

Abstract
Although translational research into autosomal dominant polycystic kidney disease (ADPKD) and its pathogenesis has made considerable progress, there is presently lack of standardized animal model for preclinical trials. In this study, we developed an orthologous mouse model of human ADPKD by cross‐mating Pkd2 conditional‐knockout mice (Pkd2 f3) to Cre transgenic mice in which Cre is driven by a spectrum of kidney‐related promoters. By systematically characterizing the mouse model, we found that Pkd2 f3/f3 mice with a Cre transgene driven by the mouse villin‐1 promoter (Vil‐Cre;Pkd2 f3/f3) develop overt cysts in the kidney, liver and pancreas and die of end‐stage renal disease (ESRD) at 4–6 months of age. To determine whether these Vil‐Cre;Pkd2 f3/f3 mice were suitable for preclinical trials, we treated the mice with the high‐dose mammalian target of rapamycin (mTOR) inhibitor rapamycin. High‐dose rapamycin significantly increased the lifespan, lowered the cystic index and kidney/body weight ratio and improved renal function in Vil‐Cre;Pkd2 f3/f3 mice in a time‐ and dose‐dependent manner. In addition, we further found that rapamycin arrested aberrant epithelial‐cell proliferation in the ADPKD kidney by down‐regulating the cell‐cycle‐associated cyclin‐dependent kinase 1 (CDK1) and cyclins, namely cyclin A, cyclin B, cyclin D1 and cyclin E, demonstrating a direct link between mTOR signalling changes and the polycystin‐2 dysfunction in cystogenesis. Our newly developed ADPKD model provides a practical platform for translating in vivo preclinical results into ADPKD therapies. The newly defined molecular mechanism by which rapamycin suppresses proliferation via inhibiting abnormally elevated CDK1 and cyclins offers clues to new molecular targets for ADPKD treatment.
Keywords: Pkd2 mouse model, ADPKD, mTOR pathway, rapamycin
Introduction
Autosomal dominant polycystic kidney disease (ADPKD) is the most common of a group of inherited kidney disorders characterized by progressive cyst development and various extrarenal manifestations 1, 2. ADPKD, which has a worldwide prevalence estimated from 1:1000 to 1:4000, is the fourth most common single cause of end‐stage renal failure worldwide 3, 4, 5.
ADPKD, which primarily affects adults, is caused by mutations in the PKD1 or PKD2 genes, which encode the proteins polycystin‐1 (PC1) and polycystin‐2 (PC2), respectively. Approximately 85% of ADPKD patients have mutations in PKD1, and the remaining 15% have mutations in PKD2 6, 7. ADPKD follows a more severe course when it is caused by a PKD1 mutation. The most common extrarenal manifestation of ADPKD is the formation of bile‐duct‐derived cysts in the liver 1, 8, 9, 10. Liver cysts occur in 83% of all ADPKD patients, and in 94% of those over 35 years of age 11, 12. Other ADPKD phenotypes include pancreatic cysts 13, 14, cerebral aneurysm 15, 16, 17, 18 and aortic root/thoracic aorta abnormalities 19, 20, 21.
There has been considerable progress in elucidating the molecular mechanisms and pathogenesis of ADPKD 3, 6, 22, 23, 24. However, there is no standardized animal model of ADPKD, and some non‐standardized ADPKD models are not systemically characterized. In addition, although several preclinical and clinical trials have reported that rapamycin has a beneficial effect on ADPKD 25, 26, no direct link between polycystin dysfunction and mTOR signalling changes or ADPKD cystogenesis has been established. In this study, we developed and well characterized a newly established ADPKD model and demonstrated that rapamycin inhibits cystic progression in the ADPKD kidney by down‐regulating the cell‐cycle‐associated CDK1 and cyclins (cyclin A, cyclin B, cyclin D1 and cyclin E), thereby arresting aberrant proliferation of the renal epithelia. The study not only generated a well‐characterized, standardized mouse model for ADPKD, but also revealed the involvement of the mTORC1–CDK1/cyclin axis in ADPKD, which leads to new molecular targets for treating this disease.
Materials and methods
Animals
This study was conducted using Vil‐Cre;Pkd2 f3/f3 mice and their normal Pkd2 f3/f3 littermates (control). All of the mouse models used in this study were from a C57BL/6J inbred background. Pkd2 f3/f3 mice 27 were bred with various Cre transgenic mice (Table 1) to generate Pkd2 conditional‐knockout mice. Mouse genotyping and rapamycin treatment are detailed in Supplementary Methods A in Appendix S1.
Table 1.
Cre transgenic mice used in this study
| Nomenclature | Promoter | Exp. Time | Expressional Distribution | JAX Stock # | Ref. |
|---|---|---|---|---|---|
| γGt‐Cre | Rat gamma‐glutamyltransferase 1 promoter | E7.5 | Cortical proximal tubules | 012841 |
84
(Table S1) |
| Nestin‐Cre | Rat nestin promoter | E11.5 | The central and peripheral nervous system, kidney, heart | 003771 |
85
(Table S1) |
| Villin‐Cre | Mouse villin 1 promoter | E12.5 |
Embryonic intestinal endoderm adult intestinal epithelium vertical and horizontal axis |
004586 | 30 |
| Ksp‐Cre | Mouse cadherin 16 (Cdh16 or Ksp‐cadherin) promoter | E15.5 |
Embryo: epithelial cells of developing nephrons, ureteric bud, mesonephric tubules, Wolffian duct, and Mullerian duct. Adult: renal tubules especially the collecting ducts, loops of Henle and distal tubules |
012237 |
86
(Table S1) |
Cell lines, antibodies and reagents
The null‐Pkd2 (E8) cell line and its maternally derived Pkd2 heterozygous (D3) cell line were described in our previous study 27. Antibodies and reagents are listed in Supplementary Methods B in Appendix S1.
Microarray analysis, quantitative PCR, histology, immunofluorescence (IF) and Western blots
Histology and IF staining procedures were as previously described 27, 28, 29. All other assays are described in Supplementary Methods C in Appendix S1.
Statistics and measurements of cystic index, proliferation, apoptosis, blood urea nitrogen (BUN) and creatinine (Cr)
Statistics and detailed procedures of cystic index calculation, proliferation and apoptosis are described in Supplementary Methods D in Appendix S1.
Results
Development of a Pkd2 mouse model that mimics human ADPKD
Standardized animal models for ADPKD would contribute greatly to the investigation of polycystin function, cystic pathogenesis and novel therapeutic compounds for treating ADPKD. Although we recently generated Pkd2 conditional‐knockout mice (Pkd2 f3/f3) 27, we still needed to find an appropriate Cre‐recombination system by which the Pkd2 f3/f3 model could be used to represent the clinical manifestations of human ADPKD and to assess the effect of various therapeutic interventions. By cross‐mating Pkd2 f3/f3 mice with Cre transgenic mice driven by various kidney‐related promoters, including γGt‐Cre, Ksp‐Cre, Nestin‐Cre and Vil‐Cre mice (Table 1), we found that the Vil‐Cre;Pkd2 f3/f3 mice developed disease phenotypes that were similar to human ADPKD (Fig. 1). Cre‐recombinase expressed in the Vil‐Cre transgenic mouse under the control of the mouse villin‐1 promoter is turned on at E12.5, when the kidney is starting to develop 30. Compared to control mice (i.e. Pkd2 f3/f3 mice without Vil‐Cre transgene) (Fig. 1A–C), Pkd2 f3/f3 mice with the Vil‐Cre transgene (Vil‐Cre;Pkd2 f3/f3) developed severe gross cysts in the kidneys, liver and pancreas (Fig. 1D–F). Anatomic and histological analyses of the Vil‐Cre;Pkd2 f3/f3 kidney (Fig. 1A versus D and G versus H) showed that renal failure was the likely cause of death in these mice, most of which died between 4 and 6 months of age (Fig. 1I). As this mouse model mimics the disease phenotype of human ADPKD patients and has an ideally temporal cystic phenotype, Vil‐Cre;Pkd2 f3/f3 mice can be used as an ADPKD model to study the pathogenesis of Pkd2‐associated ADPKD and to assess therapeutic responses.
Figure 1.
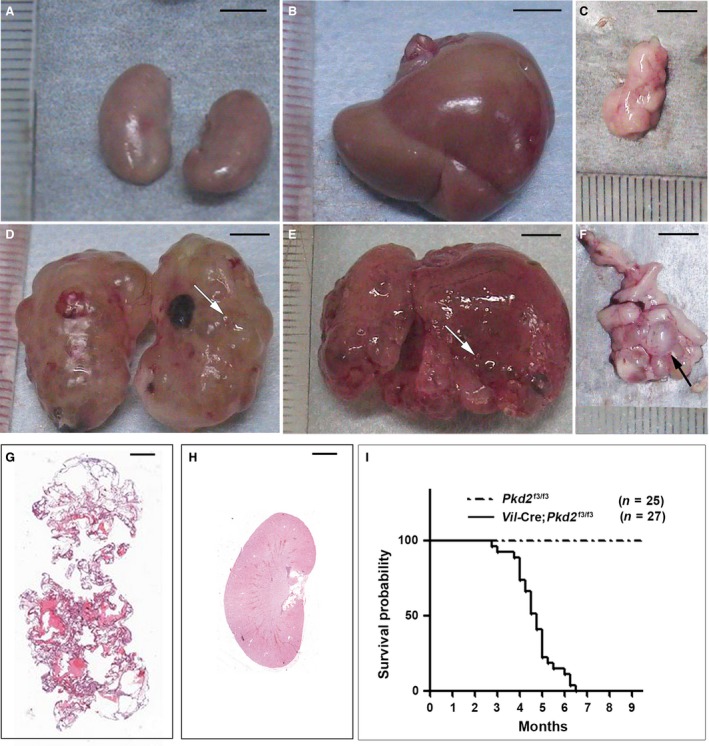
Mice with Vil‐Cre;Pkd2 f3/f3 alleles mimic the cystic phenotypes of human ADPKD. Compared to age‐matched control mice (A–C), 4‐month‐old Vil‐Cre;Pkd2 f3/f3 mice showed massive gross cysts (arrows) in the kidneys (D), liver (E) and pancreas (F). (G) H&E staining of a section from the kidney shown in (D). (H) The same H&E staining for the control kidney shown in (A). (I) Kaplan–Meier survival curves for Vil‐Cre;Pkd2 f3/f3 mice. The median survival time for Vil‐Cre;Pkd2 f3/f3 mice was about 4.5 months. Bars: 5 mm in A–F; 1 mm in G and H.
