

Summary
Ras‐associated lymphoproliferative disease (RALD) is an autoimmune lymphoproliferative syndrome (ALPS)‐like disease caused by mutations in Kirsten rat sarcoma viral oncogene homologue (KRAS) or neuroblastoma RAS viral (V‐Ras) oncogene homologue (NRAS). The immunological phenotype and pathogenesis of RALD have yet to be studied extensively. Here we report a thorough immunological investigation of a RALD patient with a somatic KRAS mutation. Patient lymphocytes were analysed for phenotype, immunoglobulin levels and T cell proliferation capacity. T and B cell receptor excision circles (TREC and KREC, respectively), markers of naive T and B cell production, were measured serially for 3 years. T and B cell receptor repertoires were studied using both traditional assays as well as next‐generation sequencing (NGS). TREC and KREC declined dramatically with time, as did T cell receptor diversity. NGS analysis demonstrated T and B clonal expansions and marked restriction of T and B cell receptor repertoires compared to healthy controls. Our results demonstrate, at least for our reported RALD patient, how peripheral T and B clonal expansions reciprocally limit lymphocyte production and restrict the lymphocyte receptor repertoire in this disease. Decreased naive lymphocyte production correlated with a clinical deterioration in our patient's immune status, suggesting that TREC and KREC may be used as an aid in monitoring disease progression. Both the methodologies used here and the conclusions regarding immune homeostasis may be applicable to the research of ALPS and other immune dysregulation syndromes.
Keywords: immunodeficiency, KRAS, next‐generation sequencing, RALD, RAS associated lymphoproliferative disease
Introduction
The hallmark of autoimmune lymphoproliferative syndrome (ALPS) is non‐malignant lymphoproliferation causing severe autoimmunity directed mainly towards blood cells 1. In a small number of patients with a distinct entity, differing from ALPS, mutations in either neuroblastoma RAS viral (V‐Ras) oncogene homologue (NRAS) or Kirsten rat sarcoma viral oncogene homologue (KRAS) have been reported. Mutations in these genes lead to a similar phenotype of non‐malignant lymphoproliferation with autoimmune cytopenias. These entities have been designated as RAS‐associated lymphoproliferative disease (RALD) 1, 2, 3, 4. RAS proteins are guanosine diphosphate/guanosine triphosphate (GDP/GTP)‐binding proteins that act as intracellular signal transducers, which play a crucial role in normal tissue signalling, including proliferation, differentiation and senescence. Due to their vitality in normal tissue function, mutated RAS genes are extremely potent oncogenes, with mutations in KRAS found in as many as 25% of all human cancers 5, solid as well as haematological. In contrast, RALD is extremely rare, with only 20 patients described in the literature to date 3, 4, 6, 7, 8. What drives some RAS mutations to induce benign, non‐malignant lymphoproliferation, while others lead to cancer development, is unknown. Some speculate that RALD is a preliminary incarnation of juvenile myelomonocytic leukaemia (JMML), a severe variant of chronic leukaemia, with which RALD shares many clinical and laboratory findings, although the two entities differ substantially in terms of disease course and prognosis 7.
In both ALPS and RALD, the pathogenesis involves an inability to halt an immune response after it had run its course. In ALPS, reduced apoptosis occurs due to malfunction of the FAS–FAS ligand (FASL) pathway. In RALD, inhibition of apoptosis is mediated by resistance to interleukin (IL)‐2 depletion‐dependent apoptosis 6. Whereas both ALPS and RALD patients present with lymphoproliferation and autoimmunity, the elevated αβ+ double‐negative T cell (DNT) counts, that are a hallmark of ALPS, are increased only minimally in RALD. Significant T and B cell immunodeficiency leading to an increased susceptibility to infections has yet to be reported and studied thoroughly in RALD.
Here we report a female patient with RALD who presented with autoimmune pancytopenia and was found to harbour a somatic missense mutation in KRAS. Using both traditional immune assays as well as high‐throughput sequencing of the immune cell receptor repertoire, we attempt to elucidate the co‐existence of autoimmunity and immune deficiency in RALD patients.
Methods
Clinical data
Patient records were obtained from the e‐record registry of our hospital. The patient was interviewed and examined by the authors. Informed consent was obtained and all procedures performed were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Exome sequencing analysis
The Burrows–Wheeler Alignment (BWA) mem algorithm (version 0.7.12) 9 was used for alignment of the sequence reads to the human reference genome (hg19). The HaplotypeCaller algorithm of the Genome Analysis Toolkit (GATK) version 3.4 was applied for variant calling, as recommended in the best practice pipeline 10 KGG‐seq version 08 11, and was used for annotation of detected variants and in‐house scripts were applied for further filtering.
