

Abstract
Chromosomal translocation occurs in some cancer cells, which results in the expression of aberrant oncogenic fusion proteins that include BCR‐ABL in chronic myelogenous leukemia (CML). Inhibitors of ABL tyrosine kinase, such as imatinib and dasatinib, exhibit remarkable therapeutic effects, although emergence of drug resistance hampers the therapy during long‐term treatment. An alternative approach to treat CML is to downregulate the BCR‐ABL protein. We have devised a protein knockdown system by hybrid molecules named Specific and Non‐genetic inhibitor of apoptosis protein [IAP]‐dependent Protein Erasers (SNIPER), which is designed to induce IAP‐mediated ubiquitylation and proteasomal degradation of target proteins, and a couple of SNIPER(ABL) against BCR‐ABL protein have been developed recently. In this study, we tested various combinations of ABL inhibitors and IAP ligands, and the linker was optimized for protein knockdown activity of SNIPER(ABL). The resulting SNIPER(ABL)‐39, in which dasatinib is conjugated to an IAP ligand LCL161 derivative by polyethylene glycol (PEG) × 3 linker, shows a potent activity to degrade the BCR‐ABL protein. Mechanistic analysis suggested that both cellular inhibitor of apoptosis protein 1 (cIAP1) and X‐linked inhibitor of apoptosis protein (XIAP) play a role in the degradation of BCR‐ABL protein. Consistent with the degradation of BCR‐ABL protein, the SNIPER(ABL)‐39 inhibited the phosphorylation of signal transducer and activator of transcription 5 (STAT5) and Crk like proto‐oncogene (CrkL), and suppressed the growth of BCR‐ABL‐positive CML cells. These results suggest that SNIPER(ABL)‐39 could be a candidate for a degradation‐based novel anti‐cancer drug against BCR‐ABL‐positive CML.
Keywords: BCR‐ABL, dasatinib, E3 ubiquitin ligase, LCL161, protein knockdown
Chronic myelogenous leukemia (CML) is a myeloproliferative disorder characterized by the Philadelphia (Ph) chromosome in cancer cells.1 The Ph chromosome results from a translocation between the long arms of chromosomes 9 and 22, t(9;22)(q34;q11),2 and a fusion gene, Bcr‐Abl, which encodes a constitutively active protein tyrosine kinase, was generated by this translocation.3, 4, 5 Several small molecule tyrosine kinase inhibitors (TKI) have been developed for CML treatment. Imatinib is the first‐generation TKI against the BCR‐ABL, and it competitively binds to the ATP‐binding site, resulting in the inhibition of cell proliferation.6, 7 Although imatinib is currently used as the first‐line therapy for CML patients, a significant number of patients develop resistance, which is commonly attributable to point mutations in the tyrosine kinase domain of BCR‐ABL protein. To overcome the imatinib resistance, second‐generation TKI such as dasatinib have been developed.8, 9 Such second generation TKI are capable of saving most CML patients; however, drug resistance also occurs against them. Therefore, novel drug candidates with different mechanisms of action are required.
Besides inhibition of tyrosine kinase activity, downregulation of the BCR‐ABL protein should have a similar therapeutic potential. Although the downregulation can be achieved by genetic knockdown technologies, such as antisense oligonucleotides and small interfering RNA, they remain clinically challenging.10, 11 An alternative approach is a protein knockdown technology, which induces the degradation of target proteins via the ubiquitin–proteasome system (UPS), and a series of active compounds, including Specific and Non‐genetic Inhibitor of apoptosis protein [IAP]‐dependent Protein Erasers (SNIPER)12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23 and Proteolysis Targeting Chimeras (PROTAC)24, 25, 26, 27, 28, 29, 30, 31, 32, 33 have been developed. They are hybrid molecules composed of two different ligands connected by a linker; one ligand is for a target protein and the other is for E3 ubiquitin ligases. Accordingly, these molecules are expected to crosslink the target protein and E3 ubiquitin ligases in cells, resulting in the ubiquitylation and subsequent degradation of the target protein via the UPS.
In this study, we synthesized a series of SNIPER(ABL) compounds by combination of various ABL inhibitors and IAP ligands, and the linker was optimized for protein knockdown activity against BCR‐ABL protein. The resulting SNIPER(ABL)‐39, in which an ABL inhibitor dasatinib is conjugated to an IAP ligand LCL161 derivative, shows a potent activity to degrade the BCR‐ABL protein, involving cIAP1 and XIAP for the degradation. We also showed that SNIPER(ABL) inhibited the phosphorylation of BCR‐ABL substrate STAT5 (signal transducer and activator of transcription 5) and CrkL (Crk like proto‐oncogene), and suppressed the growth of BCR‐ABL‐positive CML cells.
Materials and Methods
Design and synthesis of SNIPER(ABL)
We designed the hybrid molecules, SNIPER(ABL), in which an ABL inhibitor is linked to a ligand of cIAP1 via a linker containing a variable polyethylene glycol (PEG) unit. The chemical synthesis and physicochemical data on the SNIPER(ABL) are provided in the Supporting Information (Data S1 and Scheme [Link], [Link], [Link], [Link], [Link], [Link]).
Reagents
Tissue culture plastics were purchased from Greiner Bio‐One (Tokyo, Japan). Imatinib was purchased from LC Laboratories (Woburn, MA, USA). MLN7243 was purchased from Active Biochem (Maplewood, NJ, USA). Cycloheximide (CHX) was from Sigma‐Aldrich (St. Louis, MO, USA). His‐tagged ABL1 protein (full length, P3049) was from Life Technologies (Carlsbad, CA, USA). Brij(R) 35 solution was obtained from Merck Millipore (Billerica, MA, USA). Terbium‐labeled streptavidin (Tb‐SA) was from Cisbio (Codolet, France). Recombinant His‐tagged human XIAP (BIR3, Asn252‐Thr356, 895‐XB‐050) protein was purchased from R&D Systems (Minneapolis, MN, USA). Recombinant His‐tagged human cIAP1 (BIR3, Leu250‐Gly350) and cIAP2 (BIR3, Gln238‐Ser349) proteins were expressed in E. coli and purified using a Ni‐NTA column and a gel filtration chromatography. FITC‐labeled Smac peptide (FITC‐Smac, AVPIAQK(5‐FAM)‐NH2)34 was synthesized in Scrum (Tokyo, Japan). BODIPY‐FL labeled dasatinib (BODIPY‐dasatinib)35 was synthesized as described previously.
