

Summary
Forkhead box P3 (FoxP3)+ regulatory T cells (Tregs) are functionally deficient in systemic lupus erythematosus (SLE), characterized by reduced surface CD25 [the interleukin (IL)‐2 receptor alpha chain]. Low‐dose IL‐2 therapy is a promising current approach to correct this defect. To elucidate the origins of the SLE Treg phenotype, we studied its role through developmentally defined regulatory T cell (Treg) subsets in 45 SLE patients, 103 SLE‐unaffected first‐degree relatives and 61 unrelated healthy control subjects, and genetic association with the CD25‐encoding IL2RA locus. We identified two separate, uncorrelated effects contributing to Treg CD25. (1) SLE patients and unaffected relatives remarkably shared CD25 reduction versus controls, particularly in the developmentally earliest CD4+FoxP3+CD45RO–CD31+ recent thymic emigrant Tregs. This first component effect influenced the proportions of circulating CD4+FoxP3highCD45RO+ activated Tregs. (2) In contrast, patients and unaffected relatives differed sharply in their activated Treg CD25 state: while relatives as control subjects up‐regulated CD25 strongly in these cells during differentiation from naive Tregs, SLE patients specifically failed to do so. This CD25 up‐regulation depended upon IL2RA genetic variation and was related functionally to the proliferation of activated Tregs, but not to their circulating numbers. Both effects were found related to T cell IL‐2 production. Our results point to (1) a heritable, intrathymic mechanism responsible for reduced CD25 on early Tregs and decreased activation capacity in an extended risk population, which can be compensated by (2) functionally independent CD25 up‐regulation upon peripheral Treg activation that is selectively deficient in patients. We expect that Treg‐directed therapies can be monitored more effectively when taking this distinction into account.
Keywords: cytokines, regulatory T cells, systemic lupus erythematosus
Introduction
Systemic lupus erythematosus (SLE) is an autoimmune disease characterized by combined adaptive and innate‐immune aberrations that can affect diverse organs particularly through immune complex‐mediated tissue damage. However, the aetiology and pathogenic processes of SLE are not understood fully. Both genetic and environmental factors contribute to immune dysfunction 1. In the associated breakdown of tolerance, T cells play a key role by amplifying autoimmune responses 2. Many abnormalities of T lymphocytes have been described in SLE patients, including dysregulated intracellular signal transduction and imbalanced cytokine production 3, which influence B cell function and autoantibody production. While SLE T cells help B cells to generate the pathogenic autoantibodies, they also fail characteristically to produce sufficient amounts of interleukin (IL)‐2 4.
Limited amounts of IL‐2 affect primarily forkhead box P3 (FoxP3)+ regulatory T cells (Tregs), which depend greatly upon this cytokine for their development, survival and function 5. Recently, the importance of deficient IL‐2 production and its effects mediated by Tregs was supported strongly by the clinical efficacy of low‐dose IL‐2 therapy in SLE, shown to boost Treg activity and to re‐establish tolerance mechanisms that can counter autoimmunity and improve patients’ clinical outcome 6, 7. This is in line with reported Treg aberrations in SLE 8, 9, despite some discrepancies in previous studies: while some authors had reported decreased circulating Treg proportions in active disease 10, 11, 12, 13, 14, 15, others had found them normal 16, 17, 18, 19 or even increased 20, 21, 22, 23, 24. These divergences, however, can be explained largely by a population of aberrant CD4+ T cells expressing FoxP3 but low or no CD25, which is almost non‐existent in healthy individuals but present in significant numbers in SLE 19, 25, 26, 27 where it ‘dissociates’ FoxP3 from CD25‐based Treg quantitation. These CD25‐deficient cells have a natural Treg phenotype of deficient functionality 25, and their frequencies correlate with disease activity and (inversely) with Treg suppressive capacity 28. The hypothesis that they indicate an SLE‐specific Treg dysfunction is consistent with earlier reports of functional Treg defects 15, 16, 29.
Human FoxP3+ cells have been subdivided into three functionally and phenotypically distinct subsets 30: naive Tregs (FoxP3+CD45RA+), short‐lived and highly suppressive activated Tregs ( FoxP3highCD45RA–) and another CD45RA– population with low FoxP3 expression and deficient or absent functionality (FoxP3low). Activated Tregs are derived generally from naive Tregs 31, while the FoxP3low subset can, in part, represent transient FoxP3 expression by non‐regulatory T helper (Th) cells 30. While it was speculated that this could also apply to atypical CD25‐deficient Tregs in SLE 27, it is now clear that it is not the case. In fact, no evidence was found for a role of transient FoxP3 expression in SLE, as not only FoxP3highCD25high but also CD25low/negative activated Tregs bear strong markers of bona‐fide, probably thymus‐derived Tregs 7, 32, i.e. high levels of Helios expression 33, 34, and fully demethylated FoxP3–TSDR 35. Therefore, Tregs in SLE apart from FoxP3low cells can basically be seen as belonging to a lineage derived from naive Tregs.
In complex diseases, the study of unaffected relatives can reveal shared heritable factors that probably contribute to pathogenesis, and separate them from divergent phenotypes which are secondary to disease manifestation. Unaffected relatives of SLE patients frequently present IgG autoantibodies of SLE‐associated specificity 36, although they rarely develop the disease. We have reported previously that these relatives, in contrast to unrelated healthy controls or the patients themselves, showed consistent positive correlations of SLE‐associated specific IgG with CD25+ Tregs 37. This suggested a compensatory role of Tregs with the capacity to control autoantibody‐related pathogenic effects, eventually allowing the unaffected relatives to avoid manifest disease.
In order to elucidate more clearly the SLE Treg phenotype, which also plays a role in intravenous immunoglobulin (i.v.Ig) therapy 38, and the Treg‐mediated compensation in unaffected relatives, we have now studied the reduced surface CD25 quantitatively in the three classic Treg subsets 30 and tracked it through developmental stages making use of their lineage property. Our results point to two separate contributing effects: (a) a shared, probably intrathymic CD25 reduction that leads to a decreased activation capacity in developmentally early Tregs, compensated in unaffected relatives but not patients by (b) a functionally independent CD25 up‐regulation that occurs upon peripheral Treg activation.
Materials and methods
Patients and sampling
Peripheral blood samples were collected from 102 SLE patients who fulfilled current American College of Rheumatology (ACR) criteria for SLE, 197 first‐degree relatives of the patients from a total of 94 families and 141 unrelated healthy controls upon signed informed consent. Relatives were subjected to a survey questionnaire to rule out clinical SLE, and three who were, in fact, diagnosed with SLE either before or subsequent to our collection were excluded from the study that therefore included 194 unaffected relatives at the end. SLE patients underwent a detailed clinical characterization, the results of which are summarized in Table 1. Approval was obtained from the Ethics Committees of Hospital Geral de Santo Antonio, Centro Hospitalar do Porto (Porto), Centro Hospitalar Lisboa Norte/Santa Maria, Centro Hospitalar Lisboa Ocidental and Hospital de Curry Cabral, Centro Hospitalar de Lisboa Central (Lisbon).
Table 1.
General and clinical characteristics
| 1. SLE patients | Total: 102 |
|---|---|
| Gender | Male: 8%; female: 92% |
| Age | Median: 40·5 years; range 21–73 years |
| Time of SLE affection | Median: 10 years; range: 0–46 years |
| Actual disease activity (SLEDAI‐2k) | Median: 2; range: 0–30 |
| Clinical affections | |
| Malar rashes | 59% |
| Discoid rashes | 21% |
| Photosensitivity | 72% |
| Ulcers | 41% |
| Arthritis | 63% |
| Renal involvement | 47% |
| Lung involvement | 18% |
| Cardiac involvement | 16% |
| Neurological alterations | 28% |
| Haematological alterations | 54% |
| Immunological alterations | 86% |
| Anti‐phospholipid syndrome | 15% |
| Secondary Sjögren's syndrome | 12% |
| Therapy | |
| Glucocorticoids | 69% |
| Glucocorticoid dosages (mg prednisone eq.) | Median: 5·5; range: 1·25–50 |
| Antimalarials | 68% |
| Azathioprine | 21% |
| Methotrexate | 3% |
| Mycophenolate mofetil | 13% |
| Cyclosporin A | 1% |
| Oral steroids | 21% |
| 2. Unaffected 1st‐degree relatives | Total: 194 |
| Gender | Male: 39%; female: 61% |
| Age | Median: 48·5 years; range: 18–86 years |
| 3. Unrelated healthy controls | Total: 141 |
| Gender | Male: 48%; female: 52% |
| Age | Median: 44 years; range: 20–68 years |
SLE = systemic lupus erythematosus; SLEDAI = SLE Disease Activity Index.
