

Abstract
Cathepsin G (CG), a neutrophil serine protease, induces cell migration and multicellular aggregation of human breast cancer MCF‐7 cells in a process that is dependent on E‐cadherin and CG enzymatic activity. While these tumor cell aggregates can cause tumor emboli that could represent intravascular growth and extravasation into the surrounding tissues, resulting in metastasis, the molecular mechanism underlying this process remains poorly characterized. In this study, we aimed to identify the signaling pathway that is triggered during CG‐mediated stimulation of cell aggregation. Screening of a library of compounds containing approximately 90 molecular‐targeting drugs revealed that this process was suppressed by the insulin‐like growth factor‐1 (IGF‐1) receptor (IGF‐1R)‐specific kinase inhibitor OSI‐906, as well as the multikinase inhibitors axitinib and sunitinib. Antibody array analysis, which is capable of detecting tyrosine phosphorylation of 49 distinct receptor tyrosine kinases, and the results of immunoprecipitation studies indicated that IGF‐1R is phosphorylated in response to CG treatment. Notably, IGF‐1R neutralization via treatment with a specific antibody or silencing of IGF‐1R expression through siRNA transfection suppressed cell aggregation. Furthermore, CG treatment of MCF‐7 cells resulted in increased release of IGF‐1 into the medium for 24 h, while antibody‐mediated IGF‐1 neutralization partially prevented CG‐induced cell aggregation. These results demonstrate that autocrine IGF‐1 signaling is partly responsible for the cell aggregation induced by CG.
Keywords: breast neoplasm, neoplasm metastasis, neutrophil, serine protease, tumor microenvironment
Tumor invasion and metastasis frequently occur in conjunction with inflammation and leukocyte infiltration in the tumor mass.1, 2 Neutrophils that infiltrate the tumor mass, referred to as tumor‐associated neutrophils (TAN), have been widely suggested to both suppress and promote cancer‐cell proliferation and metastasis.3, 4, 5, 6 Specifically, TAN that present the antitumor phenotype (N1‐type TAN) secrete immune‐activating cytokines and chemokines, and produce reactive oxygen species, which kill the tumor cells, while N2‐type TAN promote the proliferation and migration of cancer cells through the secretion of growth factors, cytokines and chemokines. Moreover, N2‐type TAN secrete proteases, such as matrix metalloproteinase‐9 (MMP‐9), that digest the extracellular matrix (ECM) and thereby facilitate the dissemination of cancer cells into the surrounding tissues, as well as the intra/extravasation of these cells. Infiltration of neutrophils into the tumor mass indicates poor prognosis in several types of tumors.6
Neutrophils, which play an essential role in the innate immune system, engulf pathogens and secrete proteases stored in azurophil granules for the digestion of pathogens within the blood.7, 8, 9, 10 Azurophil proteases such as cathepsin G (CG) and neutrophil elastase (NE) have also been widely suggested to be associated with the aggravation of tumor development by TAN. NE was reported to accelerate tumor cell proliferation in both a loxP‐Stop‐loxP K‐rasG12D model of lung adenocarcinoma and the lung‐cancer cell line A549.11 Elevated levels of NE facilitate tumor invasion and metastasis through degradation of the ECM;12, 13 accordingly, NE levels appear to correlate with poor prognosis in breast cancer.14 In contrast to NE, the function of CG in tumor pathology remains unclear.
Cathepsin G is a serine protease secreted from activated neutrophils and a subset of monocytes.8, 9, 15, 16 Unlike other neutrophil proteases, CG exhibits chymotrypsin‐like and trypsin‐like substrate specificity, as well as a preference for large hydrophobic (Phe, Leu and Met) and positive (Lys and Arg) P1 residues, when using synthetic peptides as a substrate.17, 18, 19, 20 Moreover, CG also influences hormone activation, apoptosis, chemotaxis, blood coagulation and cardiomyocyte anoikis.21, 22, 23, 24 We previously showed that CG induces cell migration, followed by the formation of 3D‐homotypic and multicellular aggregates, through E‐cadherin‐dependent cell–cell adhesion in human breast cancer MCF‐7 cells.25, 26 The morphological and biological properties of the CG‐induced spheroids resemble those of tumor emboli observed in the lymphatic vessels of patients with inflammatory breast carcinoma.27, 28 Because tumor‐cell aggregates can cause emboli in blood and lymphatic vessels, followed by intravascular and secondary growth in target organs, these findings suggest that CG could function as a metastasis‐promoting factor. With respect to the molecular mechanism of CG‐induced cell aggregation, we have shown that this process occurs in a CG enzymatic activity‐dependent and protease‐activated receptors (PARs)‐independent manner.29 Furthermore, CG‐induced cell aggregation does not solely involve the degradation of ECM, such as fibronectin.30 These results clearly indicate that activation of intracellular signaling is involved in the stimulation of cell motility and the observed aggregation of MCF‐7 cells. However, the molecular underpinnings of CG‐induced cell aggregation, such as the binding target, proteolytic substrate and intracellular signaling pathway activated by CG, remain incompletely elucidated.
In this study, we demonstrate that CG‐induced aggregation of MCF‐7 cells is suppressed by multikinase inhibitors and by an insulin‐like growth factor‐1 (IGF‐1) receptor (IGF‐1R)‐specific inhibitor. Whereas CG stimulation evoked IGF‐1R signaling, blockage of IGF‐1R through treatment with neutralizing antibodies or siRNA hindered cell aggregation. Moreover, treatment of MCF‐7 cells with CG resulted in elevated IGF‐1 release. Therefore, we conclude that IGF‐1 signaling is responsible for the cell aggregation induced by CG.
Materials and Methods
Reagents
The following reagents were obtained from commercial sources: CG purified from human neutrophils (95% purity; BioCentrum, Kraków, Poland); anti‐Akt rabbit polyclonal antibodies, anti‐phospho Akt (S473) rabbit monoclonal antibodies (clone D9E), anti‐phospho Erk1/2 (Erk1: pT202/pY204; Erk2: pT185/pY187) rabbit monoclonal antibodies (clone D13.14.4E), and anti‐IGF‐1R β‐subunit rabbit monoclonal antibodies (clone D23H3) (Cell Signaling Technology, Danvers, MA, USA); anti‐Erk1/2 rabbit polyclonal and anti‐IGF‐1 rabbit polyclonal antibodies (Abcam, Cambridge, UK); anti‐phosphotyrosine mouse monoclonal antibodies (clone 4G10; Merck Millipore, Darmstadt, Germany); anti‐β‐actin mouse monoclonal antibodies (clone AC‐15; Sigma‐Aldrich, St. Louis, MO, USA); and anti‐IGF‐1R α‐subunit mouse monoclonal antibodies (clone #33255, R&D Systems, Minneapolis, MN, USA). The Screening Committee of Anticancer Drugs (SCADS) Inhibitor Kit was supplied by SCADS, supported by a Grant‐in‐Aid for Scientific Research on Innovative Areas, Scientific Support Programs for Cancer Research, from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan. OSI‐906 and axitinib were purchased from Shelleck Chemicals (Houston, TX, USA) and Merck Millipore, respectively, while sorafenib, sunitinib and vandetanib were purchased from Cayman Chemicals (Ann Arbor, MI, USA). Pazopanib and recombinant human IGF‐1 were obtained from SYNkinase (Parkville VIC, Australia) and Wako Pure Chemical (Osaka, Japan), respectively.
