proxy for the strength of TCR stimulation.20 We found that, as found for the reporter cell line, KLRG1 ligation during stimulation with anti‐CD3 reduces TCR signalling intensity in KLRG1+ but not in KLRG1− Treg cells (Fig. 3a). TCR signals are necessary for Treg cell accumulation,17, 18 and we found this to be the case also in our in vitro system (Fig. 3b). Accordingly, in vitro KLRG1 ligation reduced KLRG1+ Treg cell accumulation, but it did not affect KLRG1− Treg cells. (Fig. 3c). These data indicate that KLRG1 can negatively impact Treg cell accumulation.
Figure 3.
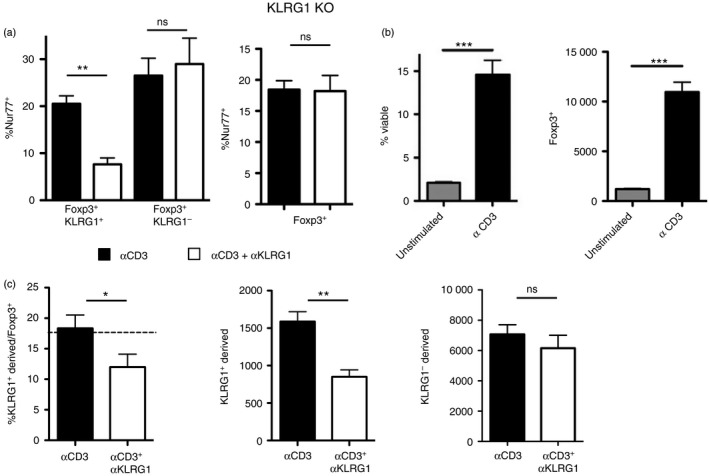
Killer cell lectin‐like receptor G1 (KLRG1) ligation reduces regulatory T (Treg) cell accumulation in vitro. (a) Frequency of Nur77+ cells among Foxp3+ CD4+ T cells after 8 hr of culture with plate‐bound anti‐CD3 (solid bars) or anti‐CD3 plus anti‐KLRG1 (empty bars). Left histogram: C57BL/6 T cells were gated on Foxp3+ KLRG1+ cells (left) or Foxp3+ KLRG1− cells (right). Right histogram: Foxp3+ cells from KLRG1 knockout (KO) mice are shown as control. Data are representative of four independent experiments for C57BL/6 cells and two experiments for KLRG1 KO cells. (b) Frequency of viable cells (left) and total numbers of Foxp3+ T cells (right) after 2 days of culture of sorted CD25+ T cells with or without plate‐bound anti‐CD3. Viability was assessed with viability dye. (c) Frequency of CD45.1− cells (derived from KLRG1+) among Foxp3+ T cells (left) and total number of CD45.1− and CD45.1+ (derived from KLRG1−) Foxp3+ T cells (centre) after 3 days of culture with plate‐bound anti‐CD3 or anti‐CD3 plus anti‐KLRG1. Sorted CD45.1− KLRG1+ and CD45.1+ KLRG1− Foxp3+ T cells were cultured for 3 days with plate‐bound anti‐CD3 or anti‐CD3 plus anti‐KLRG1 at a starting ratio of approximately 1 : 4 (dashed line). Data are representative of two independent experiments. *P < 0·05; **P < 0·01; ***P < 0·001; ns, not significant. Graph bars show mean + SD.
KLRG1 limits gut Treg cell accumulation in competitive settings
The in vitro data showing that KLRG1 ligation reduces Treg cell accumulation were at odds with our initial finding that KLRG1 KO mice do not have increased Treg cell frequencies in the gut. The size of the Treg cell pool is tightly controlled by interleukin‐2 and other factors, and Treg cells have a very high homeostatic capacity.21 We reasoned that homeostatic processes could obscure the effect of KLRG1 on Treg cells. To assess this point, we checked the competitive ability of KLRG1 KO Treg cells in mixed bone marrow chimeras where the cells could be tracked with congenic markers. To this end we generated two sets of chimeras, one with KLRG1 KO and another one with control C57BL/6 bone marrow cells, mixed with congenic bone marrow cells. KLRG1 KO and control C57BL/6 cells were CD45,2+, whereas congenic bone marrow, as well as potential residual host cells, were CD45,1+ in both chimeras. These chimeras had comparable frequencies of CD4+ T cells and Foxp3+ Treg cells among CD4+ in spleen, mesenteric lymph nodes and colon (Fig. 4a). When we checked the origin of the cells by comparing the frequency of CD45.2+ cells in the two different chimeras, we found that KLRG1 KO‐derived Treg cells accumulated better than control Treg cells in the colon, but not in the spleen or mesenteric lymph nodes (Fig. 4b). The data show that KLRG1 deficiency on Treg cells confers on them a competitive advantage to accumulate in the gut, supporting a cell‐intrinsic role for KLRG1 in gut Treg cell homeostasis. The effect was not only organ‐restricted, but also specific for Treg cells, as CD4+ Foxp3− cell accumulation was not strongly influenced by the lack of KLRG1 (Fig. 4b). Hence, KLRG1 plays a cell‐intrinsic role in gut Treg cell homeostasis, but it does not alter the size of the total Treg cell pool.