Peripheral blood samples (30 ml per individual) were collected into Vacutainer cell preparation tubes with sodium citrate [Becton‐Dickinson (BD), Franklin Lakes, NJ, USA], and plasma and peripheral blood mononuclear cells (PBMCs) were isolated by centrifugation, according to the manufacturer's protocol.
Flow cytometry
For flow cytometric analysis, PBMC were washed twice in phosphate‐buffered saline (PBS) containing 2% fetal calf serum (FCS) prior to incubation with antibodies and 106 cells were incubated for 30 min on ice with anti‐CD4‐Pacific Orange (Invitrogen, Carlsbad, CA, USA; clone S3.5), anti‐CD31‐Pacific Blue (exBio, Vestev, Czech Republic; clone MEM‐05) anti‐CD127‐allophycocyanin (APC)/eFluor780 (eBioscience, San Jose, CA, USA; clone eBioRDR5), anti‐CD25‐phycoerythrin (PE) (BD; clone 2A3), anti‐CD39‐PE/cyanin 7 (Cy7) (eBioscience; clone A1) and anti‐CD45RO‐peridinin chlorophyll (PerCP) (Invitrogen; clone UCHL1). For subsequent intracellular staining, cells were washed again, fixed and permeabilized using a fix/perm kit (eBioscience) and stained for intracellular FoxP3 and Ki67 with anti‐FoxP3‐APC (eBioscience; clone PCH101) and anti‐Ki67‐fluorescein isothiocyanate (FITC) (BD; clone 35/Ki‐67). Fluorescence was assessed using a Cyan ADP flow cytometer and analysed using FlowJo software (TreeStar Inc., Ashland, OR, USA. Adapting the scheme of Miyara et al. 30, we distinguished three FoxP3+ subsets: CD45RO– FoxP3+ naive Tregs, CD45RO+ FoxP3high activated Tregs and CD45RO+ FoxP3low cells (depicted in Fig. 1). To keep subset definitions objective and unbiased, we used identical absolute distances on the log‐fluorescence scale between the lower boundaries of FoxP3low and FoxP3high subset gates in all samples. To measure surface CD25, the PE median fluorescence intensity (MFI) was quantified. Analysis of this property was restricted to samples collected within a defined time‐period where parallel assessment of PE‐coupled cytometric beads validated the longitudinal commensurateness of MFI measures (45 SLE patients, 103 unaffected relatives and 61 controls). Also, CD127 staining was used principally for quality control (not gating; however, in 10 samples with visible separate CD127high FoxP3+ populations, these were gated out). Treg CD127 MFIs were always below 80% (median = 44%) of the MFIs measured in CD4+ FoxP3– conventional Th in the same sample with a single exception, where low CD127 as it characterizes Tregs was also found on the Th cells.
Figure 1.
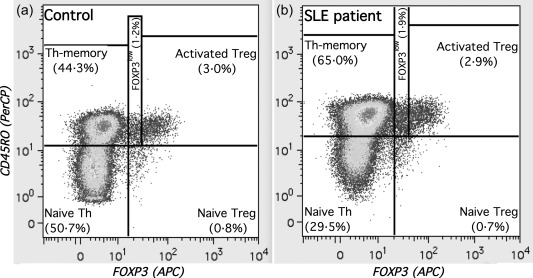
Regulatory T cell (Tregs) subset gating. The figure shows our cytometric gating of CD4+ T cell subsets. Apart from the conventional forkhead box P3 (FoxP3)– (naive and memory) T helper (Th), FoxP3+ Tregs were divided into three subsets using FoxP3 and CD45RO staining. Two examples are depicted with their T helper (Th) and Tregs subset frequencies within CD4+ cells: (a) a healthy control subject; (b) a systemic lupus erythematosus (SLE) patient. Irrespective of differences in absolute staining intensities, the distance between the lower boundaries of FoxP3l°w and activated Tregs was kept constant throughout all samples. Parallel CD127 staining (not shown) was not used for gating but for quality control.
Cell culture
Isolated 106 PBMC were stimulated with phorbol myristate acetate (PMA) (25 ng/ml) and ionomycin (1 μg/ml) for 6 h at 37°C in RPMI‐1640 medium containing 10% FCS and 10 μg/ml brefeldin A. Immediately after stimulation, the cells were stained with anti‐CD3‐APC/Cy7 (BioLegend; clone HIT3a), anti‐CD4‐PerCP (BioLegend; clone RPA‐T4), anti‐CD8‐PE/Cy7 (eBioscience; clone RPA‐T8) and anti‐CD45RO‐Pacific Blue (BioLegend; clone UCHL1). Subsequently, cells were fixed and permeabilized using Cytofix/Cytoperm (BD), washed and stained for intracellular IL‐2 in Perm buffer with anti‐IL‐2‐APC [eBioscience; clone MQ1–17H12, in parallel with interferon (IFN)‐γ and tumour necrosis factor (TNF)‐α], assessed cytometrically with a fluorescence activated cell sorter (FACS)Canto II cytometer and analysed using FlowJo software (TreeStar, Inc.).
IL2RA (CD25) genotyping
Genomic DNA was extracted from peripheral blood cells by standard salting‐out. Genotyping of 25 locus‐spanning single nucleotide polymorphisms (SNPs) (Table 2) was performed on the Sequenom™ platform, as described previously 37. All 25 typed SNPs passed quality control with call rates above 85% and Hardy–Weinberg equilibrium (P > 0·01) was fulfilled within the control subjects.
Table 2.
Genetic associations with polymorphisms in the IL2RA locus in control subjects
| SNP | Position (Chr.10) | Alleles | Association with effect 1 | Association with effect 2 | Allele assoc. with low CD25 | Allele assoc. with T1D§ | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| R * | P † | P FDR ‡ | R * | P † | P FDR ‡ | |||||
| rs12359875 | 6009144 | CT | 0·04 | 0·7438 | 0·32 | 0·0129 | 0·0647 | |||
| rs12244380 | 6011411 | AG | −0·01 | 0·9204 | −0·02 | 0·8863 | ||||
| rs9663421 | 6013641 | CT | 0·16 | 0·2213 | 0·21 | 0·1030 | ||||
| rs2076846 | 6021290 | AG | −0·18 | 0·1736 | 0·03 | 0·8338 | ||||
| rs11256369 | 6024237 | CG | −0·18 | 0·1654 | −0·10 | 0·4211 | ||||
| rs7072398 | 6037883 | AG | −0·19 | 0·1534 | −0·20 | 0·1189 | ||||
| rs4749924 | 6040433 | AC | 0·08 | 0·5316 | 0·20 | 0·1275 | ||||
| rs706781 | 6044422 | CT | 0·10 | 0·4586 | −0·12 | 0·3728 | ||||
| rs11256497 | 6045831 | AG | −0·14 | 0·2860 | −0·28 | 0·0283 | 0·1010 | |||
| rs791587 | 6046736 | AG | −0·04 | 0·7653 | −0·09 | 0·4823 | ||||
| rs791589 | 6047608 | AG | 0·04 | 0·7891 | −0·26 | 0·0506 | ||||
| rs10905669 | 6050130 | CT | 0·02 | 0·8787 | −0·01 | 0·9635 | ||||
| rs2256774 | 6055202 | CT | −0·30 | 0·0191 | 0·2384 | −0·34 | 0·0079 | 0·0656 | ||
| rs706779 | 6056861 | CT | −0·35 | 0·0056 | 0·1409 | −0·33 | 0·0112 | 0·0701 | ||
| rs706778 | 6056986 | CT | −0·14 | 0·2805 | −0·20 | 0·1375 | ||||
| rs2104286 | 6057082 | CT | 0·06 | 0·6458 | 0·13 | 0·3368 | ||||
| rs7072793 | 6064303 | CT | 0·14 | 0·2743 | 0·19 | 0·1443 | ||||
| rs7073236 | 6064589 | CT | 0·14 | 0·2743 | 0·19 | 0·1443 | ||||
| rs11597367 | 6065571 | AG | 0·11 | 0·3804 | 0·37 | 0·0035 | 0·0434 | A | A | |
| rs10795791 | 6066377 | AG | −0·14 | 0·2743 | −0·19 | 0·1443 | ||||
| rs4147359 | 6066476 | AG | 0·01 | 0·9360 | 0·07 | 0·5913 | ||||
| rs7090530 | 6068912 | AC | 0·19 | 0·1322 | 0·29 | 0·0228 | 0·0948 | |||
| rs41295061 | 6072697 | AC | −0·01 | 0·9393 | 0·02 | 0·8867 | ||||
| rs11594656 | 6080046 | AT | −0·11 | 0·3804 | −0·37 | 0·0035 | 0·0434 | T | T | |
| rs12251307 | 6081532 | CT | 0·05 | 0·7050 | −0·01 | 0·9630 | ||||
*Linear correlation coefficient. † P‐values from univariate regression (significant values < 0·05 in bold type). ‡ P‐values false‐positive discovery rate (FDR)‐corrected for multiple comparisons (significant values < 0·05 in bold type). §According to Lowe et al. 41.