Cell culture
Human breast cancer MCF‐7 cells were kindly provided by Dr Hiroshi Kosano (Teikyo University, Japan), and were maintained in RPMI 1640 medium supplemented with 10% heat‐inactivated FBS (MP Biomedicals, Solon, OH, USA) and 80 μg/mL kanamycin (Wako Pure Chemical), as described previously.30
MCF‐7 cell‐aggression assay
To quantitatively assess the degree of spheroid formation, we quantified cells that were tightly attached to culture plates after staining with crystal violet, as previously described.25 Briefly, MCF‐7 cells (1 × 104 cells/well) were seeded in 96‐well plates in RPMI 1640 medium containing 5% FBS, and then washed with serum‐free RPMI 1640 medium. After cultivation for 24 h, the cells were pre‐cultured for 1 h in RPMI 1640 containing 1% BSA and the agents whose effects on CG‐induced cell aggregation were to be tested. After pre‐culturing, the cells were cultivated in the presence of purified CG (2.5 or 40 nM) for 24 h. Subsequently, the culture plates were vigorously tapped on paper towels 10 times to eliminate loosely attached cell spheroids, and the remaining cells were dried at room temperature and stained with 0.1% crystal violet in PBS for 10 min. Finally, the plates were extensively washed with tap water and dried at room temperature, the crystal violet within the residual cells was released via lysis with 100 μL of 0.5% SDS, and the optical density at 595 nm (OD595) was measured using a microplate reader (ALVO X3; Perkin Elmer, Waltham, MA, USA).
Measurement of cathepsin G enzymatic activity
The enzymatic activity of CG was measured using N‐succinyl‐Ala‐Ala‐Pro‐Phe p‐nitroanilide (Sigma) as a substrate, as previously described.17 One unit of enzyme represented the amount needed to hydrolyze 1.0 μmol of the substrate per minute at 37°C at pH 7.5. The release rate of 4‐nitroanilide was analyzed based on OD405 values, as measured using a microplate reader.
Western blotting
SDS‐PAGE and subsequent western blotting analyses were performed as previously described.29 Cell lysates were prepared by solubilizing cells in radio‐immunoprecipitation assay (RIPA) buffer (50 mM Tris‐HCl [pH 7.5] containing 150 mM NaCl, 1% NP‐40, and 0.1% sodium deoxycholate) supplemented with protease‐inhibitor and phosphatase‐inhibitor cocktails (Sigma). Samples were then mixed at a 1:4 ration with 5 × sample buffer (300 mM Tris‐HCl [pH 6.8] containing 10% 2‐mercaptoethanol, 10% SDS, 50% glycerol and 0.05% Coomassie Brilliant Blue) and boiled for 5 min. The resulting samples (20 μg protein/lane) were subsequently separated by SDS‐PAGE using pre‐cast 5%–20% Tris‐glycine gradient gels (Atto Corporation, Tokyo, Japan) and transferred to PVDF membranes (Wako Pure Chemical). Membranes were blocked by incubation with TBS containing 0.1% Tween 20 (TBS‐T) and 5% skim milk (Megmilk Snow Bland, Tokyo, Japan) or BSA (Sigma) for 1 h, probed with the appropriate primary antibodies diluted with Can Get Signal Immunoreaction Enhancer Solution (Toyobo, Osaka, Japan) at 4°C, washed extensively with TBS‐T, and incubated with HRP‐conjugated secondary antibodies (1:5000 dilution) (GE Healthcare, Little Chalfont, UK). Finally, the membranes were washed, developed by incubation with ECL detection reagents (GE Healthcare) and exposed to Hyperfilm ECL (GE Healthcare).
Antibody array
Phosphorylation of receptor tyrosine kinases was comprehensively analyzed using Proteome Profiler Human Phospho‐RTK arrays (R&D Systems), which are capable of detecting the phosphorylation of 49 receptor tyrosine kinases, according to the manufacturer's instructions. MCF‐7 cells were seeded in RPMI 1640 containing 5% FBS and cultured for 24 h. The cells were then cultured overnight, after which the medium was replaced with RPMI 1640 containing 1% BSA for serum starvation and treated with purified CG for 5 or 15 min. Subsequently, cell lysates were prepared by solubilizing the cells (1 × 107 cells/mL) in the lysis buffer provided with the kit, but supplemented with a protease‐inhibitor cocktail (Sigma) and subjected to Phospho‐RTK array analysis (300 μg/array).
Immunoprecipitation
Whole‐cell lysates were prepared by incubating cells with Pierce IP Lysis Buffer (Thermo Fisher Scientific, Waltham, MA, USA) for 15 min on ice. The lysates (500 μg) were then incubated with the anti‐IGF‐1R β‐subunit antibody (5 μL) overnight at 4°C, following which the antibody was bound to Protein A‐Sepharose (GE Healthcare) at 4°C for 1 h. After formation of the antibody‐bead complex, the beads were washed thrice with lysis buffer, and the captured protein complex was eluted in 3 × sample buffer (180 mM Tris‐HCl [pH 6.8] containing 6% 2‐mercaptoethanol, 6% SDS, 30% glycerol and 0.03% Coomassie Brilliant Blue). Eluates were analyzed by immunoblot using the anti‐phosphotyrosine antibody.
Protein assay
Protein concentrations were determined using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific); BSA was used as the standard.
RNA interference
Silencer Select siRNA (IGF‐1R siRNA; IDs: s7211 and s7212) were purchased from Thermo Fisher Scientific. The reverse‐transfection procedure for MCF‐7 cells was performed according to the manufacturer's instruction manual. The RNA duplex was first diluted with Opti‐MEM (Thermo Fisher Scientific) to a final concentration of 5 nM, and then mixed with Lipofectamine RNAi MAX (Thermo Fisher Scientific). The cells and transfection mixture were added to RPMI 1640 medium containing 10% FBS without antibiotics. After cultivation for 24 h, the cells were stimulated with CG. Gene‐knockdown effects were evaluated via western blot analysis.
Insulin‐like growth factor‐1 quantification
The concentrations of free IGF‐1 within conditioned media were determined using an IGF‐1 ELISA Kit (R&D Systems) under denaturing conditions. To protect the released IGF‐1 and IGF‐binding proteins (IGFBP) from degradation by CG, a protease‐inhibitor cocktail (Sigma) was added to the harvested conditioned media.
Statistical analysis
Data were analyzed using Student's t‐test and are expressed as means ± SD, unless indicated otherwise. P < 0.05 was considered statistically significant for all tests.
Results
Receptor tyrosine kinase inhibitors and an insulin‐like growth factor‐1 receptor inhibitor suppress cathepsin G‐induced MCF‐7 cell aggregation
To characterize the intracellular signaling associated with CG‐induced aggregation of MCF‐7 cells, we screened for aggregation‐inhibiting agents using the SCADS Inhibitor Kit 4, which contains 79 compounds comprising molecular‐targeting anticancer drugs, including receptor tyrosine kinase inhibitors; we screened this particular library because cell migration, focal adhesion remodeling and actin rearrangement are frequently activated by extracellular stimuli, such as chemokines and growth factors.31, 32, 33, 34, 35 The inhibitory effects of these agents on CG‐induced cell aggregation were evaluated based on morphological criteria (Fig. S1). MCF‐7 cells formed spherical multicellular aggregates after incubation with 40 nM CG for 24 h. Notably, 10 and 24 compounds in the library yielded strong and moderate inhibition of this aggregation, respectively (Table S1). Intriguingly, 11 of these 34 compounds target receptor tyrosine kinases, indicating that activation of these molecules is associated with CG‐induced cell aggregation.
