

Abstract
Colorectal cancer is a common cancer and a leading cause of cancer‐related death worldwide. The liver is a dominant metastatic site for patients with colorectal cancer. Molecular mechanisms that allow colorectal cancer cells to form liver metastases are largely unknown. Activation of epithelial–mesenchymal transition is the key step for metastasis of cancer cells. We recently reported that dual‐specificity tyrosine‐regulated kinase 2 (DYRK2) controls epithelial–mesenchymal transition in breast cancer and ovarian serous adenocarcinoma. The aim of this study is to clarify whether DYRK2 regulates liver metastases of colorectal cancer. We show that the ability of cell invasion and migration was abrogated in DYRK2‐overexpressing cells. In an in vivo xenograft model, liver metastatic lesions were markedly diminished by ectopic expression of DYRK2. Furthermore, we found that patients whose liver metastases expressed low DYRK2 levels had significantly worse overall and disease‐free survival. Given the findings that DYRK2 regulates cancer cell metastasis, we concluded that the expression status of DYRK2 could be a predictive marker for liver metastases of colorectal cancer.
Keywords: Colorectal cancer, DYRK2, EMT, liver metastasis, prognostic marker
Colorectal cancer is a common malignant disease,1, 2 for which distant metastasis is the main cause of death, and less than 5% of patients with unresectable metastatic colorectal cancer are alive after 5 years.3 Colorectal cancer cells initially spread to lymph nodes and then metastasize to the liver. A substantial number of patients also develop metastases to the lung and, less frequently, to the bone or the brain. Fifteen to 25% of patients present with synchronous liver metastases at the time of diagnosis.4, 5 Liver resection is the only potentially curative treatment; however, it is feasible in only 15–20% of patients, mainly due to bi‐lobar involvement, small residual liver volume, or extrahepatic disease.6, 7, 8 Patients with multiple metastases are treated with systemic chemotherapy, mostly in a palliative manner.
Previous studies of metastasis have focused largely on traditional clinicopathologic factors such as status of primary lesion, the number of hepatic tumors or lymph node metastases, the size of largest hepatic tumors, and presence of extrahepatic disease.9, 10, 11, 12, 13 Even in the same stage or category, prognosis or response to chemotherapy varies in each patient. This means that there are unknown molecular mechanisms that determine this multiplicity.
Dual‐specificity tyrosine‐regulated kinase 2 (DYRK2) is a protein kinase that phosphorylates its substrates on serine/thereonine residues.14 Initially, DYRK2 was found to phosphorylate p53 at Ser46 to regulate apoptotic cell death in response to DNA damage.15, 16 Recent studies have shown that DYRK2‐mediated phosphorylation initiates the degradation of several proteins through the ubiquitin–proteasome system. DYRK2 functions as a scaffold for E3 ligase complex and controls mitotic transition.17, 18 In addition, it has been identified as a priming kinase for phosphorylation. For example, we have shown that c‐Jun and c‐Myc are involved in DYRK2‐mediated signaling pathways.19 Recently, we reported that DYRK2 plays an important role in epithelial–mesenchymal transition (EMT) by degrading Snail and stabilizing E‐cadherin in breast cancer,20 and that DYRK2 regulates chemosensitivity through Snail degradation in ovarian serous adenocarcinoma.21
The formation of metastasis is a multistep process, in which malignant cells disseminate from the primary tumor to colonize distant organs. One of these processes is EMT. EMT is characteristic of embryonic development, tissue remodeling, and wound healing.22 During EMT, epithelial cells show reduced intercellular adhesion.23, 24 Aberrant activation of this process has been hypothesized to promote tumor progression, especially invasion and metastasis. A hallmark of EMT is the loss of the adhesion protein E‐cadherin that is downregulated during metastasis. In this regard, its expression often correlates with tumor grade and cancer stage.25, 26 Thus, EMT has been implicated in cancer progression.
Although accumulating studies have defined multiple steps for metastasis, the mechanism by which cancer cells acquire the ability to metastasize are largely unknown. In this study, we found that DYRK2 regulates liver metastases of colorectal cancer cells through the activation of EMT, and that patients with low DYRK2‐expressing colorectal cancer liver metastases had worse outcomes than those with high DYRK2‐expressing metastases.
Materials and Methods
Cell culture and treatment
HCT116 human colorectal cancer cells were obtained from the ATCC (Manassas, Virginia, USA). The cells were maintained in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin at 37°C in a 5% CO2 incubator. HCT116 cells were grown according to standard protocols.
Cell growth assay
An MTS assay was carried out using a CellTiter 96 AQ Solution Cell Proliferation Assay Kit (Promega, Madison, WI, USA). The absorbance was measured at 490 nm with the use of a multiple counter (Infinite 200PRO, TECAN, Mennedorf, Switzerland).
Colony formation assay
Two thousand cells were plated into 6‐well culture plates and cultured for 12 days to allow colony formation. Colonies were washed with PBS and fixed with 70% ethanol for 5 min at room temperature. After fixation, the colonies were stained using 10% Giemsa Solution (Wako, Osaka, Japan) for 10 min at room temperature. The colonies were then washed twice with PBS and dried.
Cell transfection
HCT116 cells were transduced with retroviral vector encoding E2‐Crimson, Flag vector, Flag‐DYRK2‐WT, or Flag‐DYRK2‐KR. Flag and Flag‐tagged DYRK2‐WT or KR were constructed as previously described.19, 20 Briefly, E2‐Crimson cDNA was amplified from the pE2‐Crimson‐N1 vector (Clontech, Mountain View, CA, USA) and inserted into the retroviral vector (pMXs‐IRES‐Puro; Cell Biolabs, San Diego, CA, USA). Flag vector, Flag‐DYRK2‐WT, and Flag‐DYRK2‐KR were subcloned into the retroviral vector (pMXs‐IRES‐Hyg; Cell Biolabs). They were introduced into PlatA cells using polyethylenimine‐MAX (Polysciences, Warrington, PA, USA) and OptiMEM (Invitrogen, Carlsbad, CA, USA). After 48 h, virus‐containing supernatants were passed through a 0.45‐μm filter and supplemented with 10 μg/mL polybrene (Sigma, San Francisco, CA, USA). On day 2 after infection, puromycin (40 μg/mL) or hygromycin (400 μg/mL) was added to the cultures for selection.
Cell sorting
Sorting of stably E2‐Crimson‐expressing cells was undertaken using a MoFlo XDP sorter (Beckman Coulter, Brea, CA, USA) equipped with a 676‐nm Kr laser and a 700‐nm long‐pass emission filter.
Immunoblotting
Immunoblot analysis was carried out as previously described.20, 21 The membranes were incubated with antibodies against DYRK2 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), Flag (Sigma‐Aldrich, St. Louis, MO, USA), Snail (Cell Signaling Technology, Danvers, MA, USA), c‐Myc (Santa Cruz Biotechnology), E‐cadherin (BD Transduction Laboratories, Franklin, NJ, USA) or tubulin. Immune complexes were incubated with secondary antibodies and visualized using chemiluminescence (PerkinElmer, Waltham, MA, USA).