Data analysis
Data were analysed with Igor Pro software (WaveMetrics, Lake Oswego, OR, USA) and a statistical package that we have developed for it (also see 36, 37, 38). Pairwise group comparisons of continuous variables were performed by the distribution‐independent Mann–Whitney test. Our statistical approach was to compare patients and unaffected relatives separately to a control group, followed by within‐group analysis. We did not make all possible groupwise comparisons, and particularly did not directly compare patients and relatives, which were drawn from the same families and cannot be compared irrespective of their familial relations with adequate statistical power. Relations between continuous variables (including genotypes) were characterized by linear correlation coefficients (sometimes partial or semipartial) and tested by linear regression analysis. Multiple linear regression was used to consider covariates. For genetic association, univariate P‐values were corrected for multiple comparisons (i.e. the 25 SNPs tested) with the Benjamini–Hochberg false discovery rate (FDR) method. P‐values below 0·05 were considered statistically significant.
Results
CD25 reduction on SLE Tregs is shared by the patients’ first‐degree relatives
Comparing quantitative CD25 surface staining intensities in 45 SLE patients, 103 unaffected relatives and 61 unrelated healthy controls, we found CD25 MFI impressively reduced in patients versus controls not only when considering all FoxP3+ cells (Fig. 2a), but also within each of their three subsets (depicted in Fig. 1) defined by Miyara et al. 30: naive (Fig. 2b) and activated (Fig. 2c) Tregs, as well as FoxP3low cells (Fig. 2d). Patients’ median CD25 was reduced 1·7‐fold in total FoxP3+, 1·6 and 1·7‐fold in naive and activated Tregs, respectively, and 1·4‐fold in FoxP3low cells. This subset‐overarching CD25 deficiency was shared remarkably by the unaffected first‐degree relatives, where CD25 densities were also significantly lower in all CD4+FoxP3+ subpopulations than in controls (1·4‐fold in total FoxP3+ and 1·3‐fold in all three subsets), while still higher than in the patients. The intermediate position of the unaffected relatives points to shared heritable factors and therefore supports Treg CD25 as a pathogenesis‐related biomarker. Approximations of cell proportions lacking surface CD25 (although based on an arbitrary threshold definition) mirror the MFI characteristics (Supporting information, Fig. S1).
Figure 2.
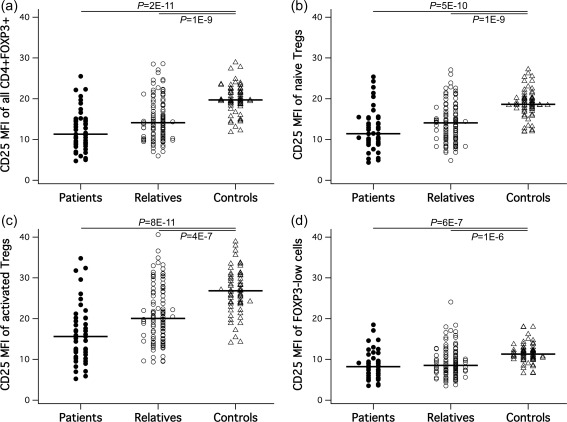
Surface CD25 in Treg subsets. Quantitative surface CD25 distributions and group differences for (a) all CD4+forkhead box P3 (FoxP3)+ cells, (b) naive regulatory T cells (Tregs), (b) activated Tregs and (d) FoxP3low cells. Significant P‐values are shown; n.s. = non‐significant.
Subsetwise CD25 levels were particularly interesting. We found that surface CD25 MFIs of the naive and activated Treg subsets were statistically independent from other measures obtained for the respective subset, i.e. CD25 was never significantly correlated with the same subset's circulating numbers, frequencies within total FoxP3+ or CD4+ cells, or Ki67+ proliferating fractions in any of our three study groups (not shown). There were also no significant relations to patients’ SLE Disease Activity Index (SLEDAI)‐2k disease activity, other clinical characteristics or treatments (Table 1) or to gender or age, except for a marginal R = 0·2/P = 0·046 age correlation with naive Treg CD25 only within unaffected relatives.
Although uncorrelated with surface CD25, circulating Treg numbers were also decreased both in patients and relatives, but less significantly (Supporting information, Fig. S2). In terms of Treg subset numbers per blood volume this was, however, restricted to the activated Tregs, which alone accounted for the differences in total FoxP3+ cells while naive Treg and FoxP3low numbers did not differ between groups (Supporting information, Fig. S2, first column). Relatively counted within total FoxP3+ cells (Supporting information, Fig. S2, second column), FoxP3low cells replaced the activated Tregs. Only when calculated within total CD4+ cells (Supporting information, Fig. S2, third column), Treg subset characteristics were no longer shared by patients and relatives – an effect that can, in fact, be explained by a relative over‐representation of Tregs in SLE patients with lymphopenia and that disappeared when only non‐lymphopenic subjects were considered (Supporting information, Fig. S3).
Effect 1: characteristic and functionally relevant CD25 reduction already in CD31+ recent thymic emigrant Tregs
Like naive Th cells, naive Tregs also contain a subpopulation of recent thymic emigrants (RTE), which can be distinguished as the developmentally earliest Tregs by their CD31 expression 39. We studied RTE Tregs with specific emphasis and found that their surface CD25 was already reduced strongly, actually showing almost identically low levels in SLE patients and unaffected relatives (1·5 and 1·4‐fold reduced, respectively) compared to healthy controls (Fig. 3a, mirrored by CD25– approximations within RTE Tregs; see Supporting information, Fig. S1).
Figure 3.

Characteristics of surface CD25 levels of recent thymic emigrant (RTE) Tregs. (a) Reduction of RTE regulatory T cells (Tregs) CD25 in systemic lupus erythematosus (SLE) patients and in unaffected relatives versus control subjects. (b,c) Significant correlation of RTE Treg CD25 with proportions of interleukin (IL)‐2 producers among phorbol myristate acetate (PMA)/I‐stimulated memory T cells in SLE patients and controls (while insignificant in unaffected relatives, not shown). (d–f) Significant correlations of RTE Treg CD25 with activated Treg frequencies in patients and relatives, but not control subjects. Linear correlation coefficients (R) and significant P‐values are shown; n.s. = non‐significant.
As human Treg development involves IL‐2‐driven intrathymic CD25 induction 40, we asked whether CD25 expression by RTE Tregs depended upon the individual capacity to produce IL‐2. The CD25 MFI of RTE Tregs was indeed found to be correlated positively with the capacity of cultured CD3+CD45RO+ T memory cells to produce IL‐2 upon PMA/ionomycin stimulation in SLE patients and also in controls (Fig. 3b,c; for IL‐2 stainings, for example, see Supporting information, Fig. S4). By contrast, RTE Treg CD25 levels were unrelated to patients’ SLEDAI‐2k disease activity, other clinical characteristics and treatment (Table 1) and to gender and age in all groups, except for a marginal increase with age within unaffected relatives, R = 0·25/P = 0·02 (not shown).