Next, we precisely characterized the inhibitory effects of six receptor tyrosine kinase inhibitors that apparently suppressed cell aggregation in the initial screen. Treatment with 2.5 nM CG for 24 h led to the formation of cell aggregates that adhered to the culture substrate on the plate, whereas treatment with 40 nM CG resulted in the formation of spheroids that floated in the medium (Fig. 1a). Meanwhile, OSI‐906, axitinib and sunitinib, markedly suppressed the cell aggregation induced by 2.5 nM CG and the spheroid formation stimulated by 40 nM CG (Fig. 1a). Conversely, treatment with vandetanib and pazopanib resulted in only slight decreases in these effects, but increases in the number of adhering elliptical cell aggregates induced by 40 nM CG, while treatment with sorafenib had little inhibitory effect on CG‐induced cell aggregation or spheroid formation. OSI‐906 is a specific inhibitor of IGF‐1R and insulin receptor,36 whereas the multikinase inhibitors, axitinib, sunitinib, vandetanib, pazopanib and sorafenib target various receptor tyrosine kinases and intracellular tyrosine kinases.37, 38, 39, 40, 41, 42, 43, 44, 45 To quantify the inhibitory effect of these compounds, we performed cell‐aggregation assays (Fig. 1b). Treatment of cells with OSI‐906, axitinib and sunitinib blocked cell detachment by >50%, as compared with vehicle treatment, while vandetanib and pazopanib, and sorafenib yielded moderate and weak inhibition of CG‐induced cell detachment, respectively. Notably, while CG enzymatic activity is essential for the induction of cell aggregation,29 none of these six compounds affected this activity (Fig. 1c). In addition to OSI‐906 and the multikinase inhibitors, several other inhibitors in the compound library (anisomycin, PAC‐1, TMPyP4, retinoids and inhibitors of mammalian target of rapamycin [mTOR]) partially suppressed CG‐induced cell aggregation (Table S1). Of these, anisomycin induces cell stress by inhibiting protein synthesis,46 PAC‐1 triggers apoptosis by cleaving procaspase‐3 to caspase‐3 in cancer cells,47 and TMPyP4 is a telomerase inhibitor that stabilizes DNA G‐quadruplexes.48 In addition, the retinoid complex and its receptor act as a transcription factor by binding to the retinoic acid‐binding element, resulting in the transcriptional regulation of a myriad of genes.49 Thus, our results indicate that CG‐induced cell aggregation might require normal cellular homeostasis and protein turnover. Moreover, they strongly support the conclusion that this process involves the activation of intracellular signaling, and not merely the hydrolysis of adhesion‐associated molecules. Finally, they suggest that activation of certain receptor tyrosine kinases is involved in CG‐induced cell aggregation, and that this activation occurs independent of the enzymatic activity of CG.
Figure 1.
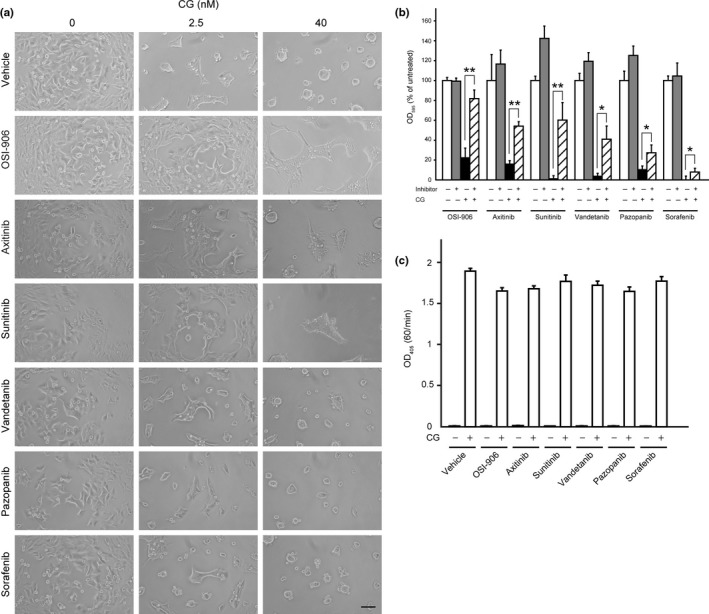
Cathepsin G (CG)‐induced MCF‐7 cell aggregation is suppressed by insulin‐like growth factor‐1 receptor (IGF‐1R)‐specific and multikinase inhibitors. (a) Morphology of cells incubated concurrently for 24 h with CG and kinase inhibitors (OSI‐906, 10 μM; axitinib, 10 μM; sunitinib, 5 μM; vandetanib, 10 μM; pazopanib, 10 μM; sorafenib, 5 μM). Scale bar = 100 μm. (b) Comparison of the inhibitory effects of various tested agents on CG‐induced MCF‐7 cell aggregation. The degree of cell aggregation was quantified using a cell‐aggregation assay. Cells were incubated with CG (40 nM) and respective inhibitors (OSI‐906, 10 μM; axitinib, 10 μM; sunitinib, 5 μM; vandetanib, 10 μM; pazopanib, 10 μM; sorafenib, 5 μM) for 24 h and then washed, following which the residual cells were stained with crystal violet. The results are expressed as means ± SD (n = 3); *P < 0.05, **P < 0.001, Student's t‐test. (c) OSI‐906 and multikinase inhibitors did not affect the enzymatic activity of CG. The enzymatic activity of CG was analyzed by measuring the rate of 4‐nitroanilide release after incubation of N‐succinyl‐Ala‐Ala‐Pro‐Phe p‐nitroanilide with CG (40 nM) and the respective inhibitors (OSI‐906, 10 μM; axitinib, 10 μM; sunitinib, 5 μM; vandetanib, 5 μM; erlotinib, 10 μM; pazopanib, 10 μM; sorafenib, 5 μM). The results are expressed as means ± SD (n = 3).
Cathepsin G activates insulin‐like growth factor‐1 receptor and its downstream signaling in MCF‐7 cells
Next, we identified the receptor tyrosine kinases whose phosphorylation is stimulated by CG in MCF‐7 cells. In the human genome, 58 genes encoding receptor tyrosine kinases have been identified, and these kinases are distributed into 20 subfamilies.34, 50 To comprehensively analyze the receptor tyrosine kinases that are phosphorylated in response to CG stimulation, we used Proteome Profiler Phospho‐RTK antibody arrays, which enable the detection of tyrosine phosphorylation of 49 receptor tyrosine kinases (www.rndsystems.com). The results of these analyses indicated that CG treatment stimulated phosphorylation of IGF‐1R and insulin receptor (Fig. 2a, Spots 1 and 2). In particular, the phosphorylation of insulin receptor was weaker than that of IGF‐1R, indicating that CG primarily induces the phosphorylation of IGF‐1R; because these two receptors and their ligands are highly homologous, insulin receptor is likely cross‐activated by IGF‐1, with a lower affinity than for IGF‐1R.51, 52 We therefore focused on IGF‐1R for subsequent experiments. First, we confirmed the observed phosphorylation of IGF‐1R through a combination of immunoprecipitation and western blot analyses (Fig. 2b). Moreover, although multikinase inhibitors inhibited CG‐induced cell aggregation (Fig. 1a,b), we did not detect phosphorylation of other receptor tyrosine kinases (Fig. 2a), suggesting that the multikinase inhibitor‐mediated suppression of cell aggregation potentially occurred through targeting of IGF‐1R.
Figure 2.
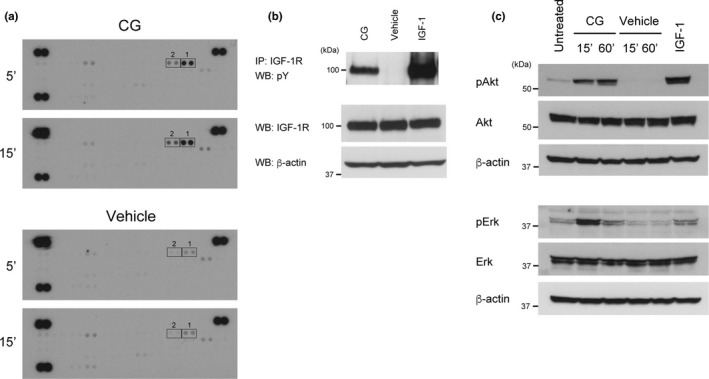
Cathepsin G (CG) induces the phosphorylation of insulin‐like growth factor‐1 receptor (IGF‐1R) and insulin receptor. (a) CG‐stimulated phosphorylation of receptor tyrosine kinases in the Proteome Profiler Human Phospho‐RTK array system. Cell lysates (300 μg/membrane) were prepared from cells treated with CG (40 nM) for 5 or 15 min. The spots were blotted with antibodies against receptor tyrosine kinases, and all antibodies were spotted on the membranes in duplicate (Spots 1: IGF‐1R, spots 2: insulin receptor). (b) Tyrosine phosphorylation of IGF‐1R stimulated by CG was detected by immunoprecipitation using an anti‐IGF‐1R antibody and western blotting with an anti‐phosphotyrosine antibody. (c) CG evokes Akt and Erk phosphorylation. Cell lysates (20 μg) prepared from cells incubated with CG (40 nM) for the indicated time periods were subjected to western blot analysis.