Real time RT‐PCR
We isolated total RNA using TRIsure (Nippon Genetics, Tokyo, Japan). cDNA was synthesized from total RNA using RevatraAce reverse transcriptase (Toyobo, Osaka, Japan) and oligonucleotides or random primers. Quantitative real‐time RT‐PCR was carried out using SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA) according to the instruction manual. The data were normalized to the input control, GAPDH. Primer sequences were as previously described.20, 21
Migration and invasion assays
Migration and invasion assays were carried out in 24‐well plate Transwell chambers with 8.0‐μm pore size polycarbonate filter inserts (migration assays, BD Biosciences, Franklin, NJ, USA; invasion assays, Corning, Corning, NY, USA). A total of 8 × 105 cells were seeded on uncoated or Matrigel‐coated inserts in 500 μL serum‐free medium for migration or invasion assays, respectively. The lower chambers were filled with 750 μL medium with 5% FBS. After 24 h of incubation at 37°C, cells on the upper side of the filter were removed and cells on the lower surface of the insert were fixed and stained with 10% Giemsa for 5 min. The number of stained cells was counted under a light microscope. Assays were carried out in triplicate.
Development of a liver metastatic model
The animal experiments were approved by the Animal Research Committee of Jikei University (Tokyo, Japan) and were undertaken in accordance with established guidelines. Four‐week‐old male nude mice (BALB/c nu/nu) were purchased from CLEA Japan (Tokyo, Japan) and were housed under specific pathogen‐free conditions. The animals were placed in the right lateral decubitus position, and the skin was disinfected with 75% alcohol. A 1‐cm incision was made on the left side of the abdominal wall to expose the spleen. The spleen was then delivered gently from the abdominal cavity and 2 × 106 colorectal cancer cells in 50 μL PBS were injected slowly into the spleen using a 29‐gauge needle. All animals underwent intrasplenic injection with visible splenic capsule swelling and whitening. After injection, a 75% ethanol cotton swab was used for hemostasis for 5 min, by which extravasated cancer cells were killed to prevent intra‐abdominal metastases. After injection, the splenic artery and vein were ligated, and the spleen was removed.
To investigate the metastatic properties of these cell lines, the animals were injected with HCT116‐E2 cells (HCT116‐E2), HCT116‐E2‐Flag cells (Flag), HCT116‐E2‐Flag‐DYRK2‐WT cells (WT), or HCT116‐E2‐Flag‐DYRK2‐KR cells (KR). All animals were first examined 4 weeks later by an In Vivo Imaging System (IVIS, Caliper Life Science, Hopkinton, MA, USA) after injection and were killed 5 weeks after injection.
Isolation and preparation of mice colorectal liver metastases
Tumor formation on intrasplenic injection was followed by IVIS spectrum. At 5 weeks after injection, the animals were killed. The animals and liver metastatic lesions were imaged using the IVIS spectrum. Liver metastatic lesions were immediately resected and minced in culture medium on ice. The lesions were put into a mixture of DMEM suspended with collagenase IV (500 μg/mL) (Sigma) and hyaluronidase (500 μg/mL) to dissociate. Samples were incubated at 37°C for 1 h on a shaker. Red blood cells were removed by NH4Cl/KHCO3/EDTA buffer at 37°C for 5 min. Undissociated tissues were removed by filtering the samples through a 40‐μm mesh (Kyoshin Rikoh, Tokyo, Japan). Suspensions were filtered and washed three times with PBS. Sorting of E2‐Crimson‐positive cells was undertaken using a MoFlo XDP cell sorter (Beckman Coulter).
Patient data
We analyzed 86 colorectal liver metastases from patients who underwent their first surgery for liver metastases at Jikei University Hospital between 2000 and 2010. They were retrospectively reviewed using the prospectively collected database. Patients who had mucinous adenocarcinoma (seven cases) or whose specimens or slides were unavailable for analysis (three cases) were excluded from analysis because of the difficulty with immunostaining. Patients who were lost to follow‐up or whose primary colorectal tumor would not be obtained were also excluded (three cases). The Jikei University School of Medicine Ethics Review Committee approved the study protocol. Colorectal cancers were staged according to the 7th edition of the American Joint Committee on Cancer/Union for International Cancer Control TNM staging system. The clinicopathologic information of all samples is described in Table 1.
Table 1.
Clinical and pathological characteristics among 86 patients with colorectal cancer liver metastases
| Factor | All (n = 86) | Expression of DYRK2 | P‐value | ||
|---|---|---|---|---|---|
| High (n = 45) | Low (n = 41) | ||||
| Patient characteristics | Age, years | 63.6 (39–84) | 63.4 (39–84) | 63.8 (43–83) | |
| ≥64 | 27 | 19 | 0.976 | ||
| <64 | 18 | 22 | |||
| Gender | Male | 32 | 29 | 0.969 | |
| Female | 13 | 12 | |||
| Past medical history | (−) | 16 | 21 | 0.143 | |
| (+) | 29 | 20 | |||
| Primary tumor characteristics | Primary site | Colon | 29 | 25 | 0.957 |
| Rectum | 16 | 16 | |||
| TNM stage | I, II | 15 | 7 | 0.084 | |
| III, IV | 30 | 34 | |||
| Colorectal liver metastases characteristics | Timing of operation | Synchronous | 9 | 12 | 0.318 |
| Metachronous | 36 | 29 | |||
| Number of liver metastases | Single | 25 | 18 | 0.280 | |
| Multiple | 20 | 23 | |||
| Size of tumor, mm | <50 | 38 | 33 | 0.890 | |
| ≥50 | 7 | 8 | |||
| Liver metastasis grade | A | 22 | 20 | 0.992 | |
| B, C | 23 | 21 | |||
| Preoperative liver function | ICG R15, % | ≤10 | 16 | 16 | 0.673 |
| >11 | 22 | 18 | |||
| Unknown | 7 | 7 | |||
| Preoperative factors | Serum CEA, ng/mL | <20 | 24 | 27 | 0.288 |
| ≥20 | 20 | 14 | |||
| Unknown | 1 | 0 | |||
| Serum CA19‐9, ng/mL | <37 | 23 | 22 | 0.791 | |
| ≥37 | 20 | 17 | |||
| Unknown | 2 | 2 | |||
CA19‐9, carbohydrate antigen 19‐9; CEA, carcinoembryonic antigen; ICG R15, indocyanine green retention at 15 min.
