

Summary
The goal of this study was to investigate the glycosylation profile of native immunoglobulin (Ig)G present in serum immune complexes in patients with rheumatoid arthritis (RA). To accomplish this, lectin binding assays, detecting the accessibility of glycans present on IgG‐containing immune complexes by biotinylated lectins, were employed. Lectins capturing fucosyl residues (AAL), fucosylated tri‐mannose N‐glycan core sites (LCA), terminal sialic acid residues (SNA) and O‐glycosidically linked galactose/N‐acetylgalactosamine (GalNac‐L) were used. Patients with recent‐onset RA at baseline and after 3‐year follow‐up were investigated. We found that native IgG was complexed significantly more often with IgM, C1q, C3c and C‐reactive protein (CRP) in RA patients, suggesting alterations of the native structure of IgG. The total accessibility of fucose residues on captured immune complexes to the respective lectin was significantly higher in patients with RA. Moreover, fucose accessibility on IgG‐containing immune complexes correlated positively with the levels of antibodies to cyclic citrullinated peptides (anti‐CCP). We also observed a significantly higher accessibility to sialic acid residues and galactose/GalNAc glyco‐epitopes in native complexed IgG of patients with RA at baseline. While sialic acid accessibility increased during treatment, the accessibility of galactose/GalNAc decreased. Hence, successful treatment of RA was associated with an increase in the SNA/GalNAc‐L ratio. Interestingly, the SNA/GalNAc‐L ratio in particular rises after glucocorticoid treatment. In summary, this study shows the exposure of glycans in native complexed IgG of patients with early RA, revealing particular glycosylation patterns and its changes following pharmaceutical treatment.
Keywords: Aleura aurantia, GalNAc‐L, immune complex, lectin ELISA, rheumatoid arthritis, Sambucus nigra
Introduction
The impact of the glycosylation status of immunoglobulins and immunoglobulin (Ig)G‐containing immune complexes on disease activity in rheumatoid arthritis (RA) has attracted increased attention in recent years 1, 2, 3. Immune complex deposition in synovial membranes or blood vessels of skin, kidney and other organs and the accompanying inflammation and tissue damage are associated mechanistically with various autoimmune diseases, such as RA and systemic lupus erythematosus (SLE) 4, 5. The formation of immune complexes containing autoreactive antibodies is discussed to play a detrimental role in the tissue damaging processes of RA 6. Deposition of immune complexes in endothelia is thought to foster atherosclerosis and thus offers an explanation for the increased risk of patients with RA to develop cardiovascular pathology 7. Moreover, there is evidence that IgM rheumatoid factor (RF) and anti‐cyclic citrullinated peptide antibodies (anti‐CCP) are independent risk factors for ischaemic heart disease 6, 8 and increased mortality rates 9. The presence of autoreactive immunoglobulins such as RF and anti‐CCP antibodies is associated with a more severe course of RA 10, 11, 12, 13, 14. Most circulating immune complexes are captured initially by the complement receptor 1 on the surfaces of erythrocytes. Erythrocytes transport immune complexes to the liver or the spleen, where they are phagocytosed by macrophages and thus cleared from the bloodstream 15.
The pathogenicity of autoantibodies is essentially influenced by their glycosylation pattern 16, 17, 18. As depicted in Supporting information, Fig. S1, several different modifications of the glycan structure are associated with native IgG complexes: (1) the canonical glycans attached to the CH2‐domains 14, 19, 20, (2) the glycans attached to some VH and CH1 domains 21, 22, (3) putative sites of hyperglycosylation, (4) increased accessibility for lectin binding of the canonical glycans attached to the CH2‐domains 23 and (5) glycans attached to non‐IgG molecules of the IgG complex 24, 25, 26.
The aim of this study was to characterize the glycan exposure of IgG‐containing immune complexes in patients with recent‐onset RA. We observed significant differences in the glycan exposure on immune complexes in patients with RA compared to healthy controls. Several of the lectins displayed an altered, mainly increased, binding to the IgG complexes. Moreover, significant differences of glycan exposure between recent‐onset RA and follow‐up have been observed.
Material and methods
Patients and control subjects
We studied 30 patients (20 women, 10 men, mean age all = 57·4 years, standard deviation (s.d.) all = ±14·5; mean age women: 54·7 years, s.d. women: ±15·6; mean men: 62·7 years, s.d. men: ±10·7) with recent‐onset RA enrolled in a prospective observational cohort designated TIRA‐1 (TIRA = a Swedish acronym for ‘timely interventions in rheumatoid arthritis’) 11. Symptom duration was at least 6 weeks, but less than 12 months. At baseline (BL), all patients fulfilled the American College of Rheumatology (ACR) criteria for RA 27. Five of 30 patients (17%) had radiographic changes at BL. The patients were treated with disease‐modifying anti‐rheumatic drugs (DMARDs) and corticosteroids, as considered appropriate by the physician 11. A 28‐joint disease activity score (DAS28) was calculated for all patients 28 and functional ability was assessed by the Health Assessment Questionnaire (HAQ) 29. Serum samples were available from BL and from the 3‐year follow‐up (FU). Serum samples from 30 age‐matched healthy blood donors (15 women, 15 men, mean age all = 53·0 years, s.d. all = 8·4, P = 0·16 compared to patients) from the same geographic area served as controls. Oral and written informed consent was obtained from all subjects, and the study protocol was approved by the local ethics committee in Linköping, Sweden.
Serological analyses
Rheumatoid factors of IgM and IgA class were analysed by enzyme immunoassay (EIA) (Autozyme RF IgM and IgA, respectively; Cambridge Life Sciences, Cambridge, UK). Anti‐CCP of IgG and IgA class were analysed as described previously 11, 30.
Analysis of IgG complexes
We employed an IgG capture enzyme‐linked immunosorbent assay (ELISA) and detected the non‐IgG proteins IgA, IgM, C3c, C1q and C‐reactive protein (CRP) in IgG complexes. F(ab')2‐fragment of goat anti‐human IgG (Jackson Laboratories Immunoresearch, West Grove, PA, USA) was diluted in coating buffer (0·1 M Na2CO3,/NaHCO3, pH 9·6) to 2 µg/ml and applied onto 96‐well MaxiSorpTM microtitre plates (Nunc, Roskilde, Denmark; F96) at 4°C overnight. After each incubation, three washing steps with phosphate‐buffered saline (PBS) containing 0·05% Tween‐20 (Roth, Bonn, Germany; PBS‐Tween) followed. We used 3% bovine serum albumin (BSA; Sigma‐Aldrich, St Louis, MO, USA) in PBS‐Tween at 37°C for 2 h as blocking step. Subsequently, the plates were incubated with sera from patients with RA or from normal healthy donors (NHD) (1 : 1000 in PBS‐Tween) at 37°C for 2 h. Saturating binding of IgG complexes to the plates was achieved (Supporting information, Fig. S2). For the detection of IgG and non‐IgG components of IgG complexes we employed horseradish peroxidase (HRP)‐labelled antibodies against human IgG (Southern Biotech), human IgA (Southern Biotech), human IgM (Southern Biotech, Birmingham, AL, USA), human C1q (Abcam, Cambridge, MA, USA), human C3c (Abcam) or human C‐reactive protein (CRP) (Abcam). The detection antibodies were diluted in PBS‐Tween with final concentrations as recommended by the manufacturer and incubated at 37°C for 1 h. The colour reaction was started by the addition of substrate solution (0·1 M Na2HPO4, 0·05 M citrate acid monohydrate, 0.02% H2O2 and 100 µg/ml tetra‐methyl benzidine, pH = 5; Merck, Kenilworth, NJ, USA). After 10 min the reaction was stopped with 25% sulphuric acid (Merck). Optical density values were measured by a plate reader (Tecan infinite F200 Pro) at 450 nm with 620 nm reference wavelength. Samples were measured in triplicates on three different plates and the values were normalized by a mean plate absorbance factor.