RTE Treg CD25 levels were also completely unrelated to their highly age‐dependent and barely group‐characteristic circulating numbers and frequencies (Supporting information, Fig. S5), as well as to Ki67+ proliferating fractions (not shown). Instead, we detected a remarkably strong correlation to proportions of activated among total Tregs in patients (R = 0·53/P = 3E‐4) as well as in unaffected relatives (R = 0·42/P = 2E‐5) (Fig. 3d–f). These activated Treg proportions were found to be correlated far less with CD25 levels when measured on all naive Tregs [patients: R = 0·29/not significant (n.s.); relatives: R = 0·32/P = 0·001], and completely unrelated to CD25 levels of the activated Tregs themselves. This suggests that the RTE Treg surface CD25 levels uniquely indicate the conditioning effect that the state of early Tregs appears to have on the individual capacity in our SLE risk population to produce high numbers of fully activated peripheral Tregs.
Effect 2: CD25 up‐regulation in activated versus naive Tregs is specifically deficient in manifest SLE
As activated Tregs are derived largely from naive Tregs 30, 31 but generally show higher surface CD25 levels, we addressed specifically their capacity to up‐regulate CD25 when differentiating from naive Tregs, measured as the MFI difference between both subsets in the same subject. While there was no significant difference between unaffected relatives and controls, CD25 up‐regulation was significantly lower and almost absent in SLE patients (Fig. 4). Thus, unaffected relatives of SLE patients appeared able to up‐regulate CD25 in activated Tregs to the same extent as healthy controls, while SLE patients had a specific deficiency in this respect.
Figure 4.
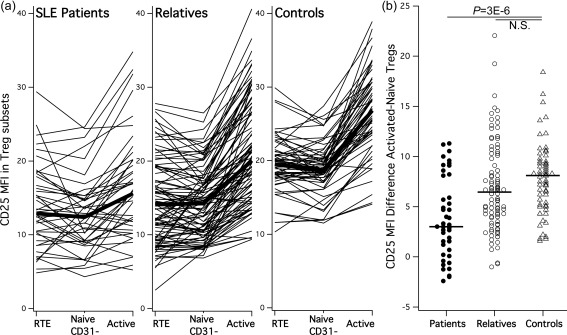
CD25 up‐regulation in activated regulatory T cells (Tregs) is selectively impaired in systemic lupus erythematosus (SLE) patients. (a) Individual trajectories of CD25 levels through the three Treg differentiation stages (thin lines) and their groupwise averages (thick lines). Reduced CD25 in naive Treg subsets appears ‘compensated’ in unaffected relatives but not in SLE patients by a subset‐specific up‐regulation in activated Tregs. (b) Quantified as CD25 mean fluorescence intensity (MFI) difference between naive and activated Tregs; this up‐regulation was equal in relatives and controls, but significantly lower in the SLE patients. The P‐value is shown; n.s. = non‐significant.
The SLE patients’ CD25 up‐regulation in activated versus naive Tregs was also found to be correlated positively with the individual capacity of their T memory cells to produce IL‐2 upon PMA/ionomycin stimulation (Fig. 5a), as was CD25 in RTE Tregs. In contrast, no such correlation was found in unaffected relatives or controls (Fig. 5b), indicating that the IL‐2 dependency of CD25 up‐regulation was limited to SLE patients where it was also particularly defective.
Figure 5.
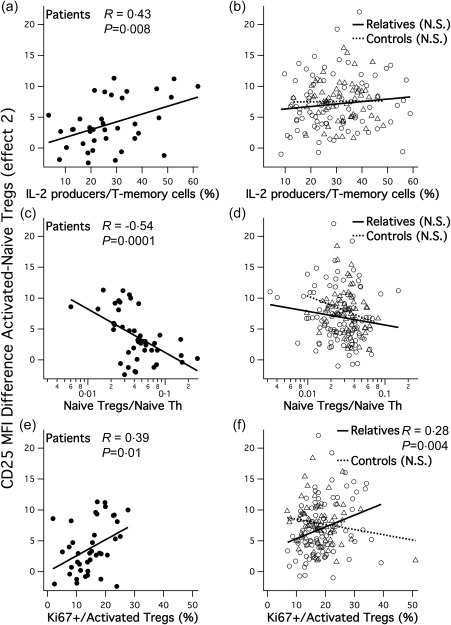
Characteristics of surface CD25 up‐regulation levels by activated regulatory T cells (Tregs). (a,b) CD25 up‐regulation correlates with proportions of interleukin (IL)‐2‐producing T memory cells upon phorbol myristate acetate/ionomycin (PMA/I) stimulation in systemic lupus erythematosus (SLE) patients but not in relatives or controls. (c,d) CD25 up‐regulation correlates with reduced naive Tregs relative to naive conventional T helper (Th) cells in SLE patients but not in relatives or controls. (e,f) CD25 up‐regulation correlates with Ki67+ proliferating fractions of activated Tregs in SLE patients and relatives but not in controls. (b,d,f) Circles and full regression lines indicate unaffected relatives; triangles and pointed lines indicate unrelated controls. Linear correlation coefficients (R) and significant P‐values are shown; n.s. = non‐significant.
CD25 up‐regulation in activated Tregs was uncorrelated with RTE Treg CD25 levels (effect 1) in relatives and in control subjects, and a positive correlation of both within patients was due entirely to their shared IL‐2 dependency (Supporting information, Fig. S6). We also detected no significant relations to age, gender, SLEDAI‐2k disease activity, clinical characteristics or treatment (not shown). Thus, CD25 up‐regulation fulfills all criteria of being an independent second effect contributing to the SLE‐associated Treg defect, reflecting peripheral but not thymic influences on Treg CD25 expression.
While CD25 up‐regulation in activated Tregs was basically uncorrelated with numbers and frequencies of activated Tregs, there were negative correlations with naive Tregs (Supporting information, Fig. S7). Interpreted as precursor consumption accompanying Treg activation, this could particularly explain that among patients, those with virtually absent CD25 up‐regulation were paradoxically the ones with most naive Tregs also in relation to naive FoxP3– T helpers (Fig. 5c,d). Furthermore, in the patients and relatives groups, CD25 up‐regulation was correlated positively with Ki67+ fractions within activated Tregs (Fig. 5e,f) suggesting that, apart from recruitment, their proliferation also influences this property.
Effect 1 shows the strongest heritability, and effect 2 is associated to IL2RA (CD25) genetic variants
In order to estimate overall genetic effects on the two described effects contributing to Treg CD25 deficiency in SLE, we addressed their probable heritability within our affected families by assessing familial correlations. Both parental and sibling correlations were indeed found highly significant for effect 1, RTE Treg CD25 (ρP = 0·73/P = 2E‐12; ρS = 0·70/P = 5E‐8), indicating a high heritability. For effect 2, CD25 MFI up‐regulation, familial correlations were clearly lower but still significant (ρP = 0·44/P = 1E‐5; ρS = 0·27/P = 0·032).
In our control group, where subjects were unrelated, we finally studied whether the two component effects were associated with genetic variation in the IL2RA locus that encodes CD25. For effect 1, RTE Treg CD25, association with two of 25 typed SNPs was univariately significant but did not retain significance upon FDR correction for multiple comparisons (Table 2). In contrast, IL2RA SNP association with effect 2, CD25 MFI up‐regulation, was significant also when FDR‐corrected. The associated SNPs were located in the IL2RA 5' portion (Table 2), corresponding to the region where genetic association primarily with type 1 diabetes (T1D) was also reported. In fact, the two SNP alleles that we found associated significantly with reduced CD25 up‐regulation were also described as T1D risk alleles 41.