Immunohistochemistry
All samples were formalin‐fixed, paraffin‐embedded, and histologically diagnosed with colorectal cancer and liver metastases by H&E staining. All samples were treated under the guidelines. Embedded blocks, 4‐μm sections cut from paraffin, were used for routine histopathology. The Discovery‐ultra autostainer (Ventana Medical Systems, Tucson, Arizona, USA) was used for all immunohistochemical (IHC) staining reactions. After deparaffinization, DYRK2 antigen retrieval was carried out at 100°C for 44 min in an autoclave with citrate buffer (pH 6.0). Then 3% hydrogen peroxidase was used for blocking. Diluted antibody DYRK2 (1:200; Sigma) was used for staining. Slides were mounted in Permount (Vector Laboratories, Burlingame, CA, USA), coverslipped, and evaluated by pathologists using a light microscope. The staining was evaluated as follows.
For DYRK2, the cytoplasmic staining intensity was scored semiquantitatively using an overall intensity score with four categories: IHC score 0, negative staining; 1, weak staining; 2, moderate staining; and 3, strong staining. The samples were divided into two groups, a high expression group (including the moderate and strong categories) and a low expression group (including the negative and weak categories).
Statistical analysis
The overall survival (OS) time was censored at the time of the last follow‐up, or death. The disease‐free survival (DFS) time was censored at the time of the last follow‐up if the patient had remained disease‐free, or at the time of cancer‐unrelated death. The calculation of OS and DFS was initiated on the date of the surgery. Statistical analyses were undertaken using StatMate III (GraphPad Software, La Jolla, California, USA). The relationship between clinical pathological factors and staining were analyzed by the χ2‐test or Fisher's exact test. Furthermore, to analyze the patients’ OS or DFS with different status of DYRK2 profiles, we divided the patients into two groups and investigated the clinical stage. Overall survival and DFS were analyzed using the Kaplan–Meier method and evaluated by log–rank test. P‐values <0.05 were considered statistically significant.
Results
Establishment of HCT116 cells expressing E2‐Crimson
To investigate the mechanisms of liver metastases of colorectal cancer cells, we introduced an expression vector with E2‐Crimson into HCT116 cells, which allows monitoring and chasing the kinetics and emergence of liver metastatic lesions by IVIS spectrum. To create stable HCT116 cells expressing E2‐Crimson, the sorting of positive cells was carried out by the MoFlo XDP cell sorter (Fig. 1a). The collected cells were maintained in the presence of puromycin. These sorting and selection procedures were undertaken twice to obtain HCT116‐E2 with substantial fluorescence intensity (Fig. 1b).
Figure 1.
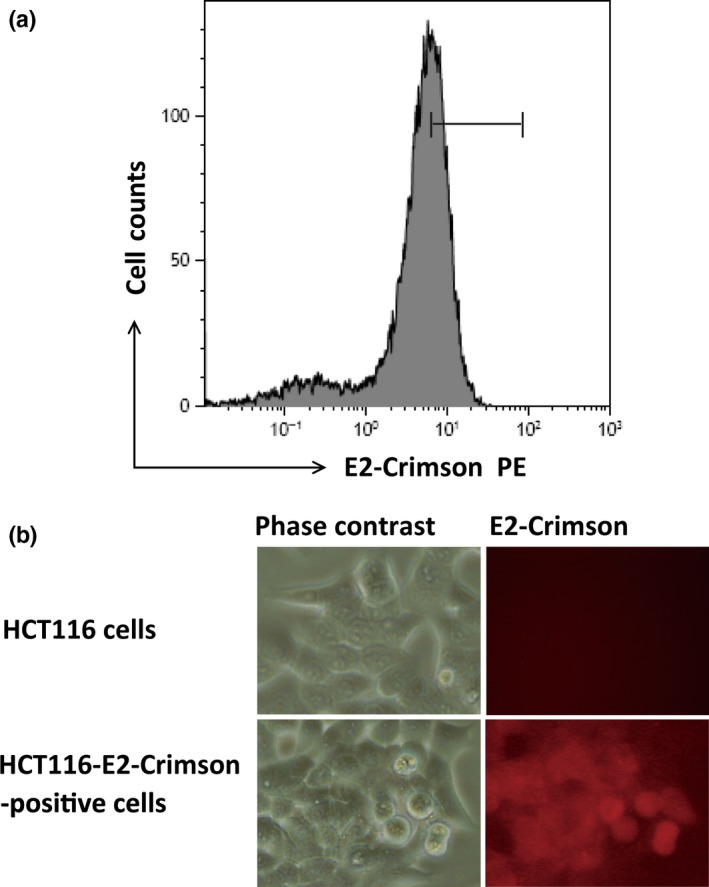
Characterization of HCT116‐E2 human colorectal cancer cells. (a) HCT116 cells were transfected with E2‐Crimson. E2‐crimson‐positive HCT116 cells were gated and sorted, and gated cells were collected. Collected cells were cultured, and this procedure was repeated twice. (b) Reflectance images of normal HCT116 cells (lower panels) and E2‐Crimson‐positive cells (upper panels) which have red excitation light.
Downregulation of DYRK2 contributes to liver metastases of colorectal cancers
Our previous studies have suggested that DYRK2 plays important roles in cancer growth, EMT, and chemosensitivity. In this study, we aimed to determine whether DYRK2 contributes to the liver metastases of colorectal cancer. To examine whether DYRK2 expression varies with liver metastases of colorectal cancer cells, we developed a xenograft mouse model (Fig. 2a). HCT116‐E2 was injected into the spleen of male nude mice (BALB/c nu/nu) to generate liver metastases as described. After injection, splenectomy was carried out. Four or 5 weeks after injection, the expression of E2‐Crimson was visible in the liver through the abdominal wall in four of six animals by IVIS spectrum (Fig. 2b). At 6 weeks after injection, the animals were killed. Four animals with liver metastasis were successfully developed in which multiple metastatic tumors were visible. Whole body imaging was undertaken by IVIS, and imaged (Fig. 2a). The tumor was excised from the liver and minced, and metastatic lesions were isolated using collagenase and hyaluronidase. Cells labeled with E2‐Crimson were isolated and collected from liver tumor cells using the MoFlo XDP cell sorter (Fig. 2c). Reverse transcription–RCR analysis was carried out to detect mRNA levels of DYRK2 in parental and metastatic HCT116‐E2 cells that were isolated from liver metastases. The results indicated that DYRK2 expression was significantly diminished in liver metastatic cells compared to parental cells (Fig. 2d). Another EMT‐associated factor, E‐cadherin, was also downregulated in liver metastases as reported (Fig. 2e). Conversely, vimentin was upregulated in liver metastases (Fig. 2d). These results imply that DYRK2 is involved in the liver metastases of colorectal cancer.
Figure 2.