Detection of lectin binding sites on IgG complexes
To detect specific glycan structures exposed on IgG complexes we conducted IgG capture lectin ELISA immobilizing IgG complexes and detecting glycan accessibility using the lectins AAL pecific for fucosyl residues, fucosylated tri‐mannose N‐glycan cores, and sialic acid, respectively 31, 32, 33. By using a N‐acetylgalactosamine‐lectin (GalNAc‐L) we detected O‐glycosidically linked (β1–3) galactose and N‐acetylgalactosamine 34. The coating of the plates with anti‐human IgG F(ab')2‐fragment was performed as described above. For blocking, a special blocking buffer (3% deglycosylated gelatin, 0·1% CaCl2, 0·1% MgCl2, 0.05% Tween‐20) was applied onto the plates at 37°C for 2 h, as described previously. Briefly, deglycosylation of gelatin was achieved by treatment with 1% periodic acid (Merck) for 24 h and subsequent dialysis against Tris‐buffered saline containing 0·1% CaCl2 and 0·1% MgCl2 (TBS‐Ca‐Mg) 23. After each incubation step, plates were washed three times with TBS‐Ca‐Mg and 0.05% Tween‐20. Sera from patients with RA or NHD were diluted 1 : 1000 in TBS‐Ca‐Mg‐Tween and incubated at 37°C for 2 h. Saturated binding of IgG complexes to the microtitre plates was confirmed by detection of human IgG (Supporting information, Fig. S2). To characterize the glycan exposure of captured IgG complexes we used biotin‐labelled lectins AAL (100 ng/ml), LCA (100 ng/ml), galactose/N‐acetylgalactosamine (GalNac‐L) (1·1 ng/ml) and SNA (100 ng/ml), respectively. Plates were incubated at room temperature for 1 h and then washed three times. In order to detect lectin binding, HRP‐conjugated streptavidin (Jackson Laboratories Immunoresearch) was employed, according to the manufacturer's instructions. After incubation for 1 h at room temperature, colorimetric analysis was performed as described above. The procedure of lectin‐based ELISA has been described previously in detail 23. Samples were measured in triplicate on three different plates and the values were normalized by a mean plate absorbance factor. The coefficients of variation for each lectin‐ELISA are shown in Supporting information, Fig. S3. All assays were performed under native conditions in order to obtain relevant information about the in‐vivo accessibility of ligand binding sites exposed under non‐denaturing conditions.
Statistical analysis
A two‐sample t‐test assuming unequal variance was used for the analysis of variations in IgG complex reactivity with detection antibodies and lectins. spss statistical software was used for statistical analysis of dependency of treatment and sex and for correlation analyses. Correlation between reactivity with detection antibodies or lectins and clinical and laboratory data were tested using Spearman's rank correlation test. Multiple sample correction by Bonferroni was applied to t‐tests and Spearman's test. The fourfold table analysis of SNA/GalNac‐L reactivity ratio was performed with the χ2 test and Yates’ correction. Differences among treatment subgroups were evaluated with the Kruskall–Wallis test with multiple sample correction by Dunn.
Results
Serum IgG derived from patients with RA was complexed with IgM‐RF, CRP and the complement proteins C1q and C3c
First, we performed sandwich ELISA catching human IgG and detected IgG‐associated proteins and glycans in the serum of 30 patients with recent‐onset RA at baseline (BL) and after 36 months (follow‐up, FU). Saturating binding of IgG complexes to the plates was achieved (Supporting information, Fig. S2). The reactivity of IgG complexes with LCA, SNA, GalNAc‐L, C3c, C1q and CRP showed no dependency on sex and age (data for age not shown) of the patients with RA, as shown in Table 1. During the 3‐year FU, IgG complex reactivity with SNA and GalNAc‐L were increased significantly (male: P = 0·008; female: P = 0·003) and decreased (male: P = 0·004; female: P = 0·014), respectively. These differences were independent of gender at each time‐point. Interestingly, only male patients presented a significant increase of reactivity with AAL at FU (P = 0·03). Expectedly, when detecting IgG‐associated IgM (equal to RF), we found significantly higher levels of IgM bound to IgG in patients compared to NHD (BL: P = 0·0007; FU: P = 0·0004; combined: P < 0·0001) (Fig. 1a). Unsurprisingly, a strong and significant correlation of IgG‐bound IgM with clinically assessed IgM‐RF analysis was found (BL: P = 0·002; FU: P < 0·0002). Similarly, we observed a correlation of IgG‐bound IgM and anti‐CCP IgG autoantibody titres (BL: P = 0·049; FU: P < 0·04) (Table 2). IgM was not the only protein bound to IgG in patients with RA. Patients also had increased reactivity for CRP complexed with IgG (baseline: P < 0·0001; FU: P < 0·0001; combined: P < 0·0001) compared to NHD. Furthermore, analysis of complement factor components revealed that C1q is bound to IgG from patients to a significantly higher extent when compared with NHD (baseline: P = 0·03; FU: P = 0·03; combined: P = 0·002) (Fig. 1b). Binding of the complement factor component C3c to IgG was also increased in RA patients (BL: P = 0·04; FU: P > 0·05; combined: P = 0·02).
Table 1.
IgG complex reactivity in ELISA (mOD) in dependency of gender and disease duration
| Male | Female | P | ||||||
|---|---|---|---|---|---|---|---|---|
| IgG bound to | BL mean ± SD | FU mean ± SD | P BL versus FU | BL mean ± SD | FU mean ± SD | P BL versus FU | BL male versus female | FU male versus female |
| IgA | 78·2 ± 12·7 | 78·0 ± 15·6 | 0·973 | 70·6 ± 17·8 | 70·3 ± 18·1 | 0·967 | 0·237 | 0·264 |
| IgM | 41·6 ± 24·1 | 43·9 ± 28·3 | 0·846 | 39·4 ± 15·7 | 37·9 ± 14·8 | 0·767 | 0·759 | 0·448 |
| CRP | 23·5 ± 10·9 | 17·1 ± 3·6 | 0·092 | 21·8 ± 5·1 | 20·7 ± 5·2 | 0·513 | 0·566 | 0·055 |
| C1q | 10·8 ± 10·2 | 8·2 ± 2·8 | 0·449 | 12·0 ± 7·8 | 10·6 ± 5·6 | 0·517 | 0·723 | 0·219 |
| C3c | 12·6 ± 11·9 | 8·6 ± 3·3 | 0·323 | 13·3 ± 8·5 | 10·2 ± 6·0 | 0·197 | 0·855 | 0·440 |
| AAL | 39·1 ± 10·3 | 42·9 ± 13·1 | 0·480 | 33·6 ± 12·4 | 32·6 ± 10·9 | 0·788 | 0·236 | 0·03 |
| LCA | 27·0 ± 24·2 | 26·6 ± 18·5 | 0·967 | 21·4 ± 25·0 | 18·8 ± 20·0 | 0·714 | 0·566 | 0·310 |
| SNA | 75·9 ± 5·4 | 95·0 ± 19·6 | 0·008 | 72·8 ± 6·6 | 88·4 ± 20·9 | 0·003 | 0·221 | 0·408 |
| GalNac‐L | 37·0 ± 7·4 | 24·9 ± 8·9 | 0·004 | 31·6 ± 11·0 | 24·5 ± 5·5 | 0·014 | 0·171 | 0·875 |
BL = baseline; FU = follow‐up; Ig = immunoglobulin; SD = standard deviation; CRP = C‐reactive protein; Ig = immunoglobulin; ELISA = enzyme‐linked immunosorbent assay; AAL = Aleuria aurantia lectin; LCA = Lens culinaris agglutinin; SNA = Sambucus nigra agglutinin; GalNAc‐L = d‐galactose, N‐acetyl galactosamine.