Cell culture and shRNA transfection
Human CML (K562, KCL‐22 and KU812), acute lymphoblastic leukemia (SK‐9), promyelocytic leukemia (HL60), acute T‐lymphoblastic leukemia (MOLT‐4) and T cell leukemia (Jurkat) were cultured in Roswell Park Memorial Institute (RPMI)‐1640 medium (Sigma‐Aldrich) containing 10% FBS (Gibco) and 50 μg/mL kanamycin (Sigma‐Aldrich). SK‐9 cells were kindly provided by Dr Okabe (Tokyo Medical University, Tokyo, Japan).36 KCL‐22 and KU812 cells were obtained from Japanese Collection of Research Bioresources (JCRB, Osaka, Japan) Cell Bank (JCRB1317 and JCRB0104). For short hairpin RNA (shRNA)‐mediated gene silencing, gene‐specific hairpin oligonucleotides were ligated into pSUPER.retro.puro vector (OrigoEngine, Seattle, WA, USA). The shRNA sequences used in this study were:
cIAP1‐#1 (5′‐CCGCCGAATTGTCTTTGGTGCTTCTCGAGAAGCACCAAAGACAATTCGGCTTTTTT‐3′);
cIAP1‐#2 (5′‐CCGCTGCGGCCAACATCTTCAAACTCGAGTTTGAAGATGTTGGCCGCAG CTTTTTT‐3′);
XIAP‐#1 (5′‐CCAGCTGTAGATAGATGGCAATACTCGAGTATTGCCATCTATCTACAGCTTTTTTT‐3′);
XIAP‐#2 (5′‐CCGCACTCCAACTTCTAATCAAACTCGAGTTTGATTAGAAGTTGGAGTGCTTTTTT‐3′);
LacZ (5′‐CCGCTACACAAATCAGCGATTTCGCTTCCTGTCACGAAATCGCTGATTTGTGTAGCTTTTTT‐3′).
K562 cells (1 × 107) were transfected by electroporation (GENE PULSER II; Bio Rad, Hercules, CA, USA) with 20 μg pSUPER/shcIAP1‐#1, shcIAP1‐#2, shXIAP‐#1, shXIAP‐#2 or shLacZ. Transfected cells were incubated in 2 mL RPMI‐1640 supplemented with 10% FBS and 100 μg/mL of kanamycin in a 6‐well dish for 24 h, and the cells were washed in PBS, and further incubated in 10 mL RPMI‐1640 supplemented with 10% FBS, 100 μg/mL of kanamycin and 2.5 μg/mL of puromycin (Sigma‐Aldrich) in a 10‐cm dish for 48 h.
Western blot analysis
Cells were collected and lysed in a lysis buffer (0.5% TritonX‐100, 0.01 M Tris‐HCl [pH 7.5], 0.15 M NaCl, Complete Mini protease inhibitor cocktail [Roche Applied Science, Indianapolis, IL, USA] and PhosStop phosphatase inhibitor cocktail [Roche Applied Science]). Protein concentration was measured by the BCA method (Thermo Scientific, Rockford, IL, USA) and an equal amount of protein lysate was separated by SDS‐PAGE, transferred to polyvinylidene difluoride membranes (Millipore), and analyzed by western blot using an appropriate antibody. The immunoreactive proteins were visualized using Clarity Western ECL substrate (Bio‐Rad), and their light emission was quantified with a LAS‐3000 lumino‐image analyzer (Fuji, Tokyo, Japan). The following antibodies were used: anti‐cAbl rabbit polyclonal antibody (pAb) (#2862), anti‐XIAP rabbit pAb (#2042), anti‐phospho‐cAbl rabbit pAb (#3009), anti‐STAT5 rabbit pAb (#9363), anti‐phospho‐STAT5 rabbit pAb (#9359), anti‐CrkL mouse monoclonal antibody (mAb) (#3182) and anti‐phospho‐CrkL rabbit pAb (#3181) (Cell Signaling Technology, Danvers, MA, USA); anti‐β‐tubulin (ab6046) rabbit pAb (Abcam, Cambridge, UK); anti‐GAPDH rabbit pAb (sc‐25778 HRP) and anti‐Cyclin B1 mouse mAb (ac‐245 HRP) (Santa Cruz, Dallas, TX, USA); anti‐MCL1 mouse mAb (559027) (BD Biosciences, San Jose, CA, USA); anti‐β‐actin mouse mAb (A2228) (Sigma‐Aldrich); and anti‐cIAP1 goat pAb (AF8181) (R&D Systems).
Time‐resolved FRET assay and data analysis
Time‐resolved FRET (TR‐FRET) assays were carried out using 384‐well white flat‐bottomed plates (Greiner Bio‐One, Frickenhausen, Germany) and the signal was measured using an EnVision Multilabel Plate Reader (PerkinElmer, Waltham, MA, USA). The solution in each well was excited with a laser (λ = 337 nm) reflected by a dichroic mirror (D400/D505 (Perkin Elmer) and fluorescence from terbium (Tb) and BODIPY or FITC were detected through two emission filters (CFP 486 [Perkin Elmer] for Tb, Emission 515 [Perkin Elmer] for BODIPY and FITC). Assay buffer utilized in this study was composed of 50 mM HEPES (pH 7.2–7.5), 10 mM MgCl2, 1 mM EGTA, 0.1 mM DTT and 0.01% (v/v) Brij(R) 35. All assays were carried out at room temperature in triplicate or quadruplicate formats.
The percentage of inhibition by test compounds was calculated according to Equation (1).
| (1) |
where T is the value of the wells containing test compounds, and μH and μL are the mean values of the 0 and 100% inhibition control wells, respectively. The half maximal inhibitory concentration (IC50) of test compounds was calculated by fitting the data with the logistic equation using GraphPad Prism 5 (GraphPad Software, La Jolla, CA, USA) or XLfit (IDBS, Guildford, UK).
Measurement of inhibitor activity of ABL1 inhibitor that bind to the ATP binding site
Before addition to the assay plate, threefold concentrations of His‐ABL1 protein, Tb‐SA and biotinylated anti‐His antibody were mixed in the assay buffer and incubated for over 1 h at room temperature. Several concentrations of test inhibitors dissolved in the assay buffer were dispensed in the assay plate. Subsequently, the ABL/antibody/Tb‐SA premix was dispensed to each well and incubated for 120 min at room temperature. Reaction was initiated by addition of assay buffer containing 13.5 nM BODIPY‐dasatinib. The plate was incubated for 30 min at room temperature and the TR‐FRET signal was measured using an EnVision Multilabel Plate Reader. The final concentrations of Tb‐SA, biotinylated anti‐His, ABL1 protein and BODIPY‐dasatinib were 0.2, 0.4, 0.38 and 4.5 nM, respectively. The values of the 0 and 100% controls were the signals obtained in the absence and presence of 3 μM dasatinib, respectively.
