

Summary
Cancer immunotherapy is an increasingly successful strategy for the treatment of patients who have advanced or conventional therapy-resistant cancers. T cells are key mediators of tumor destruction and their specificity for tumor-expressed antigens is of paramount importance, but other T cell-intrinsic qualities such as durability, longevity and functionality also play important roles in determining the efficacy of immunotherapy. The cellular energetic pathways that are utilized by T cells play a key role in regulating each of these qualities. Metabolic activity, which both regulates and is regulated by cellular signaling pathways and epigenetics, also profoundly influences the trajectories of T cell differentiation and fate. In this review, we discuss how cell metabolism influences T cell anti-tumor activity, the metabolic qualities of highly-functional T cells and strategies to modulate metabolism for improving the immune response to tumors.
Introduction
The multi-generational effort to provide better treatments for patients with cancer has resulted in clear improvements in therapeutic options and clinical outcomes. The adoption of defined chemotherapy, radiation and surgical strategies in the post-World War II years has resulted in improved patient outcomes for many types of cancer (DeVita et al., 1975; DeVita et al., 1972; Devita et al., 1970) and the advent of targeted therapies that directly inhibit oncogenic signaling pathways has also provided benefits for patients with certain cancers (Kantarjian et al., 2012). However, to date, there are very few examples of curative therapies for patients with metastatic cancer, in which cancer has disseminated throughout the body (Rosenberg, 2012; Sridhara et al., 2010). Further, treatment options are limited and often only yield short-lived responses in patients for whom frontline chemotherapeutic, radiation and surgical treatment strategies have failed to control disease (Armstrong, 2002; Oriol et al., 2010). Consequently, therapies that can successfully treat metastatic cancers or conventional treatment-resistant tumors would be of great benefit to many cancer patients.
Efforts to harness the body’s immune system to treat cancer, collectively known as cancer immunotherapy, have emerged as a powerful set of approaches to address these key needs. Many immunotherapy techniques focus on modifying the activity of T lymphocytes (T cells) to drive an anti-tumor response (Rosenberg, 2012). Encouraging results demonstrate that a number of T cell-based cancer therapies have achieved success in mediating complete, durable responses in patients with several types of metastatic cancer (Robbins et al., 2011; Rosenberg et al., 1988; Zhao et al., 2005). Further, these approaches can achieve these same responses in patients with tumors that were previously resistant to conventional treatment strategies (Maude et al., 2014; Rosenberg et al., 2011; Topalian et al., 2012).
While these clinical successes with immunotherapy approaches are promising, there are several limitations that must be considered and overcome to broaden the number of patients for whom immunotherapy can be successfully utilized. First, immunotherapy strategies are not successful in all patients. While immunotherapy can sometimes mediate complete, long-lasting responses, many patients experience partial or non-durable responses or may have no response to immunotherapy (Maude et al., 2014; Rosenberg et al., 2011; Topalian et al., 2012). Additionally, although immunotherapy can provide clinical benefit in a number of cancer types such as melanoma (Rosenberg et al., 2011; Topalian et al., 2012), acute lymphoblastic leukemia (Maude et al., 2014), synovial sarcoma (Robbins et al., 2011) and lung cancer (Brahmer et al., 2012; Topalian et al., 2012), it is not yet clear to what degree immunotherapy will benefit patients with other types of cancer. Therefore, approaches to improve and broaden the efficacy and utility of this therapy are needed.
While many factors play roles in mediating effective immune responses to cancer, including tumor-intrinsic and microenvironmental considerations, the characteristics of the T cells that are used to mount an anti-cancer immune response are likely a critical factor in determining clinical outcome (Klebanoff et al., 2012). There is mounting evidence in both human patients and in animal models suggesting that T cell replicative potential and differentiation status are important regulators of anti-tumor activity (Gattinoni et al., 2009; Klebanoff et al., 2005; Louis et al., 2011; Rosenberg et al., 2011; Zhou et al., 2005). In both settings, treatment of cancer using T cells with characteristics of heightened cellular longevity, such as increased self-renewal and replicative capacity, is associated with improved anti-tumor responses (Sukumar et al., 2017). Therefore, factors that promote these characteristics in T cells may be potential avenues through which the efficacy of immunotherapy could be improved.
The study of organismal aging and longevity has highlighted the key link between metabolic activity and longevity (Lopez-Otin et al., 2016). Cellular metabolic pathways have been shown to play key roles in regulating the fate, function (O’Neill et al., 2016) and longevity of T cells (Chang and Pearce, 2016). Additionally, the modulation of T cell metabolism has been explored as a potential therapeutic target to enhance or suppress immune responses in numerous settings, including anti-tumor immunity (O’Sullivan and Pearce, 2015). Therefore, modulating the metabolic properties of the T cells utilized in cancer immunotherapy is a promising strategy towards improving anti-tumor function and therapeutic benefit.
In this review, we discuss the key role of cellular metabolic pathways in governing the T cell response to tumor. We describe advances in the understanding of the role that T cell metabolic properties play in regulating the efficacy of the anti-tumor response. We highlight the interface between cellular metabolic pathways and anti-tumor T cell longevity and functionality against cancer, focusing on the metabolic characteristics of effective T cell responses against tumor and discussing metabolic considerations unique to the tumor microenvironment. Finally, we evaluate potential metabolic avenues through which T cell-mediated immunotherapy can be optimized for heightened clinical efficacy.
Principles of immunotherapy to treat human cancer
Cancer immunotherapy is predicated on the idea that, as a result of unique mutations or protein expression patterns, tumor cells may be recognized and eliminated by the immune system with a high degree of specificity (Lim and June, 2017; Rosenberg and Restifo, 2015). Although there are several forms of cancer immunotherapy strategies currently under study (Guillerey et al., 2016), we will limit our discussion in this review to therapies that are considered as being mediated through the action of T cells. The therapeutic strategies that seek to utilize T cells to destroy tumors can largely be divided into three categories: 1) genetic modification of T cells to confer the ability to specifically recognize cancer cells. 2) isolation of tumor-specific T cells from existing tumor masses, followed by ex vivo expansion and reinfusion into the patient. 3) treatment of patients with agents designed to activate tumor-specific T cells in situ in the tumor.
The genetic modification of T cells to impart the ability to specifically recognize and eliminate tumor cells has been achieved through transducing T cells with chimeric antigen receptors (CARs) or tumor-antigen specific T cell receptors (TCRs). A CAR is a synthetic cell surface receptor composed of an extracellular targeting element to mediate recognition of tumor-expressed surface proteins, along with signaling domains that mediate T cell function (Lim and June, 2017). Conversely, TCRs are naturally occurring T cell surface receptors that can recognize peptide fragments (or antigens) that are presented to them by the major histocompatibility complex (MHC)/human leukocyte antigen (HLA) system. TCRs within a single person are incredibly diverse and collectively have the capacity to recognize many different antigens (Nikolich-Zugich et al., 2004). In the immunotherapy setting, TCRs can recognize peptide fragments that are generated from mutated tumor proteins and may also recognize germline antigens that are sometimes aberrantly expressed in tumors (Rosenberg and Restifo, 2015). The transduction of peripheral, non-tumor specific T cells with tumor-targeting CARs or TCRs can transform these cells into potent cancer killers (Figure 1a).
Figure 1. T cell anti-tumor therapies.