Next, we confirmed the activation of downstream signaling of IGF‐1R by CG. Binding of IGF‐1 to results in autophosphorylation of IGF‐1R within the intracellular domain, followed by recruitment of substrates such as IRS‐1 and Src‐homology/collagen protein (SHC); this, in turn, results in the stimulation of diverse intracellular pathways, such as the phosphatidylinositol 3‐kinase (PI3K)/Akt and MAPK signaling pathways.33, 51, 53, 54, 55, 56 PI3K/Akt signaling plays a crucial role in cell survival and antiapoptosis, whereas the MAPK pathway regulates cell proliferation. Notably, CG treatment induced the phosphorylation of both Akt and Erk1/2 in MCF‐7 cells (Fig. 2c). Together, these data indicate that CG activates IGF‐1R and its downstream signaling.
In addition to MCF‐7 cells, CG also induced aggregation of the breast cancer cell line T47D, which expresses IGF‐1R,57 and this aggregation was suppressed by OSI‐906 (Fig. S2a,b). Furthermore, CG activated the tyrosine phosphorylation of IGF‐1R in T47D cells (Fig. S2c). These results indicate that CG‐induced cell aggregation might be universal among and specific to IGF‐1R‐positive and E‐cadherin‐positive breast cancer cells.
Insulin‐like growth factor‐1 receptor activation is required for cathepsin G‐induced cell aggregation
Given the aforementioned results, we investigated whether IGF‐1R signaling is required for CG‐induced cell aggregation. IGF‐1 signaling modulates cell motility and cell–cell adhesion in association with adhesion complexes in MCF‐7 cells.56, 58, 59, 60 Indeed, IGF‐1 stimulation resulted in increased filopodia formation, cell migration and tight intercellular adhesion, but did not induce the formation of cell spheroids in MCF‐7 cells.57 To evaluate the association between IGF‐1R activation and cell aggregation, we performed cell‐aggregation assays using MCF‐7 cells in which IGF‐1R was downregulated through siRNA transfection. To avoid off‐target effects, we used two hIGF1R‐specific siRNA that target distinct sequences within the human IGF‐1R mRNA; both siRNA reduced IGF‐1R protein expression by 95% (Fig. 3a). Notably, treatment of cells with these siRNA resulted in attenuation of the cell aggregation induced by 2.5 nM CG, as well as the spheroid formation stimulated by 40 nM CG (Fig. 3b,c). However, despite the 95% decrease in IGF‐1R protein expression in IGF‐1R siRNA‐transfected cells, morphological analysis showed that these cells exhibited slight aggregation upon treatment with 40 nM CG (Fig. 3b). These data, as well as those of a previous study,30 support the hypothesis that the cell aggregation‐inducing activity of CG results from a combination of activation of IGF‐1 signaling and hydrolysis of adhesion molecules, and that the CG‐induced cell detachment that is accompanied by spheroid formation is caused by the hydrolysis of adhesion molecules.
Figure 3.
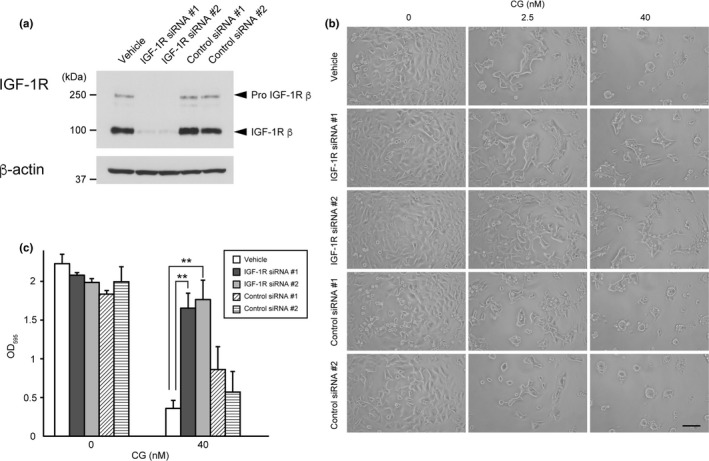
Downregulation of insulin‐like growth factor‐1 receptor (IGF‐1R) expression attenuates cathepsin G (CG)‐induced cell aggregation. (a) Western blot analysis of IGF‐1R protein expression within the lysates (20 μg) of cells transfected with IGF‐1R‐specific siRNA using an anti‐IGF‐1R β‐subunit antibody. (b) Morphology of IGF‐1R siRNA‐expressing cells following incubation with CG for 24 h. Scale bar = 100 μm. (c) Effect of IGF‐1R knockdown on CG‐induced MCF‐7 cell aggregation. The degree of cell aggregation was quantified using a cell‐aggregation assay. After washing cells that had been incubated with CG (40 nM) for 24 h, the residual cells were stained with crystal violet. The results are expressed as means ± SD (n = 3); **P < 0.001, Student's t test.
Similar to siRNA‐mediated IGF‐1R knockdown, neutralization of IGF‐1R using a monoclonal antibody specific to the extracellular domain of IGF‐1R resulted in reduced CG‐induced cell aggregation and spheroid formation (Fig. 4a,b).
Figure 4.
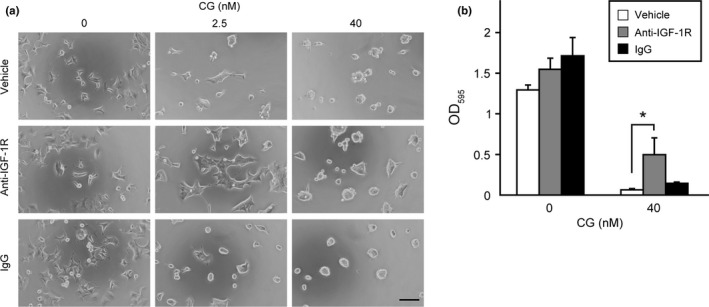
Antibody‐mediated insulin‐like growth factor‐1 receptor (IGF‐1R) neutralization attenuates cathepsin G (CG)‐induced cell aggregation. (a) Morphology of MCF‐7 cells incubated concurrently with CG and an anti‐IGF‐1R α‐subunit antibody (10 μg/mL) for 24 h. Scale bar = 100 μm. (b) Effect of an anti‐IGF‐1R α‐subunit antibody on CG‐induced MCF‐7 cell aggregation. The degree of cell aggregation was quantified using a cell‐aggregation assay. Cells were incubated with CG (40 nM) for 24 h and then washed, following which the residual cells were stained with crystal violet. The results are expressed as means ± SD (n = 3); *P < 0.05, Student's t‐test.
It was previously reported that IGF‐1R regulates intercellular adhesion, in correlation with the level of E‐cadherin expression, in several types of breast cancer cell lines.59, 60, 61 Here, we confirmed that transfection of IGF‐1R siRNA or treatment with neutralizing antibodies specific to IGF‐1R and IGF‐1 did not affect E‐cadherin expression in MCF‐7 cells (Fig. S3). Thus, we conclude that IGF‐1R activation is associated with the cell aggregation induced by CG.
Insulin‐like growth factor‐1 receptor activation induced by cathepsin G was mediated by autocrine/paracrine signaling by insulin‐like growth factor‐1 released from MCF‐7 cells
Our results suggested that CG stimulates IGF‐1 release from MCF‐7 cells, the binding of which activates IGF‐1R. To confirm this mechanism, we measured the amounts of IGF‐1 present in conditioned media harvested from CG‐treated cells. In addition, the free form of IGF‐1 was measured under non‐denaturing conditions because approximately 98% of IGF‐1 is bound to IGFBP, which interact with IGF‐1 within circulating blood and in the cellular microenvironment, thereby disrupting the binding of IGF‐1 by IGF‐1R.51, 62 The amount of free IGF‐1 in the medium was elevated at 4 h after CG addition, but was slightly reduced at 24 h post‐stimulation (Fig. 5a).
Figure 5.