Figure 4.
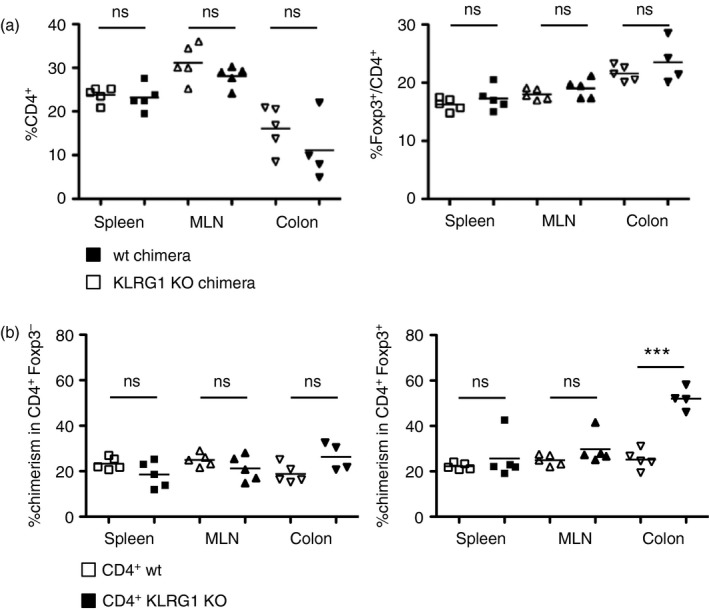
Killer cell lectin‐like receptor G1 (KLRG1) reduces regulatory T (Treg) cell competitive fitness in the gut (a) Frequencies of CD4+ and Foxp3+ among CD4+ cells in KLRG1 knockout (KO) or control mixed bone marrow chimeras 6 months after reconstitution. KLRG1 KO or KLRG1‐sufficient control bone marrow (both CD45.2+) was mixed with CD45.1+ bone marrow and injected into irradiated CD45.1+ recipients. Mice were analysed 6 months after reconstitution. Horizontal bars represent arithmetical mean. (b) Frequencies of KLRG1 KO (filled symbols) or control (empty symbols) CD45.2+ chimerism among CD4+ Foxp3− and CD4+ Foxp3+ cells in the bone marrow chimeras shown in (a). Comparison refers to frequency of CD45.2+ cells in the two different chimeras, CD45.2+ cells are either KLRG1 KO or control‐derived. Each point corresponds to an individual mouse. Horizontal bars represent arithmetic mean. Data are representative of three independent experiments. ***P < 0·001; **P < 0·01; *P < 0·05; ns, not significant.
Discussion
Inhibitory receptors are key to the functioning of the immune system. Their absence can have drastic effects such as with the inhibitory receptor CTLA‐4, where KO mice suffer from early lethal autoimmunity.22 However, in many cases the absence of the inhibitory receptors is compensated by homeostatic factors and does not lead to an obvious phenotype, making it difficult to identify the function of the involved receptors.
KLRG1 is one of these inhibitory molecules that does not lead to a severe phenotype when absent. Widely used as a marker for exhausted CD8+ T cells, it contains several inhibitory motifs and reduces TCR signals and cytotoxic activity in CD8+ and natural killer cells.14, 23 We show here that KLRG1 ligation also reduces TCR signals on freshly isolated Treg cells. In other settings, KLRG1 cross‐linking with the 2F1 antibody showed similar effects to KLRG1 ligation with membrane‐bound E‐cadherin,14, 23 suggesting that 2F1 is a good model to test KLRG1 function. In line with the reduced TCR signals, KLRG1 cross‐linking also resulted in lower cell numbers after in vitro culture. Hence, KLRG1 expression can have a significant effect on TCR‐mediated effects in Treg cells.