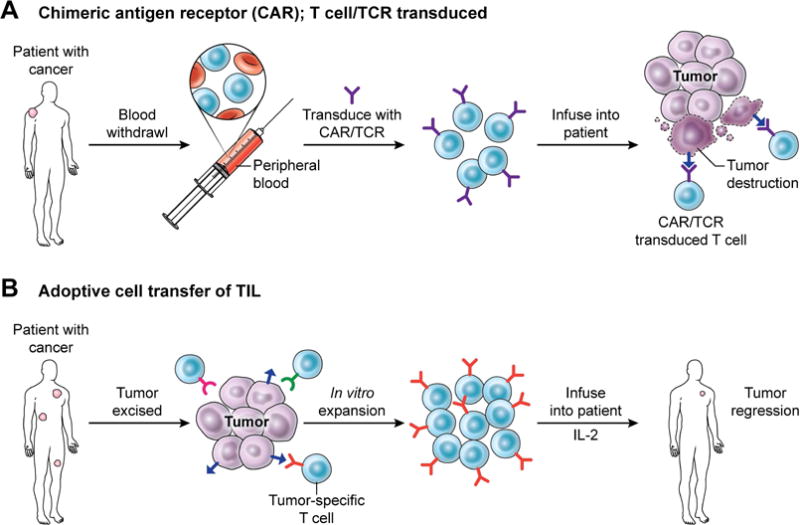
There are several clinical techniques that have been utilized to harness the immune system to treat cancer. (A) In some instances, T cells are isolated from the peripheral blood of the patient and genetically transduced to express a chimeric antigen receptor (CAR) or T cell receptor (TCR) that confers the ability to specifically recognize and destroy tumor cells when re-infused into the patient. (B) Another technique is the adoptive transfer of anti-tumor T cells that were isolated from within a patient’s tumor. Tumor-specific T cells are extracted from resected tumor samples, then expanded in vitro, followed by re-infusion into the patient and administration of the T cell growth factor IL-2.
CAR and TCR-transduced T cells have each been utilized clinically to achieve durable, complete regressions of cancer. T cells transduced with CARs recognizing CD19, a cell surface marker expressed in many B cell leukemias and lymphomas, have been shown to mediate specific destruction of cancer cells in human patients (Kochenderfer et al., 2010; Locke et al., 2017; Maude et al., 2014; Porter et al., 2011), and can yield durable responses in patients for whom frontline chemotherapy was unsuccessful (Park et al., 2016). CD19-CAR therapy is generally well tolerated by patients, but can lead to the destruction of normal cell types that express CD19, such as healthy B cells (Maude et al., 2015). Consequently, in principle, CAR therapy is restricted to targeting cell surface molecules uniquely expressed by tumor cells or those that are only expressed by dispensable normal tissues (Kakarla and Gottschalk, 2014). Due to a lack of such targets (Klebanoff et al., 2016), and perhaps a number of other factors that are incompletely elucidated (Yong et al., 2017), CAR therapy has shown extremely limited efficacy against solid tumors.
In contrast, TCR-mediated immunotherapy does not require the cell surface expression of tumor antigens and instead relies on tumor-specific mutations generating proteins that are processed and presented to T cells through the MHC/HLA system on tumor cells (Rosenberg and Restifo, 2015). A population of T cells that is capable of recognizing and mounting immune responses against cancer cells can sometimes be identified within patient tumors (Muul et al., 1987; Rosenberg et al., 1988; Rosenberg et al., 1986). These T cells likely possess TCRs that are able to recognize unique mutations expressed in tumor cells, allowing them to specifically attack malignant tissue (Robbins et al., 2013; Tran et al., 2017; van Rooij et al., 2013). In some cases, T cells have been identified that can recognize cancer-germline antigens such as MAGE-A3 and NY-ESO that can be aberrantly expressed by tumor cells and are not highly expressed in normal tissue (Scanlan et al., 2002). The identification and sequencing of TCRs that recognize these types of proteins has allowed for their transduction into patient peripheral T cells followed by expansion into large numbers and infusion into the patient. There have been clinical successes in limited trials of this therapy (Robbins et al., 2011), but the identification of TCRs that recognize epitopes generated by commonly held driver mutations in cancer such as KrasG12D may allow for future widespread applications (Tran et al., 2016; Wang et al., 2016).
A second strategy to utilize T cells in immunotherapy against tumors utilizes naturally occurring tumor-reactive T cells. This approach, known as adoptive transfer (ACT) of tumor infiltrating lymphocytes (TIL), reviewed in (Rosenberg and Restifo, 2015), involves the surgical extraction of tumor masses and the in vitro expansion of resident tumor-specific T cells. Patients are infused with expanded populations of tumor reactive T cells and dosed with high levels of interleukin 2 (IL-2) to promote anti-cancer activity (Figure 1B). This therapeutic approach has been found to mediate complete and durable responses in some patients with advanced melanoma (Rosenberg et al., 2011) and can yield durable partial responses in other solid epithelial cancer settings (Tran et al., 2016; Tran et al., 2014). Efforts are currently underway to expand the types of cancer that can be treated with adoptive transfer of TIL. In principle, any patient with a resectable tumor that contains tumor-reactive T cells may benefit from this approach.
The final type of T cell mediated immunotherapy is the restoration of immune function against cancer cells in situ. Strategies to accomplish this include the administration of cytokines such as IL-2 to promote T cell growth, survival and function (Klapper et al., 2008) and the use of immune checkpoint inhibitors that target cell surface proteins such as PD-1/PD-L1 or CTLA4 to counteract immune suppressive signals in the tumor microenvironment (Pardoll, 2012). There is also a high level of interest in utilizing vaccines against tumor antigens to drive T cell anticancer function (Carreno et al., 2015), but these strategies are yet to demonstrate clinical efficacy. Immune checkpoint inhibitor therapy currently has by far the biggest impact in the immuno-oncology sphere, and has been used to mediate complete and durable patient responses against a wide range of tumors including melanoma (Topalian et al., 2012), lung cancer (Reck et al., 2016), head and neck cancer (Ferris et al., 2016), bladder cancer (Powles et al., 2014) and even gastrointestinal tumors expressing high mutational load (such as those seen in microsatellite instability) (Le et al., 2016).
The nature of the protocols used for CAR T cell, tumor antigen specific-TCR transduced T cell and ACT therapies, in which cells are expanded or modified in an in vitro setting prior to infusion into patients, provides an opportunity for the genetic or pharmacological modulation of T cell properties (Rosenberg and Restifo, 2015) such as metabolism (Sukumar et al., 2017) to improve the functional characteristics of the cells.
Differentiation status regulates T cell lifespan
A key characteristic that determines immune functionality and longevity in T cells is the degree of differentiation of the cell. Mature T cells are categorized into two broad functional groups based on their exclusive expression of the cell surface markers CD4 or CD8 (Germain, 2002). CD4+ T cells are known as helper T cells (TH cells) and act to modulate immune responses by producing cytokines to enhance or suppress inflammation (Ziegler, 2016). CD8+ T cells are known as cytotoxic T cells (TC cells) and act to kill pathogen-infected or malignant cells by secreting inflammatory cytokines along with cell lytic molecules such as perforin and granzyme (Zhang and Bevan, 2011). Both CD4+ and CD8+ T cells can mount anti-tumor immune responses (Quezada et al., 2010).
CD4+ and CD8+ cells circulate through the body searching for antigens that stimulate their TCR, indicating the presence of infection or malignant growth (Gowans and Knight, 1964). Prior to an initial antigen encounter, T cells are referred to as naïve (TN) (Cossarizza et al., 1996). After antigen encounter, T cells are activated and undergo rapid growth and proliferation and begin to produce inflammatory cytokines and cytotoxic granules to support a sustained and robust immune response (Smith-Garvin et al., 2009). This process, known as differentiation, involves a progressive acquisition of inflammatory character and involves a host of changes to cellular functions (Gattinoni et al., 2012). Additionally, depending on the cytokine environment upon antigen encounter, T cells may further specify their lineage into a number of discrete subsets (Masopust and Schenkel, 2013) that are characterized by the expression of functionally-defining transcription factors(Zhu et al., 2010). T cells can specify their lineages into inflammatory subsets such as Th1 or Th17 for CD4+ cells (Zhu et al., 2010) or Tc1 (Sad et al., 1995) and Tc17 for CD8+ cells (Huber et al., 2009). T cells may also acquire immune-suppressive characteristics as regulatory T cells (Treg) (Smith and Kumar, 2008; Zhu et al., 2010). The differentiation and lineage subset specification of T cells drives and is driven by dramatic shifts in cellular signaling and metabolic pathways (Sukumar et al., 2017).