Characterization of cystic phenotypes in the Vil‐Cre;Pkd2 f3/f3 mice, an orthologous mouse model of ADPKD
To validate Vil‐Cre;Pkd2 f3/f3 mice as a suitable model for ADPKD, we systematically characterized the kidney, liver and pancreas by the kidney or liver/body weight ratio, cystic index and hepatorenal functions. In the kidneys, compared to control mice (i.e. Pkd2 f3/f3), the kidney/body weight ratio and the cystic index were significantly increased in Pkd2 f3/f3 mice with the Vil‐Cre transgene (Vil‐Cre;Pkd2 f3/f3). These mice had a significantly larger kidney/body weight ratio and renal cystic index by the time they were a half‐month old (P < 0.05) (Fig. 2A and B), indicating that cysts were already forming in the diseased kidneys at this early age.
Figure 2.
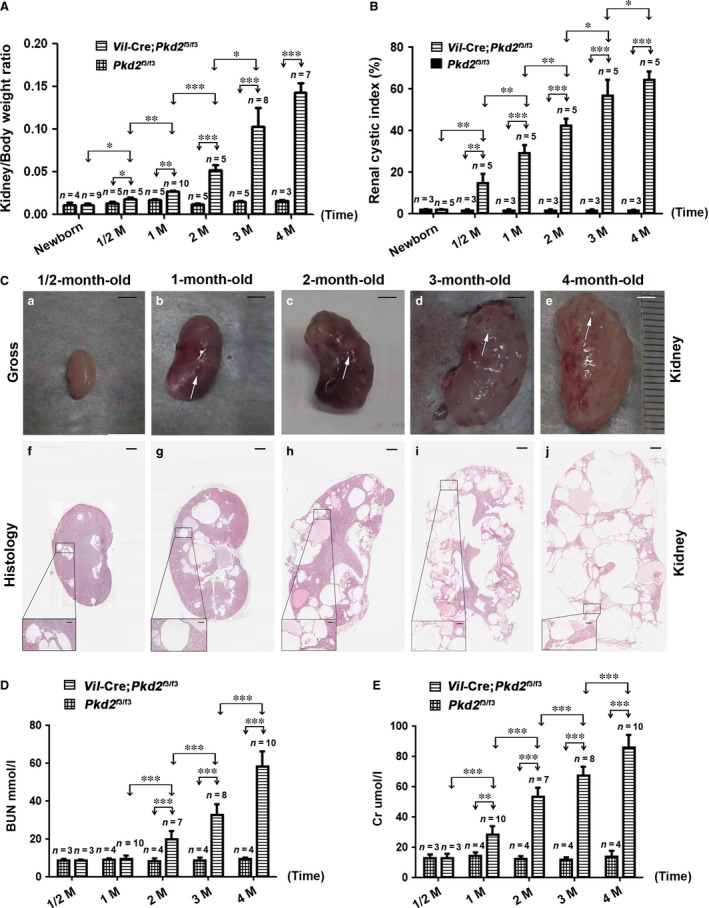
Characterization of the Vil‐Cre;Pkd2 f3/f3 kidney. (A) The kidney/body weight ratio for Vil‐Cre;Pkd2 f3/f3 newborns and for mice at ½, 1, 2, 3 and 4 months of age, compared to that of age‐matched control mice (Pkd2 f3/f3). (B) The renal cystic index of Pkd2 f3/f3 mice with or without the Vil‐Cre transgene allele at the ages shown. (C) The Vil‐Cre;Pkd2 f3/f3 kidney showed gross cysts (arrows) at 1 month of age (b) and massive gross cysts at 2–4 months (c‐e); histological results (f‐j) of the same kidneys were consistent with the gross examination. (D) BUN levels of Vil‐Cre;Pkd2 f3/f3 mice at ½, 1, 2, 3 and 4 months of age: BUN values rose significantly above those in control (Pkd2 f3/f3) mice at 2 months of age and continued to worsen with age. (E) Cr levels showed a similar pattern, starting at 1 month of age. *P < 0.05; **P < 0.01; ***P < 0.001. Bar: 3 mm in Ca‐e; 600 μm in Cf‐j; 100 μm in boxed areas. M: months.
Anatomic and histological analysis revealed that the Vil‐Cre;Pkd2 f3/f3 kidneys exhibited gross and microscopic cysts (Fig. 2C), in correlation with the kidney/body weight ratio and cystic index. By 4 months of age, normal parenchyma was almost entirely lost in the Vil‐Cre;Pkd2 f3/f3 kidney (Fig. 2Ce and j), indicating that these mice could suffer renal failure at this stage. To test renal function, we measured the blood urea nitrogen (BUN) and creatinine (Cr) levels in Pkd2 f3/f3 mice with and without the Vil‐Cre transgene. Compared to their Pkd2 f3/f3 littermates, the Vil‐Cre;Pkd2 f3/f3 mice had significantly higher BUN level at 2 months of age, but Cr level at 1 month of age (Fig. 2D and E). These results indicated that Cr might be more sensitive than BUN for detecting renal function damage in this disease model. In addition, we also used the nephron‐specific markers to clarify the renal segment of origin of the cysts in the Vil‐Cre;Pkd2 f3/f3 mice. Immunofluorescence staining revealed that the cyst‐lining cells in 3‐month‐old Vil‐Cre;Pkd2 f3/f3 kidneys were positive for all renal segments (Fig. S1), indicating that the renal cysts derived from all nephronic segments of the kidney.
By examination of the cystic liver and pancreas in Vil‐Cre;Pkd2 f3/f3 mice, we found no cysts in the liver of Vil‐Cre;Pkd2 f3/f3 mice before 1 month of age (Fig. S2Aa), or in the pancreas before 3 months of age (Fig. S2Ba‐c). However, gross cysts were observed in the liver starting at 2 months (Fig. S2Ab‐d) and in the pancreas starting at 4 months of age (Fig. S2Bd). Histological analyses revealed similar findings (Fig. S2Af‐h, Bh), but the cystic liver worsened rapidly with age. As Vil‐Cre;Pkd2 f3/f3 mice developed severe cysts in the liver, we also examined the liver/body weight ratio and measured the liver function using alanine aminotransferase (ALT). The liver/body weight ratio and liver function did not differ significantly between Pkd2 f3/f3 mice with or without the Vil‐Cre transgene (Fig. S2C‐D), indicating that as with human ADPKD patients, liver disease is not a life‐threatening factor in the mouse model. Our results showed that Vil‐Cre;Pkd2 f3/f3 mice not only exhibited a spatially and temporally appropriate cystic kidney phenotype for ADPKD, but also developed liver cysts as is often seen in human ADPKD patients.
Effect of gender on disease severity in the Vil‐Cre;Pkd2 f3/f3 mice
Recent clinical studies have demonstrated that the progression of renal cysts in ADPKD may be influenced by gender 31, 32, 33, 34; the cystic disease appears to progress more quickly in males than in females. Male patients often develop end‐stage renal failure and require haemodialysis earlier than female patients, suggesting that the prognosis for women is better than that for men 4, 35, 36. To determine whether this gender difference was also present in the ADPKD mouse model, we analysed the Kaplan–Meier survival rate, kidney/body weight ratio and renal histology and function in male and female Vil‐Cre;Pkd2 f3/f3 mice. Notably, more than 50% of the female Vil‐Cre;Pkd2 f3/f3 mice survived to 5 months of age, approximately 1 month longer than their male counterparts (P < 0.01) (Fig. S3A), indicating a better survival rate and lifespan for females. In addition, histological analysis showed that the renal cystogenesis was more severe in the kidneys from male Vil‐Cre;Pkd2 f3/f3 mice (Fig. S3B). The renal cystic index and kidney/body weight ratio differed significantly between male and female Vil‐Cre;Pkd2 f3/f3 mice after 3 months of age (P < 0.05) (Fig. S3C‐D). Furthermore, the BUN and Cr levels in 4‐month‐old Vil‐Cre;Pkd2 f3/f3 males were significantly elevated over those in age‐matched females (P < 0.05) (Fig. S3E‐F). Thus, gender might influence cystogenesis in the ADPKD kidney, with female gender being a protective factor both in the animal model and in ADPKD patients 34, 37, 38.
Validation of the Vil‐Cre;Pkd2 f3/f3 mice as an ADPKD animal model for drug treatment
Rapamycin is reported to significantly ameliorate the cystic phenotypes of diverse ADPKD animal models 10, 39, 40, 41, 42, 43, 44. To ensure that the ADPKD mouse model could be obtained therapeutic response, we treated Vil‐Cre;Pkd2 f3/f3 mice with rapamycin (50 mg/kg/day i.p.) (Protocol I, Fig. 3Aa). The Protocol I‐treated mice were given daily injections from postnatal day (P) 10 to 60; the control group was treated with the same injection and volume of DMSO solvent (placebo). The mice were killed 5 days (P65) after the end of treatment (ETR). Kaplan–Meier survival analysis showed that the Vil‐Cre;Pkd2 f3/f3 mice treated with rapamycin had a significantly longer lifespan than those treated with the placebo (P < 0.001) (Fig. 3Ab). As seen also in untreated mice (Fig. S3), the female rapamycin‐treated mice had a significantly higher survival rate than their male counterparts, with a median survival of about 7 months for females and 6 months for males (P < 0.05) (Fig. 3Ac).
Figure 3.