Measurement of inhibitory activity of IAP/peptide interaction
His‐IAP proteins (XIAP, cIAP1 or cIAP2), FITC‐Smac, Tb‐SA and biotinylated anti‐His antibody (Life Technologies) were mixed in the assay buffer and incubated for over 1 h at room temperature before addition to the assay plate. Several concentrations of test inhibitors were dispensed in the assay plate and the protein‐probe premix was dispensed to each well. All assays were carried out using 0.6 nM of IAP proteins. The concentrations of FITC‐Smac were described as follows: 27 nM for XIAP, 12 nM for cIAP1 and 19 nM cIAP2. The final concentrations of Tb‐SA and biotinylated anti‐His antibody were 0.2 and 0.4 nM, respectively. After 1 h incubation at room temperature, the TR‐FRET signal was measured using an EnVision Multilabel Plate Reader. The values of the 0 and 100% controls were the signals obtained in the presence and absence of IAP proteins, respectively.
RNA isolation and quantitative PCR
Total RNA was prepared from cells with RNeasy (Qiagen). First‐strand cDNA was synthesized from 1 μg total RNA with an oligo‐dT primer using the SuperScript First‐Strand Synthesis System (Invitrogen). Quantitative real‐time PCR was performed with an ABI Prism 7300 sequence detection system (Applied Biosystems, Foster City, CA, USA) using SYBR GreenER (Invitrogen) with gene‐specific primers. Human 36B4 mRNA was used as an invariant control. The following PCR primers were used (5′ to 3′): 36b4, GGC CCG AGA AGA CCT CCT T and CCA GTC TTG ATC AGC TGC ACA; Bcr‐Abl: TCC ACT CAG CCA CTG GAT TTA A and AAA TCA TAC AGT GCA ACG AAA AGG. The relative amounts of each mRNA were calculated using the comparative C t method.
Cell viability assay
Cell viability was determined using water‐soluble tetrazolium WST‐8 (4‐[3‐(2‐methoxy‐4‐nitrophenyl)‐2‐(4‐nitrophenyl)‐2H‐5‐tetrazolio]‐1,3‐benzene disulfonate) for the spectrophotometric assay according to the manufacturer's instructions (Dojindo, Tokyo, Japan). Cells were seeded at a concentration of 5 × 103 cells per well in a 96‐well culture plate. After 24 h, the cells were treated with the indicated compounds for 48 h. The WST‐8 reagent was added and the cells were incubated for 0.5 h at 37°C in a humidified atmosphere of 5% CO2. The absorbance at 450 nm of the medium was measured using an EnVision Multilabel Plate Reader.
Statistical analysis
Student's t‐test was used to determine the significance of differences among the experimental groups. Values of P < 0.01 were considered significant.
Results
Development of potent SNIPER(ABL) by incorporation of IAP antagonists
We previously developed SNIPER(ABL)‐2 by conjugating imatinib (ABL inhibitor) to bestatin (IAP ligand) with alkyl linker, which induced the reduction of BCR‐ABL protein in K562 cells at 100 μM.20 To improve the protein knockdown activity, we conjugated imatinib to bestatin with PEG linker (SNIPER(ABL)‐49) (Fig. 1a,b), because PEG linker is expected to improve solubility of the conjugate for improved pharmaceutical properties. However, SNIPER(ABL)‐49 showed protein knockdown activity comparable to that of SNIPER(ABL)‐2 (Fig. 1b).
Figure 1.

SNIPER(ABL) composed of various ABL inhibitors and IAP ligands. (a) Chemical structures of SNIPER(ABL). (b–e) The protein knockdown activities of imatinib‐conjugated (b), GNF5‐conjugated (c), HG‐7‐85‐01‐conjugated (d) and dasatinib‐conjugated SNIPER(ABL) (e) were evaluated. K562 cells were incubated with the indicated concentration of SNIPER or ligands mix (LM; indicated ABL inhibitor and IAP ligand) for 6 h. Numbers below the ABL panel represent BCR‐ABL/GAPDH or BCR‐ABL/β‐tubulin ratio normalized by vehicle control as 100. (f) List of SNIPER(ABL) compounds and their DC 50 values. The upper name represents the code number of the SNIPER(ABL) and the lower number shows the concentration of SNIPER(ABL) required to reduce BCR‐ABL protein by 50% (DC 50, nM).
Then, we synthesized a series of SNIPER(ABL) containing various ABL inhibitors (imatinib, GNF5,37 HG‐7‐85‐0138 and dasatinib39) and various IAP ligands (bestatin, MV1 and an LCL161 derivative) (Fig. 1a), and examined their activity to reduce BCR‐ABL protein in K562 cells for 6 h (Fig. 1b–f). We found that SNIPER(ABL)‐39 composed of dasatinib and the LCL161 derivative showed the most potent activity, representing an effective reduction of BCR‐ABL protein at 10 nM.
From SNIPER(ABL)‐39, we further synthesized analogs with various length of PEG linker chains (Fig. 2a). SNIPER(ABL) with longer PEG linker chains (SNIPER[ABL]‐39 and ‐57) exhibited more potent activity than those with shorter PEG linker chains (SNIPER[ABL]‐56 and ‐38) (Fig. 2b). On the basis of these results, we chose SNIPER(ABL)‐39 for further study. Figure 2(c) shows that the effective knockdown of BCR‐ABL by SNIPER(ABL)‐39 was observed at a concentration ≥10 nM and maximum activity was observed at around 100 nM, when the cells were incubated with SNIPER(ABL)‐39 for 24 h. Combination of dasatinib and the LCL161 derivative (1 μM each) did not effectively decrease the BCR‐ABL protein level (Fig. 2c), indicating that linking the two ligands into a single molecule is critically important for the reduction of the BCR‐ABL protein. In addition to BCR‐ABL protein, SNIPER(ABL)‐39 effectively reduced the level of cIAP1 after 6 h (Figs 1e, 2c) and XIAP after 24 h (Fig. 2c). Figure 2(d) shows that SNIPER(ABL)‐39 exhibits high affinity to bind ABL and IAP proteins as do the individual ligands.