Figure 1.
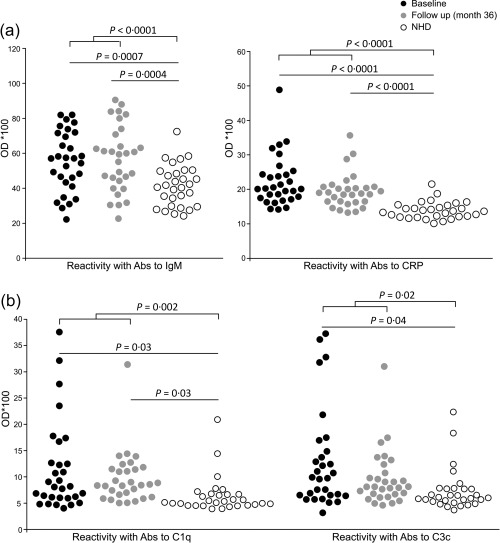
Native immunoglobulin (Ig)G complexes contain additional serum proteins detected by enzyme‐linked immunosorbent assay (ELISA). (a) Circulating native IgG complexes from patients with rheumatoid arthritis (RA) show significantly higher amounts of bound autoreactive IgM and C‐reactive protein (CRP) compared to normal healthy donors (NHD) independently of the time of assessment. (b) Binding of complement components C1q and C3c to circulating IgG immune complexes (ICs) is enhanced in patients with RA at inclusion when compared to NHD. Only the binding of C1q remained significant during the study period. Shown are optical density (OD) values assessed by capture ELISA and the significances are presented after Bonferroni correction.
Table 2.
Correlation coefficients (rho) and statistical significance (P) of reactivity of complexed immunoglobulin (Ig)G with routine laboratory and clinical parameters for baseline (BL) and follow‐up (FU) samples
| Time | α‐CCP IgA | α‐CCP IgG | IgM‐RF | IgA‐RF | ESR | CRP | DAS 28 | HAQ | |
|---|---|---|---|---|---|---|---|---|---|
| IgA | BL | −0·057 | 0·135 | 0·111 | 0·364 | −0·007 | 0·135 | 0·188 | 0·239 |
| 0·893 | > 1 | > 1 | 0·818 | > 1 | > 1 | > 1 | > 1 | ||
| FU | 0·169 | 0·134 | 0·092 | 0·442 | −0·010 | 0·122 | 0·151 | −0·041 | |
| > 1 | > 1 | > 1 | 0·246 | > 1 | > 1 | > 1 | > 1 | ||
| IgM | BL | 0·410 | 0·524 | 0·658 | 0·504 | −0·082 | −0·002 | −0·261 | −0·042 |
| 0·418 | 0·049 | 0·002 | 0·077 | > 1 | > 1 | > 1 | > 1 | ||
| FU | 0·486 | 0·534 | 0·715 | 0·423 | 0·309 | 0·236 | 0·384 | 0·273 | |
| 0·109 | 0·04 | 0·0002 | 0·336 | > 1 | > 1 | 0·617 | > 1 | ||
| CRP | BL | 0·393 | 0·349 | 0·293 | 0·072 | −0·029 | 0·100 | −0·059 | −0·363 |
| 0·535 | > 1 | > 1 | > 1 | > 1 | > 1 | > 1 | 0·822 | ||
| FU | 0·052 | 0·279 | 0·221 | 0·110 | −0·131 | 0·174 | 0·102 | 0·079 | |
| > 1 | > 1 | > 1 | > 1 | > 1 | > 1 | > 1 | > 1 | ||
| C1q | BL | 0·437 | 0·486 | 0·438 | 0·433 | −0·080 | 0·101 | −0·389 | −0·269 |
| 0·269 | 0·111 | 0·336 | 0·284 | > 1 | > 1 | 0·571 | > 1 | ||
| FU | 0·136 | 0·371 | 0·161 | 0·0002 | −0·183 | 0·131 | −0·211 | −0·123 | |
| > 1 | 0·739 | > 1 | > 1 | > 1 | > 1 | > 1 | > 1 | ||
| C3c | BL | 0·497 | 0·505 | 0·519 | 0·513 | −0·062 | 0·158 | −0·317 | −0·347 |
| 0·088 | 0·075 | 0·079 | 0·063 | > 1 | > 1 | > 1 | > 1 | ||
| FU | 0·147 | 0·294 | 0·246 | 0·016 | −0·053 | 0·070 | −0·174 | −0·042 | |
| > 1 | > 1 | > 1 | > 1 | > 1 | > 1 | > 1 | > 1 | ||
| AAL | BL | 0·629 | 0·515 | 0·339 | 0·463 | 0·205 | 0·317 | 0·116 | 0·093 |
| 0·003 | 0·062 | > 1 | 0·169 | > 1 | > 1 | > 1 | > 1 | ||
| FU | 0·478 | 0·401 | 0·454 | 0·325 | 0·395 | 0·432 | 0·460 | 0·318 | |
| 0·128 | 0·476 | 0·198 | > 1 | 0·524 | 0·289 | 0·180 | > 1 | ||
| LCA | BL | 0·583 | 0·669 | 0·548 | 0·644 | 0·344 | 0·414 | 0·119 | 0·023 |
| 0·012 | 0·001 | 0·043 | 0·002 | > 1 | 0·391 | > 1 | > 1 | ||
| FU | 0·614 | 0·624 | 0·665 | 0·619 | 0·388 | 0·388 | 0·349 | 0·129 | |
| 0·005 | 0·004 | 0·001 | 0·004 | 0·577 | 0·581 | > 1 | > 1 | ||
| SNA | BL | 0·123 | 0·019 | −0·024 | 0·264 | −0·097 | −0·036 | 0·103 | 0·315 |
| > 1 | > 1 | > 1 | > 1 | > 1 | > 1 | > 1 | > 1 | ||
| FU | 0·308 | −0·004 | −0·158 | −0·203 | 0·362 | 0·010 | 0·138 | −0·044 | |
| > 1 | > 1 | > 1 | > 1 | 0·833 | > 1 | > 1 | > 1 | ||
| GalNac‐L | BL | 0·416 | 0·027 | −0·250 | −0·025 | 0·003 | 0·051 | 0·252 | 0·029 |
| 0·378 | > 1 | > 1 | > 1 | > 1 | > 1 | > 1 | > 1 | ||
| FU | 0·183 | 0·084 | 0·145 | 0·364 | 0·192 | −0·076 | 0·161 | −0·057 | |
| > 1 | > 1 | > 1 | 0·819 | > 1 | > 1 | > 1 | > 1 |
Upper value = Spearman's correlation coefficient; lower value = P‐value; IgA‐cyclic citrullinated peptides (CCP) = IgA‐antibodies to CCP; IgG‐CCP = IgG‐antibodies to CCP, RF = rheumatoid factor; ESR = erythrocyte sedimentation rate; CRP = serum C‐reactive protein; DAS 28 = disease activity score 28 for rheumatoid arthritis; HAQ = health assessment questionnaire; AAL = Aleuria aurantia lectin; LCA = Lens culinaris agglutinin; SNA = Sambucus nigra agglutinin; GalNAc‐L = d‐galactose, N‐acetyl galactosamine. P‐values shown after Bonferroni correction (*17).