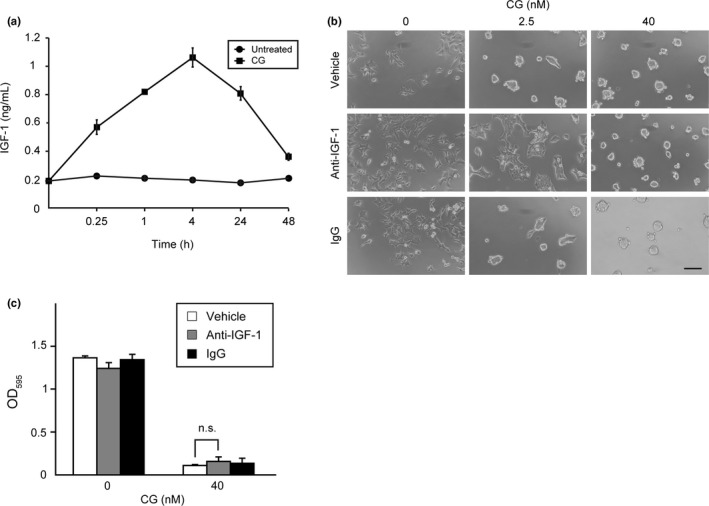
Insulin‐like growth factor‐1 (IGF‐1) released from cathepsin G (CG)‐treated MCF‐7 cells causes cell aggregation. (a) Graphic depiction of the amounts of free IGF‐1 in conditioned medium obtained from MCF‐7 cells stimulated with CG (40 nM). The results are presented as means ± SD (n = 3). (b) IGF‐1 neutralization using a specific antibody attenuates CG‐induced cell aggregation. MCF‐7 cells were incubated with CG and an anti‐IGF‐1 antibody (10 μg/mL) for 24 h. Scale bar = 100 μm. (c) Quantitative analysis of the effect of an anti‐IGF‐1 antibody on CG‐induced MCF‐7 cell aggregation. The degree of cell aggregation was quantified using a cell‐aggregation assay. After washing the cells that had been incubated with CG (40 nM) for 24 h, the residual cells were stained with crystal violet. The results are expressed as means ± SD (n = 3); n.s., not significant, Student's t test.
Meanwhile, neutralization of IGF‐1 using an anti‐IGF‐1 antibody resulted in reduced 2.5‐nM CG‐mediated cell aggregation (Fig. 5b), but did not affect the spheroid formation stimulated by 40 nM CG (Fig. 5b,c). These results indicate that direct activation of IGF‐1R by IGF‐1 is associated with the cell aggregation induced by 2.5 nM CG, but not the spheroid formation by 40 nM CG. Thus, we conclude that CG‐induced MCF‐7 cell aggregation is partially mediated by autocrine/paracrine IGF‐1 signaling. Collectively, our results support the conclusion that CG activates IGF‐1 signaling and that this signaling is required for CG‐induced cell aggregation.
Discussion
In this study, we revealed that CG induces IGF‐1 release, and that IGF‐1R and activation of its downstream signaling is required for MCF‐7 cell aggregation. IGF signaling is widely recognized to regulate not only energy metabolism and growth in normal tissues, but also cell proliferation, antiapoptosis and metastasis in tumors.(51, 52, 55, 63, 64, 65, 66) Advanced breast‐cancer cells were found to exhibit higher IGF‐1R expression, autophosphorylation and kinase activity than benign breast tumors and normal mammary epithelia.51, 55, 67 With respect to cell proliferation and antiapoptosis, overactivation of IGF‐1 signaling strongly correlates with tumor progression in cancer development. The induction of IGF‐1R activation and downstream signaling by CG suggests that this factor could accelerate cancer progression through an enhancement of cell proliferation; however, we confirmed that CG stimulation does not affect cell proliferation.25 In contrast to the function of IGF‐1R in promoting cell proliferation and survival, the roles of IGF‐1R in tumor metastasis remain incompletely understood; however, IGF‐1 signaling was shown to modulate cell motility and cell–cell adhesion in association with adhesion complexes in MCF‐7 cells.56, 58, 59, 60 Moreover, IGF‐1 treatment and IGF‐1R overactivation increased E‐cadherin‐mediated intercellular adhesion and spheroid formation in MCF‐7 cells during cultivation on semi‐solid agar.59, 61
Interestingly, the role of IGF‐1R in the regulation of intercellular adhesion appears to correlate with the level of E‐cadherin expression in breast tumors.59, 60, 61 MCF‐7 cells express estrogen receptor (ER), progesterone receptor and E‐cadherin, and are, therefore, considered highly differentiated breast cancer cells. Notably, this differentiation results in a reduction in the metastatic phenotype in intact MCF‐7 cells.68 E‐cadherin‐negative and ER‐negative MDA‐MB‐231 breast cancer cells, which exhibit a highly metastatic phenotype, express a higher level of IGF‐1R protein than do MCF‐7 cells,69 and the expression of exogenous IGF‐1R in MDA‐MB‐231 cells enabled the formation of small multicellular aggregates. Meanwhile, overexpression of both IGF‐1R and E‐cadherin allowed the cells to form larger aggregates than those of parental MDA‐MB‐231 cells.61 We found that CG induces cell detachment but does not stimulate cell migration25 or aggregation in MDA‐MB‐231 cells (unpublished observation). Therefore, the observation that CG enhances cell motility and intercellular adhesion through IGF‐1R activation is supported by these reports. In contrast, loss of E‐cadherin caused dedifferentiation and invasiveness in epithelial breast cancers.70, 71 Furthermore, in vitro invasion assay analyses using embryonic chick heart fragments indicated that the invasive potential of aggregated IGF‐1R‐overexpressing MCF‐7 cells, which were formed by cultivating cells on semi‐solid agar, was markedly lower than that of the parental cells.59, 72 However, these reports do not entirely support the view that the enhancement of cell–cell adhesion by IGF‐1R activation exerts anti‐metastatic effects, as results obtained through the in vitro invasion assay using embryonic chick heart fragment differed from those of the in vivo; for example, angiogenesis in response to VEGF did not occur in the in vitro model. Notably, IGF‐1 has been shown to stimulate angiogenesis, in part through the upregulation of vascular VEGF expression.73 IGF‐1 and IGF‐2 induced lymphangiogenesis through a vascular endothelial growth factor receptor (VEGFR) 3‐independent mechanism in an in vivo mouse cornea assay, and significantly stimulated proliferation and migration of primary lymphatic endothelial cells in an in vitro assay.74 We expect that CG accelerates breast tumor metastasis because CG induces not only IGF‐1R activation, but also the degradation of adhesion molecules and ECM, suggesting that CG might facilitate the intravasation of tumor aggregates that results from the hydrolysis of adhesion molecules and ECM, and the stimulation of angiogenesis by VEGF and lymphangiogenesis in response to IGF‐1 in vivo.
Insulin‐like growth factor‐1 bioavailability is modulated by IGFBP in the blood and the cellular microenvironment.51, 60 Approximately 98% of IGF‐1 is bound to IGFBP in the blood, and the half‐life of IGF‐1 is regulated by the cleavage of IGFBP. IGFBP cleavage is mediated by several proteases, including kallikreins, MMP, NE and CG, resulting in the release of free IGF‐1. We propose that the elevation in IGF‐1 concentration observed in response to CG is caused by the proteolysis of IGFBP, as Gibson and Cohen showed that CG hydrolyzes IGFBP, but not IGF‐1, after stimulation for at least 90 min.75 Intriguingly, this group also demonstrated that NE, a serine protease that shares a high level of sequence homology with CG, degrades both IGFBP and IGF‐1 to undetectable levels after stimulation for 20 min. Thus, in contrast to CG, NE might not increase IGF‐1 concentrations because NE degrades both IGFBP and IGF‐1; therefore, NE might not evoke cell migration and aggregation in MCF‐7 cells. We are currently testing these hypotheses.