Figure 2.
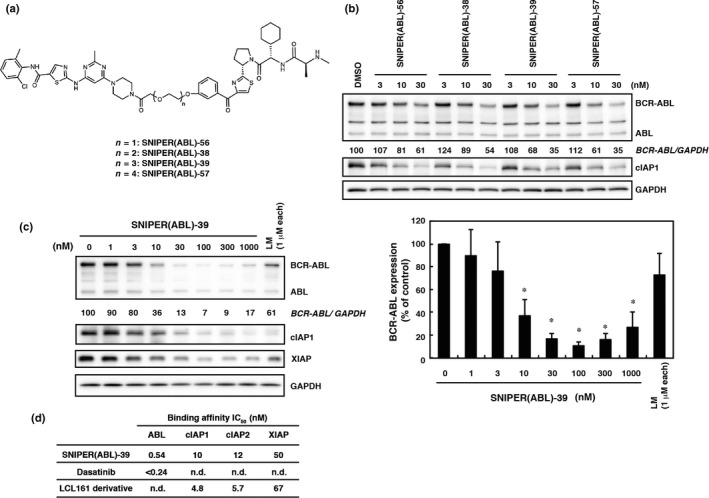
SNIPER(ABL)‐39 shows potent protein knockdown activity. (a) Chemical structures of SNIPER(ABL) with different linker length. (b) Effect of linker length on the protein knockdown activity of the SNIPER(ABL). K562 cells were incubated with the indicated concentration of SNIPER(ABL) for 6 h. (c) Dose response of the protein knockdown activity of SNIPER(ABL)‐39. K562 cells were incubated with the indicated concentration of SNIPER(ABL)‐39 or ligands mix (LM; dasatinib and the LCL161 derivative) for 24 h. Numbers below the ABL panel represent the BCR‐ABL/GAPDH ratio normalized by vehicle control as 100. Data in the bar graph (c) are means ± SD (n = 4). *P < 0.01 compared with vehicle control. (d) Binding affinities of SNIPER(ABL)‐39 to ABL and IAP. IC 50 values (concentrations of SNIPER(ABL)‐39 required to inhibit the probe binding to each protein by 50%) are presented. n.d., not determined.
Involvement of ubiquitin and IAP in SNIPER(ABL)‐39‐induced degradation of BCR‐ABL protein
To explore the mechanism of SNIPER(ABL)‐39‐induced reduction of the BCR‐ABL protein, we first examined the turnover of BCR‐ABL protein after SNIPER(ABL)‐39 treatment. When K562 cells were treated with a protein synthesis inhibitor CHX, BCR‐ABL protein was dramatically decreased within 2 h in the SNIPER(ABL)‐39‐treated cells, but was retained until 6 h in control cells (Fig. 3a). However, the reduction of other proteins, such as cyclin B1 and MCL‐1, was unaffected by SNIPER(ABL)‐39 treatment. In addition, SNIPER(ABL)‐39 did not affect the mRNA levels of Bcr‐Abl (Fig. 3b). These results indicate that SNIPER(ABL)‐39 induces degradation of BCR‐ABL protein.
Figure 3.
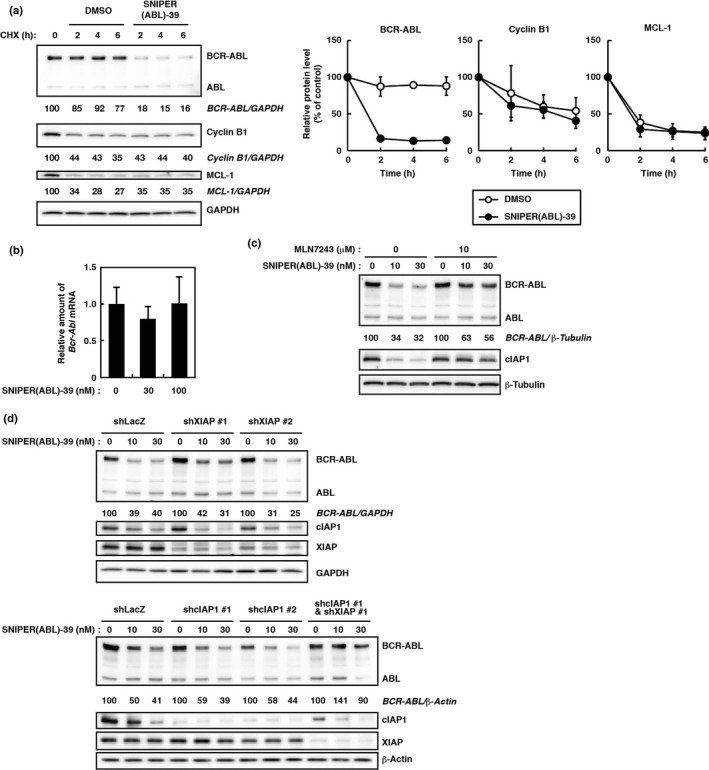
Involvement of ubiquitin and IAP in SNIPER(ABL)‐39‐induced degradation of BCR‐ABL protein. (a) Turnover of BCR‐ABL proteins after SNIPER(ABL)‐39 treatment. K562 cells were treated with 10 μg/mL of cycloheximide (CHX) in the presence or absence of 30 nM of SNIPER(ABL)‐39 for the indicated periods. Numbers below the panels represent BCR‐ABL/GAPDH, Cyclin B1/GAPDH and MCL‐1/GAPDH ratios normalized by time 0 control as 100. Data in the graphs are means ± SD (n = 3). (b) Expression of Bcr‐Abl mRNA in K562 cells. Cells were incubated with the indicated concentration of SNIPER(ABL)‐39 for 6 h. Expression levels are relative to vehicle treatment, which was arbitrarily set to 1. Data in the bar graph are means ± SD (n = 3). (c) Effect of ubiquitin activating enzyme inhibitor MLN7243 on protein knockdown activity of SNIPER(ABL)‐39 in K562 cells. Cells were incubated with the indicated concentration of SNIPER(ABL)‐39 and/or MLN7243 for 6 h. (d) Silencing of both cIAP1 and XIAP expression attenuates SNIPER(ABL)‐39‐dependent BCR‐ABL protein degradation. In K562 cells, endogenous cIAP1 and/or XIAP were depleted by shRNA for 72 h. Then cells were treated with the indicated concentration of SNIPER(ABL)‐39 for 6 h. Numbers below the ABL panel represent BCR‐ABL/GAPDH, BCR‐ABL/β‐tubulin, or BCR‐ABL/β‐actin ratio normalized by vehicle control as 100.