Fucosyl residues and fucosylated tri‐mannose N‐glycan core sites on IgG complexes were increased in patients with RA
Lectin binding of fucosyl residues by AAL and of fucosylated tri‐mannose N‐glycan by LCA was higher in IgG derived from RA patients than from NHD (AAL: BL: P = 0·02; FU: P = 0·01; combined: P = 0·002; LCA: BL: P = 0·03; FU: P = 0·01; combined: P = 0.001) (Fig. 2). Fucose accessibility, assessed by AAL binding, was altered significantly between male and female patients at follow‐up (P = 0·03). When correlating AAL and LCA signals to other clinical and laboratory parameters, we found a strong correlation between LCA and anti‐CCP IgG (P = 0·001) or anti‐CCP IgA antibodies (P = 0·012) as well as IgM‐RF (P = 0·043) or IgA‐RF (P = 0·002) at baseline (Table 2). AAL correlated significantly with anti‐CCP IgG levels (P = 0·003) (Table 2). Similar correlations for LCA were observed at follow‐up (Table 2). These results suggest that anti‐CCP antibodies and RF are responsible for the higher exposure of fucosylated tri‐mannose N‐glycan core sites on IgG complexes in patients with RA.
Figure 2.
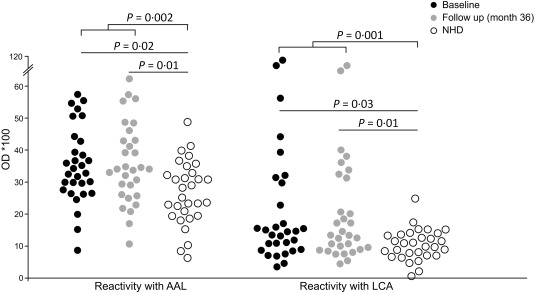
Native immunoglobulin (Ig)G complexes expose fucosyl residues and fucosylated tri‐mannose N‐glycan cores detected by lectin enzyme‐linked immunosorbent assay (ELISA). The lectins AAL and LCA show significantly higher binding to native IgG complexes from patients with RA than to those from normal healthy donors (NHD), independent of therapy. Shown are optical density (OD) values assessed by capture ELISA and the significances are presented after Bonferroni correction.
The SNA/GalNAc‐L ratio changed during anti‐rheumatic treatment
Next, we analysed binding of SNA and GalNAc‐L to IgG complexes, thereby identifying sialic acid and galactose residues, respectively. As depicted in Fig. 3, patients with RA showed a significantly higher exposure of sialic acid (BL: P < 0·0001; FU: P < 0·0001; combined: P < 0·0001) and GalNAc‐L binding sites (BL: P < 0·0001; FU: P < 0·0001; combined: P < 0·0001) on IgG complexes at baseline and follow‐up compared to NHD. Interestingly, reactivity with the SNA lectin increased significantly during treatment of RA in both genders (BL versus FU: P = 0·001). Conversely, the reactivity to GalNAc‐L decreased significantly during therapy (BL versus FU: P = 0·0003) in males and females.
Figure 3.
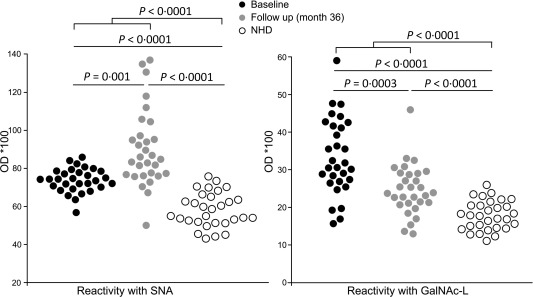
Native immunoglobulin (Ig)G complexes complexes expose sialic acids and O‐glycosidically linked (β1–3) galactose and N‐acetylgalactosamine (GalNAc‐L) detected by lectin enzyme‐linked immunosorbent assay (ELISA). Circulating native IgG complexes from patients with RA exhibit significantly increased binding of Sambucus nigra agglutinin (SNA) compared to normal healthy donors (NHD). This was increased even 36 months after start of therapy. The reactivity of native IgG complexes with GalNAc‐L compared to NHD was significantly higher for patients at inclusion and decreased significantly after 3 years of therapy. Shown are optical density (OD) values assessed by capture ELISA and the significances are presented after Bonferroni correction.
To demonstrate more clearly the changes in the exposure of sialic acid and O‐glycosidically linked (β1–3) galactose and N‐acetylgalactosamine on IgG complexes over time, we calculated the SNA/GalNAc‐L ratio (Fig. 4a,b). This ratio distinguished patients clearly before and after treatment (BL versus FU: P < 0·0001). The SNA/GalNAc‐L ratio remained either constant or increased in 26 patients with RA during therapy, whereas only four patients showed a slight decrease (BL versus FU: P < 0·0001) (Fig. 4b). Analysing associations between specific anti‐rheumatic treatment and the SNA/GalNAc‐L ratio, we found a significant increase of the SNA/GalNAc‐L ratio in patients receiving treatment with glucocorticoids (Fig. 5; P < 0·001).
Figure 4.
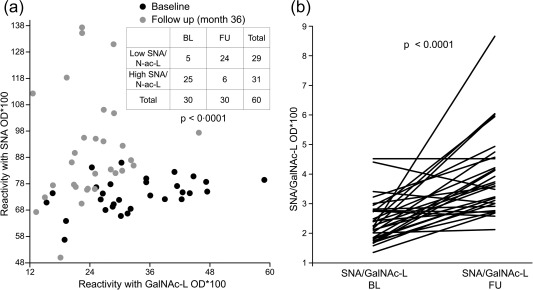
The Sambucus nigra agglutinin/d‐galactose, N‐acetyl galactosamine (SNA/GalNAc‐L) binding ratio of the native immunoglobulin (Ig)G complexes complexes changes in dependency of the therapy. (a) The SNA/GalNAc‐L reactivity ratio of native IgG complexes discriminates patients with rheumatoid arthritis (RA) before and after therapy. A contingency table analysis was performed using χ2 test and Yates’ correction. (b) The SNA/GalNAc‐L reactivity ratio increases or remains constant in course of treatment in 26 of 30 cases. Only four of 30 patients present a slight decrease in SNA/GalNAc‐L binding ratio after the inclusion visit (P < 0·0001). Shown are SNA/GalNAc‐L ratio values and P‐values presented after post‐test correction.