Discussion
In SLE Tregs, a particular surface CD25 reduction ‘dissociated’ from FoxP3 expression was described previously 25, while the details and origins of this reduction have not been assessed thoroughly. Studying CD25 in Treg subsets, we could discriminate two independent effects contributing to the reduced surface CD25 in SLE that were both related to individual IL‐2 production:
effect 1, surface CD25 of the developmentally earliest RTE Tregs, was reduced in both SLE patients and unaffected relatives in a largely shared manner; and
effect 2, the degree to which Tregs up‐regulated CD25 upon activation, was only deficient in manifest SLE but not in unaffected relatives. This CD25 up‐regulation was associated (in controls) with IL2RA genetic variants.
Effect 1 – RTE Treg surface CD25 – likely reflects intrathymic Treg development 40. It neither depended upon disease manifestation nor did it correlate with disease activity. It was also unrelated to the age‐dependent RTE Treg numbers and frequencies, but highly heritable, as suggested by familial correlations. Thus, RTE Treg CD25 clearly does not represent an effect secondary to clinical disease, treatment or age, but rather a primary, largely genetically determined and individually variable ‘baseline’ CD25 that seems to be set intrathymically, as it is best visible in the developmentally earliest Tregs. Most interestingly, RTE Treg CD25 (unlike surface CD25 of activated or overall naive Tregs) was correlated uniquely positively, thus predictive for peripheral proportions of fully differentiated activated Tregs in the individual patients and relatives. This suggests that the RTE Treg functional state reflected by their surface CD25 level is crucial for the capacity of subjects at risk for SLE to produce high proportions of activated peripheral Tregs.
Effect 2 – CD25 up‐regulation in activated versus naïve Tregs – demonstrates a specific failure of SLE Tregs to increase CD25 expression when differentiating into activated CD45RO+ Tregs in the periphery. This failure was not seen in first‐degree unaffected relatives and therefore appears secondary to SLE manifestation (although we observed no relation to clinical features or treatment). Coincident with our earlier report pointing to a compensatory T cell regulation 37, it can be said that unaffected relatives ‘compensate’ their shared baseline CD25 reduction in early non‐activated Tregs (effect 1) by activation‐dependent up‐regulation – a mechanism that could be important for subjects at risk to avoid clinical disease. Unlike effect 1, effect 2 was not associated with increased activated Treg quantities in circulation, while it was related to naive Treg consumption and to activated Treg proliferation. This can be explained by the short‐lived nature of activated Tregs 30.
Technically, our quantitation of CD25 up‐regulation (effect 2) is based upon the assumption of a common Treg lineage, i.e. that transient FoxP3 expression and peripherally induced Tregs do not need to be considered. This seems reasonable to us in this context, as SLE Foxp3+ Tregs, including CD25low and CD25– cells, have shown extraordinarily high levels of Helios expression and FoxP3–TSDR demethylation 7, 32. While Helios expression indicates a thymic origin 33, 34, cytosine–phosphate–guanine (CpG) demethylation of the FoxP3–TSDR region 35 confirms sustained bona‐fide FoxP3 expression. Both markers do not occur in transiently FoxP3‐expressing T helpers or induced Tregs. Moreover, FoxP3low cells where transient expression may occur were excluded thoroughly from our cytometric definition of activated Tregs.
Our findings add to a series of papers which showed neatly that surface CD25 (but not FoxP3) is the only marker that can reflect the SLE‐associated phenotypical 21, 28 and functional 25, 28 Treg impairment appropriately. The two effects acting on reduced Treg CD25 expression described by us here were both related to the capacity of effector cells to produce its ligand, IL‐2, which is recognized widely as the most important factor for Treg homeostasis, since the time when it was first demonstrated that IL‐2‐deficient mice harbour very few Tregs with low surface CD25 42. The crucial role of IL‐2, particularly for peripheral Treg maintenance and expansion, is also generally accepted for the human system 43, and we have recently described its effects particularly in respect to SLE‐associated autoantibody profiles 37. More recently, IL‐2 was shown to be similarly important (besides IL‐15) for the thymic generation of human Tregs 40. Particularly in SLE, T cells obviously produce insufficient amounts of IL‐2 due to altered transcriptional regulation 4. Accordingly, a recent report demonstrates that the CD25 deficiency of SLE Tregs could be reversed efficiently by IL‐2 in cell cultures, as well as in patients undergoing low‐dose IL‐2 therapy 7. This corresponds with findings that restoring IL‐2 production in lupus‐prone mice enhanced the generation of CD25+FoxP3+ Tregs 44, 45.
Our results suggest that deficient IL‐2 in SLE affects surface CD25 in two separate ways in early and in late Tregs. Both effects appear genetically influenced. While the early effect 1 showed indirect evidence of (unspecified) genetic factors, the patient‐characteristic late effect 2 was found associated with genetic variants in the region of the CD25‐encoding locus IL2RA that also conferred T1D risk 41. Located 5' upstream of the IL2RA locus, the effect linked to these variants is unlikely to alter CD25 molecular properties, but rather transcriptional control by (not well known) transcription factors 46. Among the different haplotypes that have meanwhile been identified in this region 41, 47, our findings point mainly to a functionally unexplored rs11594656 haplotype where, conversely, T1D risk variants protected from multiple sclerosis (MS) 48. For the better‐studied rs2104286 haplotype in the same region, healthy individuals bearing the risk variant for both diseases with low IL‐2 receptor signalling 49 also had fewer Tregs with reduced suppressive function and less stable FoxP3 expression under limiting IL‐2 concentrations 50. Loss of FoxP3 expression by Tregs in IL‐2 insufficient conditions is also well documented from mouse experiments 51, 52 and explained by IL‐2 mediated transcriptional stabilization 53. Ex‐Tregs convert typically into Th17 cells, can easily become pathogenic and cause lung and liver inflammation 51, arthritis 54 and autoantigen‐driven encephalitis 55. Also in human MS, IL‐2–IL2R pathway alterations were linked to decreased FoxP3 expression 56, and Tregs converting to Th17 cells probably contribute to human inflammatory bowel diseases 57, rheumatoid arthritis 58 and psoriasis 59 by shifting the paradigmatic balance between these two developmentally related cell types 60. As the relevance of this for SLE remains somewhat unclear, our discrimination of two separate component effects may help to characterize further how the probably multiple associated mechanisms affect each type of autoimmunity.
In SLE patients, defective CD25 expression by Tregs was found completely reversible under low‐dose IL‐2 therapy 7. In our context, this corresponds most probably to a boost of effect 2, most affected in SLE patients and in fact showing less overall genetic determination (i.e. heritability), thus a more ‘acquired’ character, than effect 1. However, low‐dose IL‐2 therapy increased not only CD25 expression but even more impressively the numbers of circulating FoxP3+ Tregs. Interpreted in the context of our results, this also points to the involvement of IL‐2 effects on non‐activated Tregs, possibly even inside the thymus analogously to our effect 1, in addition to peripheral stimulation (effect 2). It will be extremely interesting to determine this in future clinical studies, as well as how the clinical outcome relates to the two effect types, in order to identify optimal therapy schemes for IL‐2 and possibly other therapies seeking to improve Treg function.
Author contributions
C. F., M. L. and B. M. designed research; N. C., O. M., S. I. G., C. C., B. L., A. M. F. and C. F. performed experiments; O.M., C. V., A. M., M.F. M. F., A. G. C., C. P., R. C. M., T. C., A. R. M. and J. F. V. collected data; N. C., O. M., S. I. G. and C. F. analysed and interpreted data; N. C. and C. F. wrote the manuscript.
Disclosure
The authors declare no competing financial interests.
Supporting information
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Fig. S1. Approximation of regulatory T cell (Treg) proportions lacking surface CD25. Although we are cautious about threshold‐defined CD25 gating and do not use it for our own quantitative analysis, we have approximated proportions of CD25– cells within Tregs by a plausible but still arbitrary threshold criterion (fluorescence intensity < 5) for comparability with other published work. (a,b) CD25– cells within naive Tregs, separately for CD31+ (recent thymic emigrants) and CD31–, (c) within activated Tregs and (d) within all CD4+ forkhead box P3 (FoxP3)+ T cells. Group differences of patients or unaffected relatives, respectively, to healthy unrelated control subjects were tested. Significant P‐values are shown.