The differentiation state and subset specification of T cells can influence their relative longevity. CD8+ T cell differentiation states include TN, stem central memory T cells (TSCM), central memory T cells (TCM), effector memory T cells (TEM) and effector T cells (TEFF) and finally terminally differentiated TEMRA (Restifo and Gattinoni, 2013), with TN, TSCM and TCM cells exhibiting increased longevity and replicative capacity compared with TEM, TEFF and TEMRA. The transition from TN to TEMRA status is marked by a progressive change in the expression of transcription factors (Gattinoni et al., 2012), along with epigenetic and metabolic changes (Figure 2) that promote the acquisition of effector character (Crompton et al., 2016). Concurrently, increased T cell differentiation also leads to reduced telomere length (Weng et al., 1995), and the repression of transcription factors that promote self-renewal and cell longevity (Gattinoni et al., 2012). While increased differentiation status is characterized by reduced self-renewal capacity and longevity, these states are also correlated with increased immune functionality, including the increased production of inflammatory cytokines and cell lytic molecules such as granzyme B and interferon γ (Gattinoni et al., 2006). Subset specification also likely regulates T cell longevity, with Th17 and Tc17 cells exhibiting considerable self-renewal capacity, while Th1 and Tc1 cells tend to be short-lived terminal effectors (Muranski et al., 2011). Taken together, the process of cell differentiation and lineage specification has profound impacts on the self-renewal capacity and longevity of T cells.
Figure 2. Alterations in metabolic characteristics accompany T cell differentiation.
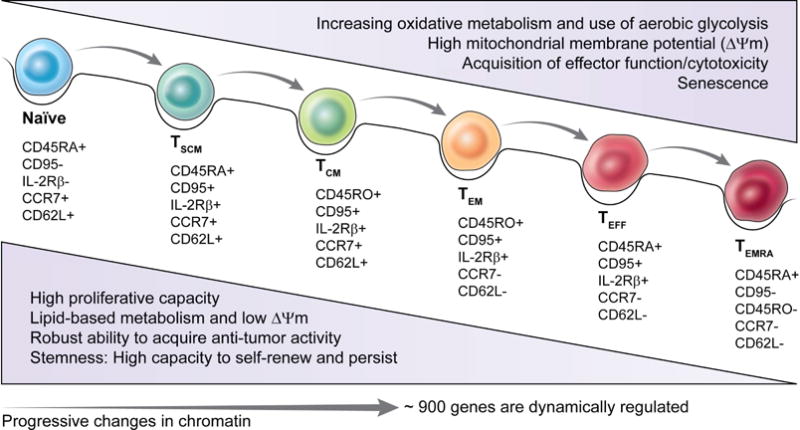
Naïve T cells, upon antigen encounter, undergo activation and differentiate into TSCM, TCM and effector T cells. This differentiation is marked by the expression of various cell surface markers and transcription factors along with profound alterations in cellular metabolic pathways. In general, T cell activation and differentiation is characterized by increased reliance on glycolysis and mitochondrial membrane potential. These metabolic pathways are critical to mediate effector function of T cells responding to infection and cancer.
T cell differentiation: Persistence is key for anti-tumor responses
T cells exhibiting characteristics of minimal differentiation and increased self-renewal capacity have proven superior to terminally differentiated effector cells in mounting immune responses against established tumors (Gattinoni et al., 2009). In human patients in the adoptive transfer of TIL setting, T cells that mount effective anti-tumor responses are characterized by increased persistence and survival in vivo (Gattinoni et al., 2011; Robbins et al., 2004). Cells with early differentiation states, including TN cells, TSCM and TCM cells are more efficacious in treating established vascularized melanoma than terminally differentiated TEM or TEFF cell subsets (Gattinoni et al., 2005). Subsets characterized by increased longevity such as Th17 cells provide superior anti-tumor activity against melanoma compared with short-lived Th1 (Muranski et al., 2008). While detailed clinical evaluations of human T cell differentiation status and anti-tumor efficacy have not been conducted in clinical trial settings, positive correlations have been found between the expression of memory cell markers along with telomere length in T cells and the degree of response against tumors (Huang et al., 2006; Huang et al., 2005; Rosenberg et al., 2011; Zhou et al., 2005). These studies indicate that T cell longevity may be an important factor in determining the efficacy of the immune response against tumor.
T cell metabolism is a dynamic aspect of immune responses
The metabolic activity of T cells is regulated, with each differentiation state and lineage subset of T cells exhibiting a unique metabolic profile (MacIver et al., 2013). The metabolic demands for T cells are not static and shift over the course of an immune response, requiring a dynamic regulation of cellular metabolic processes (Buck et al., 2015). The metabolic needs for circulating TN are low, with these cells generally utilizing low levels of mitochondrial oxidation of glucose-derived pyruvate or fatty acids to maintain cellular ATP levels (MacIver et al., 2013). However, upon encounter with antigen, T cells rapidly begin to alter their cellular signaling and metabolic pathways to prioritize proliferation and immune functionality, including the production of inflammatory cytokines and lytic molecules (O’Neill et al., 2016). This entails dramatically increased activity through anabolic signaling pathways including the PI3K/Akt/mTOR and Ras/MEK/ERK pathways to promote cell growth and replication (Buck et al., 2015). Metabolically, these activated T cells undergo a similar shift towards anabolic growth, with dramatically increased metabolic activity focused on the generation of biosynthetic intermediates such as nucleotides, proteins and membranes that are necessary for cellular growth and proliferation (MacIver et al., 2013).
In a process similar to the aerobic glycolysis (often termed the Warburg Effect) that is sometimes observed in cancer cells (Macintyre and Rathmell, 2013), activated T cells exhibit elevated rates of glycolytic activity. Also similar to what is observed in tumor cells, activated T cells also increase amino acid metabolism (Wang et al., 2011) and fatty acid synthesis (O’Neill et al., 2016). Differentiated CD8+ T cells are generally thought to become more reliant on glycolytic metabolism as they progress towards terminal differentiated effector status (Sukumar et al., 2013). Anabolic metabolic activity likely directly contributes to the inflammatory immune response, with T cell aerobic glycolysis promoting the production of inflammatory cytokines by maintaining acetyl-CoA pools substrates that are necessary for epigenetic promotion of Ifng gene expression (Peng et al., 2016). Glycolytic flux may also promote inflammation through enhancing Ifng mRNA translation into protein (Chang et al., 2013). After the antigen driving an immune response has been cleared from the body, memory T cells remain in circulation, establishing immunological memory to allow for rapid recall responses to any future encounters with the same antigen or disease (Farber et al., 2014). These memory T cells are characterized by a metabolic profile that is similar to that of naïve T cells, with a reliance on mitochondrial metabolism and fatty acid oxidation (FAO) (O’Sullivan et al., 2014).
In addition to the general metabolic program of naïve, antigen-activated and memory T cells, each lineage subset of T cells has a unique metabolic profile. In the CD4+ compartment, inflammatory effector cells such as Th1, Th2 and Th17 have been found to be glycolytic and reliant on anabolic signaling and metabolic pathways, while Treg have been characterized as utilizing an oxidative metabolism in which pyruvate and fatty acids are burned in the mitochondria (Beier et al., 2015; Gerriets et al., 2015; Liu et al., 2015; Michalek et al., 2011). Detailed studies on the metabolic programs utilized by each lineage subset of CD8+ T cells have not been conducted to date. However, there appears to be some degree of plasticity in the metabolic programs that each T cell subset utilizes. For instance, Th17 cells can extensively utilize oxidative metabolism in vivo (Franchi et al., 2017) and Treg cells can be glycolytic (Gerriets et al., 2016; Procaccini et al., 2016). It is possible that these results reflect differences between in vitro and in vivo modeling systems, indicating that further work to characterize the in vivo metabolic phenotype of T cell subsets will likely be necessary and informative.