Then, we examined the effect of a UPS inhibitor. We first tested a proteasome inhibitor, MG132, but found that MG132 itself reduced the BCR‐ABL protein (data not shown). Instead, we used an inhibitor of ubiquitin‐activating enzyme, MLN7243, as another UPS inhibitor. The SNIPER(ABL)‐39‐induced degradation of BCR‐ABL protein was abrogated by MLN7243 (Fig. 3c), suggesting that the degradation of BCR‐ABL protein requires the ubiquitin system.
SNIPER(ABL) compounds are designed to recruit IAP for degradation of BCR‐ABL protein. To understand which IAP are involved in the degradation of BCR‐ABL by SNIPER(ABL)‐39, we downregulated IAP by shRNA‐mediated gene silencing. Among IAP family members, we focused on XIAP and cIAP1, because cIAP2 is not detectable in K562 cells. Silencing of both cIAP1 and XIAP expression significantly suppressed the SNIPER(ABL)‐39‐induced BCR‐ABL protein degradation, while silencing of either cIAP1 or XIAP expression alone did not affect the degradation (Fig. 3d). This result suggests that both cIAP1 and XIAP play a role in the degradation of BCR‐ABL protein when treated with SNIPER(ABL)‐39.
SNIPER(ABL)‐39 inhibits BCR‐ABL‐related signaling pathway and proliferation of BCR‐ABL positive chronic myelogenous leukemia cells
Because BCR‐ABL is a constitutively active tyrosine kinase and drives uncontrolled cellular proliferation through signaling pathways that involve the STAT5, mitogen‐activated protein kinase, phosphatidylinositol‐3‐kinase/Akt and CrkL,40, 41 we next examined the effect of SNIPER(ABL)‐39 on BCR‐ABL‐related signaling pathways in K562 cells. In accordance with the degradation of BCR‐ABL protein, SNIPER(ABL)‐39 suppressed the phosphorylation of BCR‐ABL, STAT5 and CrkL (Fig. 4). We also examined the effect with dasatinib as a control. It reduced the phosphorylation of BCR‐ABL, STAT5 and CrkL, without affecting the levels of BCR‐ABL protein (Fig. 4).
Figure 4.
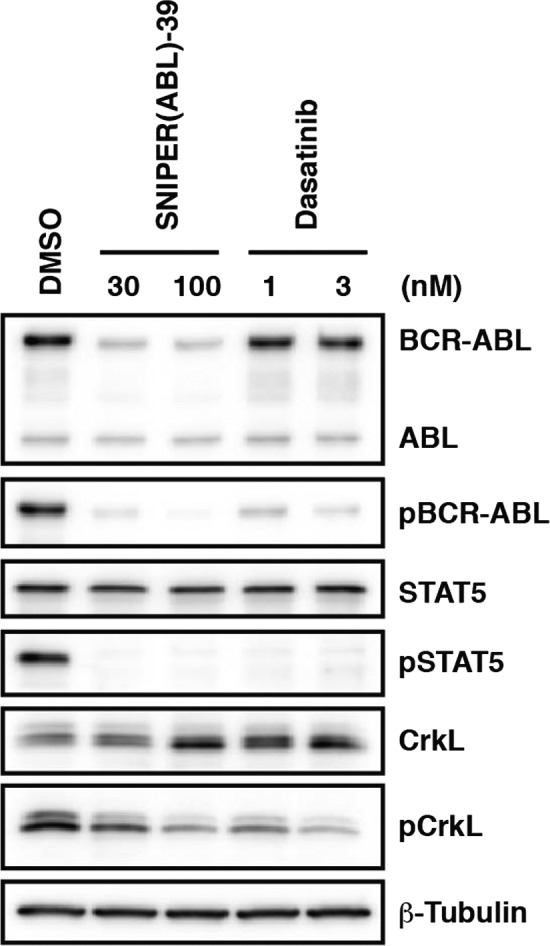
SNIPER(ABL)‐39 inhibits the BCR‐ABL‐related signaling pathway. K562 cells were incubated with the indicated concentration of SNIPER(ABL)‐39 or dasatinib for 6 h. pBCR‐ABL, pSTAT5 and pCrkL stand for phosphorylated BCR‐ABL, STAT5 and CrkL, respectively.
In line with the degradation of BCR‐ABL protein and the inhibition of the BCR‐ABL‐mediated signaling pathway, SNIPER(ABL)‐39 at 10 nM dramatically inhibits the growth of K562 cells (Fig. 5a). Similar growth inhibitory effect and protein knockdown activity of SNIPER(ABL)‐39 were also observed in other CML cell lines, KCL‐22 and KU‐812, expressing native BCR‐ABL protein (Fig. 5a–c), to which dasatinib shows a potent anti‐proliferative effect (Fig. 5c). However, SNIPER(ABL)‐39 did not inhibit the growth of the leukemia cell lines, HL‐60, MOLT‐4 and Jurkat, which do not express BCR‐ABL protein (Fig. 5a). In addition, SNIPER(ABL)‐39 did not inhibit the cell growth nor reduce the BCR‐ABL protein in SK‐9 cells that express T315I mutant BCR‐ABL protein (Fig. 5a,b) 36. These data indicate that SNIPER(ABL)‐39 can induce the degradation of native BCR‐ABL protein, but not T315I mutant, which results in the inhibition of BCR‐ABL‐mediated signaling pathways and CML proliferation in the cells expressing native BCR‐ABL protein.
Figure 5.