Fig. S2. Group differences between regulatory T cell (Treg) numbers and frequencies depend strongly upon the type of measure. The three common measures of cell abundance are shown comparatively in columns: circulating cell numbers per blood volume (first column), frequencies within total forkhead box P3 (FoxP3)+ cells (second column) and frequencies within total CD4+ T cells (third column). Rows represent the three Treg subsets and total FoxP3+. For each measure, group differences of patients or unaffected relatives, respectively, to healthy unrelated control subjects were tested. Significant P‐values are shown; n.s. = non‐significant.
Fig. S3. Lymphopenia effect on regulatory T cell (Treg) frequencies when measured within CD4+ cells. (a) Circulating lymphocyte numbers per group demonstrate that many systemic lupus erythematosus (SLE) patients (but not unaffected relatives) have low lymphocyte numbers. (b) forkhead box P3 (FoxP3)+ frequencies within total CD4+ cells correlated strongly with lymphocyte numbers, showing their overrepresentation particularly in lymphopenic SLE patients. This over‐representation also caused a distribution bias of Treg subset measures within CD4+ cells: when only non‐lymphopenic subjects were considered, the previously detected increase in patients’ naive Tregs/CD4+ was abrogated (c), and activated Treg differences corresponded to those found for absolute cell numbers (d). Linear correlation coefficients (R) and significant P‐values are shown; n.s. = non‐significant.
Fig. S4. Intracellular interleukin (IL)‐2 staining examples. Gated on CD3+CD45RO+ memory T cells, representative cytograms for intracellular IL‐2 [vertically versus horizontal interferon (IFN)‐γ] staining are shown for (a) a systemic lupus erythematosus (SLE) patient, (b) a first‐degree relative, (c) an unrelated healthy control and (d) without anti‐IL‐2 antibody. Percentages of IL‐2 positive cells are annotated.
Fig. S5. Characteristics of different measures of recent thymic emigrants (RTE) regulatory T cell (Treg) numbers and frequencies. (a–d) Four different measures of RTE Tregs were analysed for their relation to age and groups. All measures showed a significant decrease with age in all three groups. Accordingly, group comparisons were calculated with age as a covariate. None of the four measures showed a significant difference between relatives and controls. Patients had significant reductions in absolute numbers (a) and frequencies within naive Tregs (d) but not when measured within total forkhead box P3 (FoxP3)+ (b) or CD4+ (b). (e,f) Relations of RTE Treg measures to RTE Treg surface CD25. No significant relations were found, as all groupwise direct or semipartial correlations (numbers/frequencies regressed against age) were found univariately insignificant.
Fig. S6. The correlation between recent thymic emigrants (RTE) regulatory T cell (Treg) CD25 and CD25 up‐regulation in activated Tregs is due entirely to their shared interleukin (IL)‐2 dependency. (a) The direct correlation between the two CD25 component effects was found significant only for patients but not relatives or controls. (b) As both CD25 component effects depended upon IL‐2 producers in patients, we further calculated their partial correlation (i.e. of both regressed against our measures of IL‐2 producer proportions). The absence of significance demonstrates that the previously detected correlation was due to an IL‐2 effect. Linear correlation coefficients (R) and significant P‐values are shown; n.s. = non‐significant.
Fig. S7. Relations of CD25 up‐regulation in activated regulatory T cells (Tregs) (effect 2) to Treg subset measures. We studied the possible relation of CD25 up‐regulation in activated Tregs (effect 2) to three common measures of naive and activated Treg numbers and frequencies, respectively (analogously to Fig. S2). (a–c) None of the activated Treg measures showed a significant relation when assessed by multiple regression, differentiating the three subject groups by two contrast covariates. Univariately, only activated Tregs/CD4+ measured within patients showed a significant correlation (c) which was, however, ascribable to lymphopenia bias as abrogated when considering only non‐lymphopenic subjects analogously to Fig. S3. (d–f) In contrast, all three measures of naive recent thymic emigrants (RTE) regulatory T cell (Treg) numbers or frequencies were found related significantly to decreasing CD25 up‐regulation in activated Tregs (effect 2) using the same multiple regression approach. Univariate groupwise correlation (although not significant throughout) also remained significant with naive Tregs/CD4+ in patients when considering only non‐lymphopenic patients (f). Linear correlation coefficients (R) and significant P‐values are shown; n.s. = non‐significant.
Acknowledgements
This study was supported by Fundação para a Ciência e a Tecnologia (Portugal) through the research grant PIC/IC/82746/2007 and fellowships SFRH/BPD/34648/2007 and SFRH/BPD/101836/2014 for C. F. We thank IGC and ICBAS technical services for their invaluable assistance, and V. Martins and I. Caramalho for critical reading.
References
- 1. Deane KD, El‐Gabalawy H. Pathogenesis and prevention of rheumatic disease: focus on preclinical RA and SLE. Nat Rev Rheumatol 2014; 10:212–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shlomchik MJ, Craft JE, Mamula MJ. From T to B and back again: positive feedback in systemic autoimmune disease. Nat Rev Immunol 2001; 1:147–53. [DOI] [PubMed] [Google Scholar]
- 3. Moulton VR, Tsokos GC. Abnormalities of T cell signaling in systemic lupus erythematosus. Arthritis Res Ther 2011; 13:207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Comte D, Karampetsou MP, Tsokos GC. T cells as a therapeutic target in SLE. Lupus 2015; 24:351–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fehervari Z, Yamaguchi T, Sakaguchi S. The dichotomous role of IL‐2: tolerance versus immunity. Trends Immunol 2006; 27:109–11. [DOI] [PubMed] [Google Scholar]
- 6. Humrich JY, von Spee‐Mayer C, Siegert E et al Rapid induction of clinical remission by low‐dose interleukin‐2 in a patient with refractory SLE. Ann Rheum Dis 2015; 74:791–2. [DOI] [PubMed] [Google Scholar]
- 7. von Spee‐Mayer C, Siegert E, Abdirama D et al Low‐dose interleukin‐2 selectively corrects regulatory T cell defects in patients with systemic lupus erythematosus. Ann Rheum Dis 2016; 75:1407–15. [DOI] [PubMed] [Google Scholar]
- 8. Scheinecker C, Bonelli M, Smolen JS. Pathogenetic aspects of systemic lupus erythematosus with an emphasis on regulatory T cells. J Autoimmun 2010; 35:269–75. [DOI] [PubMed] [Google Scholar]
- 9. Crispin JC, Kyttaris VC, Terhorst C, Tsokos GC. T cells as therapeutic targets in SLE. Nat Rev Rheumatol 2010; 6:317–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lee HY, Hong YK, Yun HJ, Kim YM, Kim JR, Yoo WH. Altered frequency and migration capacity of CD4+CD25+ regulatory T cells in systemic lupus erythematosus. Rheumatology (Oxford) 2008; 47:789–94. [DOI] [PubMed] [Google Scholar]
- 11. Mellor‐Pita S, Citores MJ, Castejon R et al Decrease of regulatory T cells in patients with systemic lupus erythematosus. Ann Rheum Dis 2006; 65:553–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Miyara M, Amoura Z, Parizot C et al Global natural regulatory T cell depletion in active systemic lupus erythematosus. J Immunol 2005; 175:8392–400. [DOI] [PubMed] [Google Scholar]
- 13. Yang J, Chu Y, Yang X et al Th17 and natural Treg cell population dynamics in systemic lupus erythematosus. Arthritis Rheum 2009; 60:1472–83. [DOI] [PubMed] [Google Scholar]
- 14. Barreto M, Ferreira RC, Lourenco L et al Low frequency of CD4+CD25+ Treg in SLE patients: a heritable trait associated with CTLA4 and TGFbeta gene variants. BMC Immunol 2009; 10:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lyssuk EY, Torgashina AV, Soloviev SK, Nassonov EL, Bykovskaia SN. Reduced number and function of CD4+CD25highFoxP3+ regulatory T cells in patients with systemic lupus erythematosus. Adv Exp Med Biol 2007; 601:113–9. [PubMed] [Google Scholar]