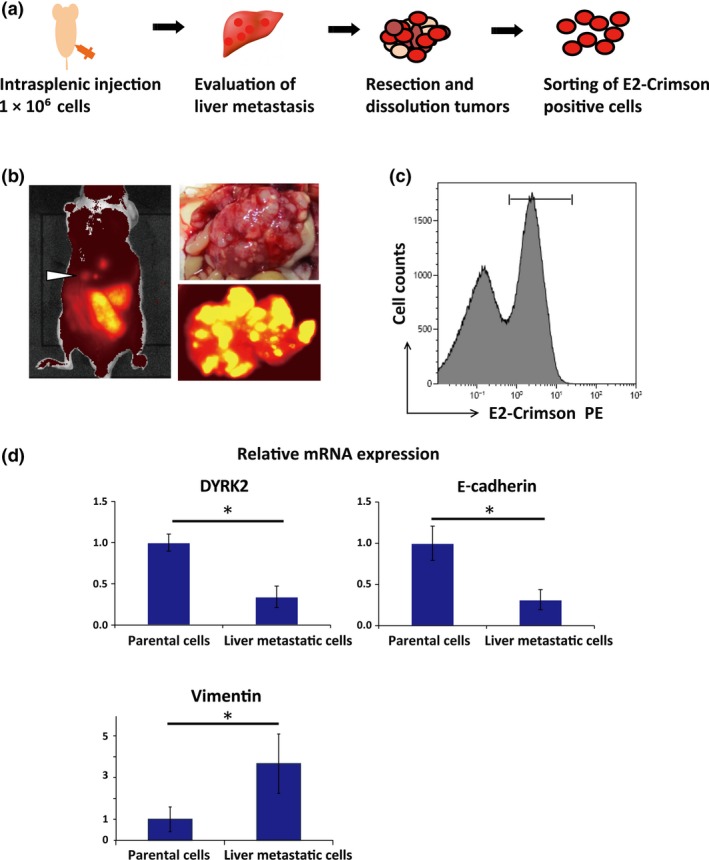
Development and assessment of liver metastases in a mouse model of colorectal cancer. (a) Schematic representation of in vivo selection and isolation of liver metastatic cells. Cells were injected into the spleen of nude mice to generate liver metastases. At 5 weeks after injection, the liver metastatic lesions were resected and minced. After cells were dissociated, E2‐crimson‐positive cells were sorted and collected. (b) Representative image of liver metastases and In Vivo Imaging System luminescent images in the animals and liver inoculated with colorectal cancer cells (HCT116‐E2). Liver metastatic lesions were identified as white masses. (c) Flow cytometric analysis and isolation of E2‐Crimson‐positive HCT116 cells from liver metastases. Cells were gated by fluorescence intensity (as E2‐Crimson‐positive) and cells derived from mice were removed. E2‐Crimson‐positive cells were isolated and collected from the liver tumors. (d) Expression of dual‐specificity tyrosine‐regulated kinase 2 (DYRK2) and E‐cadherin in cell lines. Relative mRNA expression of DYRK2, E‐cadherin, and vimentin was analyzed by real‐time RT‐PCR in parental cells and isolated cells from liver metastases. Data are displayed as mean ± SD (n = 3). *P < 0.05. n.s., not significant.
Stable expression of DYRK2 leads to E‐cadherin accumulation
To define a role of DYRK2 in controlling colorectal cancer invasion, we generated HCT116‐E2 cells that stably expressed DYRK2. We used Flag‐tagged WT DYRK2 (Flag‐DYRK2‐WT), kinase‐dead DYRK2 mutant (K178R) (Flag‐DYRK2‐KR), or Flag vector as a vehicle control. Cancer cell invasion requires substantial changes in cell adhesion and migration properties, known as EMT. In EMT, the downregulation of E‐cadherin is a key step to achieve the invasion phase of carcinoma. In this regard, Mimoto et al. reported that DYRK2 regulates E‐cadherin and Snail expression in breast cancer cells.20 As expected, we similarly observed an increase or decrease of E‐cadherin or Snail in Flag‐DYRK2‐WT, but not in KR or control cells, respectively (Fig. 3a). To confirm this result, we evaluated IHC staining and obtained comparable results (Fig. 3b). These results indicated that DYRK2 induces upregulation of E‐cadherin.
Figure 3.
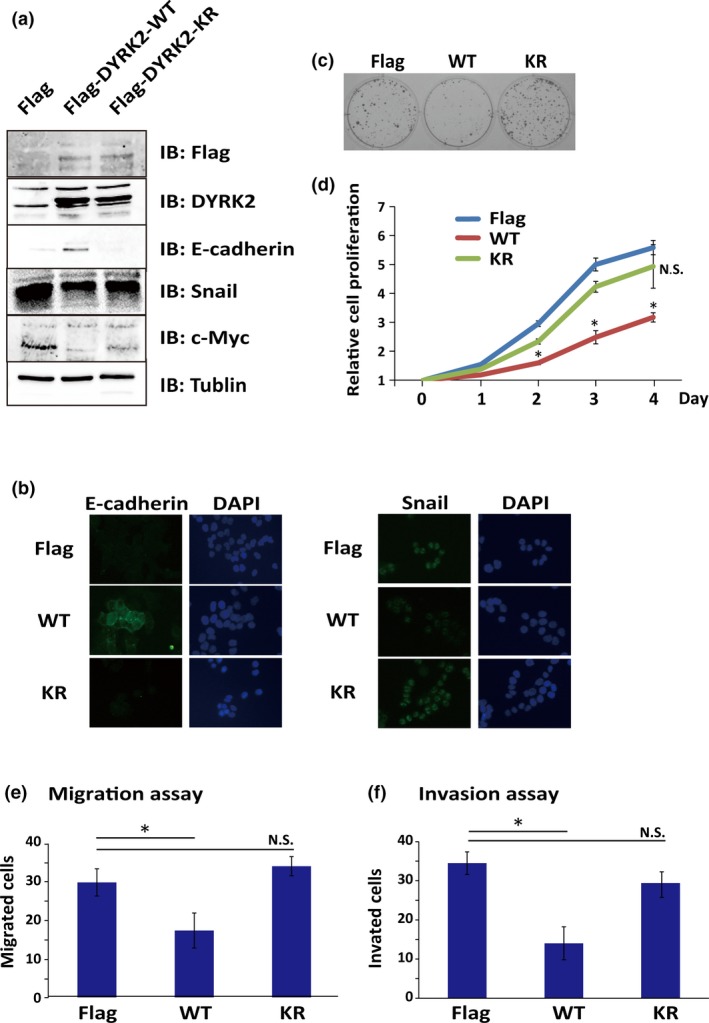
Overexpression of dual‐specificity tyrosine‐regulated kinase 2 (DYRK2) inhibits cell activity in HCT116 colorectal cancer cell line. (a) HCT116‐E2 cells were transfected with WT DYRK2 (Flag‐DYRK2‐WT [WT]), kinase‐dead DYRK2 mutant (K178R) (Flag‐DYRK2‐KR [KR]), or Flag vector as control (Flag). Expression levels of DYRK2, E‐cadherin, Snail, and c‐Myc were analyzed by Western blotting. (b) Flag, WT, or KR cells were immunostained with anti‐Snail or anti‐E‐cadherin antibodies. (c,d) Association between DYRK2 expression and cell proliferation motility of colorectal cancer. Colony formation assay (c) and cell growth analysis by MTS (d) were carried out. Data are shown as relative values based on the first day and are expressed as the mean ± SD (n = 5). *P < 0.05. (e,f) Stable overexpression of DYRK2 leads to inhibition of invasion and migration. Migration (e) and invasion (f) potentials were analyzed by a migration assay and an invasion assay, respectively. Numbers of migrating and invading cells were counted. Data are displayed as mean ± SD (n = 3). *P < 0.05.