These results are in contrast to the normal frequencies and suppressive activity of Treg cells in KLRG1 KO mice, as Treg cell homeostasis and function are dependent on TCR signals. It may be argued that KLRG1 is only expressed on a minor fraction of Treg cells under steady‐state conditions, which may render its effects less apparent. However, KLRG1 KO mice also have normal levels of effector Treg cells, a population that includes a sizeable number of KLRG1+ cells. Differences between KLRG1 KO and control Treg cells are only found under competitive settings, where KLRG1‐deficiency confers an advantage to Treg cells. Interestingly, this advantage was observed in the gut, but not in the spleen or in the mesenteric lymph nodes. Together, our data show that the size of the Treg cell pool and the frequency of effector Treg cells in the tissue are independent of KLRG1. However, KLRG1 ligation diminishes the competitive fitness of individual Treg cells and so modulates the composition of the effector Treg cell pool.
An outstanding question is which antigen‐presenting cells provide a ligand for KLRG1. Biochemical analyses have shown that KLRG1 can bind several cadherins, and it might potentially distinguish between ligands, as not all binding regions are conserved between the cadherins.24 Both E‐cadherin and N‐cadherin have been reported to act as ligands for KLRG1 in reporter cell assays, but only E‐cadherin has been shown to induce KLRG1‐mediated inhibition in functional assays so far. E‐cadherin is typically expressed by epithelial cells, which represent the most abundant source of E‐cadherin in the gut. They can also express MHC‐II, so they could potentially act as antigen presenters to Treg cells. However, populations of MHC‐II+ myeloid cells expressing E‐cadherin that could ligate both TCR and KLRG1 are also present in the intestine.25 The relevance of different antigen‐presenting populations in controlling gut Treg cells remains to be determined.
Treg cells are key supervisors of appropriate immune responses, and their role is especially important in the intestine. The local composition of the Treg cell population in the gut is not only controlled by food and commensal microbiota,26 but also by intrinsic factors. Our data here show that KLRG1 affects the competitive fitness of individual intestinal Treg cells, suggesting that KLRG1 could be used as a target to manipulate gut Treg cells.
Author contributions
H.M., A.B. and M.B. performed the experiments involving KLRG1 KO mice, A.B. and A.I. performed the in vitro cultures, K.S. prepared histology samples for colitis scoring; H.P. analysed data and contributed essential reagents, A.I. directed research and wrote the paper with A.B. and input from all the authors.
Disclosures
The authors signal no conflicting interest.
Acknowledgements
We thank members of the Izcue laboratory for helpful discussions and comments on the manuscript, P. Rauf for help during initial work, and K. Maloy for critical reading of the manuscript. We are indebt to Rebecca Hussong for outstanding technical help. We thank Inke Krupka‐Dyachenko, Melanie Pfunder, Uta Stauffer, Senta Burkart, Armstrong Ogboro and the rest of the mouse facility staff for excellent animal care, and A. Wuerch, S. Hobitz and K. Schuldes for cell sorting. This work was supported by the Bundesministerium für Bildung und Forschung (BMBF 01 EO 0803) and by the Max Planck Society. H.M. was supported by a Walter Hitzig fellowship of the Centre of Chronic Immunodeficiency, University Hospital Freiburg. A.B. is a member of the IMPRS‐MCB, a joint international PhD programme of the Max Planck Institute of Immunobiology and Epigenetics and the University of Freiburg, Germany.
H.M. and A.B. contributed equally to this work.
References
- 1. Nishikawa H, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Curr Opin Immunol 2014; 27:1–7. [DOI] [PubMed] [Google Scholar]
- 2. Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly YM, et al The microbial metabolites, short‐chain fatty acids, regulate colonic Treg cell homeostasis. Science 2013; 341():569–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Barnes MJ, Griseri T, Johnson AM, Young W, Powrie F, Izcue A. CTLA‐4 promotes Foxp3 induction and regulatory T cell accumulation in the intestinal lamina propria. Mucosal Immunol 2013; 6:324–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Izcue A, Hue S, Buonocore S, Arancibia‐Carcamo CV, Ahern PP, Iwakura Y, et al Interleukin‐23 restrains regulatory T cell activity to drive T cell‐dependent colitis. Immunity 2008; 28:559–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ohnmacht C, Park JH, Cording S, Wing JB, Atarashi K, Obata Y, et al The microbiota regulates type 2 immunity through RORγt+ T cells. Science 2015; 349:989–93. [DOI] [PubMed] [Google Scholar]