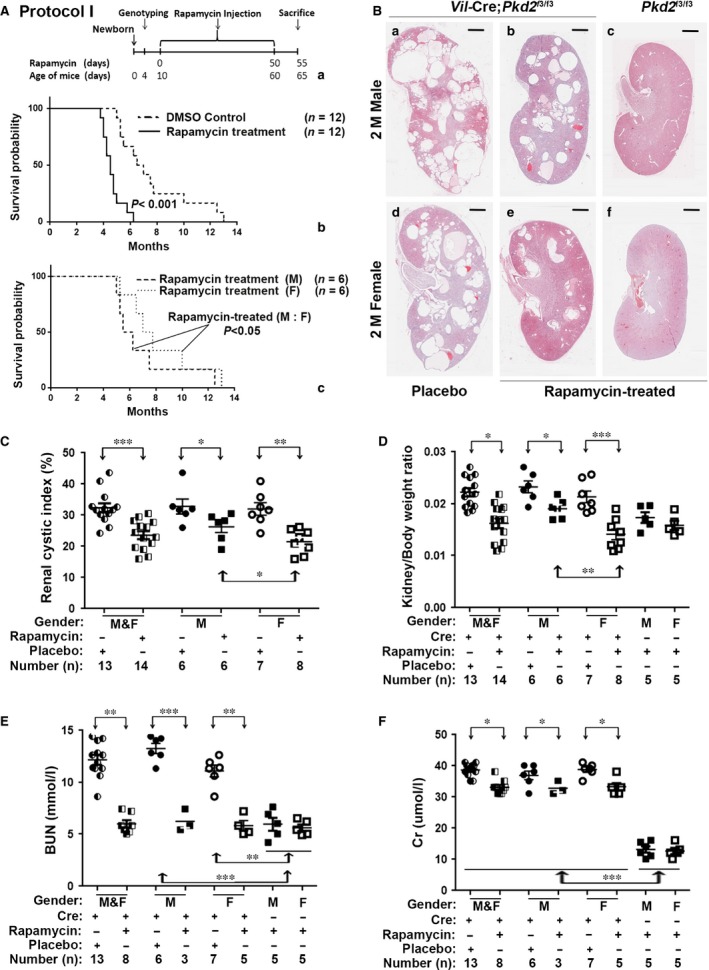
Rapamycin delayed cyst formation in Vil‐Cre;Pkd2 f3/f3 mice. (A) A diagram showing the initial treatment schedule for rapamycin injection (Protocol I) (a) in this study. Kaplan–Meier survival curves showed that compared to a placebo treatment, rapamycin significantly prolonged the lifespan in Vil‐Cre;Pkd2 f3/f3 mice (b) (P < 0.001), and that the lifespan of rapamycin‐treated Vil‐Cre;Pkd2 f3/f3 females was significantly longer than that of their male counterparts (c) (P < 0.05). (B) Histological examination of kidney sections from 2‐month‐old mice showed that cysts were reduced in the rapamycin‐treated compared to DMSO (placebo)‐treated Vil‐Cre;Pkd2 f3/f3 mice, and that kidneys from the rapamycin‐treated Vil‐Cre;Pkd2 f3/f3 females appeared less cystic than those of their male counterparts (b versus e). (C) The cystic index was significantly lower in rapamycin‐treated than placebo‐treated Vil‐Cre;Pkd2 f3/f3 mice (P < 0.001). Rapamycin produced a significantly stronger therapeutic response in the females than in the males (P < 0.05). (D) Kidney/body weight ratios for the mice in (C) were consistent with the cyst‐index findings. (E) BUN levels were significantly suppressed in the rapamycin‐treated compared to placebo‐treated Vil‐Cre;Pkd2 f3/f3 mice (P < 0.01). In both male and female Vil‐Cre;Pkd2 f3/f3 mice, rapamycin reduced the BUN to the level found in control (Pkd2 f3/f3) mice. (F) In the same mice, rapamycin significantly suppressed the Cr level (P < 0.05) but did not rescue Cr to the control level (P < 0.001). Bars: 600 μm in B. M: Male; F: Female.
Besides improving survival, intensive rapamycin treatment also significantly reduced the growth of renal cysts (Fig. 3B), the renal cystic index (Fig. 3C) and the kidney/body weight ratio (Fig. 3D) in Vil‐Cre;Pkd2 f3/f3 mice. The treatment response to rapamycin was significantly better in females (Fig. 3C and D), consistent with the survival analyses (Fig. 3Ac). We also investigated renal function and found that BUN and Cr levels were significantly improved in rapamycin‐treated as compared to placebo‐treated Vil‐Cre;Pkd2 f3/f3 mice (P < 0.01 and P < 0.05, respectively) (Fig. 3E and F). Improvements in BUN and Cr were similar in males and females. Interestingly, although rapamycin rescued the BUN level in Vil‐Cre;Pkd2 f3/f3 mice to that in control Pkd2 f3/f3 mice (Fig. 3E), rapamycin significantly reduced Cr (P < 0.05) but could not bring it down to the normal control levels (P < 0.001) (Fig. 3F). Thus, the intensive rapamycin protocol (Protocol I, Fig. 3Aa) could significantly improve the cystic phenotype of the Vil‐Cre;Pkd2 f3/f3 kidney, but might not fully rescue renal function.
Enhanced rapamycin treatment improved the therapeutic response in our ADPKD animal model
Although preclinical studies have reported that rapamycin is an effective treatment for ADPKD in animal models 10, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, clinical trials of rapamycin treatment for ADPKD patients have been less successful 50, 51, 52, 53, 54, 55, 56, 57. These poor clinical results might be due to insufficient rapamycin 48, 55, 58. To verify this possibility in a cohort study in vivo, we treated Vil‐Cre;Pkd2 f3/f3 mice using three additional rapamycin protocols (Fig. 4A, protocols II–IV) that used the same rapamycin dose as in Protocol I (50 mg/kg/day i.p.) but varied the duration or timing of treatment. Protocol II extended the duration of treatment; mice received rapamycin injections from P10 to P110. In protocols III and IV, mice received rapamycin injections from P10 to P60 and from P60 to P110, respectively. Mice treated with protocols II–IV were killed at P115.
Figure 4.
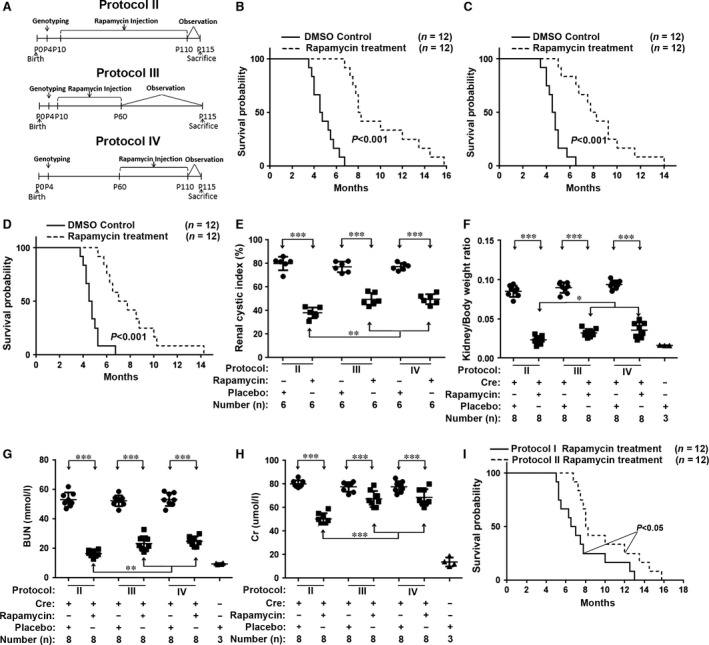
Increasing the dosage or duration of rapamycin treatment significantly improved the prognosis for the Vil‐Cre;Pkd2 f3/f3 mice. (A) Diagram showing the rapamycin treatment protocols (protocols II–IV) used in this study. (B) Kaplan–Meier survival curves showed that the lifespan was significantly prolonged in the Vil‐Cre;Pkd2 f3/f3 mice treated by Protocol II compared with those treated with DMSO (placebo) (P < 0.001); and that (C–D) survival was similarly improved for Vil‐Cre;Pkd2 f3/f3 mice treated with Protocol III versus IV (P < 0.001). (E) The cystic index for kidneys from Vil‐Cre;Pkd2 f3/f3 mice treated with Protocol II, III or IV. Protocol II provided the greatest improvement in the cystic index value (P < 0.01); (F) Kidney/body weight ratios for the same mice were consistent with the cystic index. Protocol II yielded the greatest improvement in the kidney/body weight ratio (P < 0.05). (G) In Vil‐Cre;Pkd2 f3/f3 mice, Protocol II significantly decreased the BUN level compared to that in DMSO (placebo)‐treated mice (P < 0.001). Protocol II decreased the BUN level significantly more than did Protocol III or IV (P < 0.01), returning the BUN nearly to the normal control level. (H) Similarly, Cr analysis of the same mice showed that Protocol II suppressed the abnormal Cr much more effectively than did Protocol III or IV (P < 0.001). (I) Kaplan–Meier survival curves for Vil‐Cre;Pkd2 f3/f3 mice treated with Protocol I or II, showing that Protocol II significantly prolonged the lifespan (P < 0.05).
Kaplan–Meier survival analyses showed that the lifespans were significantly improved in Vil‐Cre;Pkd2 f3/f3 mice treated with protocols II–IV compared to those treated with the placebo (P < 0.001) (Fig. 4B–D). Rapamycin also significantly reduced the renal cyst growth (Fig. 4E) and kidney/body weight ratios (Fig. 4F) and improved BUN (Fig. 4G) and Cr values (Fig. 4H). Statistical analyses showed that Protocol II, with the longest duration of rapamycin treatment (100 days), yielded better results than did protocols III and IV, in which mice received rapamycin for the first or second 50 treatment days, respectively. Protocol II significantly reduced renal cyst growth (P < 0.01) (Fig. 4E), lowered the kidney/body weight ratios (P < 0.05) (Fig. 4F) and improved the BUN (P < 0.01) (Fig. 4G) and Cr (P < 0.001) (Fig. 4H) levels compared to protocols III and IV. We also compared the survival rates in the Protocol II group with those of other protocols (I, III and IV) (Figs 3Aa and 4A). Protocol II prolonged the lifespan in Vil‐Cre;Pkd2 f3/f3 mice to a median survival of about 8 months, compared to 6.5 months (P < 0.05) for Protocol I (Fig. 4I), and about 7 months for protocols III and IV (P < 0.001) (data not shown). Thus, extending the duration of rapamycin treatment significantly improved the prognosis of Vil‐Cre;Pkd2 f3/f3 mice.