Immunological evaluation
Lymphocyte subset determination. Cell surface markers of peripheral blood mononuclear cells (PBMCs) were determined by immunofluorescent staining and flow cytometry (Epics V; Coulter Electronics, Hialeah, FL, USA) using anti‐CD3, anti‐CD19, anti‐CD16 and anti‐CD56 from BD Biosciences (San Jose, CA, USA) and anti‐CD4 and anti‐CD8 from Beckman Coulter Immunotech (Brea, CA, USA).
Quantification of signal joint T cell receptor excision circles (sjTRECs), signal joint kappa deleting receptor excision circles (sjKRECs) and intronic recombination signal sequence–kappa‐deleting element( iRSS–Kde) rearrangements. sjTREC copy numbers were determined using quantitative real‐time polymerase chain reaction (qRT–PCR). PCR reactions were performed as described previously 12 using as template 0·5 µg genomic DNA (gDNA) extracted from the patient's PBMCs. qRT–PCR was carried out using an ABI PRISM 7900 Sequence Detector System (Applied Biosystems, Foster City, CA, USA). A standard curve was constructed using serial dilutions containing 103–106 copies of a known sjTREC plasmid. Patient and control samples were tested in triplicate, and the number of sjTRECs in a given sample was calculated by comparing the obtained cycle threshold (Ct) value of the sample to the standard curve using an absolute quantification algorithm. The amount of sjKRECs and iRSS–Kde rearrangements was determined by qRT–PCR, as described previously 12. The threshold for Ct determination was positioned at the same level each time. Amplification of RNAseP (TaqMan assay; Applied Biosystems) served as a quality control of DNA amplification for both the sjTREC and the sjKREC/iRSS–Kde assays.
Cell surface analysis of T cell receptor (TCR) Vβ expression. Surface expression of individual TCR‐Vβ gene families was analysed using flow cytometry and a set of 24 Vβ‐specific fluorochrome‐labelled monoclonal antibodies (Beckman Coulter), as described previously 13. Normal control values were obtained from the IOTest Beta Mark‐Quick Reference Card (Beckman Coulter).
B cell receptor (BCR) repertoire. Complementarity determining region 3 (CDR3) length distribution for a cluster of BCR heavy chain (IGH) V and J gene segment pairings was assessed by means of PCR amplification according to the standardized BIOMED‐2 protocol 14. Fluorescence‐labelled PCR products (1 µl) were added to a mixture of 8·5 µl of formamide and 0·5 µl of GeneScan 500 Rox internal lane standard (Applied Biosystems) and separated using the 3100 Genetic Analyzer (Applied Biosystems). Results were analysed with Gene Mapper software (Applied Biosystems).
Next‐generation sequencing (NGS). TCR and BCR libraries were generated from patient and control genomic DNA using primers for conserved regions of V and J genes in the TRG (TCR‐γ) and IGH loci, respectively, according to the manufacturer's protocol (Lymphotrack; Invivoscibe Technologies, Carlsbad, CA, USA). Quantified libraries were pooled and sequenced using Mi‐Seq Illumia technology. FASTA files from the filtered sequences were submitted to ImMunoGeneTics (IMGT) HighV‐QUEST webserver (http://www.imgt.org), filtered for productive sequences only (no stop codons or frame shifts) and analysed 15. Analyses were performed on CDR3 amino acid sequences. For both BCR and TCR repertoires, V and J gene usage patterns were analysed and repertoire diversity was calculated using several diversity indices, as follows:
Results
Clinical description
The patient, a 6‐year‐old girl, was born to second‐degree consanguineous parents with a negative family history of immunodeficiency and/or autoimmunity. She was clinically immune deficient from the age of 4 months, suffering from recurrent infections including otitis media, pneumonia, urinary tract infection and herpetic skin infection requiring hospitalizations. At the age of 9 months, she developed hepatosplenomegaly and lymphadenopathy followed by Coombs positive haemolytic anaemia and thrombocytopaenia (Table 1). Repeated bone marrow aspirations revealed normal cellularity. At the age of 1·5 years, she developed severe cytomegalovirus (CMV) disease that manifested in severe pneumonia requiring mechanical ventilation for 2 weeks in the paediatric intensive care unit. Following recovery, she continued to suffer from pancytopaenia requiring multiple blood product transfusions. Due to protracted thrombocytopaenia, she was treated with intravenous immunoglobulin, steroids and rapamycin and eventually underwent splenectomy due to a large splenic infarct. An extensive diagnostic work‐up to rule out malignancy, rheumatological and infectious aetiologies was performed and resulted negative.