Cell proliferation and colony formation affected by DYRK2
We next investigated the impact of DYRK2 expression on cell proliferation motility of colorectal cancer. In this regard, as previously shown in breast cancer cells, c‐Myc was suppressed by the expression of DYRK2 in a kinase activity‐dependent manner (Fig. 3a), suggesting its function on the suppression of proliferation. To generate the colony formation, cells were seeded and cell colonies were counted after 12 days. Expression of WT DYRK2 was correlated with a lower ability to form cell colonies compared with that of kinase‐dead DYRK2 or the vector control, indicating that the expression of WT DYRK2 downregulates cell growth (Fig. 3c). To confirm this result, we evaluated the growth rate of cells with the Flag vector, Flag‐DYRK2‐WT, or Flag‐DYRK2‐KR by MTS assay. Analysis of the growth curves indicated that, in Flag‐DYRK2‐WT cells, the growth was decreased compared with control cells or Flag‐DYRK2‐KR cells (Fig. 3d). These results showed that the expression of DYRK2 inhibits cell proliferation and colony formation in a kinase activity‐dependent manner.
Migration, invasion, and metastasis ability in vitro attenuated by DYRK2
Given the finding that DYRK2 expression was downregulated in liver metastases, it is plausible that DYRK2 inhibits cell migration and invasion. Cell migration and invasion are processes that offer rich targets for intervention in key physiologic and pathologic phenomena such as cancer metastasis. We assessed a potential role of DYRK2 in colon cancer cell migration and invasion by Matrigel invasion assay. The migratory potential in ectopic expression of DYRK2 was examined by migration assays. The results revealed that Flag‐DYRK2‐WT cells showed a lower migratory ability compared to DYRK2‐KR cells or control cells (Fig. 3e). The influence of DYRK2 on cell invasion was determined with Matrigel‐coated Transwell chambers. The invasive ability of Flag‐DYRK2‐WT cells was lower than DYRK2‐KR or control cells (Fig. 3f). These findings indicated that DYRK2 modulates the invasive and migratory potential of colorectal cancer cells in a kinase activity‐dependent manner.
Liver metastases regulated by DYRK2 in a xenograft model
Next, we investigated the metastatic potential of DYRK2 in vivo. Flag vector or Flag‐DYRK2‐WT cells were injected into the spleens of six male mice to develop liver metastases. After 4 weeks, the animals were monitored by IVIS. After 5 weeks, the animals were killed. The liver was removed and photographs of the organs were taken followed by IVIS. All six animals developed liver metastases (Fig. 4a). We measured the proportion of tumor weights in the liver, which was significantly suppressed in Flag‐DYRK2‐WT cells (Fig. 4b). To extend this finding, Flag vector or Flag‐DYRK2‐KR cells formed liver metastases in three animals. After injected through the spleen, the animals were killed. There was no significant difference among them (Fig. 4c,d). These data indicated that, compared to the control, overexpression of DYRK2 suppressed liver metastases in a kinase activity‐dependent manner.
Figure 4.
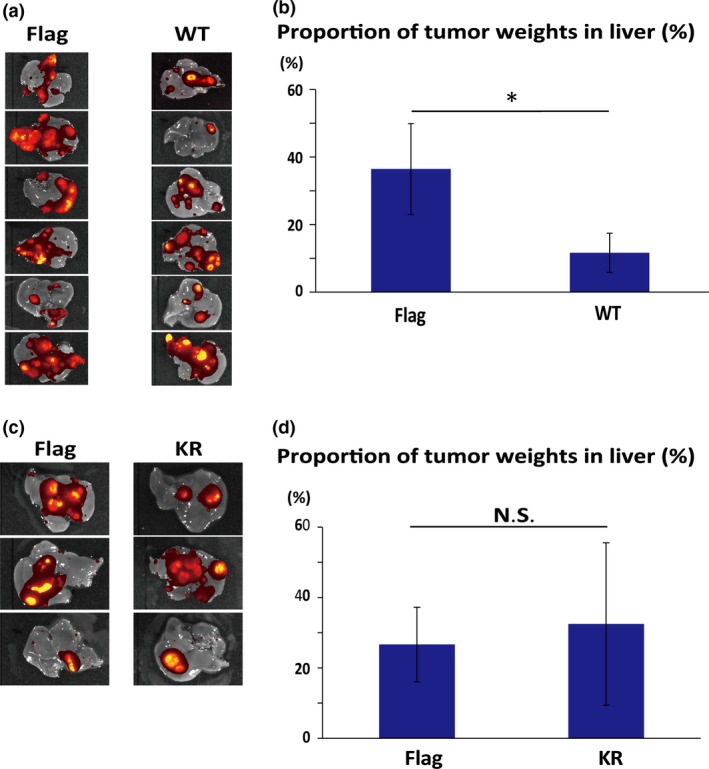
Overexpression of dual‐specificity tyrosine‐regulated kinase 2 (DYRK2) inhibits liver metastases in HCT116‐E2 human colorectal cancer cells. (a) Flag vector (control) or (Flag‐DYRK2‐WT [WT]) cells were injected into the spleen of six mice, and the formation of liver tumors was determined by In Vivo Imaging System. (b) Tumor formation efficiency of Flag and WT cells was assessed by measuring tumor weight. After injection, the percentage of tumor weight in the liver was measured. Data are displayed as mean ± SD (n = 6). *P < 0.05. (c) Flag or kinase‐dead DYRK2 mutant (K178R) (Flag‐DYRK2‐KR [KR]) was injected into the spleen of nude mice. The formation of liver tumors was determined by In Vivo Imaging System. Orange signals show tumors in the liver. (d) Tumor formation efficiency of Flag and KR cells was assessed by measuring tumor weight. After injection, the percentage of tumor weight in the liver was measured. The data are displayed as mean ± SD (n = 3).