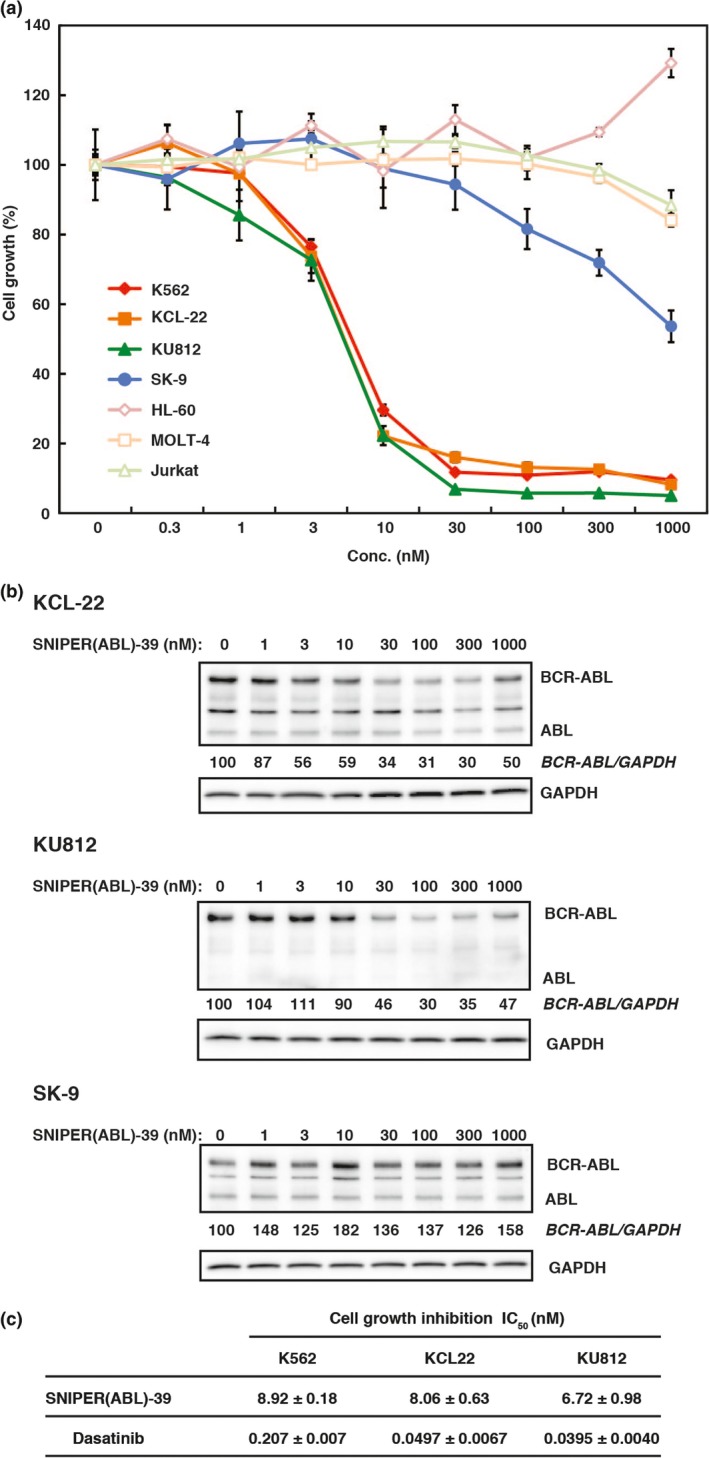
SNIPER(ABL)‐39 inhibits proliferation of chronic myelogenous leukemia (CML) cells expressing native BCR‐ABL. (a) Effect of SNIPER(ABL)‐39 on cell growth in various CML and leukemia cells. Cells were incubated with the indicated concentration of SNIPER(ABL)‐39 for 48 h and subjected for WST assay. Data in the graphs are means ± SD (n = 3). (b) Degradation of BCR‐ABL protein in various CML cells. Cells were incubated with the indicated concentration of SNIPER(ABL)‐39 for 6 h, and the cell lysates were analyzed by western blot. (c) Growth inhibitory effect of SNIPER(ABL)‐39 and dasatinib in various CML cells. Cells were incubated with the SNIPER(ABL)‐39 and dasatinib for 48 h and subjected for WST assay. IC 50 values (half‐maximal inhibitory concentration of cell growth) are presented as means ± SD (n = 3).
Discussion
In this study, we tested various combinations of ABL inhibitors and IAP ligands to develop SNIPER(ABL) that induces the degradation of oncogenic kinase BCR‐ABL, and the linker length was optimized for the activity. We found that SNIPER(ABL)‐39, in which dasatinib is conjugated to an LCL161 derivative with PEG × 3 linker, shows the most potent activity to degrade the BCR‐ABL protein. SNIPER(ABL)‐39 showed an effective protein knockdown activity at 10 nM and the maximum activity was observed at 100 nM. Notably, the protein knockdown activity was rather attenuated at higher concentrations of SNIPER(ABL)‐39 (Fig. 2c). This is known as a high‐dose hook effect, where a certain pharmaceutical activity is interfered with higher concentration of drugs. We speculate that formation of ternary complex consisting of BCR‐ABL/SNIPER(ABL)‐39/IAP required for the protein knockdown activity would be suppressed by higher concentrations of SNIPER(ABL)‐39, and, therefore, the protein knockdown activity is attenuated.
With respect to the small compounds that induce the degradation of BCR‐ABL protein, PROTAC against BCR‐ABL were reported by conjugation of dasatinib to a von Hippel‐Lindau (VHL) E3 ligase ligand or a thalidomide derivative, pomalidomide, that is a ligand for Cereblon (CRBN) E3 ligase.32 Interestingly, CRBN‐based PROTAC can reduce BCR‐ABL protein at 25 nM, whereas VHL‐based PROTAC cannot. Because IAP‐based SNIPER(ABL) can induce the degradation of BCR‐ABL protein, it is suggested that IAP and CRBN are appropriate E3 ligases to degrade BCR‐ABL protein when dasatinib is incorporated as a ligand for BCR‐ABL protein. Probably, SNIPER(ABL)‐39 and CRBN‐based PROTAC can recruit E3 ligases to an appropriate position so that the lysine residues on the surface of BCR‐ABL can be ubiquitylated. Thus, the combination of an E3 ligase ligand and a target ligand is critically important to develop the degradation inducers such as SNIPER and PROTAC.
Consistent with the degradation of BCR‐ABL protein, SNIPER(ABL)‐39 inhibited the BCR‐ABL‐related signaling pathway and proliferation of BCR‐ABL positive CML, such as K562, KCL‐22 and KU812 cell, expressing native BCR‐ABL protein. However, in SK‐9 cells expressing T315I mutant BCR‐ABL protein, SNIPER(ABL)‐39 did not reduce the BCR‐ABL protein nor inhibit cell proliferation. This may be attributed that SNIPER(ABL)‐39 could not bind to the T315I mutant BCR‐ABL protein, because the T315I is a gatekeeper mutation that prevents the binding of dasatinib.39, 42 However, BCR‐ABL protein has multiple domains, such as pleckstrin homology, Src homology (SH) 2 and SH3 domains, to which novel ligands could be developed. Incorporation of such ligands into SNIPER would allow us to develop a novel SNIPER(ABL) that can induce the degradation of BCR‐ABL proteins resistant to kinase inhibitors, which could be a novel strategy to overcome drug resistance against kinase inhibitors.