Table 1.
Laboratory characteristics of the patient at diagnosis
| Variable | Patient at diagnosis (cells per microlitre ×10−3) | Normal range* (cells per microlitre ×10 −3) |
|---|---|---|
| CBC | ||
| WBC | 7 | (5·2–11) |
| Monocytes | 1·61 | (0·2–1) |
| Lymphocytes | 2·378 | (1·5–6·5) |
| Haemoglobin (mg/dl) | 7·59 | (11–14) |
| Platelets | 31 | (150–400) |
| Lymphocyte subsets | ||
| Lymphocytes | 2·378 | (2·3–5·4) |
| T (CD3+) | 1·237 | (1·4–3·7) |
| T helper (CD4+) | 0·595 | (0·7–2·2) |
| T cytotoxic (CD8+) | 0·666 | (0·49–1·3) |
| Double‐negative (CD4–CD8–CD3+αβ+) | 2·6% | (<1%) |
| CD20+ | 0·927 (39%) | (5–25%) |
| T cell proliferation (cpm†) | Patient at diagnosis | Control |
| PHA 6 µg/ml | 94 000 | 85 400 |
| PHA 25 µg/ml | 93 300 | 64 500 |
| Anti‐CD3 | 40 800 | 28 900 |
| Serum Ig (mg/dl) | Patient at diagnosis | Normal range |
| IgM | 57 mg/dl | (normal 56–208 mg/dl) |
| IgA | 265 mg/dl | (normal 38–222 mg/dl) |
| IgG | 1650 mg/dl | (normal 590–1430 mg/dl) |
*Healthy donors, aged 1–2 years, with percentages/counts presented as median (10th and 90th percentiles) 16. †[3H]‐thymidine uptake in response to mitogens. PHA = phytohaemagglutinin; cpm = counts per min; Ig = immunoglobulin; CBC = complete blood count; WBC = white blood cell count.
Genetic evaluation
Whole exome sequencing (WES) of DNA obtained from PBMCs of the patient and her parents was performed, yielding 4106 homozygous variants which affect protein sequences. This list of variants was reduced subsequently to 43 rare variants by filtering out variants present in ≥ 0·01 of our in‐house exomes (n = 250) and variants present with a minor allele frequency (MAF) ≥ 0·01 in either the 1000 Genomes Project (1KG; http://browser.1000genomes.org/index.html) or dbSNP 135 database, or the NHLBI Exome Sequencing Project (ESP) (http://evs.gs.washington.edu/EVS/). Parental consanguinity suggested autosomal recessive inheritance. Recessive analysis reduced the variant list of candidate genes to 18; nevertheless, none of the genes was found to be associated with autoimmunity or malignant processes. Filtering for de‐novo variants revealed 12 candidate genes (Table 2), including a mutation in KRAS, which was reported previously to be associated with RALD 6. Dideoxy Sanger sequencing confirmed the presence of a heterozygote mutation in the patient's PBMCs (c.38G > A; p. Gly13Asp, GGC→GAC). This mutation was not detected in her parents and healthy siblings. Sequencing of DNA from the patient's nails revealed that the genotype at this genomic position was homozygote wild‐type, indicating a somatic mutation in the haematopoietic lineage (Fig. 1).
Table 2.