Expression level of DYRK2 as a predictor of outcome among patients with colorectal cancer liver metastases
To determine the clinical relevance of DYRK2, we analyzed DYRK2 expression in colorectal liver metastases and follow‐up information. This study included 86 patients who underwent their first surgery for colorectal liver metastases. Representative expression patterns of DYRK2 are shown in Figure 5 (a). Positive cases showed strong granular staining in the cytoplasm of colorectal cancer cells. In order to assess the relationship between DYRK2 and clinicopathological factors at the time of the first operation for liver metastases, the χ2‐test was used for our follow‐up database (Table 1). Expression of DYRK2 in liver metastases was high in 45 samples (51.7%) and low in 41 samples (48.3%). Table 1 indicates that there was no evidence that DYRK2 regulated clinical factors among patients with colorectal cancer liver metastases. The 5‐year OS rate was 55.6% in the high expression group and 39.0% in the low expression group. The 5‐year DFS was 31.1% and 22.0% in the high and low expression groups, respectively. Patients with high DYRK2 expression in their liver metastasis showed significantly better OS and DFS in the univariate analysis (Fig. 5b,c, Tables 2 and 3; P < 0.05). In multivariate analyses, the association between DYRK2 expression and prognosis was not statistically significant (Table S1). These findings indicate that the expression level of DYRK2 is a predictor of outcome among patients with colorectal cancer liver metastases.
Figure 5.
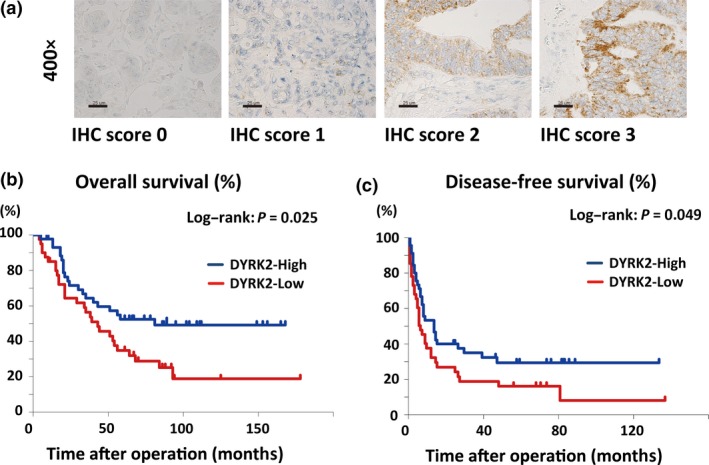
Immunohistochemical (IHC) analysis of dual‐specificity tyrosine‐regulated kinase 2 (DYRK2) expression in human colorectal liver metastases predicts clinical outcome. (a) Tissue specimens of colorectal cancer liver metastases were immunostained with anti‐DYRK2 antibodies. Scale bar = 25 μm. (b,c) Overall survival (b) and disease‐free survival (c) are shown. The groups with high and low expression of DYRK2 were compared.
Table 2.
Univariate analyses of predictors of overall survival among 86 patients with colorectal cancer liver metastases
| Factor | Univariate analysis | |
|---|---|---|
| Hazard ratio (95% CI) | P‐value | |
| Age, years | ||
| ≥64 | 1.059 (0.605–1.857) | 0.839 |
| <64 | ||
| Gender | ||
| Female | 1.007 (0.532–1.906) | 0.983 |
| Male | ||
| Primary site | ||
| Rectum | 1.304 (0.741–2.349) | 0.347 |
| Colon | ||
| TNM stage | ||
| III, IV | 1.786 (0.895–3.102) | 0.107 |
| I, II | ||
| Timing of operation | ||
| Metachronous | 1.202 (0.644–2.300) | 0.546 |
| Synchronous | ||
| Number of liver metastases | ||
| Multiple | 2.423 (1.387–4.284) | 0.002 |
| Single | ||
| Size of tumor, mm | ||
| ≥50 | 1.873 (0.940–5.327) | 0.069 |
| <50 | ||
| Liver metastasis grade | ||
| B, C | 3.531 (2.166–6.988) | <0.001 |
| A | ||
| Serum CEA, ng/mL | ||
| ≥20 | 1.521 (0.869–2.807) | 0.136 |
| <20 | ||
| Serum CA19‐9, ng/mL | ||
| ≥37 | 1.900 (1.121–3.504) | 0.019 |
| <37 | ||
| DYRK2 expression | ||
| Low | 1.879 (1.088–3.258) | 0.025 |
| High | ||
CA19‐9, carbohydrate antigen 19‐9; CEA, carcinoembryonic antigen; CI, confidence interval; DYRK2, dual‐specificity tyrosine‐regulated kinase 2.
Table 3.
Univariate analyses of predictors of disease‐free survival among 86 patients with colorectal cancer liver metastases
| Factor | Univariate analysis | |
|---|---|---|
| Hazard ratio (95% CI) | P‐value | |
| Age, years | ||
| ≥64 | 1.364 (0.842–2.305) | 0.197 |
| <64 | ||
| Gender | ||
| Female | 1.222 (0.703–2.211) | 0.451 |
| Male | ||
| Primary site | ||
| Rectum | 1.119 (0.673–1.886) | 0.65 |
| Colon | ||
| TNM stage | ||
| III, IV | 1.407 (0.809–2.411) | 0.231 |
| I, II | ||
| Timing of operation | ||
| Metachronous | 0.846 (0.504–1.391) | 0.493 |
| Synchronous | ||
| Size of tumor, mm | ||
| ≥50 | 1.440 (0.748–3.207) | 0.239 |
| <50 | ||
| Number of liver metastases | ||
| Multiple | 1.851 (1.164–3.189) | 0.011 |
| Single | ||
| Liver metastasis grade | ||
| B, C | 1.943 (1.233–3.400) | 0.006 |
| A | ||
| Serum CEA, ng/mL | ||
| ≥20 | 1.300 (0.791–2.235) | 0.283 |
| <20 | ||
| Serum CA19‐9, ng/mL | ||
| ≥37 | 1.709 (1.067–3.066) | 0.028 |
| <37 | ||
| DYRK2 expression | ||
| Low | 1.560 (1.003–2.765) | 0.049 |
| High | ||
CA19‐9, carbohydrate antigen 19‐9; CEA, carcinoembryonic antigen; CI, confidence interval; DYRK2, dual‐specificity tyrosine‐regulated kinase 2.
Discussion
Metastasis is a complex series of steps in which cancer cells leave the original tumor site and migrate to distant organs. Many studies have revealed that certain genes and signaling pathways might play a role in colon cancer liver metastases. However, the mechanisms are not completely understood. Recently, our studies have suggested that DYRK2 plays an important role in cancer growth, invasion, and chemosensitivity.19, 20, 21 In this study, we aimed to determine whether DYRK2 contributes to liver metastases of colorectal cancer, and showed that the invasion and migration abilities of cancer cells are attenuated by DYRK2 (Fig. 3e,f). In a mouse model, liver metastatic lesions were decreased by the ectopic expression of DYRK2 (Fig. 4).