- 16. Alvarado‐Sanchez B, Hernandez‐Castro B, Portales‐Perez D et al Regulatory T cells in patients with systemic lupus erythematosus. J Autoimmun 2006; 27:110–8. [DOI] [PubMed] [Google Scholar]
- 17. Yates J, Whittington A, Mitchell P, Lechler RI, Lightstone L, Lombardi G. Natural regulatory T cells: number and function are normal in the majority of patients with lupus nephritis. Clin Exp Immunol 2008; 153:44–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Azab NA, Bassyouni IH, Emad Y, Abd El‐Wahab GA, Hamdy G, Mashahit MA. CD4+CD25+ regulatory T cells (TREG) in systemic lupus erythematosus (SLE) patients: the possible influence of treatment with corticosteroids. Clin Immunol 2008; 127:151–7. [DOI] [PubMed] [Google Scholar]
- 19. Zhang B, Zhang X, Tang FL, Zhu LP, Liu Y, Lipsky PE. Clinical significance of increased CD4+CD25‐Foxp3+ T cells in patients with new‐onset systemic lupus erythematosus. Ann Rheum Dis 2008; 67:1037–40. [DOI] [PubMed] [Google Scholar]
- 20. Lin SC, Chen KH, Lin CH, Kuo CC, Ling QD, Chan CH. The quantitative analysis of peripheral blood FOXP3‐expressing T cells in systemic lupus erythematosus and rheumatoid arthritis patients. Eur J Clin Invest 2007; 37:987–96. [DOI] [PubMed] [Google Scholar]
- 21. Bonelli M, von Dalwigk K, Savitskaya A, Smolen JS, Scheinecker C. Foxp3 expression in CD4+ T cells of patients with systemic lupus erythematosus: a comparative phenotypic analysis. Ann Rheum Dis 2008; 67:664–71. [DOI] [PubMed] [Google Scholar]
- 22. Suarez A, Lopez P, Gomez J, Gutierrez C. Enrichment of CD4+ CD25high T cell population in patients with systemic lupus erythematosus treated with glucocorticoids. Ann Rheum Dis 2006; 65:1512–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Venigalla RK, Tretter T, Krienke S et al Reduced CD4+,CD25‐ T cell sensitivity to the suppressive function of CD4+, CD25high, CD127‐/low regulatory T cells in patients with active systemic lupus erythematosus. Arthritis Rheum 2008; 58:2120–30. [DOI] [PubMed] [Google Scholar]
- 24. Yan B, Ye S, Chen G, Kuang M, Shen N, Chen S. Dysfunctional CD4+,CD25+ regulatory T cells in untreated active systemic lupus erythematosus secondary to interferon‐alpha‐producing antigen‐presenting cells. Arthritis Rheum 2008; 58:801–12. [DOI] [PubMed] [Google Scholar]
- 25. Bonelli M, Savitskaya A, Steiner CW, Rath E, Smolen JS, Scheinecker C. Phenotypic and functional analysis of CD4+ CD25‐ Foxp3+ T cells in patients with systemic lupus erythematosus. J Immunol 2009; 182:1689–95. [DOI] [PubMed] [Google Scholar]
- 26. Yang HX, Zhang W, Zhao LD et al Are CD4+CD25‐Foxp3+ cells in untreated new‐onset lupus patients regulatory T cells?. Arthritis Res Ther 2009; 11:R153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Horwitz DA. Identity of mysterious CD4+CD25‐Foxp3+ cells in SLE. Arthritis Res Ther 2010; 12:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bonelli M, Savitskaya A, von Dalwigk K et al Quantitative and qualitative deficiencies of regulatory T cells in patients with systemic lupus erythematosus (SLE). Int Immunol 2008; 20:861–8. [DOI] [PubMed] [Google Scholar]
- 29. Valencia X, Yarboro C, Illei G, Lipsky PE. Deficient CD4+CD25high T regulatory cell function in patients with active systemic lupus erythematosus. J Immunol 2007; 178:2579–88. [DOI] [PubMed] [Google Scholar]
- 30. Miyara M, Yoshioka Y, Kitoh A et al Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity 2009; 30:899–911. [DOI] [PubMed] [Google Scholar]
- 31. Booth NJ, McQuaid AJ, Sobande T et al Different proliferative potential and migratory characteristics of human CD4+ regulatory T cells that express either CD45RA or CD45RO. J Immunol 2010; 184:4317–26. [DOI] [PubMed] [Google Scholar]
- 32. Alexander T, Sattler A, Templin L et al Foxp3+ Helios+ regulatory T cells are expanded in active systemic lupus erythematosus. Ann Rheum Dis 2013; 72:1549–58. [DOI] [PubMed] [Google Scholar]
- 33. Thornton AM, Korty PE, Tran DQ et al Expression of Helios, an Ikaros transcription factor family member, differentiates thymic‐derived from peripherally induced Foxp3+ T regulatory cells. J Immunol 2010; 184:3433–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zabransky DJ, Nirschl CJ, Durham NM et al Phenotypic and functional properties of Helios+ regulatory T cells. PLOS ONE 2012; 7:e34547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Baron U, Floess S, Wieczorek G et al DNA demethylation in the human FOXP3 locus discriminates regulatory T cells from activated FOXP3(+) conventional T cells. Eur J Immunol 2007; 37:2378–89. [DOI] [PubMed] [Google Scholar]
- 36. Ferreira R, Barreto M, Santos E et al Heritable factors shape natural human IgM reactivity to Ro60/SS‐A and may predispose for SLE‐associated IgG anti‐Ro and anti‐La autoantibody production. J Autoimmun 2005; 25:155–63. [DOI] [PubMed] [Google Scholar]
- 37. Fesel C, Barreto M, Ferreira RC et al Compensatory T‐cell regulation in unaffected relatives of SLE patients, and opposite IL‐2/CD25‐mediated effects suggested by coreferentiality modeling. PLOS ONE 2012; 7:e33992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Costa N, Pires AE, Gabriel AM et al Broadened T‐cell repertoire diversity in ivIg‐treated SLE patients is also related to the individual status of regulatory T‐cells. J Clin Immunol 2014; 33:349–60. [DOI] [PubMed] [Google Scholar]
- 39. Kohler S, Thiel A. Life after the thymus: CD31(+) and CD31(–) human naive CD4(+) T‐cell subsets. Blood 2009; 113:769–74. [DOI] [PubMed] [Google Scholar]
- 40. Caramalho I, Nunes‐Silva V, Pires AR et al Human regulatory T‐cell development is dictated by interleukin‐2 and −15 expressed in a non‐overlapping pattern in the thymus. J Autoimmun 2015; 56:98–110. [DOI] [PubMed] [Google Scholar]
- 41. Lowe CE, Cooper JD, Brusko T et al Large‐scale genetic fine mapping and genotype‐phenotype associations implicate polymorphism in the IL2RA region in type 1 diabetes. Nat Genet 2007; 39:1074–82. [DOI] [PubMed] [Google Scholar]
- 42. Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A function for interleukin 2 in Foxp3‐expressing regulatory T cells. Nat Immunol 2005; 6:1142–51. [DOI] [PubMed] [Google Scholar]
- 43. Zorn E, Nelson EA, Mohseni M et al IL‐2 regulates FOXP3 expression in human CD4+CD25+ regulatory T cells through a STAT‐dependent mechanism and induces the expansion of these cells in vivo . Blood 2006; 108:1571–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Koga T, Ichinose K, Mizui M, Crispin JC, Tsokos GC. Calcium/calmodulin‐dependent protein kinase IV suppresses IL‐2 production and regulatory T cell activity in lupus. J Immunol 2012; 189:3490–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ohl K, Wiener A, Schippers A, Wagner N, Tenbrock K. Interleukin‐2 treatment reverses effects of cAMP‐responsive element modulator alpha‐over‐expressing T cells in autoimmune‐prone mice. Clin Exp Immunol 2015; 181:76–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Butter F, Davison L, Viturawong T et al Proteome‐wide analysis of disease‐associated SNPs that show allele‐specific transcription factor binding. PLoS Genet 2012; 8:e1002982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dendrou CA, Plagnol V, Fung E et al Cell‐specific protein phenotypes for the autoimmune locus IL2RA using a genotype‐selectable human bioresource. Nat Genet 2009; 41:1011–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Maier LM, Lowe CE, Cooper J et al IL2RA genetic heterogeneity in multiple sclerosis and type 1 diabetes susceptibility and soluble interleukin‐2 receptor production. PLOS Genet 2009; 5:e1000322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cerosaletti K, Schneider A, Schwedhelm K et al Multiple autoimmune‐associated variants confer decreased IL‐2R signaling in CD4(+)CD25(hi) T cells of type 1 diabetic and multiple sclerosis patients. PLOS ONE 2013; 8:e83811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Garg G, Tyler JR, Yang JH et al Type 1 diabetes‐associated IL2RA variation lowers IL‐2 signaling and contributes to diminished CD4+CD25+ regulatory T cell function. J Immunol 2012; 188:4644–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Duarte JH, Zelenay S, Bergman ML, Martins AC, Demengeot J. Natural Treg cells spontaneously differentiate into pathogenic helper cells in lymphopenic conditions. Eur J Immunol 2009; 39:948–55. [DOI] [PubMed] [Google Scholar]