Rapamycin down‐regulates renal tubule proliferation in Vil‐Cre;Pkd2 f3/f3 mice by inhibiting cell‐cycle‐associated cyclins and cyclin‐dependent kinase
Previous studies demonstrated that rapamycin can effectively inhibit mTORC1 and its downstream factors 59, 60. Recent studies have shown that the mTOR pathway is dysregulated in ADPKD patients and in ADPKD animal models 45, 61. Rapamycin significantly inhibits cyst formation and greatly improves the prognosis for several ADPKD animal models 10, 40, 41, 42, 43, 44, 46, 47, 48, 49. However, the molecular link between polycystin dysfunction and mTOR signalling changes has not been clarified. In this study, we initially investigated the mTORC1 downstream indicators S6K1, S6rp, 4E‐BP1 and eIF4E in the Vil‐Cre;Pkd2 f3/f3 kidney with or without rapamycin treatment. Rapamycin significantly lowered the p‐S6K1 (T389), p‐S6rp (S235/236), p‐4E‐BP1 (S65) and p‐eIF4E (S209) levels in the Vil‐Cre;Pkd2 f3/f3 kidney (Fig. S4A‐H). However, Akt, a downstream marker of the mTORC2 complex in the mTOR pathway, did not appear to be affected (Fig. S4I‐J), indicating that rapamycin inhibited ADPKD cyst formation via the mTORC1 pathway, which might not be through mTORC2 40, 48.
The abnormal proliferation and apoptosis of cystic epithelial cells are hallmarks of ADPKD 62, 63, 64, 65. Consistent with previous studies 40, 41, 53, we found that although rapamycin did not affect apoptosis in the cyst‐lining epithelia of the Vil‐Cre;Pkd2 f3/f3 kidney (Fig. S5A and C‐D), rapamycin significantly suppressed the dramatic proliferation seen in the Vil‐Cre;Pkd2 f3/f3 renal epithelia (Fig. S5B and E‐F), indicating that rapamycin impeded cystic progression in the ADPKD model by inhibiting the proliferation of cyst‐lining epithelia.
To explore the molecular mechanism by which rapamycin suppresses epithelial proliferation, we have searched our previously established cDNA microarrays data between our E8 cell line (Pkd2‐null, Pkd2 −/−) and its maternally derived D3 cell line (Pkd2 f/−, Pkd2 +/−) 27. Of the signature genes, Ccnd1 mRNA level encoding cyclin D1, whose function closely associates cell proliferation, showed a fourfold higher elevation in E8 than in D3 cells. To validate the microarray data, we performed quantitative PCR using mRNAs of E8 and D3 cells, as well as renal tissue mRNA from Vil‐Cre;Pkd2 f3/3 and its normal control Pkd2 f3/f3. We found that down‐regulating PC2 not only significantly increased Ccnd1 expression, but also elevated all cell‐cycle‐associated cyclins and CDK1 (Fig. S5G‐H).
To confirm the finding from these mRNA assays was also affected in the mouse kidney tissue or in their epithelial cells with and without rapamycin treatment, we performed Western blot analyses of kidneys from control (Pkd2 f3/f3) mice and from rapamycin‐ or placebo‐treated Vil‐Cre;Pkd2 f3/f3 mice. The results revealed that all of the cell‐cycle‐associated cyclins (A, B, D1 and E) were abnormally up‐regulated in the Vil‐Cre;Pkd2 f3/f3 kidney tissue (Fig. 5A). These aberrantly up‐regulated cyclins were still further significantly suppressed by rapamycin to levels approaching those in the control kidney (Fig. 5B–E), indicating that rapamycin suppressed epithelial proliferation in the Vil‐Cre;Pkd2 f3/f3 kidney by inhibiting cell‐cycle‐associated cyclins. Interestingly, although rapamycin brought the cyclins A, B and E down almost to normal control levels (Fig. 5B,C and E), cyclin D1 was reduced to only approximately half of the excess level found in the untreated Vil‐Cre;Pkd2 f3/f3 kidney (Fig. 5D). The result suggested that rapamycin could not fully rescue all of the abnormally cell‐cycle‐associated cyclins in the Vil‐Cre;Pkd2 f3/f3 kidney.
Figure 5.
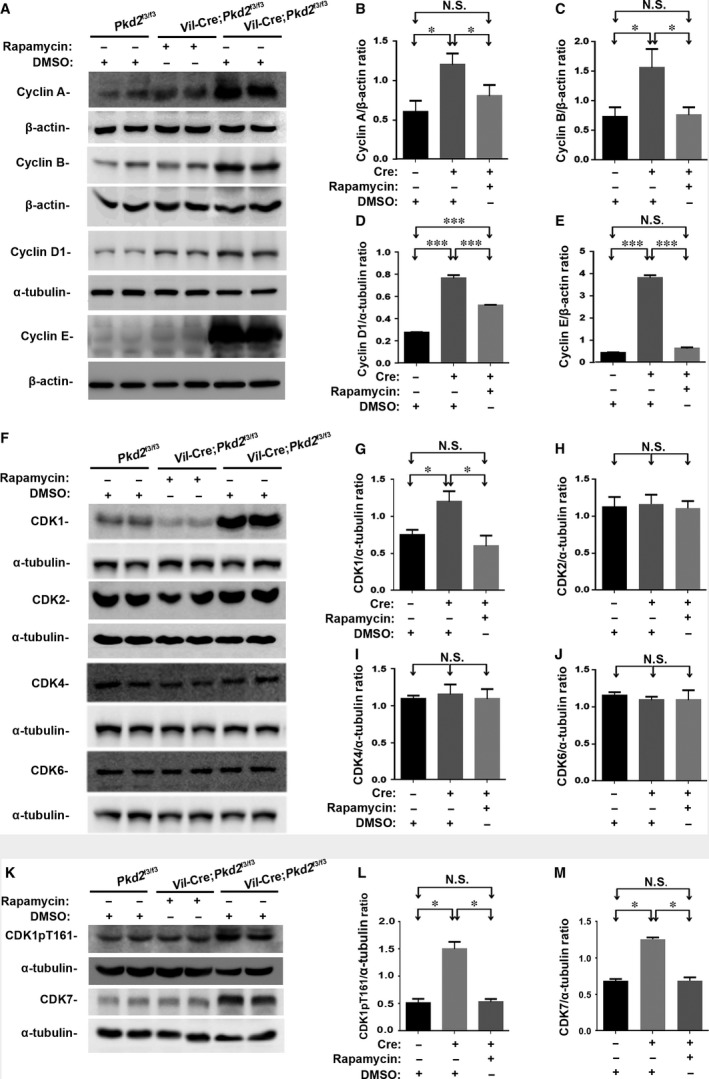
Rapamycin down‐regulates cell‐cycle‐associated cyclins and CDKs in Vil‐Cre;Pkd2 f3/f3 mice with or without Protocol II treatment. (A) Representative Western blots of kidney tissue lysates, showing that the aberrant elevation of cyclins A, B, D1 and E in Vil‐Cre;Pkd2 f3/f3 mice was suppressed by rapamycin treatment. (B–E) Normalized quantitative analyses using the densitometry values from the Western blots for cyclin A (B), cyclin B (C), cyclin D1 (D) and cyclin E (E). (F) Similar Western blots of the cell‐cycle‐associated CDK1, CDK2, CDK4 and CDK6 showed that only CDK1 was significantly elevated and suppressed by rapamycin treatment. (G–J) Normalized quantitative analyses using the densitometry values from the Western blots for CDK1 (G), CDK2 (H), CDK4 (I) and CDK6 (J). (K) Western blot analyses of the same tissues were performed with antibodies to phospho‐CDK1 (CDK1pT161), and its upstream factor CDK7. Both phospho‐CDK1 (CDK1pT161) and CDK7 were significantly elevated and suppressed by rapamycin treatment. (L–M) Normalized quantitative analysis using the densitometry values from the Western blots for CDK1pT161/α‐tubulin and CDK7/α‐tubulin. Statistical analysis indicated that compared to placebo treatment, rapamycin treatment significantly down‐regulated cyclins A, B, D1 and E and CDK1/CDK7 (including its activated form CDK1pT161) in the kidneys of Vil‐Cre;Pkd2 f3/f3 mice. N.S. = No significance; *P < 0.05; ***P < 0.001.
Most cyclins couple with CDKs to form CDK/cyclin complexes which control kinase activity and substrate specificity 66, 67. CDK/cyclin complexes exert important role in driving cell cycle transition and regulating cell cycle progression. Our mRNA data showed that only CDK1, but not other cell‐cycle‐associated CDK2/4/6, was significantly affected (Fig. S5G‐H). We therefore tested all these CDKs in the same tested kidneys. Compared to the kidneys from control and rapamycin‐ or placebo‐treated Vil‐Cre;Pkd2 f3/f3 mice, the similar result to mRNA finding was obtained in the Vil‐Cre;Pkd2 f3/f3 kidney (Fig. 5F–J). This aberrantly up‐regulated CDK1 could also be significantly suppressed by rapamycin to level of the control kidney (Fig. 5G). As the tested CDKs’ function was usually activated by their phosphorylation sites, we therefore performed Western blot analyses to investigate all activated CDK forms, including CDK1pT161, CDK2pT160, CDK4pT172 and CDK6pY13. The same finding was seen in the tested renal tissues (Fig. 5K and L and Fig. S5I‐L). Because CDK‐activating kinase (CAK) phosphorylates CDK1pT161, we tested expressional level of a CAK catalytic subunit CDK7 in the same kidney. Similar results to CDK1pT161 were also seen in the tested renal tissues (Fig. 5K and M).