The expression of DYRK2 in liver metastatic cells was reduced compared to that in parental cells (Fig. 2d). The mechanism remains unclear; however, there are two possibilities. One is that downregulation of DYRK2 could occur after metastasis. The other is that only the parental cells that low levels of DYRK2 acquire oncogene products and such cells could transfer to liver. Further studies are necessary to uncover the mechanism.
In expression of WT DYRK2, the protein level of E‐cadherin was increased in HCT116 cells (Fig. 3a,b). This result supports that DYRK2 inhibits EMT ability in colorectal cancer cells. Moreover, migration and invasion assays support that DYRK2 suppresses migration and invasion ability. We reported that DYRK2 regulates Snail and promotes EMT in breast cancer and ovarian serous adenocarcinoma, and that DYRK2 expression inversely or positively correlates with Snail or E‐cadherin expression, respectively.20, 21 As a member of the Snail regulatory kinases, DYRK2 phosphorylates Ser104 as a priming phosphorylation for glycogen synthase kinase‐3β in breast cancer.20
Recently, EMT was shown to be associated with cancer stem cells in colorectal cancer cells.4, 5, 6, 7, 8, 27 However, in our study, there was no significant difference in cancer stem cell markers, such as CD24, CD26, CD44, CD133, and CD166 (data not shown). This implies that the regulation of invasion, migration, and EMT by DYRK2 is a different pathway from cancer cell stemness.
Stable expression of WT DYRK2 inhibited cell proliferation and colony formation (Fig. 3c,d). We reported that DYRK2 regulates tumor progression through modulation of c‐Jun and c‐Myc.19 Knockdown of DYRK2 in human cancer cells was also shown to shorten the G1 phase and accelerate cell proliferation.28 Based on these findings, we speculate that overexpressed DYRK2 lengthens the G1 phase to suppress cell proliferation and colony formation.
In a mouse model, stable expression of DYRK2 inhibited liver metastases (Fig. 4). In contrast, the stable expression of the DYRK2 kinase‐dead mutant did not affect liver metastases (Fig. 4c,d). These results suggest that DYRK2 kinase activity is one of the key factors in the process of colorectal cancer metastasis.
In this study, we showed that DYRK2 could be a potential prognostic marker of liver metastases originated from colorectal cancer. By analyzing the prognosis of such patients, we discovered that patients with low DYRK2‐expressing metastatic tumors had worse outcomes than those with high‐expressing tumors in the univariate analysis. A recent report by Yan et al. supports this finding.29 In the multivariate analysis, we could not show the significance (Table S1). Higher numbers of patients should be investigated to determine this relationship. We previously reported that nuclear DYRK2 was ubiquitinated by murine double minute 2 to allow its constitutive degradation under normal conditions.30 Thus, DYRK2 expression was maintained at a low level by the proteolytic machinery in disease progression. Recent studies have reported that DYRK2 is downregulated in breast, lung, colon, and prostate cancer, and that low DYRK2 expression was associated with a poor prognosis in lung adenocarcinoma and hepatocellular carcinoma.20, 31, 32, 33, 34 Moreover, low DYRK2 expression was associated with chemoresistance in ovarian serous adenocarcinoma and bladder cancer.21, 34 In contrast, no data have been reported on the role of DYRK2 with regard to chemoresistance in colorectal cancer. In addition, there has been no report on treatment. The clinical application of DYRK2 regulation for treatment of colorectal liver metastasis, like gene therapy, should be researched in the future.
In conclusion, we reported for the first time that overexpression of DYRK2 inhibits colorectal cancer liver metastasis. Dual‐specificity tyrosine‐regulated kinase 2 plays an important role in inhibiting the migration and invasion of colorectal cancer cells. The level of DYRK2 expression could thus be a predictive marker of prognosis in liver metastases of colorectal cancer. Patients undergoing resection of colorectal cancer liver metastases who have lower levels of DYRK2 may require closer surveillance compared to those with high levels of DYRK2. The common strategy for treatment of cancer is to inhibit oncogenes. Stabilized or forced expression of tumor suppressor genes, such as DYRK2, may lead to the development of a novel treatment for colorectal cancer.
Disclosure Statement
The authors have no conflict of interest.
Supporting information
Table S1a. Multivariate analyses of predictors of overall survival among 86 patients with colorectal cancer liver metastases.
Table S1b. Multivariate analyses of predictors of disease‐free survival among 86 patients with colorectal cancer liver metastases.
Acknowledgments
The authors appreciate all patients who provided clinical samples for this study. This work was supported by grants from the Japan Society for the Promotion of Science (KAKENHI Grant Nos. JP26290041, JP17H03584, and JP26861056), Takeda Science Foundation, the Vehicle Racing Commemorative Foundation, and a research grant from the Princess Takamatsu Cancer Research Fund.
Cancer Sci 108 (2017) 1565–1573
Funding Information
Princess Takamatsu Cancer Research Fund, Takeda Science Foundation, JSPS KAKENHI, (Grant / Award Number: ‘JP26290041, JP17H03584, and JP26861056‘), Vehicle Racing Commemorative Foundation
References
- 1. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 2015; 136(5): E359–86. [DOI] [PubMed] [Google Scholar]
- 2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin 2015; 65(1): 5–29. [DOI] [PubMed] [Google Scholar]
- 3. Cejas P, Lopez‐Gomez M, Aguayo C, Madero R, de Castro Carpeno J, Belda‐Iniesta C, et al KRAS mutations in primary colorectal cancer tumors and related metastases: a potential role in prediction of lung metastasis. PLoS ONE 2009; 4(12): e8199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Manfredi S, Lepage C, Hatem C, Coatmeur O, Faivre J, Bouvier AM. Epidemiology and management of liver metastases from colorectal cancer. Ann Surg 2006; 244: 254–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Leporrier J, Maurel J, Chiche L, Bara S, Segol P, Launoy G. A population‐based study of the incidence, management and prognosis of hepatic metastases from colorectal cancer. Br J Surg 2006; 93: 465–74. [DOI] [PubMed] [Google Scholar]