Whole exome sequencing analysis, filtered for de‐novo variants, revealing 12 candidate genes, including Kirsten rat sarcoma viral oncogene homologue (KRAS)
| Chr | Pos | Gene | Description | RefAlt | Mutation | MaxFreq | Father | Mother | Clinvar |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 35251040 | GJB3 | Gap junction protein, beta 3, 31 kDa (approved) | G/T | Missense | 0 | 5 | 9 | N |
| 1 | 26582243 | CEP85 | Centrosomal protein 85 kDa (approved) | T/G | Missense | 0 | 5 | 7 | N |
| 2 | 170664996 | SSB | Sjögren syndrome antigen B (autoantigen La) (approved) | G/A | Missense | 0 | 9 | 10 | N |
| 4 | 54362563 | LNX1 | Ligand of numb‐protein X 1, E3 ubiquitin protein ligase (approved) | T/G | Splicing | 0 | 5 | 2 | N |
| 8 | 39468070 | ADAM18 | ADAM metallopeptidase domain 18 (approved) | A/C | Missense | 0·00706 | 4 | 4 | N |
| 9 | 127994901 | RABEPK | Rab9 effector protein with Kelch motifs (approved) | C/A | Missense | 0 | 9 | 8 | N |
| 9 | 139413124 | NOTCH1 | Notch 1 (approved) | C/G | Missense | 0 | 10 | 13 | N |
| 11 | 71850734 | FOLR3 | Folate receptor 3 (gamma) (approved) | C/T | Stopgain | 0·00524 | 5 | 3 | N |
| 12 | 25398281 | KRAS | Kirsten rat sarcoma viral oncogene homologue (approved) | C/T | Missense | 0 | 29 | 26 | Y |
| 16 | 46934693 | GPT2 | Glutamic pyruvate transaminase (alanine aminotransferase) 2 (approved) | A/G | Missense | 0 | 5 | 4 | N |
| 17 | 41352498 | NBR1 | Neighbour of BRCA1 gene 1 (approved) | G/A | Missense | 0 | 12 | 3 | N |
| X | 90691540 | PABPC5 | Poly(A) binding protein, cytoplasmic 5 (approved) | T/G | Missense | 0 | 5 | 6 | N |
Chromosomal location, gene description, nucleotide change, type of mutation, frequency in both parents and clinical relevancy (Y = yes, N = no) are presented.
Figure 1.
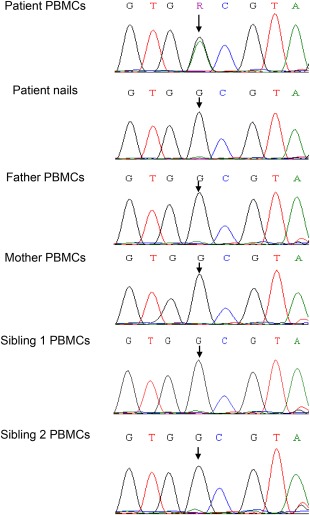
Validation of a somatic Kirsten rat sarcoma viral oncogene homologue (KRAS) mutation. Sanger sequencing confirmed the presence of a somatic missense mutation in the patient's peripheral blood mononuclear cells (PBMCs), but not in the patient's nails, her parents and healthy siblings (c.38G>A; p. Gly13Asp). [Colour figure can be viewed at wileyonlinelibrary.com]
Immunological assessment
Immunological work‐up at the age of 2 years revealed prominent monocytosis and lymphocytosis. Normal numbers of T and natural killer (NK) cells but increased B cells with an inverted ratio of CD4+/CD8+ in peripheral blood were noted (Table 1), as described previously for ALPS 17. DNT cells were only mildly increased (2.6%) (normal below 1%). The patient's IgG and IgA levels were elevated with normal IgM. T cell proliferation in response to mitogen stimulation was normal.
Production of new T and B cells decreased in a RALD patient over time in concordance with clinical deterioration. To assess whether the process of non‐malignant proliferation in our patient affected lymphocyte homeostasis, the amounts of sjTRECs and sjKRECs, markers of de‐novo T and B cell production, respectively, were determined. Serial measurements of these markers in peripheral blood during 3 years revealed a significant decrease with time (Table 3, Fig. 2), suggesting a decreasing production of new B and T cells. Similarly, the ratio between B cells containing sjKREC and total B cells (cjKREC), declined over time (Table 3). The sjKREC/cjKREC ratio estimates the homeostasis between naive B cell production and peripheral B cell replication and when it favours cjKREC, as it did in our patient, this suggests diminished B cell production, increased peripheral proliferation, or both. The decline in B and T cell production over time was also evident clinically, as the patient suffered from severe infections requiring prolonged and recurrent hospitalizations.
Table 3.
Quantification of sjTREC, sjKREC and cjKREC and the sjKREC/cjKREC ratio is presented for the patient at different ages (4–6 years) compared to healthy donors
| Age (years) | 4 | 5 | 6 | HD* |
|---|---|---|---|---|
| sjTREC | 375 | 89 | 23 | 1065 |
| sjKREC | 5579 | 2088 | 106 | 447 |
| cjKREC | 15 960 | 5566 | 1099 | 2083 |
| sjKREC/cjKREC | 0·34 | 0·37 | 0·09 | 0·21 |
*Thirty‐five healthy pediatric donors, median (controls were healthy children not taking chronic medications who came in for a routine paediatric visit and blood work. Their ages ranged between 8 and 12 years and 40% were female). TREC = T cell receptor excision circle; KREC = kappa deleting receptor excision circle; sj = signal joint; cj = coding joint; HD = healthy donors.