To further validate this finding, we conducted cell‐based experiments to detect the cyclins (A, B, D1 and E) and CDKs (CDK1, CDK2, CDK4 and CDK6) in cells. We treated E8 (Pkd2‐null) and its maternally derived D3 (Pkd2 +/−) cells with rapamycin at different concentrations (non‐treated and treated with 1 nM or 10 nM for 12 hrs) (Fig. 6A and C–F and Fig. 7A–C) and durations (0, 6 and 12 hrs at 10 nM) (Fig. 6B and G–J and Fig. 7D–F). Increasing the rapamycin concentration appeared to produce a trend towards the delayed expression of the cell‐cycle‐associated cyclins (A, B, D1 and E) and CDK1/CDK1pT161 in both D3 and E8 cells. The highest rapamycin concentration (10 nM) brought all of these cyclins and CDK1/CDK1pT161 down to their normal control levels (Figs 6C–F and 7B‐C). Extending the duration of rapamycin treatment also increased its effect (Figs 6B and 7D). Treating Pkd2‐null cells (E8) with rapamycin at a concentration of 10 nM for the longest treatment duration (12 hrs) reduced the cyclins and CDK1/CDK1pT161 to the normal control levels (Figs 6G–J and 7E and F). In parallel, we also applied CDK1 inhibitor Dinaciclib to these cells with different concentrations and durations. The result showed that the highest concentration and the longest duration brought CDK1/CDK1pT161 down to their normal control levels (Fig. 7G–I and J–L). Thus, rapamycin significantly repressed the aberrant activation of cell‐cycle‐associated cyclins and CDK1/CDK1pT161 in Pkd2‐null cells in a dose‐ and time‐dependent manner.
Figure 6.
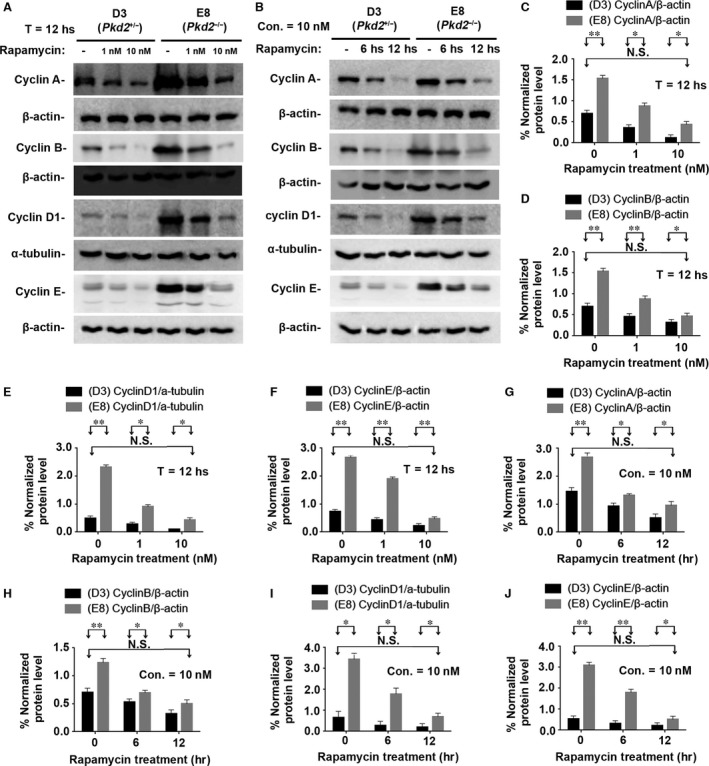
Rapamycin suppresses cell‐cycle‐associated cyclins in vitro. (A) Cell line E8 and its material derived cell line D3 were incubated with 0, 1 or 10 nM rapamycin for 12 hrs. Western blots of the lysates of D3 and E8 cells showed that rapamycin decreased the expression of cyclins A, B, D1 and E in a dose‐dependent manner. (B) D3 and E8 cells were incubated with 10 nM rapamycin for 0, 6 or 12 hrs. Western blots of the cell lysates showed that rapamycin significantly decreased the expression of cyclins A, B, D1 and E in a manner dependent on the duration of incubation. (C–F) Normalized quantitative analysis using the densitometry values from the Western blots in (A). (G–J) Normalized quantitative analysis using the densitometry values from the Western blots presented in (B). N.S. = No significance; *P < 0.05; **P < 0.01.
Figure 7.
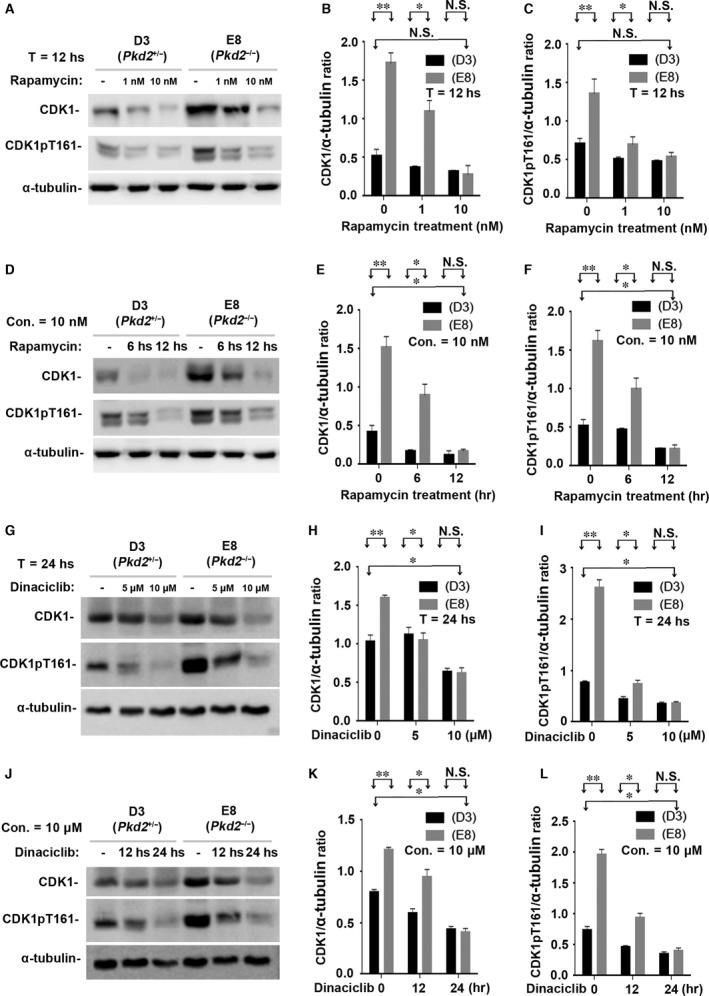
Rapamycin suppresses CDK1 and its activated form CDK1pT161 in vitro. (A) The same cell lines (E8 and D3) were also incubated with 0, 1, or 10 nM rapamycin for 12 hrs. Western blots of the lysates of E8 and D3 cells showed that rapamycin decreased the expression of CDK1 and its activated form CDK1pT161 in a dose‐dependent manner. (B–C) Normalized quantitative analysis using the densitometry values from the Western blots in (A). (D) The cell lines were also incubated with 10 nM rapamycin for 0, 6 or 12 hrs. Western blots of the cell lysates showed that rapamycin significantly decreased the expression of CDK1 and its activated forms CDK1pT161 in a manner dependent on the duration of incubation. (E–F) Normalized quantitative analysis using the densitometry values from the Western blots presented in (D). Other cell‐cycle‐associated CDKs (such as CDK2, CDK4 and CDK6) were not affected by PC2 expressional levels and rapamycin treatment (data not shown). (G) The same cell lines were cultured with CDK inhibitor Dinaciclib with 0, 5 and 10 μM for 24 hrs. Western blots of the cell lysates showed that Dinaciclib significantly inhibited the expression of CDK1 and its activated forms CDK1pT161 in a dose‐dependent manner. (H–I) Normalized quantitative analysis using the densitometry values from the Western blots presented in (G). (J) The cell lines were also cultured with CDK inhibitor Dinaciclib with 10 μM for 0, 12 or 24 hrs. Similar results to different concentrations were seen in Western blot assays on the different durations of Dinaciclib incubation. (K–L) Normalized quantitative analysis using the densitometry values from the Western blots presented in (J). N.S. = No significance; *P < 0.05; **P < 0.01.
Discussion
We used a Pkd2 conditional‐knockout model 27 to screen a spectrum of Cre transgenic mice driven by various kidney‐related promoters (Table 1) and identified the Vil‐Cre;Pkd2 f3/f3 ADPKD mouse model, which spatially and temporally mimics human ADPKD phenotypes. To validate Vil‐Cre;Pkd2 f3/f3 mice as a useful ADPKD model for preclinical trials, we used a rapamycin‐based treatment that is known to be effective for ADPKD mice 40, 41, 42. The response of the Vil‐Cre;Pkd2 f3/f3 mice to rapamycin was consistent with that reported for other ADPKD models, and the progression and timeline of the cystic phenotype and of renal failure in the Vil‐Cre;Pkd2 f3/f3 mice, along with the ease of evaluating the progression of the disease, make these mice a suitable model for evaluating and conducting preclinical trials of therapeutic interventions for ADPKD.