- 6. De Simone M, Arrigoni A, Rossetti G, Gruarin P, Ranzani V, Politano C, et al Transcriptional landscape of human tissue lymphocytes unveils uniqueness of tumor‐infiltrating T Regulatory cells. Immunity 2016; 45:1135–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lehmann J, Huehn J, de la Rosa M, Maszyna F, Kretschmer U, Krenn V, et al Expression of the integrin αEβ7 identifies unique subsets of CD25+ as well as CD25– regulatory T cells. Proc Natl Acad Sci USA 2002; 99:13031–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Beyersdorf N, Ding X, Tietze JK, Hanke T. Characterization of mouse CD4 T cell subsets defined by expression of KLRG1. Eur J Immunol 2007; 37:3445–54. [DOI] [PubMed] [Google Scholar]
- 9. Feuerer M, Hill JA, Kretschmer K, von Boehmer H, Mathis D, Benoist C. Genomic definition of multiple ex vivo regulatory T cell subphenotypes. Proc Natl Acad Sci USA 2010; 107:5919–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schiering C, Krausgruber T, Chomka A, Frohlich A, Adelmann K, Wohlfert EA, et al The alarmin IL‐33 promotes regulatory T‐cell function in the intestine. Nature 2014; 513:564–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lupar E, Brack M, Garnier L, Laffont S, Rauch KS, Schachtrup K, et al Eomesodermin expression in CD4+ T cells restricts peripheral Foxp3 induction. J Immunol 2015; 195:4742–52. [DOI] [PubMed] [Google Scholar]
- 12. Manetti M, Ibba‐Manneschi L, Liakouli V, Guiducci S, Milia AF, Benelli G, et al The IL1‐like cytokine IL33 and its receptor ST2 are abnormally expressed in the affected skin and visceral organs of patients with systemic sclerosis. Ann Rheum Dis 2010; 69:598–605. [DOI] [PubMed] [Google Scholar]
- 13. Ito M, Maruyama T, Saito N, Koganei S, Yamamoto K, Matsumoto N. Killer cell lectin‐like receptor G1 binds three members of the classical cadherin family to inhibit NK cell cytotoxicity. J Exp Med 2006; 203:289–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rosshart S, Hofmann M, Schweier O, Pfaff AK, Yoshimoto K, Takeuchi T, et al Interaction of KLRG1 with E‐cadherin: new functional and structural insights. Eur J Immunol 2008; 38:3354–64. [DOI] [PubMed] [Google Scholar]
- 15. Grundemann C, Schwartzkopff S, Koschella M, Schweier O, Peters C, Voehringer D, et al The NK receptor KLRG1 is dispensable for virus‐induced NK and CD8+ T‐cell differentiation and function in vivo . Eur J Immunol 2010; 40:1303–14. [DOI] [PubMed] [Google Scholar]
- 16. Sanos SL, Diefenbach A. Isolation of NK cells and NK‐like cells from the intestinal lamina propria. Methods Mol Biol 2010; 612:505–17. [DOI] [PubMed] [Google Scholar]
- 17. Levine AG, Arvey A, Jin W, Rudensky AY. Continuous requirement for the TCR in regulatory T cell function. Nat Immunol 2014; 15:1070–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vahl JC, Drees C, Heger K, Heink S, Fischer JC, Nedjic J, et al Continuous T cell receptor signals maintain a functional regulatory T cell pool. Immunity 2014; 41:722–36. [DOI] [PubMed] [Google Scholar]
- 19. Worthington JJ, Kelly A, Smedley C, Bauche D, Campbell S, Marie JC, et al Integrin αvβ8‐mediated TGF‐β activation by effector regulatory T cells is essential for suppression of T‐cell‐mediated inflammation. Immunity 2015; 42:903–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bremser A, Brack M, Izcue A. Higher sensitivity of Foxp3+ Treg compared to Foxp3– conventional T cells to TCR‐independent signals for CD69 induction. PLoS One 2015; 10:e0137393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gene Ontology Consortium . Gene Ontology Consortium: going forward. Nucleic Acids Res 2015;43(Database issue):D1049–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, et al Lymphoproliferative disorders with early lethality in mice deficient in CTLA‐4. Science 1995; 270:985–8. [DOI] [PubMed] [Google Scholar]
- 23. Plitas G, Konopacki C, Wu K, Bos PD, Morrow M, Putintseva EV, et al Regulatory T cells exhibit distinct features in human breast cancer. Immunity 2016; 45:1122–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li Y, Hofmann M, Wang Q, Teng L, Chlewicki LK, Pircher H, et al Structure of natural killer cell receptor KLRG1 bound to E‐cadherin reveals basis for MHC‐independent missing self recognition. Immunity 2009; 31:35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Siddiqui KR, Laffont S, Powrie F. E‐cadherin marks a subset of inflammatory dendritic cells that promote T cell‐mediated colitis. Immunity 2010; 32:557–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hegazy AN, Powrie F. MICROBIOME. Microbiota RORgulates intestinal suppressor T cells. Science 2015; 349:929–30. [DOI] [PubMed] [Google Scholar]