- 52. Zhou XY, Bailey‐Bucktrout SL, Jeker LT et al Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo . Nat Immunol 2009; 10:1000–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Feng YQ, Arvey A, Chinen T, Van der Veeken JD, Gasteiger G, Rudensky AY. Control of the inheritance of regulatory T cell identity by a cis element in the Foxp3 locus. Cell 2014; 158:749–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sato K, Suematsu A, Okamoto K et al Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med 2006; 203:2673–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bailey‐Bucktrout SL, Martinez‐Llordella M, Zhou X et al Self‐antigen‐driven activation induces instability of regulatory T cells during an inflammatory autoimmune response. Immunity 2013; 39:949–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Carbone F, De Rosa V, Carrieri PB et al Regulatory T cell proliferative potential is impaired in human autoimmune disease. Nat Med 2013; 20:69–74. [DOI] [PubMed] [Google Scholar]
- 57. Ueno A, Jijon H, Chan R et al Increased prevalence of circulating novel IL‐17 secreting Foxp3 expressing CD4+T cells and defective suppressive function of circulating Foxp3+ regulatory cells support plasticity between Th17 and regulatory T cells in inflammatory bowel disease patients. Inflamm Bowel Dis 2013; 19:2522–34. [DOI] [PubMed] [Google Scholar]
- 58. Wang T, Sun XL, Zhao J et al Regulatory T cells in rheumatoid arthritis showed increased plasticity toward Th17 but retained suppressive function in peripheral blood. Ann Rheum Dis 2015; 74:1293–301. [DOI] [PubMed] [Google Scholar]
- 59. Bovenschen HJ, van de Kerkhof PC, van Erp PE, Woestenenk R, Joosten I, Koenen H. Foxp3+ regulatory T cells of psoriasis patients easily differentiate into IL‐17A‐producing cells and are found in lesional skin. J Invest Dermatol 2011; 131:1853–60. [DOI] [PubMed] [Google Scholar]
- 60. Noack M, Miossec P. Th17 and regulatory T cell balance in autoimmune and inflammatory diseases. Autoimmun Rev 2014; 13:668–77. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Fig. S1. Approximation of regulatory T cell (Treg) proportions lacking surface CD25. Although we are cautious about threshold‐defined CD25 gating and do not use it for our own quantitative analysis, we have approximated proportions of CD25– cells within Tregs by a plausible but still arbitrary threshold criterion (fluorescence intensity < 5) for comparability with other published work. (a,b) CD25– cells within naive Tregs, separately for CD31+ (recent thymic emigrants) and CD31–, (c) within activated Tregs and (d) within all CD4+ forkhead box P3 (FoxP3)+ T cells. Group differences of patients or unaffected relatives, respectively, to healthy unrelated control subjects were tested. Significant P‐values are shown.
Fig. S2. Group differences between regulatory T cell (Treg) numbers and frequencies depend strongly upon the type of measure. The three common measures of cell abundance are shown comparatively in columns: circulating cell numbers per blood volume (first column), frequencies within total forkhead box P3 (FoxP3)+ cells (second column) and frequencies within total CD4+ T cells (third column). Rows represent the three Treg subsets and total FoxP3+. For each measure, group differences of patients or unaffected relatives, respectively, to healthy unrelated control subjects were tested. Significant P‐values are shown; n.s. = non‐significant.
Fig. S3. Lymphopenia effect on regulatory T cell (Treg) frequencies when measured within CD4+ cells. (a) Circulating lymphocyte numbers per group demonstrate that many systemic lupus erythematosus (SLE) patients (but not unaffected relatives) have low lymphocyte numbers. (b) forkhead box P3 (FoxP3)+ frequencies within total CD4+ cells correlated strongly with lymphocyte numbers, showing their overrepresentation particularly in lymphopenic SLE patients. This over‐representation also caused a distribution bias of Treg subset measures within CD4+ cells: when only non‐lymphopenic subjects were considered, the previously detected increase in patients’ naive Tregs/CD4+ was abrogated (c), and activated Treg differences corresponded to those found for absolute cell numbers (d). Linear correlation coefficients (R) and significant P‐values are shown; n.s. = non‐significant.
Fig. S4. Intracellular interleukin (IL)‐2 staining examples. Gated on CD3+CD45RO+ memory T cells, representative cytograms for intracellular IL‐2 [vertically versus horizontal interferon (IFN)‐γ] staining are shown for (a) a systemic lupus erythematosus (SLE) patient, (b) a first‐degree relative, (c) an unrelated healthy control and (d) without anti‐IL‐2 antibody. Percentages of IL‐2 positive cells are annotated.
Fig. S5. Characteristics of different measures of recent thymic emigrants (RTE) regulatory T cell (Treg) numbers and frequencies. (a–d) Four different measures of RTE Tregs were analysed for their relation to age and groups. All measures showed a significant decrease with age in all three groups. Accordingly, group comparisons were calculated with age as a covariate. None of the four measures showed a significant difference between relatives and controls. Patients had significant reductions in absolute numbers (a) and frequencies within naive Tregs (d) but not when measured within total forkhead box P3 (FoxP3)+ (b) or CD4+ (b). (e,f) Relations of RTE Treg measures to RTE Treg surface CD25. No significant relations were found, as all groupwise direct or semipartial correlations (numbers/frequencies regressed against age) were found univariately insignificant.
Fig. S6. The correlation between recent thymic emigrants (RTE) regulatory T cell (Treg) CD25 and CD25 up‐regulation in activated Tregs is due entirely to their shared interleukin (IL)‐2 dependency. (a) The direct correlation between the two CD25 component effects was found significant only for patients but not relatives or controls. (b) As both CD25 component effects depended upon IL‐2 producers in patients, we further calculated their partial correlation (i.e. of both regressed against our measures of IL‐2 producer proportions). The absence of significance demonstrates that the previously detected correlation was due to an IL‐2 effect. Linear correlation coefficients (R) and significant P‐values are shown; n.s. = non‐significant.
Fig. S7. Relations of CD25 up‐regulation in activated regulatory T cells (Tregs) (effect 2) to Treg subset measures. We studied the possible relation of CD25 up‐regulation in activated Tregs (effect 2) to three common measures of naive and activated Treg numbers and frequencies, respectively (analogously to Fig. S2). (a–c) None of the activated Treg measures showed a significant relation when assessed by multiple regression, differentiating the three subject groups by two contrast covariates. Univariately, only activated Tregs/CD4+ measured within patients showed a significant correlation (c) which was, however, ascribable to lymphopenia bias as abrogated when considering only non‐lymphopenic subjects analogously to Fig. S3. (d–f) In contrast, all three measures of naive recent thymic emigrants (RTE) regulatory T cell (Treg) numbers or frequencies were found related significantly to decreasing CD25 up‐regulation in activated Tregs (effect 2) using the same multiple regression approach. Univariate groupwise correlation (although not significant throughout) also remained significant with naive Tregs/CD4+ in patients when considering only non‐lymphopenic patients (f). Linear correlation coefficients (R) and significant P‐values are shown; n.s. = non‐significant.