Metabolic dysfunction in T cells can lead to the loss of proper immune function and homeostasis. For instance, imposing increased anabolic metabolism in quiescent, antigen-naïve T cells through transgenic expression of the glucose transporter Glut1 (Jacobs et al., 2008) or by genetically increasing the activity of the mTOR pathway (Pollizzi et al., 2015) results in the loss of immune quiescence and elevated production of inflammatory cytokines. Conversely, reducing the ability of activated T cells to engage anabolic metabolic programs with the genetic deletion of Glut1 (Macintyre et al., 2014), or by impairing amino acid uptake (Nakaya et al., 2014), or reducing fatty acid synthesis (Berod et al., 2014) results in compromised immune responses to antigens that are reflected by reduced T cell numbers and inflammatory cytokine production. Similarly, inhibiting pyruvate dehydrogenase kinase (PDHK), a positive regulator of aerobic glycolysis, can reduce the inflammatory capacity of Th17 cells and promote the generation and suppressive function of Treg cells (Gerriets et al., 2015). Memory and regulatory T cell functionality is also influenced by cell metabolism, as the inhibition of mitochondrial metabolism and FAO may prevent memory cell formation (van der Windt et al., 2012) and reduce Treg induction (Michalek et al., 2011) and functionality (Beier et al., 2015; Gerriets et al., 2015). Taken together, it appears that the coupling of the metabolic activity of T cells with the functional needs of each T cell lineage is an important aspect of maintaining proper immune homeostasis and functionality.
Metabolism impacts cellular signaling and epigenetics to govern longevity
In the setting of organismal longevity, it has been conclusively demonstrated that metabolic interventions can provide a therapeutic strategy to enhance lifespan (Lopez-Otin et al., 2016). Caloric restriction (CR) along with pharmacological mimetics such as metformin, are linked to increased longevity (Lin et al., 2002; Martin-Montalvo et al., 2013; Weindruch and Walford, 1982). At a cellular level, CR (and pharmacological or genetic mimetics) acts through the regulation of cellular signaling pathways that are key players in modulating the activities of cell intrinsic metabolic pathways (Figure 3) (Johnson et al., 2013). Mechanistically, CR and mimetics inhibit the activity of anabolic signaling pathways including the PI3K-Akt-mTORC1 pathway, while promoting the activity of signaling pathways such as the AMPK signaling cascade and autophagy, which serve to promote catabolic metabolism (Galluzzi et al., 2014; Marino et al., 2014). Showing the key link between the regulation of anabolic growth and longevity, the pharmacological inhibition of mTORC1 with rapamycin has been demonstrated to extend lifespan in aged mice (Harrison et al., 2009). The inhibition of anabolic cellular signaling pathways results in the suppression of pro-growth metabolic pathways including glycolysis, amino acid metabolism and fatty acid synthesis, while simultaneously promoting catabolic metabolic pathways such as FAO (Buck et al., 2015). Intriguingly, there is some evidence that metabolic pathways can directly influence organismal longevity, with glycolysis having been demonstrated to negatively regulate lifespan (Priebe et al., 2013; Schulz et al., 2007). In elegant studies, Ristow and colleagues found that increased glucose availability decreased C. elegans lifespan, while the genetic and pharmacological inhibition of glycolytic metabolism extended lifespan in an AMPK-dependent fashion (Schulz et al., 2007). Conversely, the whole-body genetic overexpression of genes involved in FAO (Paik et al., 2012) extended lifespan (Lee et al., 2012) in D. melanogaster. Organismal and cellular aging is associated with profound alterations in epigenetic modifications and consequently, changes in gene expression (Lopez-Otin et al., 2016; Sun et al., 2014). Cellular metabolic pathways may also regulate longevity by controlling the availability of substrates for epigenetic modifications such as acetyl-CoA for acetylation and methionine for methylation (Sharma and Rando, 2017). Overall, it is clear that the activities of cellular signaling and metabolic pathways play key roles in determining the biological aging of individual cells and organisms. The principles that have been elucidated in the field of aging research may hold value in considering methods to improve the T cell response to cancer.
Figure 3. Cellular signaling and metabolism interface to regulate longevity.
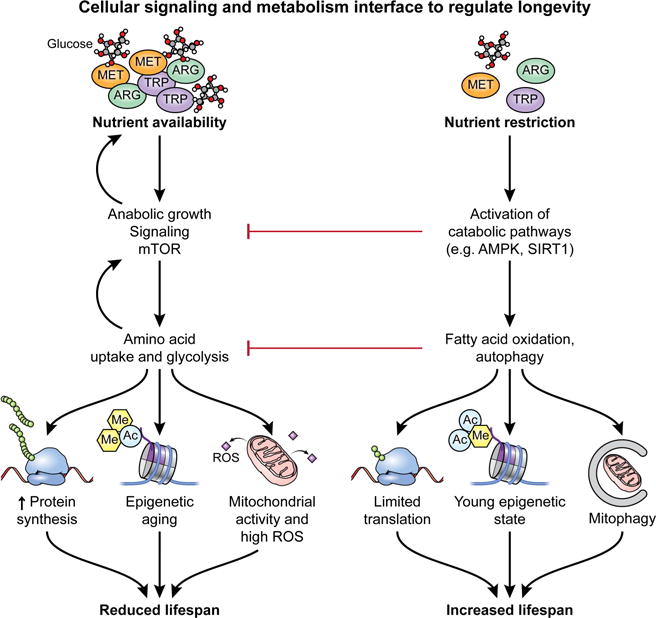
At an organismal and cellular level, the availability of key nutrients such as glucose and amino acids strongly influences longevity. Under conditions of nutrient abundance, anabolic growth signaling pathways including the PI3K-Akt-mTORC1 axis are active, resulting in a feed forward loop promoting greater uptake and usage of nutrients. This ultimately results in increased cell growth through elevated synthesis of proteins and other cellular building blocks. However, increased anabolic signaling is also associated with epigenetic aging and elevated mitochondrial activity and increased ROS production. Ultimately, these factors result in reduced lifespan. On the other hand, nutrient restriction activates catabolic signaling pathways such as AMPK or SIRT1, which in addition to actively inhibiting anabolic growth pathways, also act to promote catabolic metabolism including FAO and autophagy. Catabolic signaling and metabolism limits protein translation and biosynthesis, while promoting a young epigenetic state and increased mitochondrial turnover. Ultimately, nutrient restriction is a key promoter of increased longevity.
Metabolic activity is coupled to T cell longevity
Many of the metabolic principles that regulate cell biology are conserved across species and various cell types (Caspi et al., 2006). There is now mounting evidence that metabolic pathways, as in other cell settings, play a key role in the regulation of T cell longevity (Chang and Pearce, 2016). Increased activity in anabolic cell signaling pathways such as the PI3K-Akt-mTOR pathway, through genetic loss of negative regulators of the pathway such as PTEN (Huynh et al., 2015; Shrestha et al., 2015), tuberous sclerosis complex (TSC) proteins (Shrestha et al., 2014), AMPK (MacIver et al., 2011), or key regulators of autophagy (Wei et al., 2016) imposes increased activity in anabolic metabolic pathways in T cells. T cells with heightened mTORC1 activity are characterized by increased glycolytic metabolism, along with high levels of pentose phosphate pathway (PPP) and amino acid metabolism pathway activities (Pollizzi et al., 2016; Verbist et al., 2016). These alterations drive rapid proliferation, but can also result in decreased cell persistence (Pollizzi et al., 2016; Verbist et al., 2016) and an increased susceptibility to cell death (MacIver et al., 2011). Similarly, the catabolic autophagy process is essential for T cell memory and persistence, as genetic deletion of Atg5 or Atg7 results in impaired memory cell formation and long-term survival of CD8+ T cells (Xu et al., 2014). Conversely, the inhibition of the mTORC1 signaling pathway using the pharmacological inhibitor rapamycin has been demonstrated to increase T cell persistence in the context of infection (Araki et al., 2009; Pearce et al., 2009). Signaling pathways that drive catabolic metabolic programs, such as IL-15, which promotes mitochondrial metabolism, have been found to promote T cell longevity in vivo (van der Windt et al., 2012). Likewise, AMPK pathway activity can promote the persistence and survival of normal and malignantly-transformed T cells through regulating the activity of metabolic pathways (Blagih et al., 2015; Kishton et al., 2016). Beyond survival, AMPK signaling has been demonstrated to be required for antigen recall responses (Rolf et al., 2013). The pharmacological activation of AMPK signaling via metformin treatment has also been demonstrated to promote T cell persistence in vivo (Pearce et al., 2009).