- 6. Nordlinger B, Sorbye H, Glimelius B, Poston GJ, Schlag PM, Rougier P, et al Perioperative chemotherapy with FOLFOX4 and surgery versus surgery alone for resectable liver metastases from colorectal cancer (EORTC Intergroup trial 40983): a randomised controlled trial. Lancet 2008; 371: 1007–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nanji S, Cleary S, Ryan P, Guindi M, Selvarajah S, Al‐Ali H, et al Up‐front hepatic resection for metastatic colorectal cancer results in favorable long‐term survival. Ann Surg Oncol 2013; 20(1): 295–304. [DOI] [PubMed] [Google Scholar]
- 8. Minagawa M, Makuuchi M, Imamura H, et al Extension of the frontiers of surgical indications in the treatment of liver metastases from colorectal cancer. Ann Surg 2000; 4: 487–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. D'Angelica M, Kornprat P, Gonen M, DeMatteo RP, Fong Y, Blumgart LH, et al Effect on outcome of recurrence patterns after hepatectomy for colorectal metastases. Ann Surg Oncol 2011; 18: 1096–103. [DOI] [PubMed] [Google Scholar]
- 10. Fong Y, Fortner J, Sun RL, Brennan MF, Blumgart LH. Clinical score for predicting recurrence after hepatic resection for metastatic colorectal cancer: analysis of 1001 consecutive cases. Ann Surg 1999; 230: 309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nordlinger B, Guiguet M, Vaillant JC, Balladur P, Boudjema K, Bachellier P, et al Surgical resection of colorectal carcinoma metastases to the liver: a prognostic scoring system to improve case selection, based on 1568 patients. Cancer 1996; 77: 1254–62. [PubMed] [Google Scholar]
- 12. Hughes K, Simon R, Songhorabodi S, Adson M, Ilstrup D, Fortner J, et al Resection of the liver for colorectal carcinoma metastases: a multi‐institutional study of patterns of recurrence. Surgery 1986; 100: 278–84. [PubMed] [Google Scholar]
- 13. Ueno H, Mochizuki H, Hashiguchi Y, Hatsuse K, Fujimoto H, Hase K. Predictors of extrahepatic recurrence after resection of colorectal liver metastases. BrJ Surg 2004; 91: 327–33. [DOI] [PubMed] [Google Scholar]
- 14. Yoshida K. Role for DYRK family kinases on regulation of apoptosis. Biochem Pharmacol 2008; 76: 1389–94. [DOI] [PubMed] [Google Scholar]
- 15. Taira N, Nihira K, Yamaguchi T, Miki Y, Yoshida K. DYRK2 is targeted to the nucleus and controls p53 via Ser46 phosphorylation in the apoptotic response to DNA damage. Mol Cell 2007; 25: 725–38. [DOI] [PubMed] [Google Scholar]
- 16. Yoshida K. Nuclear trafficking of pro‐apoptotic kinases in response to DNA damage. Trends Mol Med 2008; 14: 305–13. [DOI] [PubMed] [Google Scholar]
- 17. Becker W. Emerging role of DYRK family protein kinases as regulators of protein stability in cell cycle control. Cell Cycle 2012; 11: 3389–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Maddika S, Chen J. Protein kinase DYRK2 is a scaffold that facilitates assembly of an E3 ligase. Nature Cell Biol 2009; 11: 409–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Taira N, Mimoto R, Kurata M, Yamaguchi T, Kitagawa M, Miki Y, et al DYRK2 priming phosphorylation of c‐Jun and c‐Myc modulates cell cycle progression in human cancer cells. J Clin Invest 2012; 122: 859–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mimoto R, Taira N, Takahashi H, Yamaguchi T, Okabe M, Uchida K, et al DYRK2 controls the epithelial‐mesenchymal transition in breast cancer by degrading Snail. Cancer Lett 2013; 339: 214–25. [DOI] [PubMed] [Google Scholar]
- 21. Yamaguchi N, Mimoto R, Yanaihara N, Imawari Y, Hirooka S, Okamoto A, et al DYRK2 regulates epithelial‐mesenchymal‐transition and chemosensitivity through Snail degradation in ovarian serous adenocarcinoma. Tumor Biol 2015; 36: 5913–23. [DOI] [PubMed] [Google Scholar]
- 22. Radisky DC. Epithelial‐mesenchymal transition. J Cell Sci 2005; 118: 4325–6. [DOI] [PubMed] [Google Scholar]
- 23. Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial‐mesenchymal transitions in development and disease. Cell 2009; 139: 871–90. [DOI] [PubMed] [Google Scholar]
- 24. Thiery JP. Epithelial–mesenchymal transitions in tumour progression. Nat Rev Cancer 2002; 2: 442–54. [DOI] [PubMed] [Google Scholar]
- 25. Hemler ME. Tetraspanin proteins promote multiple cancer stages. Nat Rev Cancer 2014; 14(1): 49–60. [DOI] [PubMed] [Google Scholar]
- 26. Van Roy F. Beyond E‐cadherin: roles of other cadherin superfamily members in cancer. Nat Rev Cancer 2014; 14(2): 121–34. [DOI] [PubMed] [Google Scholar]
- 27. Gavert N, Vivanti A, Hazin J, Brabletz T, Ben‐Ze'ev A. L1‐mediated colon cancer cell metastasis does not require changes in EMT and cancer stem cell markers. Mol Cancer Res: MCR 2011; 9(1): 14–24. [DOI] [PubMed] [Google Scholar]
- 28. Zhang X, Xu P, Ni W, Fan H, Xu J, Chen Y, et al Downregulated DYRK2 expression is associated with poor prognosis and Oxaliplatin resistance in hepatocellular carcinoma. Pathol Res Pract 2016; 212: 162–70. [DOI] [PubMed] [Google Scholar]
- 29. Yan H, Hu K, Wu W, et al Low expression of DYRK2 (Dual Specificity Tyrosine Phosphorylation Regulated Kinase 2) correlates with poor prognosis in colorectal cancer. PLoS ONE 2016; 11: e0159954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Taira N, Yamamoto H, Yamaguchi T, Miki Y, Yoshida K. ATM augments nuclear stabilization of DYRK2 by inhibiting MDM2 in the apoptotic response to DNA damage. J Biol Chem 2010; 285: 4909–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yamashita S, Chujo M, Moroga T, Anami K, Tokuishi K, Miyawaki M, et al DYRK2 expression may be a predictive marker for chemotherapy in non‐small cell lung cancer. Anticancer Res 2009; 29: 2753–7. [PubMed] [Google Scholar]
- 32. Wang Y, Wu Y, Miao X, Zhu X, Miao X, He Y, et al Silencing of DYRK2 increases cell proliferation but reverses CAM‐DR in non‐Hodgkin's lymphoma. Int J Biol Macromol 2015; 81: 809–17. [DOI] [PubMed] [Google Scholar]
- 33. Enomoto Y, Yamashita S, Yoshinaga Y, Fukami Y, Miyahara S, Nabeshima K, et al Downregulation of DYRK2 can be a predictor of recurrence in early stage breast cancer. Tumor Biol 2014; 35: 11021–5. [DOI] [PubMed] [Google Scholar]
- 34. Nomura S, Suzuki Y, Takahashi R, Terasaki M, Kimata R, Terasaki Y, et al Dual‐specificity tyrosine phosphorylation‐regulated kinase 2 (DYRK2) as a novel marker in T1 high‐grade and T2 bladder cancer patients receiving neoadjuvant chemotherapy. BMC Urol 2015; 15(1): 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1a. Multivariate analyses of predictors of overall survival among 86 patients with colorectal cancer liver metastases.
Table S1b. Multivariate analyses of predictors of disease‐free survival among 86 patients with colorectal cancer liver metastases.