Disclosure Statement
N. Miyamoto, K. Nagai, K. Shimokawa, T. Sameshima, Y. Imaeda, H. Nara and N. Cho are employees of Takeda Pharmaceutical (Osaka, Japan). M. Naito received a research fund from Takeda Pharmaceutical. The other authors have no conflict of interest to declare.
Supporting information
Scheme S1. Synthesis of SNIPER(ABL)‐020 and ‐019.
Scheme S2. Synthesis of SNIPER(ABL)‐056, ‐038, ‐039 and ‐057.
Scheme S3. Synthesis of SNIPER(ABL)‐049, ‐050 and ‐058.
Scheme S4. Synthesis of SNIPER(ABL)‐013, ‐015 and ‐024.
Scheme S5. Synthesis of SNIPER(ABL)‐044 and ‐047.
Scheme S6. Synthesis of SNIPER(ABL)‐033
Data S1. The chemical synthesis and physicochemical data.
Acknowledgments
This study was supported, in part, by the Japan Society for the Promotion of Science (KAKENHI Grant Numbers 26860050 to N.S., 26860049 to N.O., 16H05090 to T.H. and M.N. and 16K15121 to N.O. and M.N.), by the Project for Cancer Research And Therapeutic Evolution (P‐CREATE) (16cm0106124j0001 to N.O.) and the Research on Development of New Drugs (15ak0101029h1402 and 16ak0101029j1403 to M.N.) from the Japan Agency for Medical Research and Development (AMED), by the Ministry of Health and Labor Welfare, Japan (to M.N.), by Takeda Science Foundation (to N.O.) and by Takeda Pharmaceutical Co. Ltd. (to M.N.). The authors thank Dr S. Okabe for kindly providing SK‐9 cells and M. Seki for measurement of the protein knockdown activities.
Cancer Sci 108 (2017) 1657–1666
Funding information
Japan Society for the Promotion of Science (26860050, 26860049, 16H05090, 16K15121); Japan Agency for Medical Research and Development (AMED) (16cm0106124j0001, 15ak0101029h1402, 16ak0101029j1403); Ministry of Health and Labor Welfare, Japan; Takeda Science Foundation; Takeda Pharmaceutical Co. Ltd.
References
- 1. Rudkin CT, Hungerford DA, Nowell PC. DNA contents of chromosome Ph1 and chromosome 21 in human chronic granulocytic leukemia. Science 1964; 144: 1229–31. [DOI] [PubMed] [Google Scholar]
- 2. Rowley JD. Letter: A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature 1973; 243: 290–3. [DOI] [PubMed] [Google Scholar]
- 3. Konopka JB, Watanabe SM, Witte ON. An alteration of the human c‐abl protein in K562 leukemia cells unmasks associated tyrosine kinase activity. Cell 1984; 37: 1035–42. [DOI] [PubMed] [Google Scholar]
- 4. Shtivelman E, Lifshitz B, Gale RP, Canaani E. Fused transcript of abl and bcr genes in chronic myelogenous leukaemia. Nature 1985; 315: 550–4. [DOI] [PubMed] [Google Scholar]
- 5. Heisterkamp N, Stam K, Groffen J, de Klein A, Grosveld G. Structural organization of the bcr gene and its role in the Ph′ translocation. Nature 1985; 315: 758–61. [DOI] [PubMed] [Google Scholar]
- 6. Druker BJ, Tamura S, Buchdunger E et al Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr‐Abl positive cells. Nat Med 1996; 2: 561–6. [DOI] [PubMed] [Google Scholar]
- 7. Druker BJ. Translation of the Philadelphia chromosome into therapy for CML. Blood 2008; 112: 4808–17. [DOI] [PubMed] [Google Scholar]
- 8. Talpaz M, Shah NP, Kantarjian H et al Dasatinib in imatinib‐resistant Philadelphia chromosome‐positive leukemias. N Engl J Med 2006; 354: 2531–41. [DOI] [PubMed] [Google Scholar]
- 9. Puttini M, Coluccia AM, Boschelli F et al In vitro and in vivo activity of SKI‐606, a novel Src‐Abl inhibitor, against imatinib‐resistant Bcr‐Abl+ neoplastic cells. Cancer Res 2006; 66: 11314–22. [DOI] [PubMed] [Google Scholar]
- 10. Kim DH, Rossi JJ. Strategies for silencing human disease using RNA interference. Nat Rev Genet 2007; 8: 173–84. [DOI] [PubMed] [Google Scholar]
- 11. Davidson BL, McCray PB Jr. Current prospects for RNA interference‐based therapies. Nat Rev Genet 2011; 12: 329–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Itoh Y, Ishikawa M, Naito M, Hashimoto Y. Protein knockdown using methyl bestatin‐ligand hybrid molecules: design and synthesis of inducers of ubiquitination‐mediated degradation of cellular retinoic acid‐binding proteins. J Am Chem Soc 2010; 132: 5820–6. [DOI] [PubMed] [Google Scholar]
- 13. Okuhira K, Ohoka N, Sai K et al Specific degradation of CRABP‐II via cIAP1‐mediated ubiquitylation induced by hybrid molecules that crosslink cIAP1 and the target protein. FEBS Lett 2011; 585: 1147–52. [DOI] [PubMed] [Google Scholar]
- 14. Demizu Y, Okuhira K, Motoi H et al Design and synthesis of estrogen receptor degradation inducer based on a protein knockdown strategy. Bioorg Med Chem Lett 2012; 22: 1793–6. [DOI] [PubMed] [Google Scholar]
- 15. Itoh Y, Ishikawa M, Kitaguchi R, Okuhira K, Naito M, Hashimoto Y. Double protein knockdown of cIAP1 and CRABP‐II using a hybrid molecule consisting of ATRA and IAPs antagonist. Bioorg Med Chem Lett 2012; 22: 4453–7. [DOI] [PubMed] [Google Scholar]
- 16. Okuhira K, Demizu Y, Hattori T et al Development of hybrid small molecules that induce degradation of estrogen receptor‐alpha and necrotic cell death in breast cancer cells. Cancer Sci 2013; 104: 1492–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ohoka N, Nagai K, Hattori T et al Cancer cell death induced by novel small molecules degrading the TACC3 protein via the ubiquitin‐proteasome pathway. Cell Death Dis 2014; 5: e1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tomoshige S, Naito M, Hashimoto Y, Ishikawa M. Degradation of HaloTag‐fused nuclear proteins using bestatin‐HaloTag ligand hybrid molecules. Org Biomol Chem 2015; 13: 9746–50. [DOI] [PubMed] [Google Scholar]