Although cellular signaling pathways can influence many cell phenotypes beyond metabolism, direct alterations in the activities of metabolic pathways alone are sufficient to modulate the longevity of T cells. T cells that take up low levels of glucose are metabolically fit for long term survival and memory cell formation (Sukumar et al., 2013; Verbist et al., 2016). Further, inhibiting T cell glycolysis by pharmacological inhibition with 2-deoxyglucose results in increased cell lifespan and memory cell induction (Sukumar et al., 2013). Conversely, the imposition of glycolytic metabolism through genetic overexpression of Pgam1 decreases the long term survival of T cells (Sukumar et al., 2013). These results suggest that glycolysis itself, while crucial for enabling T cell inflammatory character, imposes a penalty of reduced longevity on the cell.
Altering the metabolic phenotype of long-lived memory cells also results in reduced functionality. The reduction of memory T cell FAO through pharmacological or genetic inhibition of Cpt1a or lysosomal acid lipase (LAL) results in significant reductions in T cell persistence (O’Sullivan et al., 2014), indicating that usage of mitochondrial oxidation is an important aspect of T cell longevity. Another common feature of T cells that exhibit increased longevity and persistence is increased mitochondrial spare respiratory capacity (SRC), as measured upon treatment of cells with the uncoupling agent FCCP (van der Windt et al., 2012). SRC may be reflective of the capacity of the cell to utilize FAO, but uncertainty remains in the biological meaning of this measurement (Pfleger et al., 2015). However, it is unclear whether SRC is causative of increased T cell persistence, or perhaps is a feature that is simply correlated with increased longevity. The stabilization of hypoxia inducible factor 1α (HIF1α) through the loss of the tumor suppressor Von Hippel-Lindau (VHL) results in T cells with increased in vivo persistence, along with elevated glycolysis and no increase in SRC (Phan et al., 2016). Consequently, further work is likely necessary to definitively understand the role of fatty acid oxidation and mitochondrial metabolism in the promotion of T cell longevity.
Additional layers of nuance are indicated by studies suggesting that the overall rate of cellular metabolic activity, rather than that of specific pathways, may be important in determining cell longevity. Indeed, T cells and hematopoietic stem cells (HSCs) with increased mitochondrial membrane potential, indicative of high rates of glycolysis and basal mitochondrial oxidative metabolism, have reduced persistence and self-renewal capacity when compared with cells exhibiting low mitochondrial membrane potential (Sukumar et al., 2016). The reduction of mitochondrial mass through the pharmacological promotion of mitophagy was similarly found to promote HSC self-renewal and longevity (Vannini et al., 2016). High Δψm in T cells is characterized by reduced expression of memory-associated genes that are associated with cell longevity and self-renewal potential, indicating that increased mitochondrial activity may drive T cells towards a terminally differentiated state (Sukumar et al., 2016). Interestingly, high Δψm in T cells is associated with increased reactive oxygen species (ROS) production and DNA damage (Sukumar et al., 2016). While a clear role has been established for ROS in promoting T cell response to antigens (Sena et al., 2013), it is possible that elevated mitochondrial activity also causes lifespan-reducing DNA damage. Taken together, there appears to increasing evidence that T cells (and HSCs) with high rates of metabolic activity may be characterized by reduced self-renewal capacity and longevity, while moderated metabolic activity can promote increased longevity and memory cell generation (Figure 4A).
Figure 4. Metabolism regulates T cell functionality and longevity.
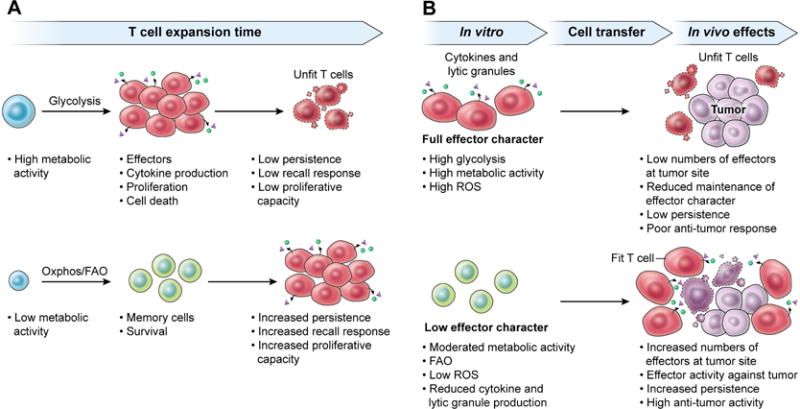
The activity of metabolic pathways in T cells exerts a profound influence on immune function and lifespan. (A) In general, T cells that exhibit high metabolic activity, including elevated glycolysis, mitochondrial activity, and anabolic growth activity are characterized by inflammatory effector character. While these cells are capable of high levels of cytokine production and rapid proliferation, they exhibit poor long-term persistence. Conversely, cells that maintain low levels of metabolic activity are favored for long lifespans and memory formation, with the ability to expand and enact robust immune responses at later times. (B) The metabolic activity of T cells that are used for adoptive transfer to mediate anti-tumor responses also plays a key role in governing the efficacy of the response. Cells that achieve full effector character, with heightened metabolic activity, during in vitro expansion exhibit poor persistence and anti-tumor efficacy upon in vivo transfer. However, cells that exhibit moderated metabolic activity during in vitro expansion mediate efficacious anti-tumor responses, characterized by increased proliferation, production of inflammatory cytokines and lytic granules and ultimately improved tumor clearance.
Two-stage metabolism for T cell longevity in immunotherapy
There has been considerable research effort to understand the metabolic properties of highly effective anti-tumor T cells, along with how metabolism can be targeted as a possible approach to improve the anti-tumor efficacy of immunotherapy (Chang and Pearce, 2016). An emerging narrative that can be found in many studies is that optimal anti-tumor T cell metabolism is bi-phasic. During the in vitro growth and priming phase of immunotherapy, in which T cells are transduced with a CAR or tumor-specific TCR or TIL are grown up to large numbers prior to infusion into the patient, the maintenance of a program of moderated metabolic activity yields improved anti-cancer activity (Sukumar et al., 2017). Conversely, at the site of the tumor in vivo, an effective anti-tumor T cell response requires robust metabolic activity to support the production of biosynthetic intermediates for cell proliferation along with the expression of key inflammatory proteins such as interferon γ and granzyme B (Figure 4B).