Figure 2.
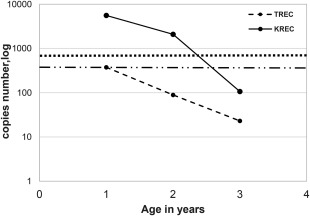
Signal joint T cell receptor excision circle (sjTREC) and signal joint kappa deleting receptor excision circle (sjKREC) copy numbers over time. Excision circle copy numbers (in log scale) as detected by reverse transcription–polymerase chain reaction (RT–PCR) in sequential measurements throughout disease progression compared to healthy controls.
TCR and BCR repertoires in a RALD patient are restricted. In order to examine peripheral clonality, TCR and BCR repertoires were analysed using several modalities. The TCR profile was assessed with the traditional TCR‐Vβ assay using flow cytometry. At the age of 3 years, the initial TCR analysis revealed a relatively normal Vβ usage distribution (Fig. 3, upper panel). However, 2 years later the TCR repertoire was abnormal, demonstrating several over‐ and under‐represented Vβ genes, and displaying overall a mildly skewed representation (over‐expression was seen in VB1, VB3, VB9 and under‐expression was seen in VB2, VB4, VB14, VB16,VB17 and VB21.3, Fig. 3, lower panel). The BCR profile, examined by measurement of junctional diversity for a mixture of Vh–Jh pairings 18, consistently exhibited a Gaussian‐like distribution in the patients' peripheral blood performed at the ages of 4, 5 and 6 years (Fig. 4). In order to characterize further the TCR and BCR repertoires, high‐throughput immunosequencing of the TRG and IGH repertoires was performed on PBMCs from the patient and two healthy age‐matched controls.
Figure 3.
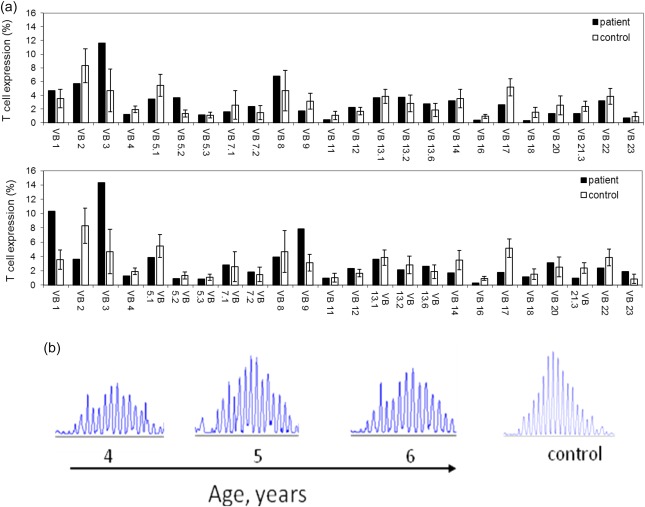
T and B cell receptor repertoire analyses. (a) Flow cytometry analysis of surface membrane expression of 24 T cell receptor β variable gene families in our Ras‐associated lymphoproliferative disease (RALD) patient (black bars) compared with healthy controls (white bars, n = 85) at ages 4 (upper panel) and 6 (lower panel) years. (b) Polyclonal (Gaussian) distribution of CDR3 length at the immunoglobulin heavy chain (IGH) locus in RALD patients at different time‐points (aged 4–6) compared to a healthy 3‐year‐old girl. [Colour figure can be viewed at wileyonlinelibrary.com]
Figure 4.