Figure 5.
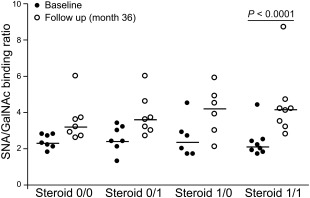
The Sambucus nigra agglutinin/d‐galactose, N‐acetyl galactosamine (SNA/GalNAc‐L) binding ratio reflects glucocorticoid therapy. The studied cohort was divided into four groups according to the period of time receiving glucocorticoid therapy. The first group did not receive glucocorticoids during the whole observation period (steroid 0/0), patients starting glucocorticoid therapy after enrolment (steroid 0/1), patients who were treated with glucocorticoids at the time of inclusion but were withdrawn later (steroid 1/0) and patients receiving glucocorticoids during the whole study period (steroid 1/1). A significant increase of the SNA/GalNAc‐L binding ratio was observed only for the patients who were treated with glucocorticoids over the entire period.
For control purposes we tested whether GalNAc‐L binding is related to IgG‐IgA complexes 35. We neither observed a correlation between IgG‐bound IgA molecules and the reactivity with GalNAc‐L nor significant differences in the amount of IgA molecules present in IgG complexes (Table 1, Supporting information, Fig. S4). Rather, therefore, the reactivity of IgG complexes with GalNAc‐L can be explained by the changes of O‐glycosidically linked (β1–3) galactose and N‐acetylgalactosamine exposure on IgG complex constituents other than IgA.
Discussion
Herein, we characterized native IgG complexes from RA patients and determined whether they are associated with other immunoglobulins such IgM or other proteins such as complement factor components C1q and C3c and the pentraxin CRP. Furthermore, we characterized the binding of lectins to native complexed IgG to test for accessible fucosyl, galactosyl and sialic acid residues. We analysed these structural features of native IgG complexes in RA patients at the start of their disease and after 3 years of treatment. Recently, we showed that patients with SLE exhibit IgG complexes with increased AAL and LCA reactivity and that AAL accessibility is associated with disease activity 23.
A complex formation of native IgG with IgM was expected to be found in RA patients representing IgM‐RF. Indeed, results on IgM complexed with IgG reflected the data obtained by standard IgM‐RF testing and also correlated with anti‐CCP IgG levels, which together with RF are associated strongly with a severe disease course in RA 36. In addition, we found complexes of IgG with C1q and C3c. This observation may reflect the constant activation of the complement system in patients with RA. Clearance of immune complexes is mediated via C3 fragments and their binding to the erythrocyte‐borne C3b/C4b receptor CR1. The circulating erythrocyte‐bound immune complexes are cleared subsequently in the liver or spleen, thus preventing immune complex deposition and tissue damage 37. Therefore, in patients with RA, circulating IgG complexes might expose enough C3b for their efficient clearance from the bloodstream, which is not the case in patients with SLE 23. Despite the role of classical complement activation in RA remaining unclear, the presence of complement cleavage products bound to circulating immune complexes suggests a certain level of complement activation in RA.
IgG also forms complexes with CRP in RA patients. Similarly, complexes consisting of antibodies against CRP have been described in lupus nephritis 38. In contrast to SLE 23, CRP itself appears to associate with IgG in RA. CRP is elevated chronically in RA and has shown to contribute to bone destruction 39. Nielen et al. reported that patients with RA have higher levels of serum CRP during the 2 years before diagnosis and that patients carrying anti‐CCP autoantibodies show slightly higher concentrations of serum CRP than patients lacking these autoantibodies 36.
Glycosylation of IgG essentially impacts its function and pathogenicity 40. Several studies demonstrated that variations in the galactosylation and sialylation status of IgG influence disease activity in RA 1, 2, 3. However, most of these studies analysed the presence of glycans on denatured and proteolytically cleaved fragments of IgG. Additionally, in these studies IgG was purified and therefore not complexed to other serum proteins, thus not reflecting the in‐vivo situation. In this study, we characterized the carbohydrate pattern of native IgG resembling the in‐vivo accessibility of glycans on IgG molecules to effector cells. We showed recently that patients with SLE exhibit IgG complexes with increased AAL and LCA reactivity and that AAL accessibility is associated with disease activity 23. Here, we show that patients with RA also exhibit stronger binding of AAL and LCA to IgG complexes than NHD. This observation suggests an increased exposure of fucosyl residues and the fucosylated tri‐mannose N‐glycan core on immune complexes in RA, which might be a result of the higher protein content of immune complexes in patients. It has been shown previously using denaturing methods that IgG heavy chains exhibit increased fucosylation 41 in RA and that fucose determines the binding to FcγRIIIA (CD16a), and hence antibody‐dependent cellular cytotoxicity (ADCC) 42, 43. As these findings were achieved using mass spectrometry or electrophoretic separation, they do not necessarily represent the in‐vivo exposure of certain glycans by immune complexes 23. Our results provide evidence that fucose is indeed exposed and may affect the interaction of immune complexes with effector cells.
We also detected a significant increase of AAL reactivity after 3 years FU between male versus female RA patients. This is particularly interesting in the light of previous findings that the FcγRIIIA‐158VV genotype, which is associated with higher IgG immune complex binding affinity, is also associated with a more severe disease course in male, but not female, RA patients 44, 45. FcγRIIIA, expressed on natural killer cells, monocytes, macrophages and neutrophils, is known to primarily bind IgG‐containing immune complexes 46. Furthermore, copy number variation (CNV) of FCGR3B, the gene encoding FcγRIIIB receptor, also expressed on neutrophils 47, has been associated with glomerulonephritis in SLE 48 and with the development of systemic autoimmunity 49. In this context, the increased accessibility of fucose residues may modify the activation modus of FcγRIIIA and FcγRIIIB receptors on effector cells such as neutrophils, monocytes and macrophages.
IgG complexes from patients with RA exhibited a significantly elevated exposure of both sialic acid (SNA) and GalNAc (GalNAc‐L) residues compared to healthy individuals. Interestingly, the exposure of sialylated sites on IgG complexes increased significantly during follow‐up. The association of increased SNA and GalNAc‐L reactivity and a clinical amelioration of the disease during the study period support the protective and anti‐inflammatory impact of immunoglobulin sialylation in autoimmunity and suggests a pathogenetic role of IgG O‐glycosylation. A protective effect of increased sialylation of autoreactive and total IgG has been described in anti‐phospholipid syndrome 16 and RA 2, 3, respectively. A recent study revealed that sialic acid residues on IgG facilitate its binding to the C‐type lectin receptor SIGN‐R1 on regulatory macrophages in a RA mouse model 50. This resulted in an up‐regulation of the inhibitory FcγRIIB on effector macrophages, thus dampening the proinflammatory actions of IgG. Furthermore, it has been shown that the production of antigen‐specific IgG rich in sialic acid is driven by the induction of tolerance to the specific antigen and that this sialylated IgG prevents proinflammatory T and B cell responses 51. Erythrocyte‐bound IgG complexes are not present in sera and therefore are not analysed in our study. Nevertheless, we suggest that an altered sialylation of IgG complexes may modify its interaction with macrophages, proinflammatory T and B cells and erythrocytes, and consequently alter the pathogenic impact of IgG immune complexes and erythrocyte clearance 52.