Moderated in vitro metabolism promotes increased anti-tumor activity in vivo
The notion that moderating metabolic activity in the in vitro setting is useful to promote cell longevity and proliferative capacity is supported by the results of several studies. Repressing the activity of the glycolytic pathway during in vitro priming and expansion of anti-tumor T cells by treating them with 2-deoxyglucose resulted in increased and sustained T cell infiltration of melanoma after infusion in a murine model, yielding improved tumor clearance (Sukumar et al., 2013). T cells that were treated with this protocol exhibited increased expression of genes encoding for glycolytic proteins at the tumor site, and were also able to produce increased levels of inflammatory cytokines in vivo (Sukumar et al., 2013). Similarly, the pharmacological inhibition of Akt, a key positive regulator of glycolytic metabolism, during in vitro T cell expansion also resulted in decreased in vitro glycolytic activity that corresponded with improved subsequent in vivo anti-tumor activity (Crompton et al., 2015). In each of these studies, the observed increase in anti-tumor activity as a consequence of inhibiting glycolysis was also correlated with increased T cell longevity and persistence in antigen-recognition models. Further supporting the principle that glycolytic metabolism may be a negative regulator of T cell longevity in the anti-tumor setting, CD19-CAR T cells expressing a CD28 co-stimulatory domain that promoted glycolysis were found to exhibit reduced in vivo persistence when compared with cells transduced with a 4-1BB co-stimulatory domain that drove oxidative metabolism (Kawalekar et al., 2016). While the anti-tumor efficacy of each construct was not evaluated in the study, anti-tumor T cell persistence has previously been shown to correlate with anti-tumor efficacy in human patients (Robbins et al., 2004). It has also been observed that the supplementation of in vitro culture medium with exogenous arginine inhibited T cell glycolytic metabolism while promoting oxidative pathways, leading to increased cell persistence and anti-tumor activity in vivo (Geiger et al., 2016). These results suggest that detailed studies on the effects of various concentrations of in vitro culture media components such as amino acids may yield novel strategies to modify anti-tumor T cell function. The modulation of T cell metabolism to promote anti-tumor activity has also been accomplished through altering the mitochondrial dynamics of the cell. Pearce and colleagues observed that the process of mitochondrial fission promotes the usage of glycolytic metabolism, while mitochondrial fusion drives mitochondrial metabolism and FAO (Buck et al., 2016). The pharmacological promotion of mitochondrial fusion in tumor-specific T cells during in vitro culturing limited glycolytic metabolism and promoted anti-tumor activity in vivo. Taken together, these studies indicate that limiting the glycolytic activity of T cells during in vitro culturing can preserve their subsequent replicative capacity and longevity to mediate a successful immune response against tumors in vivo.
Moderation of the activity of additional metabolic pathways in anti-tumor T cells can also be considered to improve in vivo efficacy. T cells that exhibit high mitochondrial activity as measured by Δψm and ROS production during in vitro expansion were found to have reduced longevity and anti-tumor functionality upon in vivo transfer (Sukumar et al., 2016). Reducing the activity of fatty acid and cholesterol biosynthesis pathways during in vitro culturing may also improve the in vivo anti-tumor activity of T cells (Yang et al., 2016). Future studies examining the role of additional metabolic pathways, such as the pentose phosphate pathway and the metabolism of additional amino acids will result in a more complete understanding of how various metabolic pathways interface with T cell longevity and anti-tumor function.
Elevated metabolic activity at the tumor site promotes T cell function
In contrast to the metabolic moderation of in vitro T cell metabolism that improves subsequent in vivo activity, efficacious T cell responses to cancer at the tumor site are characterized by high rates of metabolic activity (Sukumar et al., 2017). Glycolytic metabolism can directly contribute to the production of inflammatory cytokines such as interferon gamma through epigenetic promotion of gene transcription (Peng et al., 2016), and the elevated expression of genes that encode for proteins that promote glycolytic metabolism such as Slc2a1 (Glut1) or Pkm2 in T cells that have localized to a tumor are associated with increased cytokine production and anti-tumor response (Sukumar et al., 2013). The imposition of glycolytic metabolism in anti-tumor T cells in vivo through the genetic loss of negative regulators of HIF signaling also results in improved tumor clearance (Clever et al., 2016). Similarly, transgenic expression of Glut1 or constitutively active Akt mediated increased effector function in T cells in a murine model of ALL (Siska et al., 2016). There are also indications that elevated mitochondrial metabolic activity in T cells in vivo may promote anti-tumor activity. Consistent with the idea that ROS activity plays a key role in mediating T cell signaling after antigen recognition (Sena et al., 2013), cells that exhibit high Δψm have elevated ROS levels and increased production of inflammatory cytokines in vitro (Sukumar et al., 2016). A recent report demonstrated that the pharmacological enhancement of mitochondrial metabolic activity to increase ROS production in T cells can mediate increased T cell activity against tumors. In this study, treatment of melanoma bearing mice with mitochondrial activating chemicals synergized with checkpoint inhibitor blockade to improve T cell anti-tumor response in a ROS mediated process (Chamoto et al., 2017). Supporting the idea that elevated T cell metabolism at the tumor site is necessary for a robust immune response to cancer, tumor mediated inhibition of T cell metabolic activity via repression of the PI3K pathway by PD-L1 ligation (Chang et al., 2015; Patsoukis et al., 2015) or increased potassium in the tumor microenvironment (Eil et al., 2016) have been shown to suppress anti-tumor immunity. Likewise, reduced mitochondrial biogenesis mediated through the loss of PGC1α expression in T cells in the tumor microenvironment has been implicated as a mechanism of reduced anti-tumor activity (Scharping et al., 2016). Conversely, a recent report indicated that neo-antigen reactive T cells that are capable of mediating tumor regression in mice upon vaccination against tumor antigens or checkpoint blockade are characterized by increased metabolic activity (Gubin et al., 2014).
Taken together, these studies indicate that after infusion into the body and localization to the tumor site, an effective T cell response to cancer is characterized by robust metabolic activity, with high rates of glycolysis and mitochondrial activity to support the proliferative and immunological demands of a sustained attack on the tumor.
T cells face a hostile tumor environment with fierce competition for nutrients
However, there are several factors in the tumor microenvironment that inhibit the ability of anti-tumor T cells to fully engage in robust metabolic activity. Indeed, tumor metabolic properties along with inhibitory factors in the tumor microenvironment play an active role in repressing T cell metabolism and functionality (Siska and Rathmell, 2015). The idea of metabolic competition between T cells and tumor cells has been appreciated as a possible mechanism that may govern how effectively T cells eliminate tumor. Two studies found that tumors that are highly glycolytic can deplete glucose levels in the tumor microenvironment, dampening the ability of T cells to maintain anabolic growth signaling and produce inflammatory cytokines (Chang et al., 2015; Ho et al., 2015). Increasing T cell competitiveness in acquiring nutrients through overexpression of PCK1 (Ho et al., 2015) or by checkpoint blockade therapy targeting CTLA-4 and PD-1 (Chang et al., 2015) results in increased T cell metabolic activity, anabolic signaling and response to tumor. Interestingly, PD-L1 expression in tumor cells may promote tumor cell glycolysis in an autocrine signaling loop (Chang et al., 2015). Additional studies highlighting the importance of amino acids in promoting T cell growth and biosynthesis (Hosios et al., 2016), along with proliferation and effector character (Sinclair et al., 2013) suggest that the high rates of amino acid uptake commonly exhibited by tumor cells (Wang and Holst, 2015) could potentially also act to inhibit the anti-tumor T cell response. There is also emerging evidence that some tumors may engage in high rates of fatty acid uptake (Beloribi-Djefaflia et al., 2016). Given that effector T cells have been found to take up fatty acids at high rates (O’Sullivan et al., 2014), it is possible that this may be another environmental nutrient that T cells must compete for in the tumor microenvironment. There are also indications that metabolic alterations found in some tumors as a consequence of oncogenic mutations may also directly influence T cell function. Treatment of T cells with the “oncometabolite” 2-hydroxyglutarate (2-HG), generated at high levels by tumors expressing mutant forms of IDH (Dang et al., 2009), results in reduced effector character (Tyrakis et al., 2016). Mutant IDH tumors generate an immunosuppressive environment through high concentrations of 2-HG at the tumor site that can be overcome by the pharmacological inhibition of 2-HG production by tumor cells (Kohanbash et al., 2017). Taken together, these results suggest that the inhibition of T cell metabolism by the metabolic activity of tumor cells is an important mechanism of immunosuppression in the tumor.