Rapamycin has been very effective for treating ADPKD mice, but has been less successful in clinical trials with ADPKD patients 26, 52, 53, 54, 57. Several explanations have been suggested for the different responses of mice and humans to rapamycin 41, 42, one being that to prevent adverse events, ADPKD patients have received lower doses (2–3 mg/kg) of rapamycin than those used in mouse models (5–10 or 100 mg/kg) 41, 42. Another possible reason for this discrepancy is that rapamycin has not been used as a primary treatment for humans in the early stages of ADPKD cyst development 52, 55, 68. To explore whether intensive rapamycin treatment could significantly improve disease phenotypes in an orthologous mouse model of ADPKD under similar conditions as those in human trials, we designed four rapamycin treatment protocols that varied the duration and timing of rapamycin treatment. Our results indicated that a longer duration of rapamycin treatment (Protocol II, from P10 to P110) provided the strongest therapeutic response than any of the other protocols (protocols I, III and IV, each with a duration of 50 days) in the ADPKD animal model. Protocol II yielded the largest increase in lifespan, significantly reduced the growth of renal cysts and significantly improved kidney function. Western blot analyses showed that rapamycin inhibited the activated forms of p‐S6K1, p‐S6rp, p‐4E‐BP1 and p‐eIF4E (indicators of mTORC1 activity; Fig S6) in a dose‐dependent manner, and this inhibition coincided with a significant improvement in the renal cystic phenotype in the rapamycin‐treated Vil‐Cre;Pkd2 f3/f3 mouse model. Thus, it is possible that an extended rapamycin treatment protocol would greatly benefit ADPKD patients with a PKD2 mutation.
Interestingly, our results showed that early (P10‐60, Protocol I)‐ and late‐treatment (P60‐110, Protocol IV) protocols using the same rapamycin dosage provided similar improvements in survival, cystic phenotype and renal function. This finding indicated that starting rapamycin treatment during the early stages of cyst development might not be critical for a better prognosis in the ADPKD model. In addition, we also compared the effects of Protocol I with those of Protocol III, which used the same rapamycin dosage and treatment period as Protocol I (P10‐60) but includes a non‐treated observation period ending at P110. The responses to Protocol I and Protocol III did not differ significantly in the ADPKD model, indicating that rapamycin effect on cystogenesis of the animal model could be such sustained long after the last dose of rapamycin is administered.
Although rapamycin has shown clear benefits in preclinical experiments and clinical trials for ADPKD, the precise molecular mechanism by which PC2 regulates cell growth via mTOR and how rapamycin inhibits cyst growth and epithelial‐cell proliferation have not been clarified. To investigate the molecular mechanism by which rapamycin prevents proliferation, we examined the serial factors most likely to be involved in cell proliferation. Based on our microarray data and qPCR assay, we identified cyclin D1 was significantly up‐regulated. Subsequently, we also examined other cyclin‐family members and their cofactors CDKs, by which cell cycling was driven. We found that all of the cell‐cycle‐associated cyclins (A, B, D1 and E), activated CDK1 (CDK1pT161) and CDK7 (which involves in the phosphorylation of CDK1pT161) were significantly up‐regulated in Pkd2‐null renal cells and in the tissue of our ADPKD model mice and that these cyclins and CDK7/CDK1 were down‐regulated by rapamycin treatment. This finding indicated that the increased epithelial proliferation observed in the ADPKD kidney might result from abnormal activation of the mTORC1 pathway and the subsequent up‐regulation of cell‐cycle‐associated cyclins and activated CDK1.
Taking our findings together with those of other distinguished reports, we deduced the molecular mechanism of rapamycin effect on ADPKD as follows: PC2 dysfunction causes PC1 C‐terminal region (Cterm) to mislocalize and significantly down‐regulate PC1 expression (Fig. S7) 69, 70, 71. The down‐regulation of PC1 disrupts interactions between PC1 and tuberous sclerosis type 2 (TSC2) 45, 72, 73, 74; this abolishes the ability of the TSC complex to inhibit the mTORC1 pathway and thereby activates the mTORC1 downstream factors S6K1, S6rp, 4E‐BP1 and eIF4E (Figs S4 and S6) 75, 76. Activated eIF4E binds to the translation initiation site of the cyclin D1 gene, causing it to overexpress cyclin D1, a key regulator of cell growth (Figs 5D and 6E and I) 77, 78, 79. Cyclin D1 forms a complex with CDK4 or CDK6 to further phosphorylate the retinoblastoma protein (RB) and release E2F from the RB/E2F‐gene‐suppressing domain 80 (Fig. S8). E2F is a transcriptional factor that can initiate and enhance the expression of all of the other cell‐cycle‐associated cyclins, cyclin E (Figs 5E and 6F and J), cyclin A (Figs 5B and 6C and G) and cyclin B (Figs 5C and 6D and H) 66, 67, 81, 82, 83. These activated cyclins cooperate with their corresponding CDKs to promote the renal epithelial proliferation and induce cystogenesis in the ADPKD model (Fig. 8A). When rapamycin was applied, the PC2 inactivation‐induced overactivation of mTORC1 can be inhibited, and subsequently suppress all downstream factors of the mTORC1 pathway (Fig. 8B), thereby finally arresting abnormal cell proliferation in the orthologous mouse model of ADPKD.
Figure 8.
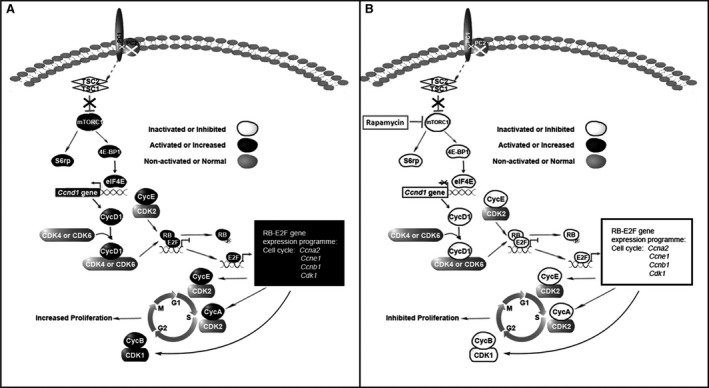
The molecular mechanism of the mTOR pathway in Vil‐Cre;Pkd2 f3/f3 mice, with or without rapamycin treatment. Based on our study and other group finding, we deduced the molecular link between mTOR signalling changes and the polycystin‐2 dysfunction in cystogenesis. (A) The loss of PC2 impairs normal PC1 function, which may in turn disrupt PC1's inhibitory effect on the mTORC1 pathway by disturbing interactions between PC1 and the tuberous sclerosis 2 gene product (TSC2). Activated mTORC1 promotes the phosphorylation of 4E‐BP1 and eIF4E, which activates cyclin D1 gene expression. Cyclin D1 couples with CDK4/6 and phosphorylates retinoblastoma protein (RB), thereby releasing (E2F), which promotes the expression of the other cell‐cycle‐associated cyclins (A, B and E) and CDK1, eventually leading to increased cell proliferation. (B) Rapamycin treatment suppressed all of the mTORC1 downstream factors, including cyclins D1, A, B, E and CDK1, thereby arresting aberrant cell proliferation.
In the light of our previous studies 27, 28, we identified and established a new ADPKD mouse model bearing Pkd2 mutant alleles (Vil‐Cre;Pkd2 f3/f3). We systematically characterized the Vil‐Cre;Pkd2 f3/f3 mouse and verified its suitability as a model that mimics the phenotype and progression of human ADPKD. Using this new mouse system, we demonstrated that these animals responded to rapamycin in a time‐ and dose‐dependent manner: increasing the dosage or duration of treatment improved the therapeutic effect. We also demonstrated the molecular mechanism by which the mTOR inhibitor rapamycin arrests the aberrant epithelial proliferation in the ADPKD kidney, which resulted de novo from PC2 dysfunction. This newly developed ADPKD model will promote translational medicine from in vivo preclinical trials to ADPKD therapies and will also provide a platform for identifying new molecular targets for treating ADPKD based on the newly defined mTORC1–CDK1/cyclin axis.
Conflict of interest
All the authors disclose no conflict.
Author contributions
GW conceived and designed the experiments. AL, JM, XS and JM performed the experiments. AL, YX, LZ, XZ, DW, GM, CL and GW analysed and interpreted the data. AL, CL and GW wrote the manuscript.
Supporting information
Figure S1 Segmental origin of tubular cysts in the Vil‐Cre;Pkd2 f3/f3 kidney
Figure S2 Extrarenal cystic phenotypes in Vil‐Cre;Pkd2 f3/f3 mice
Figure S3 Gender affects disease severity in Vil‐Cre;Pkd2 f3/f3 mice
Figure S4 Western blot analyses for mTOR downstream factors in the kidneys of 4‐month‐old Vil‐Cre;Pkd2 f3/f3 mice with or without Protocol II treatment
Figure S5 Rapamycin decreases proliferation in renal cells in Vil‐Cre;Pkd2 f3/f3 mice. Apoptosis and proliferation in kidneys from DMSO‐treated and rapamycin‐treated (Protocol II) 4‐month‐old Vil‐Cre;Pkd2 f3/f3 mice were analysed by IF staining
Figure S6 Rapamycin suppresses mTORC1 downstream indictors: phospho‐S6K1 (S6K1pT389), phospho‐S6rp (S6rppS235/236), phospho‐4E‐BP1 (4E‐BP1pS65) and phospho‐eIF4E (eIF4EpS209) in a dose‐dependent manner
Figure S7 Lacking of PC2 down‐regulates PC1 expression in vivo and in vitro
Figure S8 Rapamycin suppresses the up‐regulated RB/E2F pathway in the kidneys of 4‐month‐old Vil‐Cre;Pkd2 f3/f3 mice with or without Protocol II treatment
Table S1 Genotyping and survival analyses of γGt‐, Nestin‐ and Ksp‐Cre;Pkd2 f3/f3 mice
Appendix S1 Supplementary Method A‐D
Acknowledgements
We deeply thank Dr. S. Somlo's (Yale University) suggestion and comments on this study. This work was supported by the National 973 Program (2012CB517700) and the National Nature Science Foundation of China (81173114, 81072688, 81473282 and 81673402) (to G.W.).
Contributor Information
Guanqing Wu, Email: guanqing.wu@ahmu.edu.cn.
Chaozhao Liang, Email: liang_chaozhao@ahmu.edu.cn.