Curiously, while the phosphorylation of Akt and Erk evoked by CG peaked at 60 and 15 min, respectively, and decreased thereafter, the levels of IGF‐1 within the conditioned medium reached a peak at 4 h (Fig. 2c and 5a and Fig. S4). Therefore, we expect that the immediate activation of IGF‐1R signaling results from the elevation of free IGF‐1 in the vicinity of the cell surface upon release from membrane‐bound IGFBP digested by CG.
In summary, we provide the first evidence that CG activates IGF‐1R by stimulating IGF‐1 release from MCF‐7 cells, and that IGF‐1R activation is required for CG‐induced cell aggregation. However, our results also indicate that IGF‐1 signaling contributes only partially to the cell aggregation phenotype observed in response to CG, suggesting that another mechanism is involved in this process.
Disclosure Statement
The authors have no conflicts of interest to declare.
Supporting information
Fig. S1 Morphological criteria used for assessing the inhibitory effect of various agents on cathepsin G (CG)‐induced MCF‐7 cell aggregation. Scale bar = 100 μm.
Fig. S2 Cathepsin G (CG) induces cell aggregation and insulin‐like growth factor‐1 receptor (IGF‐1R) activation in other breast cancer cell lines. (a and b) Cathepsin G (CG)‐induced cell aggregation of T47D cells is also suppressed by IGF‐1R‐specific inhibitors. (a) Morphology of T47D cells incubated concurrently for 24 h with CG and OSI‐906 (10 μM). Scale bar = 100 μm. (b) Effects of OSI‐906 on CG‐induced T47D cell aggregation. The degree of cell aggregation was quantified using a cell‐aggregation assay. Cells were incubated with CG (40 nM) and OSI‐906 (10 μM) for 24 h and then washed, following which the residual cells were stained with crystal violet. The results are expressed as means ± SD (n = 3); *P < 0.05, Student's t‐test. (c) Cathepsin G (CG) induces the phosphorylation of IGF‐1R. Tyrosine phosphorylation of IGF‐1R stimulated by CG in T47D cells was detected by immunoprecipitation using an anti‐IGF‐1R antibody and western blotting with an anti‐phosphotyrosine antibody.
Fig. S3 Transfection of insulin‐like growth factor‐1 receptor (IGF‐1R) siRNA or treatment with neutralizing antibodies specific to IGF‐1R and IGF‐1 did not affect with E‐cadherin expression in MCF‐7 cells. (a) Western blot analysis of E‐cadherin and IGF‐1R protein expression within the lysates (20 μg) of cells transfected with IGF‐1R‐specific siRNA. (b) Western blot analysis of E‐cadherin protein expression within the lysates (20 μg) of cells treated with anti‐IGF‐1 and anti‐IGF‐1Rα‐subunit antibody.
Fig. S4 The phosphorylation status of Akt and Erk in MCF‐7 cells stimulated with cathepsin G (CG) for 24 h. Cell lysates (20 μg) prepared from cells incubated with CG (40 nM) for the indicated time periods were analyzed by western blot analysis.
Table S1 Screening of compounds included in the Screening Committee of Anticancer Drugs (SCADS) Inhibitor Kit 4 revealed novel inhibitors of cathepsin G (CG)‐induced MCF‐7 cell aggregation. Cells were incubated with CG (40 nM) and the tested compounds (1, 3 or 10 μM) for 24 h. The inhibitory effect was judged based on morphological criteria (Fig. S1).
Acknowledgments
This work was supported by a KAKENHI Grant (JP15K18417 to RMK) from MEXT of Japan. The SCADS Inhibitor Kit was kindly supplied by SCADS, supported by a Grant‐in‐Aid for Scientific Research on Innovative Areas, Scientific Support Programs for Cancer Research, from MEXT, Japan.
Cancer Sci 108 (2017) 1574–1583
Funding Information
Ministry of Education, Culture, Sports, Science and Technology, (Grant/Award Number: ‘KAKENHI Grant (JP15K18417 to RMK)’)
References
- 1. Coussens LM, Werb Z. Inflammation and cancer. Nature 2002; 420: 860–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Elinav E, Nowarski R, Thaiss CA, Hu B, Jin C, Flavell RA. Inflammation‐induced cancer: crosstalk between tumours, immune cells and microorganisms. Nat Rev Cancer 2013; 13: 759–71. [DOI] [PubMed] [Google Scholar]
- 3. Fridlender ZG, Sun J, Kim S et al Polarization of tumor‐associated neutrophil phenotype by TGF‐β: “N1” versus “N2” TAN. Cancer Cell 2009; 16: 183–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gregory AD, Houghton AM. Tumor‐associated neutrophils: new targets for cancer therapy. Cancer Res 2011; 71: 2411–6. [DOI] [PubMed] [Google Scholar]
- 5. Fridlender ZG, Albelda SM. Tumor‐associated neutrophils: friend or foe? Carcinogenesis 2012; 33: 949–55. [DOI] [PubMed] [Google Scholar]
- 6. Uribe‐Querol E, Rosales C. Neutrophils in cancer: two sides of the same coin. J Immunol Res 2015; 2015: 983698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Salvesen G, Farley D, Shuman J, Przybyla A, Reilly C, Travis J. Molecular cloning of human cathepsin G: structural similarity to mast cell and cytotoxic T lymphocyte proteinases. Biochemistry 1987; 26: 2289–93. [DOI] [PubMed] [Google Scholar]
- 8. Owen CA, Campbell EJ. The cell biology of leukocyte‐mediated proteolysis. J Leukoc Biol 1999; 65: 137–50. [DOI] [PubMed] [Google Scholar]
- 9. Korkmaz B, Horwitz MS, Jenne DE, Gauthier F. Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human diseases. Pharmacol Rev 2010; 62: 726–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Meyer‐Hoffert U, Wiedow O. Neutrophil serine proteases: mediators of innate immune responses. Curr Opin Hematol 2011; 18: 19–24. [DOI] [PubMed] [Google Scholar]
- 11. Houghton AM, Rzymkiewicz DM, Ji H et al Neutrophil elastase‐mediated degradation of IRS‐1 accelerates lung tumor growth. Nat Med 2010; 16: 219–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Doi K, Horiuchi T, Uchinami M et al Neutrophil elastase inhibitor reduces hepatic metastases induced by ischaemia‐reperfusion in rats. Eur J Surg 2002; 168: 507–10. [DOI] [PubMed] [Google Scholar]
- 13. Sun Z, Yang P. Role of imbalance between neutrophil elastase and α1‐antitrypsin in cancer development and progression. Lancet Oncol 2004; 5: 182–90. [DOI] [PubMed] [Google Scholar]
- 14. Sato T, Takahashi S, Mizumoto T et al Neutrophil elastase and cancer. Surg Oncol 2006; 15: 217–22. [DOI] [PubMed] [Google Scholar]
- 15. Burster T, Macmillan H, Hou T, Boehm BO, Mellins ED. Cathepsin G: roles in antigen presentation and beyond. Mol Immunol 2010; 47: 658–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Heutinck KM, ten Berge IJ, Hack CE, Hamann J, Rowshani AT. Serine proteases of the human immune system in health and disease. Mol Immunol 2010; 47: 1943–55. [DOI] [PubMed] [Google Scholar]
- 17. Nakajima K, Powers JC, Ashe BM, Zimmerman M. Mapping the extended substrate binding site of cathepsin G and human leukocyte elastase. Studies with peptide substrates related to the α1‐protease inhibitor reactive site. J Biol Chem 1979; 254: 4027–32. [PubMed] [Google Scholar]
- 18. McRae B, Nakajima K, Travis J, Powers JC. Studies on reactivity of human leukocyte elastase, cathepsin G, and porcine pancreatic elastase toward peptides including sequences related to the reactive site of α1‐protease inhibitor (α1‐antitrypsin). Biochemistry 1980; 19: 3973–8. [DOI] [PubMed] [Google Scholar]