Immunosuppressive, non-tumor cells in the tumor microenvironment also regulate T cell metabolism to inhibit anti-tumor function. Treg may have a metabolic advantage to survive and function in the tumor microenvironment (Angelin et al., 2017), and can act to promote the expression of indoleamine 2,3-dioxygenase (IDO) in dendritic cells, resulting in tryptophan depletion (Fallarino et al., 2003) and inhibited immune activity in anti-tumor T cells (Munn et al., 1999). Additionally, Treg cells may express CD39, allowing them to hydrolyze extracellular ATP in a process that is also known to reduce the functionality of an immune response (Borsellino et al., 2007). Additional subsets of suppressive cell types exist in the tumor microenvironment, including myeloid-derived suppressor cells (MDSCs), which can also act to reduce the availability of key amino acids including arginine (Rodriguez et al., 2004) and tryptophan (Yu et al., 2013), resulting in the suppression of anti-tumor T cell responses. Further suppressive elements can be generated by tumor-derived lactate, which can act to promote the polarization of immunosuppressive macrophages that act to promote tumor growth (Colegio et al., 2014) and can increase Treg induction under in vitro polarizing conditions (Angelin et al., 2017), indicating that tumor production of lactate is one mechanism through which the tumor microenvironment acquires immunosuppressive characteristics.
These studies lead to a model in which anti-tumor T cells are in competition for essential metabolic substrates with not only tumor cells, but also with additional suppressive immune cells subsets (Figure 5). To mount a successful and sustained immune response against tumor cells, anti-tumor T cells must be able to overcome the difficulties of a metabolically hostile environment.
Figure 5. The tumor microenvironment fosters a struggle for T cells to acquire key nutrients.
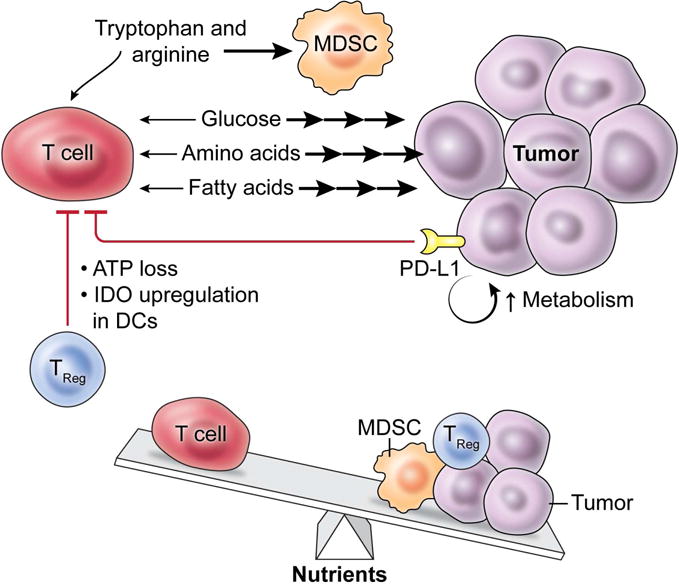
At the tumor site, several factors act in opposition to anti-tumor T cells acquiring sufficient nutrients to support immune function. Tumor cells are known to take up large amounts of key nutrients that are required for optimal T cell activity, including glucose, amino acids and fatty acids. Tumor expression of the inhibitory PD-L1 molecule acts to both directly inhibit T cell metabolism and to further promote the metabolic activity of the tumor. Anti-tumor T cells must also manage the impact of tumor-localized suppressive cell subsets such as MDSCs and Treg, which can act to deplete key nutrients such as arginine and tryptophan. Taken together, anti-tumor T cells must face and overcome a challenging metabolic microenvironment in order to successfully mount an immune response against tumors.
Strategies to optimize T cell metabolism for immunotherapy
How, then, can researchers leverage cellular metabolic pathways to improve the efficacy of tumor immunotherapy? Numerous approaches have been taken towards modulating T cell metabolism with this goal in mind. As previously discussed, the clinical protocols that are used to produce anti-tumor T cells, in which there is extensive culturing and in vitro manipulation of cells prior to infusion into the patient, provide an excellent opportunity for metabolic intervention to promote increased in vivo anti-tumor activity (Rosenberg and Restifo, 2015). Currently utilized protocols for anti-tumor T cell expansion rely on high dose IL-2 and can drive T cells towards a terminal effector differentiation status characterized by high rates of metabolic activity, particularly glycolysis. Modifications to culturing techniques may allow for an improved T cell product for infusion into patients (Figure 6). A number of studies in model organisms have indicated that the modulation of T cell metabolism during in vitro culture and expansion can improve subsequent anti-tumor efficacy in vivo. Limiting the ability of T cells to engage glycolysis during in vitro culture through direct inhibition of hexokinase by 2-DG results in improved anti-tumor efficacy against established and vascularized melanoma (Sukumar et al., 2013). Elevated Δψm is indicative of highly active mitochondria and is associated with increased mTORC1 activity and poor anti-tumor efficacy (Sukumar et al., 2016). Therefore, targeting mTORC1 with rapamycin may be one method to control Δψm to promote a more favorable mitochondrial energetic profile for improved anti-tumor response. Similarly, pharmacological modulation of mitochondrial dynamics with the small molecule Mdivi has been utilized in T cells to promote fused mitochondrial structures and inhibit mitochondrial fission during in vitro culture, resulting in improved anti-tumor activity in vivo (Buck et al., 2016). Targeting Akt with pharmacological inhibitors has also been utilized in vitro to reduce T cell glycolysis and promote mitochondrial metabolism, yielding cells that had heightened anti-tumor properties in vivo (Crompton et al., 2015). c-Myc signaling activity has previously been observed to drive anabolic growth and metabolism in activated T cells (Wang et al., 2011), and the pharmacological inhibition of c-Myc with the BRD inhibitor JQ1 during in vitro culture of T cells has been found to improve in vivo persistence and anti-cancer function (Kagoya et al., 2016). Additional approaches that have been evaluated include the activation of the Wnt signaling cascade (Gattinoni et al., 2009) and the inclusion of additional cytokines such as IL-15 (Klebanoff et al., 2004) and IL-21 (Hinrichs et al., 2008) in culturing media. These signals are known to promote catabolic metabolism (Scholz et al., 2016; van der Windt et al., 2012), and each resulted in reduced cell differentiation during ex vivo expansion and a consequent improvement in anti-tumor activity in vivo.
Figure 6. Modifications to cell metabolism can boost T cell anti-tumor function.
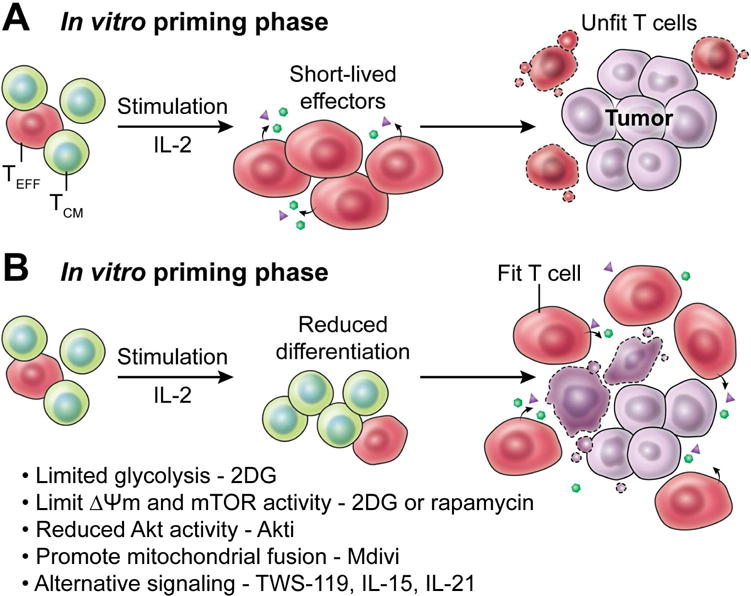
(A) The protocols currently used to prime and expand anti-tumor T cells in vitro can lead to the generation of terminally differentiated effector cells that are unable to mount a robust response in vivo at the tumor site. However, (B) alterations to in vitro expansion protocols that maintain anti-tumor T cells in a state of reduced differentiation may allow for the generation of T cells that have increased fitness to engage in a sustained immune response against tumors in vivo. Adjustments to culturing conditions that have been shown to achieve this in pre-clinical settings include the limitation of anabolic growth pathways including glycolysis, mitochondrial activity and mTORC1 signaling with the glycolytic inhibitor 2-deoxyglucose or the mTORC1 inhibitor rapamycin. Additional interventions include the pharmacological inhibition of Akt activity, the promotion of mitochondrial fusion, and the activation of alternative signaling cascades such as the Wnt pathway that act to maintain T cells in a state of reduced differentiation.