References
- 1. Gallagher AR, Germino GG, Somlo S. Molecular advances in autosomal dominant polycystic kidney disease. Adv Chronic Kidney Dis. 2010; 17: 118–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Torres VE, Harris PC, Pirson Y. Autosomal dominant polycystic kidney disease. Lancet. 2007; 369: 1287–301. [DOI] [PubMed] [Google Scholar]
- 3. Harris PC, Torres VE. Genetic mechanisms and signaling pathways in autosomal dominant polycystic kidney disease. J Clin Invest. 2014; 124: 2315–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ong AC, Devuyst O, Knebelmann B, et al; on behalf of the ERA‐EDTA Working Group for Inherited Kidney Diseases . Autosomal dominant polycystic kidney disease: the changing face of clinical management. Lancet. 2015; 385: 1993–2002. [DOI] [PubMed] [Google Scholar]
- 5. Willey CJ, Blais JD, Hall AK, et al Prevalence of autosomal dominant polycystic kidney disease in the European Union. Nephrol Dial Transplant. 2016; doi: 10.1093/ndt/gfw240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fedeles SV, Gallagher AR, Somlo S. Polycystin‐1: a master regulator of intersecting cystic pathways. Trends Mol Med. 2014; 20: 251–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Igarashi P, Somlo S. Polycystic kidney disease. J Am Soc Nephrol. 2007; 18: 1371–3. [DOI] [PubMed] [Google Scholar]
- 8. Torres VE, Harris PC. Autosomal dominant polycystic kidney disease: the last 3 years. Kidney Int. 2009; 76: 149–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Milutinovic J, Fialkow PJ, Rudd TG, et al Liver cysts in patients with autosomal dominant polycystic kidney disease. Am J Med. 1980; 68: 741–4. [DOI] [PubMed] [Google Scholar]
- 10. Spirli C, Okolicsanyi S, Fiorotto R, et al Mammalian target of rapamycin regulates vascular endothelial growth factor‐dependent liver cyst growth in polycystin‐2‐defective mice. Hepatology. 2010; 51: 1778–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bae KT, Zhu F, Chapman AB, et al Magnetic resonance imaging evaluation of hepatic cysts in early autosomal‐dominant polycystic kidney disease: the Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease cohort. Clin J Am Soc Nephrol. 2006; 1: 64–9. [DOI] [PubMed] [Google Scholar]
- 12. Bae KT, Grantham JJ. Imaging for the prognosis of autosomal dominant polycystic kidney disease. Nat Rev Nephrol. 2010; 6: 96–106. [DOI] [PubMed] [Google Scholar]
- 13. Torra R, Nicolau C, Badenas C, et al Ultrasonographic study of pancreatic cysts in autosomal dominant polycystic kidney disease. Clin Nephrol. 1997; 47: 19–22. [PubMed] [Google Scholar]
- 14. Yazdanpanah K, Manouchehri N, Hosseinzadeh E, et al Recurrent acute pancreatitis and cholangitis in a patient with autosomal dominant polycystic kidney disease. Int J Prev Med. 2013; 4: 233–6. [PMC free article] [PubMed] [Google Scholar]
- 15. Ring T, Spiegelhalter D. Risk of intracranial aneurysm bleeding in autosomal‐dominant polycystic kidney disease. Kidney Int. 2007; 72: 1400–2. [DOI] [PubMed] [Google Scholar]
- 16. Ong AC. Screening for intracranial aneurysms in ADPKD. BMJ. 2009; 339: b3763. [DOI] [PubMed] [Google Scholar]
- 17. Xu HW, Yu SQ, Mei CL, et al Screening for intracranial aneurysm in 355 patients with autosomal‐dominant polycystic kidney disease. Stroke. 2011; 42: 204–6. [DOI] [PubMed] [Google Scholar]
- 18. Rozenfeld MN, Ansari SA, Mohan P, et al Autosomal Dominant Polycystic Kidney Disease and Intracranial Aneurysms: Is There an Increased Risk of Treatment? AJNR Am J Neuroradiol. 2016; 37: 290–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Adeola T, Adeleye O, Potts JL, et al Thoracic aortic dissection in a patient with autosomal dominant polycystic kidney disease. J Natl Med Assoc. 2001; 93: 282–7. [PMC free article] [PubMed] [Google Scholar]
- 20. Liu D, Wang CJ, Judge DP, et al A Pkd1‐Fbn1 Genetic Interaction Implicates TGF‐beta Signaling in the Pathogenesis of Vascular Complications in Autosomal Dominant Polycystic Kidney Disease. J Am Soc Nephrol. 2014; 25: 81–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Perrone RD, Malek AM, Watnick T. Vascular complications in autosomal dominant polycystic kidney disease. Nat Rev Nephrol. 2015; 11: 589–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Watnick TJ, Germino GG. Polycystic kidney disease: polycystin‐1 and polycystin‐2–it's complicated. Nat Rev Nephrol. 2013; 9: 249–50. [DOI] [PubMed] [Google Scholar]
- 23. Ong AC, Harris PC. A polycystin‐centric view of cyst formation and disease: the polycystins revisited. Kidney Int. 2015; 88: 699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kim B, King BF Jr, Vrtiska TJ, et al Inherited renal cystic diseases. Abdom Radiol (NY). 2016; 41: 1035–51. [DOI] [PubMed] [Google Scholar]
- 25. Lieberthal W, Levine JS. Mammalian target of rapamycin and the kidney. II. Pathophysiology and therapeutic implications. Am J Physiol Renal Physiol. 2012; 303: F180–91. [DOI] [PubMed] [Google Scholar]
- 26. Braun WE, Schold JD, Stephany BR, et al Low‐dose rapamycin (sirolimus) effects in autosomal dominant polycystic kidney disease: an open‐label randomized controlled pilot study. Clin J Am Soc Nephrol. 2014; 9: 881–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kim I, Ding T, Fu Y, et al Conditional mutation of Pkd2 causes cystogenesis and upregulates beta‐catenin. J Am Soc Nephrol. 2009; 20: 2556–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li A, Tian X, Zhang X, et al Human polycystin‐2 transgene dose‐dependently rescues ADPKD phenotypes in Pkd2 mutant mice. Am J Pathol. 2015; 185: 2843–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang MZ, Mai W, Li C, et al PKHD1 protein encoded by the gene for autosomal recessive polycystic kidney disease associates with basal bodies and primary cilia in renal epithelial cells. Proc Natl Acad Sci USA. 2004; 101: 2311–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Madison BB, Dunbar L, Qiao XT, et al Cis elements of the villin gene control expression in restricted domains of the vertical (crypt) and horizontal (duodenum, cecum) axes of the intestine. J Biol Chem. 2002; 277: 33275–83. [DOI] [PubMed] [Google Scholar]
- 31. Neugarten J, Acharya A, Silbiger SR. Effect of gender on the progression of nondiabetic renal disease: a meta‐analysis. J Am Soc Nephrol. 2000; 11: 319–29. [DOI] [PubMed] [Google Scholar]
- 32. Stringer KD, Komers R, Osman SA, et al Gender hormones and the progression of experimental polycystic kidney disease. Kidney Int. 2005; 68: 1729–39. [DOI] [PubMed] [Google Scholar]
- 33. Grzegorczyk K, Krajewska M, Weyde W, et al Gender and kidney diseases: the clinical importance and mechanisms of modifying effects. Postepy Hig Med Dosw (Online). 2011; 65: 849–57. [DOI] [PubMed] [Google Scholar]
- 34. Heyer CM, Sundsbak JL, Abebe KZ, et al Predicted mutation strength of nontruncating PKD1 mutations aids genotype‐phenotype correlations in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2016; 9: 2872–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ishikawa I, Maeda K, Nakai S, et al Gender difference in the mean age at the induction of hemodialysis in patients with autosomal dominant polycystic kidney disease. Am J Kidney Dis. 2000; 35: 1072–5. [DOI] [PubMed] [Google Scholar]
- 36. Silbiger S, Neugarten J. Gender and human chronic renal disease. Gend Med. 2008; 5: S3–10. [DOI] [PubMed] [Google Scholar]
- 37. Menezes LF, Germino GG. Systems biology of polycystic kidney disease: a critical review. Wiley Interdiscip Rev Syst Biol Med. 2015; 7: 39–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Boehn SN, Spahn S, Neudecker S, et al Inhibition of Comt with tolcapone slows progression of polycystic kidney disease in the more severely affected PKD/Mhm (cy/+) substrain of the Hannover Sprague‐Dawley rat. Nephrol Dial Transplant. 2013; 28: 2045–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sarbassov DD, Ali SM, Sengupta S, et al Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006; 22: 159–68. [DOI] [PubMed] [Google Scholar]
- 40. Zafar I, Ravichandran K, Belibi FA, et al Sirolimus attenuates disease progression in an orthologous mouse model of human autosomal dominant polycystic kidney disease. Kidney Int. 2010; 78: 754–61. [DOI] [PubMed] [Google Scholar]
- 41. Shillingford JM, Piontek KB, Germino GG, et al Rapamycin ameliorates PKD resulting from conditional inactivation of Pkd1. J Am Soc Nephrol. 2010; 21: 489–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shillingford JM, Leamon CP, Vlahov IR, et al Folate‐conjugated rapamycin slows progression of polycystic kidney disease. J Am Soc Nephrol. 2012; 23: 1674–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Stayner C, Shields J, Slobbe L, et al Rapamycin‐mediated suppression of renal cyst expansion in del34 Pkd1‐/‐ mutant mouse embryos: an investigation of the feasibility of renal cyst prevention in the foetus. Nephrology. 2012; 17: 739–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ravichandran K, Zafar I, Ozkok A, et al An mTOR kinase inhibitor slows disease progression in a rat model of polycystic kidney disease. Nephrol Dial Transplant. 2015; 30: 45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shillingford JM, Murcia NS, Larson CH, et al The mTOR pathway is regulated by polycystin‐1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci USA. 2006; 103: 5466–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wahl PR, Serra AL, Le Hir M, et al Inhibition of mTOR with sirolimus slows disease progression in Han:SPRD rats with autosomal dominant polycystic kidney disease (ADPKD). Nephrol Dial Transplant. 2006; 21: 598–604. [DOI] [PubMed] [Google Scholar]