We also observed a significant increase in the exposure of O‐glycosidically linked (β1–3) galactose and N‐acetylgalactosamine in the serum of patients with RA. The three major components of GalNAc‐L‐binding O‐glycosidically linked oligosaccharides are N‐acetylgalactosamine, N‐acetylglucosamine and galactose. Agalactosylated IgG N‐glycans are always associated with a lack of terminal sialic acids 13. Whether this is cause or consequence of the ongoing inflammation in RA remains to be clarified. Similar information about O‐glycan variations in RA and other autoimmune diseases is scarce. Wada et al. reported that not only N‐glycans but also O‐glycans on IgG display a significant reduction in their GalNAc content in RA patients 53. Altered O‐glycosylation of IgA molecules is suspected to be an antigen for endogenous IgG in IgA nephropathy leading to immune complex formation 35. In contrast to our results, these data were generated applying denaturing conditions and, therefore, were not reflecting the in‐vivo accessibility of glycans for surface lectins of effector cells. In our assays, the increased reactivity of IgG complexes with GalNAc‐L and its decline during therapy represents the actual alterations of the exposure of GalNAc, GlcNAc and galactose on IgG O‐glycans. It has also been demonstrated that Ig O‐glycosylation protects the glycoprotein from proteolytic cleavage 54. According to this, therapy of patients with RA might lead to a reduction of the accessibility of IgG O‐glycans, making it less vulnerable to proteolytic cleavage.
We also discovered a significant increase of the SNA/GalNAc‐L ratio for patients who received glucocorticoid therapy between inclusion and follow up (P < 0·0001). Previous studies have also shown increased protein sialylation in patients treated with hydrocortisone 55. We suggest that the alteration of the IgG complex glycan in patients with RA might be related to glucocorticoid therapy. Decline of the GalNAc‐L reactivity by glucocorticoid therapy may increase the susceptibility of the IgG complex to be cleared and may therefore contribute to successful abrogation of inflammation in RA. Hence, some of the sustained anti‐inflammatory properties of glucocorticoids may be due to glycan alterations, i.e. increase in IgG sialylation and decrease in GalNAc‐L binding glycans.
In conclusion, we report that patients with RA exhibit a particular glycan composition of IgG complexes including associated non‐IgG molecules. We found that the increased exposure of fucosylated tri‐mannose N‐glycan core sites in the IgG of RA patients is related to higher autoantibody titres. We also demonstrate that native complexed IgG of RA patients shows substantial changes in the exposure of sialic acid and O‐linked galactosylic residues and that these changes fluctuate over time following pharmacotherapy. Specifically, the SNA/GalNAc‐L ratio appears to be an indicator for the efficacy of glucocorticoid therapy. Taken together, our study provides important information about the composition and accessibility of specific carbohydrate residues on native IgG complexes showing that glycans can be recognized by soluble lectins in the circulation, thereby modulating the complement cascade and activation of myeloid effector cells. This ex‐ vivo analysis of native IgG complexes complements the molecular deciphering of the glycosylation patterns of denatured IgG heavy chain fragments.
Author contributions
J. S., M. H. C. B., J. K., I. M., A. S., C. S. and S. W. planned and performed experiments, conducted data analysis and wrote the manuscript. A. K., A. S. and C. S. collected serum samples and clinical data, performed routine laboratory analysis and wrote the manuscript. C. J., R. B., G. S., M. H. and L. E. M. supervised the project, planned experiments, performed data analysis and wrote the manuscript. All authors read and approved the manuscript.
Disclosure
The authors declare that there are no conflicts of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web‐site:
Fig. S1. Composition of native immunoglobulin (Ig)G complexes and their putative glycosylation sites. This scheme shows (1) the canonical glycans attached to the IgG–CH2‐domain, (2) the glycans attached to some VH and CH1 domains, (3) putative sites of hyper‐glycosylation, (4) increased accessibility for lectin binding of the canonical glycans attached to the CH2‐domains 23 and (5) that glycans can also be exposed by IgG‐associated molecules exemplified by IgA, IgM, C1q, C3d and C‐reactive protein (CRP).
Fig. S2. Saturated binding of human immunoglobulin (Ig)G on microtitre plates. No difference in binding of IgG on the microtitre plate between patients with rheumatoid arthritis (RA) and normal healthy donors (NHD) [baseline (BL): P = 0.051; follow‐up (FU): P = 0.51; BL/FU combined: P = 0.15] was observed. This confirms saturation of the microtitre plate with IgG complexes in all groups.
Fig. S3. Coefficients of variation of lectin enzyme‐linked immunosorbent assays (ELISAs). The coefficients of variation for each ELISA were calculated from at least three replicates from each sample.
Fig. S4. The reactivities of native immunoglobulin (Ig)G complexes with IgA or d‐galactose, N‐acetyl galactosamine (GalNAc‐L) do not correlate. Non‐parametric (Spearman's) correlation analysis between the binding of IgA antibodies and the binding of GalNAc‐L to IgG immune complexes (ICs) is shown. Note: this makes it unlikely that the reactivity of the native IgG complexes (ICs) with GalNAc‐L is due to its known reactivity with IgA. Correlation coefficients are presented for each time‐point and P‐values are shown after post‐test correction.
Acknowledgements
This work was funded partially by the Volkswagen‐foundation, the Thyssen‐Stiftung, the German Research Foundation (DFG, CRC1181‐C03, KFO257, GK1660), the EU H2020‐MSCE‐RISE‐2015 project no. 690836 PANG and the County Council of Östergötland, the Swedish Society for Medical Research, the Swedish Rheumatism Association, the Swedish Society of Medicine, the Professor Nanna Svartz Foundation the Svenska Sällskapet för Medicinsk Forskning, the Svenska Läkaresällskapet and the King Gustaf V 80‐year Foundation.
The present work was performed in fulfilment of the requirements for obtaining the degree ‘Dr med.’ for Judith Stümer.