- 19. Tanaka T, Minematsu Y, Reilly CF, Travis J, Powers JC. Human leukocyte cathepsin G. Subsite mapping with 4‐nitroanilides, chemical modification and effect of possible cofactors. Biochemistry 1985; 24: 2040–7. [DOI] [PubMed] [Google Scholar]
- 20. Réhault S, Brillard‐Bourdet M, Juliano MA, Juliano L, Gauthier F, Moreau T. New, sensitive fluorogenic substrates for human cathepsin G based on the sequence of serpin‐reactive site loops. J Biol Chem 1999; 274: 13810–7. [DOI] [PubMed] [Google Scholar]
- 21. Sabri A, Alcott SG, Elouardighi H et al Neutrophil cathepsin G promotes detachment‐induced cardiomyocyte apoptosis via a protease‐activated receptor‐independent mechanism. J Biol Chem 2003; 278: 23944–54. [DOI] [PubMed] [Google Scholar]
- 22. Rafiq K, Kolpakov MA, Abdelfettah M et al Role of protein‐tyrosine phosphatase SHP2 in focal adhesion kinase down‐regulation during neutrophil cathepsin G‐induced cardiomyocytes anoikis. J Biol Chem 2006; 281: 19781–92. [DOI] [PubMed] [Google Scholar]
- 23. Rafiq K, Hanscom M, Valerie K, Steinberg SF, Sabri A. Novel mode for neutrophil protease cathepsin G‐mediated signaling: membrane shedding of epidermal growth factor is required for cardiomyocyte anoikis. Circ Res 2008; 102: 32–41. [DOI] [PubMed] [Google Scholar]
- 24. Rafiq K, Guo J, Vlasenko L et al c‐Cbl ubiquitin ligase regulates focal adhesion protein turnover and myofibril degeneration induced by neutrophil protease cathepsin G. J Biol Chem 2012; 287: 5327–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yui S, Tomita K, Kudo T, Ando S, Yamazaki M. Induction of multicellular 3‐D spheroids of MCF‐7 breast carcinoma cells by neutrophil‐derived cathepsin G and elastase. Cancer Sci 2005; 96: 560–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kudo T, Kigoshi H, Hagiwara T et al A neutrophil protease, induces compact cell–cell adhesion in MCF‐7 human breast cancer cells. Mediators Inflamm 2009; 2009: 850940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tomlinson JS, Alpaugh ML, Barsky SH. An intact overexpressed E‐cadherin/α, β‐catenin axis characterizes the lymphovascular emboli of inflammatory breast carcinoma. Cancer Res 2001; 61: 5231–41. [PubMed] [Google Scholar]
- 28. Alpaugh ML, Tomlinson JS, Kasraeian S, Barsky SH. Cooperative role of E‐cadherin and sialyl‐Lewis X/A‐deficient MUC1 in the passive dissemination of tumor emboli in inflammatory breast carcinoma. Oncogene 2002; 21: 3631–43. [DOI] [PubMed] [Google Scholar]
- 29. Morimoto‐Kamata R, Mizoguchi S, Ichisugi T, Yui S. Cathepsin G induces cell aggregation of human breast cancer MCF‐7 cells via a 2‐step mechanism: catalytic site‐independent binding to the cell surface and enzymatic activity‐dependent induction of the cell aggregation. Mediators Inflamm 2012; 2012: 456462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yui S, Osawa Y, Ichisugi T, Morimoto‐Kamata R. Neutrophil cathepsin G, but not elastase, induces aggregation of MCF‐7 mammary carcinoma cells by a protease activity‐dependent cell‐oriented mechanism. Mediators Inflamm 2014; 2014: 971409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Griffith JW, Sokol CL, Luster AD. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol 2014; 32: 659–702. [DOI] [PubMed] [Google Scholar]
- 32. Lauffenburger DA, Horwitz AF. Cell migration: a physically integrated molecular process. Cell 1996; 84: 359–69. [DOI] [PubMed] [Google Scholar]
- 33. Brunton VG, MacPherson IR, Frame MC. Cell adhesion receptors, tyrosine kinases and actin modulators: a complex three‐way circuitry. Biochim Biophys Acta 2004; 1692: 121–44. [DOI] [PubMed] [Google Scholar]
- 34. Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell 2010; 141: 1117–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Balkwill F. Cancer and the chemokine network. Nat Rev Cancer 2004; 4: 540–50. [DOI] [PubMed] [Google Scholar]
- 36. Mulvihill MJ, Cooke A, Rosenfeld‐Franklin M et al Discovery of OSI‐906: a selective and orally efficacious dual inhibitor of the IGF‐1 receptor and insulin receptor. Future Med Chem 2009; 1: 1153–71. [DOI] [PubMed] [Google Scholar]
- 37. Mendel DB, Laird AD, Xin X et al In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet‐derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res 2003; 9: 327–37. [PubMed] [Google Scholar]
- 38. O'Farrell AM, Abrams TJ, Yuen HA et al SU11248 is a novel FLT3 tyrosine kinase inhibitor with potent activity in vitro and in vivo. Blood 2003; 101: 3597–605. [DOI] [PubMed] [Google Scholar]
- 39. Rugo HS, Herbst RS, Liu G et al Phase I trial of the oral antiangiogenesis agent AG‐013736 in patients with advanced solid tumors: pharmacokinetic and clinical results. J Clin Oncol 2005; 23: 5474–83. [DOI] [PubMed] [Google Scholar]
- 40. Ratain MJ, Eisen T, Stadler WM et al Phase II placebo‐controlled randomized discontinuation trial of sorafenib in patients with metastatic renal cell carcinoma. J Clin Oncol 2006; 24: 2505–12. [DOI] [PubMed] [Google Scholar]
- 41. Escudier B, Gore M. Axitinib for the management of metastatic renal cell carcinoma. Drugs R D 2011; 11: 113–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hennequin LF, Stokes ES, Thomas AP et al Novel 4‐anilinoquinazolines with C‐7 basic side chains: design and structure activity relationship of a series of potent, orally active, VEGF receptor tyrosine kinase inhibitors. J Med Chem 2002; 45: 1300–12. [DOI] [PubMed] [Google Scholar]
- 43. Podar K, Tonon G, Sattler M et al The small‐molecule VEGF receptor inhibitor pazopanib (GW786034B) targets both tumor and endothelial cells in multiple myeloma. Proc Natl Acad Sci USA 2006; 103: 19478–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Morabito A, Piccirillo MC, Falasconi F et al Vandetanib (ZD6474), a dual inhibitor of vascular endothelial growth factor receptor (VEGFR) and epidermal growth factor receptor (EGFR) tyrosine kinases: current status and future directions. Oncologist 2009; 14: 378–90. [DOI] [PubMed] [Google Scholar]
- 45. Garuti L, Roberti M, Bottegoni G. Multi‐kinase inhibitors. Curr Med Chem 2015; 22: 695–712. [DOI] [PubMed] [Google Scholar]
- 46. Grollman AP. Inhibitors of protein synthesis. II. Mode of action of anisomycin. J Biol Chem 1967; 242: 3226–33. [PubMed] [Google Scholar]
- 47. Putt KS, Chen GW, Pearson JM et al Small‐molecule activation of procaspase‐3 to caspase‐3 as a personalized anticancer strategy. Nat Chem Biol 2006; 2: 543–50. [DOI] [PubMed] [Google Scholar]
- 48. Anantha NV, Azam M, Sheardy RD. Porphyrin binding to quadrupled T4G4 . Biochemistry 1998; 37: 2709–14. [DOI] [PubMed] [Google Scholar]
- 49. Blomhoff R, Blomhoff HK. Overview of retinoid metabolism and function. J Neurobiol 2006; 66: 606–30. [DOI] [PubMed] [Google Scholar]
- 50. Robinson DR, Wu YM, Lin SF. The protein tyrosine kinase family of the human genome. Oncogene 2000; 19: 5548–57. [DOI] [PubMed] [Google Scholar]
- 51. Pollak MN, Schernhammer ES, Hankinson SE. Insulin‐like growth factors and neoplasia. Nat Rev Cancer 2004; 4: 505–18. [DOI] [PubMed] [Google Scholar]
- 52. Pollak M. Insulin and insulin‐like growth factor signalling in neoplasia. Nat Rev Cancer 2008; 8: 915–28. [DOI] [PubMed] [Google Scholar]