- 19. Ohoka N, Shibata N, Hattori T, Naito M. Protein knockdown technology: application of ubiquitin ligase to cancer therapy. Curr Cancer Drug Targets 2016; 16: 136–46. [DOI] [PubMed] [Google Scholar]
- 20. Demizu Y, Shibata N, Hattori T et al Development of BCR‐ABL degradation inducers via the conjugation of an imatinib derivative and a cIAP1 ligand. Bioorg Med Chem Lett 2016; 26: 4865–9. [DOI] [PubMed] [Google Scholar]
- 21. Okuhira K, Shoda T, Omura R et al Targeted degradation of proteins localized in subcellular compartments by hybrid small molecules. Mol Pharmacol 2017; 91: 159–66. [DOI] [PubMed] [Google Scholar]
- 22. Ohoka N, Okuhira K, Ito M et al In vivo knockdown of pathogenic proteins via specific and nongenetic IAP‐dependent protein erasers (SNIPERs). J Biol Chem 2017; 292: 4556–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ohoka N, Nagai K, Shibata N et al SNIPER(TACC3) induces cytoplasmic vacuolization and sensitizes cancer cells to Bortezomib. Cancer Sci 2017;. https://doi.org/10.1111/cas.13198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sakamoto KM, Kim KB, Verma R et al Development of Protacs to target cancer‐promoting proteins for ubiquitination and degradation. Mol Cell Proteomics 2003; 2: 1350–8. [DOI] [PubMed] [Google Scholar]
- 25. Schneekloth JS Jr, Fonseca FN, Koldobskiy M et al Chemical genetic control of protein levels: selective in vivo targeted degradation. J Am Chem Soc 2004; 126: 3748–54. [DOI] [PubMed] [Google Scholar]
- 26. Schneekloth AR, Pucheault M, Tae HS, Crews CM. Targeted intracellular protein degradation induced by a small molecule: en route to chemical proteomics. Bioorg Med Chem Lett 2008; 18: 5904–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Puppala D, Lee H, Kim KB, Swanson HI. Development of an aryl hydrocarbon receptor antagonist using the proteolysis‐targeting chimeric molecules approach: a potential tool for chemoprevention. Mol Pharmacol 2008; 73: 1064–71. [DOI] [PubMed] [Google Scholar]
- 28. Rodriguez‐Gonzalez A, Cyrus K, Salcius M et al Targeting steroid hormone receptors for ubiquitination and degradation in breast and prostate cancer. Oncogene 2008; 27: 7201–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bondeson DP, Mares A, Smith IE et al Catalytic in vivo protein knockdown by small‐molecule PROTACs. Nat Chem Biol 2015; 11: 611–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lu J, Qian Y, Altieri M et al Hijacking the E3 ubiquitin ligase cereblon to efficiently target BRD4. Chem Biol 2015; 22: 755–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Winter GE, Buckley DL, Paulk J et al DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015; 348: 1376–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lai AC, Toure M, Hellerschmied D et al Modular PROTAC design for the degradation of oncogenic BCR‐ABL. Angew Chem Int Ed Engl 2016; 55: 807–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Toure M, Crews CM. Small‐molecule PROTACS: new approaches to protein degradation. Angew Chem Int Ed Engl 2016; 55: 1966–73. [DOI] [PubMed] [Google Scholar]
- 34. Nikolovska‐Coleska Z, Wang R, Fang X et al Development and optimization of a binding assay for the XIAP BIR3 domain using fluorescence polarization. Anal Biochem 2004; 332: 261–73. [DOI] [PubMed] [Google Scholar]
- 35. Vetter ML, Zhang Z, Liu S et al Fluorescent visualization of Src by using dasatinib‐BODIPY. ChemBioChem 2014; 15: 1317–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Okabe S, Tauchi T, Ohyashiki K. Establishment of a new Philadelphia chromosome‐positive acute lymphoblastic leukemia cell line (SK‐9) with T315I mutation. Exp Hematol 2010; 38: 765–72. [DOI] [PubMed] [Google Scholar]
- 37. Zhang J, Adrian FJ, Jahnke W et al Targeting Bcr‐Abl by combining allosteric with ATP‐binding‐site inhibitors. Nature 2010; 463: 501–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Weisberg E, Choi HG, Ray A et al Discovery of a small‐molecule type II inhibitor of wild‐type and gatekeeper mutants of BCR‐ABL, PDGFRalpha, Kit, and Src kinases: novel type II inhibitor of gatekeeper mutants. Blood 2010; 115: 4206–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shah NP, Tran C, Lee FY, Chen P, Norris D, Sawyers CL. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science 2004; 305: 399–401. [DOI] [PubMed] [Google Scholar]
- 40. Quintas‐Cardama A, Cortes J. Molecular biology of bcr‐abl1‐positive chronic myeloid leukemia. Blood 2009; 113: 1619–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hantschel O, Warsch W, Eckelhart E et al BCR‐ABL uncouples canonical JAK2‐STAT5 signaling in chronic myeloid leukemia. Nat Chem Biol 2012; 8: 285–93. [DOI] [PubMed] [Google Scholar]
- 42. Tokarski JS, Newitt JA, Chang CY et al The structure of Dasatinib (BMS‐354825) bound to activated ABL kinase domain elucidates its inhibitory activity against imatinib‐resistant ABL mutants. Cancer Res 2006; 66: 5790–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Scheme S1. Synthesis of SNIPER(ABL)‐020 and ‐019.
Scheme S2. Synthesis of SNIPER(ABL)‐056, ‐038, ‐039 and ‐057.
Scheme S3. Synthesis of SNIPER(ABL)‐049, ‐050 and ‐058.
Scheme S4. Synthesis of SNIPER(ABL)‐013, ‐015 and ‐024.
Scheme S5. Synthesis of SNIPER(ABL)‐044 and ‐047.
Scheme S6. Synthesis of SNIPER(ABL)‐033
Data S1. The chemical synthesis and physicochemical data.