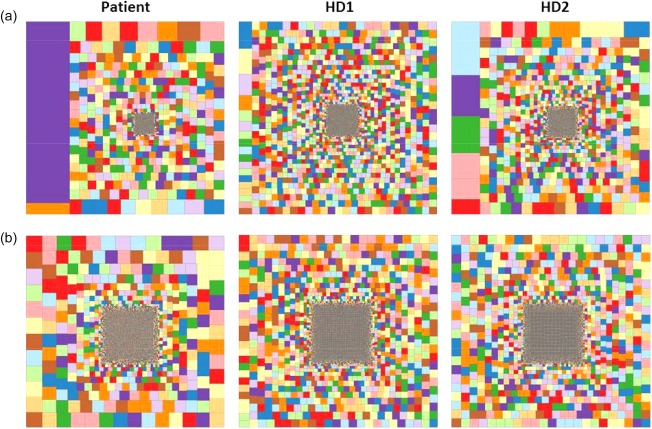
Immune repertoire high‐throughput sequencing analysis. Hierarchical tree map representation of T cell receptor gamma (TRG) (a) and immunoglobulin heavy chain (IGH) (b) repertoires in blood samples from the Ras‐associated lymphoproliferative disease (RALD) patient and two healthy controls (males, aged 4 and 11 years). Each square represents a unique CDR3 sequence and the size of the square represents its relative frequency. [Colour figure can be viewed at wileyonlinelibrary.com]
As seen in Fig. 4a, the patient's T cells displayed a more significantly restricted TRG repertoire than controls. The patient's calculated diversity indices (Table 4) clearly show increased clonality with decreased diversity in the patient's TCR repertoire compared to the two healthy controls. Shannon's entropy measures the uncertainty in predicting the identity of a single receptor sequence selected at random from the repertoire. The more diverse the repertoire, the higher Shannon's entropy. Our patient's Shannon's entropy index was lower than both controls for both TRB and IGH repertoires, reflecting restriction. Gini–Simpson's and Simpson's diversity indices calculate the evenness of distribution of individual receptors within a repertoire. The more evenly distributed the repertoire, the larger these indices are. For both TRG and IGH, the patient's repertoires were distributed less evenly than those of the controls. The top 100 clones in the patient's repertoire comprised 57% of all TRG clonotypes, whereas in the control samples the top 100 clones comprised 20 and 44% of all clonotypes. The patient's BCR repertoire (Fig. 4b) demonstrated a similarly restricted repertoire compared to the healthy control, albeit to a lesser extent than the TCR. The diversity indices showed moderate restriction compared to healthy, age‐matched controls. The top 100 B cell clones in the patient's sample comprised slightly more than 50% of all IGH clonotypes, a substantially larger share than that comprised by the top 100 clones in either control (19 and 21%). Vγ and Vh gene usage profiles, harvested from the NGS data, demonstrated an abnormal repertoire when compared to the two healthy controls, similarly to the Vβ usage profile depicted above (data not shown).
Table 4.
Receptor repertoire diversity indices
| Shannon's entropy | Gini–Simpson's evenness | Simpson's diversity | Top 100/total clonotypes | |
|---|---|---|---|---|
| TRG | ||||
| Patient | 5·27 | 0·960 | 25 | 0·57 |
| HD1 | 7·20 | 0·999 | 893 | 0·20 |
| HD2 | 6·30 | 0·994 | 177 | 0·44 |
| IGH | ||||
| Patient | 6·33 | 0·997 | 295 | 0·51 |
| HD1 | 7·38 | 0·999 | 894 | 0·19 |
| HD2 | 7·25 | 0·999 | 802 | 0·21 |
Shannon's entropy is a measurement of the richness and diversity of a repertoire, whereas Gini–Simpson's, Simpson's and Top 100 clonotypes provide different views into clone frequency distribution within a repertoire. HD = healthy donor.
Discussion
RALD is an extremely rare disease characterized by monocytosis and non‐malignant lymphoproliferation, which manifests with lymphadenopathy, massive splenomegaly, increased circulating B cells, hypergammaglobulinaemia and autoimmunity.
In some RALD patients, recurrent and at times invasive infections have been documented, suggestive of immune deficiency 7. Hidden behind normal to elevated lymphocyte counts and hypergammaglobulinemia, this component of RALD has yet to be studied extensively. A deficiency in TCR and BCR repertoires is a critical factor in the pathogenesis, evolution and progression of many autoimmune diseases 10. Indeed, abnormalities of antigen receptor repertoire diversity and complexity have been demonstrated in various autoimmune diseases 19. The development of NGS technologies enabled analysis of these immune repertoires in various pathological conditions, including immunodeficiency 18, 19 and autoimmune disorders 20, 21. The obtained analyses reached a depth that could not be achieved with previously available techniques. Applying high‐throughput sequencing to our patient's lymphocytes, we found that the TCR repertoire in our RALD patient was constricted substantially compared to two healthy age‐matched controls. A small group of expended clones occupy a considerable portion of the total receptor repertoire, constricting the remaining share reciprocally. Diversity indices, such as Shannon's entropy index and Simpson's diversity index, showed that our patient's TCR repertoire is significantly less diverse and more restricted compared with healthy age‐matched controls.