References
- 1. Van Beneden K, Coppieters K, Laroy W et al Reversible changes in serum immunoglobulin galactosylation during the immune response and treatment of inflammatory autoimmune arthritis. Ann Rheum Dis 2009; 68:1360–5. [DOI] [PubMed] [Google Scholar]
- 2. van de Geijn FE, Wuhrer M, Selman MH et al Immunoglobulin G galactosylation and sialylation are associated with pregnancy‐induced improvement of rheumatoid arthritis and the postpartum flare: results from a large prospective cohort study. Arthritis Res Ther 2009; 11:R193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kaneko Y, Nimmerjahn F, Ravetch JV. Anti‐inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science 2006; 313:670–3. [DOI] [PubMed] [Google Scholar]
- 4. Carter PM. Immune complex disease. Ann Rheum Dis 1973; 32:265–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Weissmann G. Rheumatoid arthritis and systemic lupus erythematosus as immune complex diseases. Bull NYU Hosp Jt Dis 2009; 67:251–3. [PubMed] [Google Scholar]
- 6. Edwards CJ, Syddall H, Goswami R et al The autoantibody rheumatoid factor may be an independent risk factor for ischaemic heart disease in men. Heart 2007; 93:1263–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shoenfeld Y, Gerli R, Doria A et al Accelerated atherosclerosis in autoimmune rheumatic diseases. Circulation 2005; 112:3337–47. [DOI] [PubMed] [Google Scholar]
- 8. Lopez‐Longo FJ, Oliver‐Minarro D, de la Torre I et al Association between anti‐cyclic citrullinated peptide antibodies and ischemic heart disease in patients with rheumatoid arthritis. Arthritis Rheum 2009; 61:419–24. [DOI] [PubMed] [Google Scholar]
- 9. Humphreys JH, van Nies JA, Chipping J et al Rheumatoid factor and anti‐citrullinated protein antibody positivity, but not level, are associated with increased mortality in patients with rheumatoid arthritis: results from two large independent cohorts. Arthritis Res Ther 2014; 16:483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Berglin E, Padyukov L, Sundin U et al A combination of autoantibodies to cyclic citrullinated peptide (CCP) and HLA‐DRB1 locus antigens is strongly associated with future onset of rheumatoid arthritis. Arthritis Res Ther 2004; 6:R303–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kastbom A, Strandberg G, Lindroos A, Skogh T. Anti‐CCP antibody test predicts the disease course during 3 years in early rheumatoid arthritis (the Swedish TIRA project). Ann Rheum Dis 2004; 63:1085–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nielen MM, van Schaardenburg D, Reesink HW et al Specific autoantibodies precede the symptoms of rheumatoid arthritis: a study of serial measurements in blood donors. Arthritis Rheum 2004; 50:380–6. [DOI] [PubMed] [Google Scholar]
- 13. Scherer HU, van der Woude D, Ioan‐Facsinay A et al Glycan profiling of anti‐citrullinated protein antibodies isolated from human serum and synovial fluid. Arthritis Rheum 2010; 62:1620–9. [DOI] [PubMed] [Google Scholar]
- 14. Scherer HU, Wang J, Toes RE et al Immunoglobulin 1 (IgG1) Fc‐glycosylation profiling of anti‐citrullinated peptide antibodies from human serum. Proteomics Clin Appl 2009; 3:106–15. [DOI] [PubMed] [Google Scholar]
- 15. Rojko JL, Evans MG, Price SA et al Formation, clearance, deposition, pathogenicity, and identification of biopharmaceutical‐related immune complexes: review and case studies. Toxicol Pathol 2014; 42:725–64. [DOI] [PubMed] [Google Scholar]
- 16. Fickentscher C, Magorivska I, Janko C et al The pathogenicity of anti‐beta2GP1‐IgG autoantibodies depends on Fc glycosylation. J Immunol Res 2015; 2015:638129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Imafuku Y, Yoshida H, Yamada Y. Reactivity of agalactosyl IgG with rheumatoid factor. Clin Chim Acta 2003; 334:217–23. [DOI] [PubMed] [Google Scholar]
- 18. Lin CW, Tsai MH, Li ST et al A common glycan structure on immunoglobulin G for enhancement of effector functions. Proc Natl Acad Sci USA 2015; 112:10611–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rombouts Y, Ewing E, van de Stadt LA et al Anti‐citrullinated protein antibodies acquire a pro‐inflammatory Fc glycosylation phenotype prior to the onset of rheumatoid arthritis. Ann Rheum Dis 2015; 74:234–41. [DOI] [PubMed] [Google Scholar]
- 20. Biermann MH, Griffante G, Podolska MJ et al Sweet but dangerous – the role of immunoglobulin G glycosylation in autoimmunity and inflammation. Lupus 2016; 25:934–42. [DOI] [PubMed] [Google Scholar]
- 21. Rombouts Y, Willemze A, van Beers JJ et al Extensive glycosylation of ACPA‐IgG variable domains modulates binding to citrullinated antigens in rheumatoid arthritis. Ann Rheum Dis 2016; 75:578–85. [DOI] [PubMed] [Google Scholar]
- 22. Bondt A, Wuhrer M, Kuijper TM, Hazes JM, Dolhain RJ. Fab glycosylation of immunoglobulin G does not associate with improvement of rheumatoid arthritis during pregnancy. Arthritis Res Ther 2016; 18:274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sjowall C, Zapf J, von Lohneysen S et al Altered glycosylation of complexed native IgG molecules is associated with disease activity of systemic lupus erythematosus. Lupus 2015; 24:569–81. [DOI] [PubMed] [Google Scholar]
- 24. Bondt A, Nicolardi S, Jansen BC et al Longitudinal monitoring of immunoglobulin A glycosylation during pregnancy by simultaneous MALDI‐FTICR‐MS analysis of N‐ and O‐glycopeptides. Sci Rep 2016; 6:27955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Franc V, Yang Y, Heck AJ. Proteoform profile mapping of the human serum complement component C9 revealing unexpected new features of N‐, O‐, and C‐glycosylation. Anal Chem 2017; 89:3483–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kratz EM, Borysewicz K, Katnik‐Prastowska I. Terminal monosaccharide screening of synovial immunoglobulins G and A for the early detection of rheumatoid arthritis. Rheumatol Int 2010; 30:1285–92. [DOI] [PubMed] [Google Scholar]
- 27. Arnett FC, Edworthy SM, Bloch DA et al The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum 1988; 31:315–24. [DOI] [PubMed] [Google Scholar]
- 28. Prevoo ML, van't Hof MA, Kuper HH, van Leeuwen MA, van de Putte LB, van Riel PL. Modified disease activity scores that include twenty‐eight‐joint counts. Development and validation in a prospective longitudinal study of patients with rheumatoid arthritis. Arthritis Rheum 1995; 38:44–8. [DOI] [PubMed] [Google Scholar]
- 29. Ekdahl C, Eberhardt K, Andersson SI, Svensson B. Assessing disability in patients with rheumatoid arthritis. Use of a Swedish version of the Stanford Health Assessment Questionnaire. Scand J Rheumatol 1988; 17:263–71. [DOI] [PubMed] [Google Scholar]
- 30. Svard A, Kastbom A, Reckner‐Olsson A, Skogh T. Presence and utility of IgA‐class antibodies to cyclic citrullinated peptides in early rheumatoid arthritis: the Swedish TIRA project. Arthritis Res Ther 2008; 10:R75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fujihashi M, Peapus DH, Kamiya N, Nagata Y, Miki K. Crystal structure of fucose‐specific lectin from Aleuria aurantia binding ligands at three of its five sugar recognition sites. Biochemistry 2003; 42:11093–9. [DOI] [PubMed] [Google Scholar]