- 47. Belibi F, Ravichandran K, Zafar I, et al mTORC1/2 and rapamycin in female Han:SPRD rats with polycystic kidney disease. Am J Physiol Renal Physiol. 2011; 300: F236–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Novalic Z, van der Wal AM, Leonhard WN, et al Dose‐dependent effects of sirolimus on mTOR signaling and polycystic kidney disease. J Am Soc Nephrol. 2012; 23: 842–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sabbatini M, Russo L, Cappellaio F, et al Effects of combined administration of rapamycin, tolvaptan, and AEZ‐131 on the progression of polycystic disease in PCK rats. Am J Physiol Renal Physiol. 2014; 306: F1243–50. [DOI] [PubMed] [Google Scholar]
- 50. Serra AL, Kistler AD, Poster D, et al Clinical proof‐of‐concept trial to assess the therapeutic effect of sirolimus in patients with autosomal dominant polycystic kidney disease: SUISSE ADPKD study. BMC Nephrol. 2007; 8: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Edelstein CL. Therapeutic interventions for autosomal dominant polycystic kidney disease. Nephrol News Issues. 2008; 22: 25–6. [PubMed] [Google Scholar]
- 52. Walz G, Budde K, Mannaa M, et al Everolimus in patients with autosomal dominant polycystic kidney disease. N Engl J Med. 2010; 363: 830–40. [DOI] [PubMed] [Google Scholar]
- 53. Serra AL, Poster D, Kistler AD, et al Sirolimus and kidney growth in autosomal dominant polycystic kidney disease. N Engl J Med. 2010; 363: 820–9. [DOI] [PubMed] [Google Scholar]
- 54. Perico N, Antiga L, Caroli A, et al Sirolimus therapy to halt the progression of ADPKD. J Am Soc Nephrol. 2010; 21: 1031–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Soliman A, Zamil S, Lotfy A, et al Sirolimus produced S‐shaped effect on adult polycystic kidneys after 2‐year treatment. Transplant Proc. 2012; 44: 2936–9. [DOI] [PubMed] [Google Scholar]
- 56. Xue C, Dai B, Mei C. Long‐term treatment with mammalian target of rapamycin inhibitor does not benefit patients with autosomal dominant polycystic kidney disease: a meta‐analysis. Nephron Clin Pract. 2013; 124: 10–6. [DOI] [PubMed] [Google Scholar]
- 57. Ruggenenti P, Gentile G, Perico N, et al ; SIRENA 2 Study Group . Effect of sirolimus on disease progression in patients with autosomal dominant polycystic kidney disease and CKD stages 3b‐4. Clin J Am Soc Nephrol. 2016; 11: 785–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Leonhard WN, Zandbergen M, Veraar K, et al Scattered deletion of PKD1 in kidneys causes a cystic snowball Effect and recapitulates polycystic kidney disease. J Am Soc Nephrol. 2015; 26: 1322–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Huang J, Manning BD. The TSC1‐TSC2 complex: a molecular switchboard controlling cell growth. Biochem J. 2008; 412: 179–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006; 124: 471–84. [DOI] [PubMed] [Google Scholar]
- 61. Rowe I, Chiaravalli M, Mannella V, et al Defective glucose metabolism in polycystic kidney disease identifies a new therapeutic strategy. Nat Med. 2013; 19: 488–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ong AC, Harris PC. Molecular pathogenesis of ADPKD: the polycystin complex gets complex. Kidney Int. 2005; 67: 1234–47. [DOI] [PubMed] [Google Scholar]
- 63. Takiar V, Caplan MJ. Polycystic kidney disease: pathogenesis and potential therapies. Biochim Biophys Acta. 2011; 1812: 1337–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhou X, Fan LX, Sweeney WE Jr, et al Sirtuin 1 inhibition delays cyst formation in autosomal‐dominant polycystic kidney disease. J Clin Invest. 2013; 123: 3084–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kuo IY, DesRochers TM, Kimmerling EP, et al Cyst formation following disruption of intracellular calcium signaling. Proc Natl Acad Sci USA. 2014; 111: 14283–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Asghar U, Witkiewicz AK, Turner NC, et al The history and future of targeting cyclin‐dependent kinases in cancer therapy. Nat Rev Drug Discov. 2015; 14: 130–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lim S, Kaldis P. Cdks, cyclins and CKIs: roles beyond cell cycle regulation. Development. 2013; 140: 3079–93. [DOI] [PubMed] [Google Scholar]
- 68. Stallone G, Infante B, Grandaliano G, et al Rapamycin for treatment of type I autosomal dominant polycystic kidney disease (RAPYD‐study): a randomized, controlled study. Nephrol Dial Transplant. 2012; 27: 3560–7. [DOI] [PubMed] [Google Scholar]
- 69. Li A, Hu B, Liang D, et al PC2 regulates canonical wnt signaling requiring PC1 expression. J Am Soc Nephrol. 2010; 21: 18A.19917780 [Google Scholar]
- 70. Cai Y, Fedeles SV, Dong K, et al Altered trafficking and stability of polycystins underlie polycystic kidney disease. J Clin Invest. 2014; 124: 5129–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Gainullin VG, Hopp K, Ward CJ, et al Polycystin‐1 maturation requires polycystin‐2 in a dose‐dependent manner. J Clin Invest. 2015; 125: 607–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hartman TR, Liu D, Zilfou JT, et al The tuberous sclerosis proteins regulate formation of the primary cilium via a rapamycin‐insensitive and polycystin 1‐independent pathway. Hum Mol Genet. 2009; 18: 151–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Boletta A. Emerging evidence of a link between the polycystins and the mTOR pathways. Pathogenetics. 2009; 2: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Pema M, Drusian L, Chiaravalli M, et al mTORC1‐mediated inhibition of polycystin‐1 expression drives renal cyst formation in tuberous sclerosis complex. Nat Commun. 2016; 7: 10786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Gingras AC, Raught B, Gygi SP, et al Hierarchical phosphorylation of the translation inhibitor 4E‐BP1. Genes Dev. 2001; 15: 2852–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Steelman LS, Chappell WH, Abrams SL, et al Roles of the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways in controlling growth and sensitivity to therapy‐implications for cancer and aging. Aging (Albany NY). 2011; 3: 192–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Mamane Y, Petroulakis E, LeBacquer O, et al mTOR, translation initiation and cancer. Oncogene. 2006; 25: 6416–22. [DOI] [PubMed] [Google Scholar]
- 78. Guan L, Song K, Pysz MA, et al Protein kinase C‐mediated down‐regulation of cyclin D1 involves activation of the translational repressor 4E‐BP1 via a phosphoinositide 3‐kinase/Akt‐independent, protein phosphatase 2A‐dependent mechanism in intestinal epithelial cells. J Biol Chem. 2007; 282: 14213–25. [DOI] [PubMed] [Google Scholar]
- 79. Grech G, von Lindern M. The role of translation initiation regulation in haematopoiesis. Comp Funct Genomics. 2012; 2012: 576540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Burkhart DL, Sage J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat Rev Cancer. 2008; 8: 671–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Besson A, Yong VW. Mitogenic signaling and the relationship to cell cycle regulation in astrocytomas. J Neurooncol. 2001; 51: 245–64. [DOI] [PubMed] [Google Scholar]
- 82. Simmons Kovacs LA, Orlando DA, Haase SB. Transcription networks and cyclin/CDKs: the yin and yang of cell cycle oscillators. Cell Cycle. 2008; 7: 2626–9. [DOI] [PubMed] [Google Scholar]
- 83. Orlando DA, Lin CY, Bernard A, et al Global control of cell‐cycle transcription by coupled CDK and network oscillators. Nature. 2008; 453: 944–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Rajagopalan S, Park JH, Patel PD, et al Cloning and analysis of the rat gamma‐glutamyltransferase gene. J Biol Chem. 1990; 265: 11721–5. [PubMed] [Google Scholar]
- 85. Dubois NC, Hofmann D, Kaloulis K, et al Nestin‐Cre transgenic mouse line Nes‐Cre1 mediates highly efficient Cre/loxP mediated recombination in the nervous system, kidney, and somite‐derived tissues. Genesis. 2006; 44: 355–60. [DOI] [PubMed] [Google Scholar]
- 86. Shao X, Somlo S, Igarashi P. Epithelial‐specific Cre/lox recombination in the developing kidney and genitourinary tract. J Am Soc Nephrol. 2002; 13: 1837–46. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Segmental origin of tubular cysts in the Vil‐Cre;Pkd2 f3/f3 kidney
Figure S2 Extrarenal cystic phenotypes in Vil‐Cre;Pkd2 f3/f3 mice
Figure S3 Gender affects disease severity in Vil‐Cre;Pkd2 f3/f3 mice
Figure S4 Western blot analyses for mTOR downstream factors in the kidneys of 4‐month‐old Vil‐Cre;Pkd2 f3/f3 mice with or without Protocol II treatment
Figure S5 Rapamycin decreases proliferation in renal cells in Vil‐Cre;Pkd2 f3/f3 mice. Apoptosis and proliferation in kidneys from DMSO‐treated and rapamycin‐treated (Protocol II) 4‐month‐old Vil‐Cre;Pkd2 f3/f3 mice were analysed by IF staining
Figure S6 Rapamycin suppresses mTORC1 downstream indictors: phospho‐S6K1 (S6K1pT389), phospho‐S6rp (S6rppS235/236), phospho‐4E‐BP1 (4E‐BP1pS65) and phospho‐eIF4E (eIF4EpS209) in a dose‐dependent manner
Figure S7 Lacking of PC2 down‐regulates PC1 expression in vivo and in vitro
Figure S8 Rapamycin suppresses the up‐regulated RB/E2F pathway in the kidneys of 4‐month‐old Vil‐Cre;Pkd2 f3/f3 mice with or without Protocol II treatment
Table S1 Genotyping and survival analyses of γGt‐, Nestin‐ and Ksp‐Cre;Pkd2 f3/f3 mice
Appendix S1 Supplementary Method A‐D