Sequential measurements of sjTREC showed marked deterioration in de‐novo T cell production, while sequential repertoire analyses demonstrated a worsening in TCR Vβ gene usage abnormality over time. Together, these results provide further evidence that an ongoing process of peripheral oligoclonal T lymphoproliferation constricts and distorts the TCR repertoire as a whole in RALD.
In B cells, while BCR spectratyping (a crude measurement of junctional diversity within all B cells using the same V–J pairings) failed to demonstrate repertoire abnormalities in our patient, high‐throughput sequencing revealed a constricted BCR repertoire. Unlike the TCR repertoire, which is dominated by a few massively expanded clones, the BCR repertoire appears to be restricted due to moderate expansions of many separate clones, resulting in a ‘top–heavy’ repertoire.sjKREC copy numbers, like sjTREC, declined dramatically over time in our patient, indicating a decrease in de‐novo B cell production. In addition, the patient displayed a decreased sjKREC/cjKREC ratio with time, further supporting the concept of peripheral clonal proliferation skewing B cell homeostasis.
As reported above, this decline in immune competence, as manifested in both constrictions of the TCR and BCR repertoires as well as decreased de‐novo lymphocyte production, was accompanied clinically by an increase in both the frequency and severity of infections suffered by our patient.
In this paper, we report for the first time, to our knowledge, the use of NGS to analyse the T and B cell receptor repertoires in a RALD patient harbouring a somatic KRAS mutation. Due to the extreme rarity of this syndrome, we could evaluate only one patient. In classical ALPS, patients are known to exhibit autoimmune clonal expanded cells, infiltrating haematopoietic organs 17. Nevertheless, receptor diversity analysis of these cells was reported to be relatively normal 22. Recently, it was shown that ALPS patients display a disturbed B lymphocytes selection that may trigger clonal expansion 23, although this has yet to be studied in RALD. Immunophenotyping of peripheral blood of RALD patients showed characteristic polyclonal B cells 7; however, with the use of NGS we were able to show a restricted and clonal expansion of both TCR and BCR repertoires in one RALD patient. Our findings suggest that clonal expansion in RALD reciprocally restricts the remaining lymphocyte receptor repertoire, contributing to an immunodeficient state. This concept may be applicable to any syndrome that is characterized by autoimmunity.
No study to date, to our knowledge, has explored TREC or KREC markers of T and B cell production in RALD patients. We found that these two markers decreased gradually with time in the patient and were associated with both clinical immunodeficiency and clonal expansion. Some RALD patients have lived to the age of 50 years, whereas others succumbed to severe sepsis at an early age. TREC and KREC measurement, readily accessible to most immunodeficiency laboratories, may help distinguishing between severe and mild RALD phenotypes, and allow monitoring of disease progression.
TCR and BCR repertoires using advanced genomic technologies, such as NGS as well as serial assays quantifying TRECs and KRECs, may help in understating the underlying immune dysfunction in RALD, ALPS and similar non‐malignant lymphoproliferative conditions. In addition, using these methodologies will enable more accurate classification of RALD patients 24, 25 and understanding of their homeostatic imbalance which, in turn, may pave the way to developing tailor‐made follow‐up and therapeutic approaches.
Disclosure
The authors declare that they have no conflict of interests.
Acknowledgements
The authors wish to thank the patient and her family for their collaboration. R. S. is supported by the Jeffrey Modell Foundation (JMF).