- 32. Kornfeld K, Reitman ML, Kornfeld R. The carbohydrate‐binding specificity of pea and lentil lectins. Fucose is an important determinant. J Biol Chem 1981; 256:6633–40. [PubMed] [Google Scholar]
- 33. Shibuya N, Goldstein IJ, Broekaert WF, Nsimba‐Lubaki M, Peeters B, Peumans WJ. The elderberry (Sambucus nigra L.) bark lectin recognizes the Neu5Ac(alpha 2–6)Gal/GalNAc sequence. J Biol Chem 1987; 262:1596–601. [PubMed] [Google Scholar]
- 34. Kabir S, Gerwig GJ. The structural analysis of the O‐glycans of the jacalin‐bound rabbit immunoglobulin G. Biochem Mol Biol Int 1997; 42:769–78. [DOI] [PubMed] [Google Scholar]
- 35. Tomana M, Novak J, Julian BA, Matousovic K, Konecny K, Mestecky J. Circulating immune complexes in IgA nephropathy consist of IgA1 with galactose‐deficient hinge region and antiglycan antibodies. J Clin Invest 1999; 104:73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nielen MM, van Schaardenburg D, Reesink HW et al Increased levels of C‐reactive protein in serum from blood donors before the onset of rheumatoid arthritis. Arthritis Rheum 2004; 50:2423–7. [DOI] [PubMed] [Google Scholar]
- 37. Hepburn AL, Mason JC, Wang S et al Both Fcgamma and complement receptors mediate transfer of immune complexes from erythrocytes to human macrophages under physiological flow conditions in vitro . Clin Exp Immunol 2006; 146:133–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sjowall C, Olin AI, Skogh T et al C‐reactive protein, immunoglobulin G and complement co‐localize in renal immune deposits of proliferative lupus nephritis. Autoimmunity 2013; 46:205–14. [DOI] [PubMed] [Google Scholar]
- 39. Kim KW, Kim BM, Moon HW, Lee SH, Kim HR. Role of C‐reactive protein in osteoclastogenesis in rheumatoid arthritis. Arthritis Res Ther 2015; 17:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pfeifle R, Rothe T, Ipseiz N et al Regulation of autoantibody activity by the IL‐23‐TH17 axis determines the onset of autoimmune disease. Nat Immunol 2017; 18:104–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gornik I, Maravic G, Dumic J, Flogel M, Lauc G. Fucosylation of IgG heavy chains is increased in rheumatoid arthritis. Clin Biochem 1999; 32:605–8. [DOI] [PubMed] [Google Scholar]
- 42. Peipp M, Lammerts van Bueren JJ, Schneider‐Merck T et al Antibody fucosylation differentially impacts cytotoxicity mediated by NK and PMN effector cells. Blood 2008; 112:2390–9. [DOI] [PubMed] [Google Scholar]
- 43. Shields RL, Lai J, Keck R et al Lack of fucose on human IgG1 N‐linked oligosaccharide improves binding to human Fcgamma RIII and antibody‐dependent cellular toxicity. J Biol Chem 2002; 277:26733–40. [DOI] [PubMed] [Google Scholar]
- 44. Robinson JI, Barrett JH, Taylor JC et al Dissection of the FCGR3A association with RA: increased association in men and with autoantibody positive disease. Ann Rheum Dis 2010; 69:1054–7. [DOI] [PubMed] [Google Scholar]
- 45. Kastbom A, Ahmadi A, Soderkvist P, Skogh T. The 158V polymorphism of Fc gamma receptor type IIIA in early rheumatoid arthritis: increased susceptibility and severity in male patients (the Swedish TIRA project). Rheumatology (Oxford) 2005; 44:1294–8. [DOI] [PubMed] [Google Scholar]
- 46. Mayadas TN, Tsokos GC, Tsuboi N. Mechanisms of immune complex‐mediated neutrophil recruitment and tissue injury. Circulation 2009; 120:2012–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. van der Heijden J, Nagelkerke S, Zhao X et al Haplotypes of FcgammaRIIa and FcgammaRIIIb polymorphic variants influence IgG‐mediated responses in neutrophils. J Immunol 2014; 192:2715–21. [DOI] [PubMed] [Google Scholar]
- 48. Aitman TJ, Dong R, Vyse TJ et al Copy number polymorphism in Fcgr3 predisposes to glomerulonephritis in rats and humans. Nature 2006; 439:851–5. [DOI] [PubMed] [Google Scholar]
- 49. Fanciulli M, Norsworthy PJ, Petretto E et al FCGR3B copy number variation is associated with susceptibility to systemic, but not organ‐specific, autoimmunity. Nat Genet 2007; 39:721–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Collin M, Ehlers M. The carbohydrate switch between pathogenic and immunosuppressive antigen‐specific antibodies. Exp Dermatol 2013; 22:511–4. [DOI] [PubMed] [Google Scholar]
- 51. Oefner CM, Winkler A, Hess C et al Tolerance induction with T cell‐dependent protein antigens induces regulatory sialylated IgGs. J Allergy Clin Immunol 2012; 129:1647–55.e13. [DOI] [PubMed] [Google Scholar]
- 52. Shkandina T, Herrmann M, Bilyy R. Sweet kiss of dying cell: sialidase activity on apoptotic cell is able to act toward its neighbors. Autoimmunity 2012; 45:574–8. [DOI] [PubMed] [Google Scholar]
- 53. Wada Y, Tajiri M, Ohshima S. Quantitation of saccharide compositions of O‐glycans by mass spectrometry of glycopeptides and its application to rheumatoid arthritis. J Proteome Res 2010; 9:1367–73. [DOI] [PubMed] [Google Scholar]
- 54. Plomp R, Dekkers G, Rombouts Y et al Hinge‐region O‐glycosylation of human immunoglobulin G3 (IgG3). Mol Cell Proteomics 2015; 14:1373–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rouiller Y, Perilleux A, Marsaut M, Stettler M, Vesin MN, Broly H. Effect of hydrocortisone on the production and glycosylation of an Fc‐fusion protein in CHO cell cultures. Biotechnol Prog 2012; 28:803–13. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article at the publisher's web‐site:
Fig. S1. Composition of native immunoglobulin (Ig)G complexes and their putative glycosylation sites. This scheme shows (1) the canonical glycans attached to the IgG–CH2‐domain, (2) the glycans attached to some VH and CH1 domains, (3) putative sites of hyper‐glycosylation, (4) increased accessibility for lectin binding of the canonical glycans attached to the CH2‐domains 23 and (5) that glycans can also be exposed by IgG‐associated molecules exemplified by IgA, IgM, C1q, C3d and C‐reactive protein (CRP).
Fig. S2. Saturated binding of human immunoglobulin (Ig)G on microtitre plates. No difference in binding of IgG on the microtitre plate between patients with rheumatoid arthritis (RA) and normal healthy donors (NHD) [baseline (BL): P = 0.051; follow‐up (FU): P = 0.51; BL/FU combined: P = 0.15] was observed. This confirms saturation of the microtitre plate with IgG complexes in all groups.
Fig. S3. Coefficients of variation of lectin enzyme‐linked immunosorbent assays (ELISAs). The coefficients of variation for each ELISA were calculated from at least three replicates from each sample.
Fig. S4. The reactivities of native immunoglobulin (Ig)G complexes with IgA or d‐galactose, N‐acetyl galactosamine (GalNAc‐L) do not correlate. Non‐parametric (Spearman's) correlation analysis between the binding of IgA antibodies and the binding of GalNAc‐L to IgG immune complexes (ICs) is shown. Note: this makes it unlikely that the reactivity of the native IgG complexes (ICs) with GalNAc‐L is due to its known reactivity with IgA. Correlation coefficients are presented for each time‐point and P‐values are shown after post‐test correction.