- 53. Hubbard SR, Till JH. Protein tyrosine kinase structure and function. Annu Rev Biochem 2000; 69: 373–98. [DOI] [PubMed] [Google Scholar]
- 54. Schlessinger J. Cell Signaling by receptor tyrosine kinases. Cell 2000; 103: 211–25. [DOI] [PubMed] [Google Scholar]
- 55. Surmacz E. Function of the IGF‐I receptor in breast cancer. J Mammary Gland Biol Neoplasia 2000; 5: 95–105. [DOI] [PubMed] [Google Scholar]
- 56. Mauro L, Surmacz E. IGF‐I receptor, cell‐cell adhesion, tumor development and progression. J Mol Histol 2004; 35: 247–53. [DOI] [PubMed] [Google Scholar]
- 57. Pacher M, Seewald MJ, Mikula M et al Impact of constitutive IGF1/IGF2 stimulation on the transcriptional program of human breast cancer cells. Carcinogenesis 2007; 28: 49–59. [DOI] [PubMed] [Google Scholar]
- 58. Guvakova MA, Surmacz E. The activated insulin‐like growth factor I receptor induces depolarization in breast epithelial cells characterized by actin filament disassembly and tyrosine dephosphorylation of FAK, Cas, and paxillin. Exp Cell Res 1999; 251: 244–55. [DOI] [PubMed] [Google Scholar]
- 59. Mauro L, Salerno M, Morelli C, Boterberg T, Bracke ME, Surmacz E. Role of the IGF‐I receptor in the regulation of cell–cell adhesion: implications in cancer development and progression. J Cell Physiol 2003; 194: 108–16. [DOI] [PubMed] [Google Scholar]
- 60. Cox OT, O'Shea S, Tresse E, Bustamante‐Garrido M, Kiran‐Deevi R, O'Connor R. IGF‐1 receptor and adhesion signaling: an important axis in determining cancer cell phenotype and therapy resistance. Front Endocrinol (Lausanne) 2015; 6: 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mauro L, Bartucci M, Morelli C, Andò S, Surmacz E. IGF‐I receptor‐induced cell–cell adhesion of MCF‐7 breast cancer cells requires the expression of junction protein ZO‐1. J Biol Chem 2001; 276: 39892–7. [DOI] [PubMed] [Google Scholar]
- 62. Firth SM, Baxter RC. Cellular actions of the insulin‐like growth factor binding proteins. Endocr Rev 2002; 23: 824–54. [DOI] [PubMed] [Google Scholar]
- 63. Clemmons DR, Maile LA. Minireview: integral membrane proteins that function coordinately with the insulin‐like growth factor I receptor to regulate intracellular signaling. Endocrinology 2003; 144: 1664–70. [DOI] [PubMed] [Google Scholar]
- 64. Pollak M. The insulin and insulin‐like growth factor receptor family in neoplasia: an update. Nat Rev Cancer 2012; 12: 159–69. [DOI] [PubMed] [Google Scholar]
- 65. Christopoulos PF, Msaouel P, Koutsilieris M. The role of the insulin‐like growth factor‐1 system in breast cancer. Mol Cancer 2015; 14: 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zielinska HA, Bahl A, Holly JM, Perks CM. Epithelial‐to‐mesenchymal transition in breast cancer: a role for insulin‐like growth factor I and insulin‐like growth factor‐binding protein 3? Breast Cancer (Dove Med Press) 2015; 7: 9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Resnik JL, Reichart DB, Huey K, Webster NJ, Seely BL. Elevated insulin‐like growth factor I receptor autophosphorylation and kinase activity in human breast cancer. Cancer Res 1998; 58: 1159–64. [PubMed] [Google Scholar]
- 68. Soule HD, Vazguez J, Long A, Albert S, Brennan M. A human cell line from a pleural effusion derived from a breast carcinoma. J Natl Cancer Inst 1973; 51: 1409–16. [DOI] [PubMed] [Google Scholar]
- 69. Bartucci M, Morelli C, Mauro L, Andò S, Surmacz E. Differential insulin‐like growth factor I receptor signaling and function in estrogen receptor (ER)‐positive MCF‐7 and ER‐negative MDA‐MB‐231 breast cancer cells. Cancer Res 2001; 61: 6747–54. [PubMed] [Google Scholar]
- 70. Berx G, Van Roy F. The E‐cadherin/catenin complex: an important gatekeeper in breast cancer tumorigenesis and malignant progression. Breast Cancer Res 2001; 3: 289–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Singhai R, Patil VW, Jaiswal SR, Patil SD, Tayade MB, Patil AV. E‐Cadherin as a diagnostic biomarker in breast cancer. N Am J Med Sci 2011; 3: 227–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Bracke ME, Vyncke BM, Bruyneel EA et al Insulin‐like growth factor I activates the invasion suppressor function of E‐cadherin in MCF‐7 human mammary carcinoma cells in vitro. Br J Cancer 1993; 68: 282–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Oh JS, Kucab JE, Bushel PR et al Insulin‐like growth factor‐1 inscribes a gene expression profile for angiogenic factors and cancer progression in breast epithelial cells. Neoplasia 2002; 4: 204–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Björndahl M, Cao R, Nissen LJ et al Insulin‐like growth factors 1 and 2 induce lymphangiogenesis in vivo. Proc Natl Acad Sci USA 2005; 102: 15593–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Gibson TL, Cohen P. Inflammation‐related neutrophil proteases, cathepsin G and elastase, function as insulin‐like growth factor binding protein proteases. Growth Horm IGF Res 1999; 9: 241–53. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Morphological criteria used for assessing the inhibitory effect of various agents on cathepsin G (CG)‐induced MCF‐7 cell aggregation. Scale bar = 100 μm.
Fig. S2 Cathepsin G (CG) induces cell aggregation and insulin‐like growth factor‐1 receptor (IGF‐1R) activation in other breast cancer cell lines. (a and b) Cathepsin G (CG)‐induced cell aggregation of T47D cells is also suppressed by IGF‐1R‐specific inhibitors. (a) Morphology of T47D cells incubated concurrently for 24 h with CG and OSI‐906 (10 μM). Scale bar = 100 μm. (b) Effects of OSI‐906 on CG‐induced T47D cell aggregation. The degree of cell aggregation was quantified using a cell‐aggregation assay. Cells were incubated with CG (40 nM) and OSI‐906 (10 μM) for 24 h and then washed, following which the residual cells were stained with crystal violet. The results are expressed as means ± SD (n = 3); *P < 0.05, Student's t‐test. (c) Cathepsin G (CG) induces the phosphorylation of IGF‐1R. Tyrosine phosphorylation of IGF‐1R stimulated by CG in T47D cells was detected by immunoprecipitation using an anti‐IGF‐1R antibody and western blotting with an anti‐phosphotyrosine antibody.
Fig. S3 Transfection of insulin‐like growth factor‐1 receptor (IGF‐1R) siRNA or treatment with neutralizing antibodies specific to IGF‐1R and IGF‐1 did not affect with E‐cadherin expression in MCF‐7 cells. (a) Western blot analysis of E‐cadherin and IGF‐1R protein expression within the lysates (20 μg) of cells transfected with IGF‐1R‐specific siRNA. (b) Western blot analysis of E‐cadherin protein expression within the lysates (20 μg) of cells treated with anti‐IGF‐1 and anti‐IGF‐1Rα‐subunit antibody.
Fig. S4 The phosphorylation status of Akt and Erk in MCF‐7 cells stimulated with cathepsin G (CG) for 24 h. Cell lysates (20 μg) prepared from cells incubated with CG (40 nM) for the indicated time periods were analyzed by western blot analysis.
Table S1 Screening of compounds included in the Screening Committee of Anticancer Drugs (SCADS) Inhibitor Kit 4 revealed novel inhibitors of cathepsin G (CG)‐induced MCF‐7 cell aggregation. Cells were incubated with CG (40 nM) and the tested compounds (1, 3 or 10 μM) for 24 h. The inhibitory effect was judged based on morphological criteria (Fig. S1).