References
- 1. Oliveira J, Bleesing JJ, Dianzani U et al Revised diagnostic criteria and classification for the autoimmune lymphoproliferative syndrome (ALPS): report from the 2009 NIH International Workshop. Blood 2010; 116:e35–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Maecker HT, Lindstrom TM, Robinson WH et al New tools for classification and monitoring of autoimmune diseases. Nat Rev Rheumatol 2015; 8:317–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Niemela JE, Lu L, Fleisher TA et al Somatic KRAS mutations associated with a human nonmalignant syndrome of autoimmunity and abnormal leukocyte homeostasis. Blood 2011; 117:2883–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Oliveira JB, Bidere N, Niemela JE et al NRAS mutation causes a human autoimmune lymphoproliferative syndrome. Proc Natl Acad Sci USA 2017; 104:8953–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kranenburg O. The KRAS oncogene: past, present, and future. Biochim Biophys Acta 2015; 1756:81–2. [DOI] [PubMed] [Google Scholar]
- 6. Takagi M, Shinoda K, Piao J et al Autoimmune lymphoproliferative syndrome‐like disease with somatic KRAS mutation. Blood 2011; 117:2887–90. [DOI] [PubMed] [Google Scholar]
- 7. Calvo KR, Price S, Braylan RC et al JMML and RALD (Ras‐associated autoimmune leukoproliferative disorder): common genetic etiology yet clinically distinct entities. Blood 2015; 125:2753–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shiota M, Yang X, Kubokawa M et al Somatic mosaicism for a NRAS mutation associates with disparate clinical features in RAS‐associated leukoproliferative disease: a report of two cases. J Clin Immunol 2015; 35:454–8. [DOI] [PubMed] [Google Scholar]
- 9. Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009; 25:1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McKenna A, Hanna M, Banks E et al The genome analysis toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Res 2010; 20:1297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li MX, Gui HS, Kwan JS, Bao SY, Sham PC. A comprehensive framework for prioritizing variants in exome sequencing studies of Mendelian diseases. Nucleic Acids Res 2012; 40:e53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lev A, Simon AJ, Broides A et al Thymic function in MHC class II‐deficient patients. J Allergy Clin Immunol 2013; 131:831–9. [DOI] [PubMed] [Google Scholar]
- 13. Lev A, Simon AJ, Bareket M et al The kinetics of early T and B cell immune recovery after bone marrow transplantation in RAG‐2‐deficient SCID patients. PLoS One 2012; 7:e30494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kraus M, Lev A, Simon A et al Disturbed B and T cell homeostasis and neogenesis in patients with ataxia telangiectasia. J Clin Immunol 2014; 34:561–72. [DOI] [PubMed] [Google Scholar]
- 15. Alamyar E, Giudicelli V, Li S, Duroux P, Lefranc MP. IMGT/HighV‐QUEST: the IMGT® web portal for immunoglobulin (IG) or antibody and T cell receptor (TR) analysis from NGS high throughput and deep sequencing. Immunome Res 2012; 8:1–2. [Google Scholar]
- 16. Shearer WT, Rosenblatt HM, Gelman RS et al Lymphocyte subsets in healthy children from birth through 18 years of age: the pediatric AIDS clinical trials group P1009 study. J Allergy Clin Immunol 2013; 112:973–80. [DOI] [PubMed] [Google Scholar]
- 17. Lim MS, Straus SE, Dale JK et al Pathological findings in human autoimmune lymphoproliferative syndrome. Am J Pathol 1998; 153:1541–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. O'Connell AE, Volpi S, Dobbs K et al Next generation sequencing reveals skewing of the T and B cell receptor repertoires in patients with Wiskott–Aldrich syndrome. Front Immunol 2014;5:340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ramesh M, Hamm D, Simchoni N, Cunningham‐Rundles C. Clonal and constricted T cell repertoire in common variable immune deficiency. Clin Immunol 2017;178:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Henderson LA, Volpi S, Frugoni F et al Next‐generation sequencing reveals restriction and clonotypic expansion of Treg cells in juvenile idiopathic arthritis. Arthritis Rheumatol 2016; 68:1758–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dziubianau M, Hecht J, Kuchenbecker L et al TCR repertoire analysis by next generation sequencing allows complex differential diagnosis of T cell‐related pathology. Am J Transplant 2013; 13:2842–54. [DOI] [PubMed] [Google Scholar]
- 22. Lev A, Simon AJ, Amariglio N, Rechavi G, Somech R. Thymic functions and gene expression profile distinct double‐negative cells from single positive cells in the autoimmune lymphoproliferative syndrome. Autoimmun Rev 2012; 11:723–30. [DOI] [PubMed] [Google Scholar]
- 23. Janda A, Schwarz K, Van der Burg M et al Disturbed B‐lymphocyte selection in autoimmune lymphoproliferative syndrome. Blood 2016; 127:2193–202. [DOI] [PubMed] [Google Scholar]
- 24. Li P, Huang P, Yang Y, Hao M, Peng H, Li F. Updated understanding of autoimmune lymphoproliferative syndrome (ALPS). Clin Rev Allergy Immunol 2015; 50:55–63. [DOI] [PubMed] [Google Scholar]
- 25. Shah S, Wu E, Rao VK, Tarrant TK. Autoimmune lymphoproliferative syndrome: an update and review of the literature. Curr Allergy Asthma Rep 2014; 14:462. [DOI] [PMC free article] [PubMed] [Google Scholar]