An important open question is whether metabolism can be modulated in vivo to boost anti-tumor T cell functionality. Attempts to improve anti-tumor T cell function through the modulation of metabolism in vivo have also shown some promise in pre-clinical models. For instance, genetically enhancing T cell metabolic activity in vivo through the overexpression of PCK1 (Ho et al., 2015) or Glut1 (Siska et al., 2016) may allow for improved functionality and metabolic competitiveness in the tumor microenvironment. The use of sophisticated techniques for genetic modification of primary human T cells through CRISPR technology (Su et al., 2016) may allow for genetically modifying patient T cell metabolism in the clinical setting. Additionally, strategies to target tumor PD-L1 mediated inhibition of T cell functionality may act in part through changing the metabolic balance in the tumor. Anti-PD-L1 therapy has been shown to directly inhibit tumor cell glycolytic activity, resulting in a boost to T cell metabolic competitiveness (Chang et al., 2015). Immune checkpoint inhibitor therapy may act to promote T cell anabolic metabolism directly (Patsoukis et al., 2015). The targeting of immunosuppressive macrophages and Treg in the tumor microenvironment may also allow for more robust T cell responses against tumors. Suppressive macrophages (O’Neill and Pearce, 2016) and Treg (Liu et al., 2015) each have distinguishing cellular signaling and metabolic characteristics that may potentially allow for specific inhibition of their suppressive capacity while sparing the effector function of anti-tumor T cells.
While it is important to keep in mind that the metabolic similarities between tumor cells and immune cells may result in difficulties in achieving tumor-specific metabolic inhibition (Kishton and Rathmell, 2015; Macintyre and Rathmell, 2013), it may be possible to achieve a therapeutic window where T cell function is not compromised by metabolic inhibition. Interestingly, recent work in the tumor metabolism field has indicated that, in contrast to highly glycolytic cancer cell lines, primary tumor cells may be reliant on oxidative metabolic pathways in vivo (Davidson et al., 2016; Kishton et al., 2016), highlighting that further understanding of the in vivo metabolic characteristics of both tumor and T cells is critical. There are indications that the targeting of tumor oncogenic signaling pathways may allow for the inhibition of tumor metabolic pathways while sparing or even improving T cell immune function (Baudy et al., 2012; Ho et al., 2014). However, the in vivo targeting of certain oncogenic signaling pathways that are also critical to T cell function, such as the Myc pathway, could exert profound inhibitory effects on T cells (Wang et al., 2011).
Additional insight may be gained from the field of nutritional immunology. Compelling evidence indicates that systemic nutritional states of malnutrition (Lord et al., 1998; Saucillo et al., 2014) or obesity (Yang et al., 2010) can have dramatic effects on the functional characteristics of the immune system, including T cells (Gerriets and MacIver, 2014). These findings indicate that the nutritional status of cancer patients, who can have greatly reduced nutrient intake and sometimes experience a wasting syndrome known as cachexia (Nitenberg and Raynard, 2000), may play a role in determining the efficacy of anti-tumor T cell therapies. Studies to parse out the role of systemic nutritional status in regulating the anti-tumor immune response may allow for nutritional interventions to improve T cell function.
Key challenges remain in devising appropriate strategies to use metabolic interventions to promote T cell anti-tumor function in vivo. In addition to the further work required to characterize the in vivo metabolic dependencies of tumor cells and T cells, the role of metabolic pathways in the function of key players in the tumor microenvironment such as tumor stromal and vascular cells remain to be elucidated. Studies making use of cell-specific genetic modifications, which have previously demonstrated that a competition for nutrients in the tumor environment between tumor and T cells likely plays a role in influencing immune responses to cancer(Chang et al., 2015; Ho et al., 2015), are likely to play an important role in further understanding this complex biological issue. Further, the role of many key metabolic pathways such as mitochondrial oxidation, fatty acid synthesis and oxidation, along with amino acid metabolic pathways in tumor and T cells remains largely unknown in the context of tumor immunology. Studies that broaden the understanding of these complex interactions will likely generate novel metabolic targets to enhance the immune response against cancer cells.
Summary and Conclusions
The field of tumor immunotherapy is currently rapidly growing in the clinical and research settings. While immunotherapy techniques such as CAR T cell therapy, the adoptive transfer of tumor infiltrating lymphocytes and immune checkpoint inhibitor therapies have been shown to be capable of mediating complete and durable responses in patients with a number of cancer types, further advances may increase the number of patients that can benefit from these techniques. Pre-clinical and clinical studies indicate that the longevity and durability of the T cells utilized in immunotherapy are likely to be important factors in determining the efficacy of therapy. The balance of T cell metabolic activity is a key regulator of these characteristics, and genetic and pharmacological interventions to modulate anti-tumor T cell metabolic properties have the potential to generate improved cell products for patient treatment.
Does metabolism determine the fate of T cells? Kishton et al. reviews the relationships between metabolism, aging and epigenetics in T cells, which are at the heart of new cancer immunotherapies. By manipulating key metabolic pathways, the efficacy of T cells can be improved in the setting of cancer immunotherapy.
Acknowledgments
The authors of this review were supported by the Center for Cell-Based Therapy, NCI, NIH (Bethesda, MD), NIH-Center for Regenerative Medicine, the Milstein Family Foundation and by the Intramural Research Program of the NCI (ZIA BC010763). We would like to thank Ethan Tyler and Alan Hoofring for their efforts designing and creating the illustrations used in this review. We would also like to thank the members of the Restifo laboratory for their insights into the topics of this review.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
R.J.K., M.S. and N.P.R. each developed, wrote and edited the manuscript.
References
- Angelin A, Gil-de-Gomez L, Dahiya S, Jiao J, Guo L, Levine MH, Wang Z, Quinn WJ, 3rd, Kopinski PK, Wang L, et al. Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab. 2017 doi: 10.1016/j.cmet.2016.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, Larsen CP, Ahmed R. mTOR regulates memory CD8 T-cell differentiation. Nature. 2009;460:108–112. doi: 10.1038/nature08155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong DK. Relapsed ovarian cancer: challenges and management strategies for a chronic disease. The oncologist. 2002;7(Suppl 5):20–28. doi: 10.1634/theoncologist.7-suppl_5-20. [DOI] [PubMed] [Google Scholar]
- Baudy AR, Dogan T, Flores-Mercado JE, Hoeflich KP, Su F, van Bruggen N, Williams SP. FDG-PET is a good biomarker of both early response and acquired resistance in BRAF(V600) mutant melanomas treated with vemurafenib and the MEK inhibitor GDC-0973. Ejnmmi Res. 2012;2 doi: 10.1186/2191-219X-2-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beier UH, Angelin A, Akimova T, Wang L, Liu Y, Xiao H, Koike MA, Hancock SA, Bhatti TR, Han R, et al. Essential role of mitochondrial energy metabolism in Foxp3(+) T-regulatory cell function and allograft survival. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2015;29:2315–2326. doi: 10.1096/fj.14-268409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beloribi-Djefaflia S, Vasseur S, Guillaumond F. Lipid metabolic reprogramming in cancer cells. Oncogenesis. 2016;5:e189. doi: 10.1038/oncsis.2015.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berod L, Friedrich C, Nandan A, Freitag J, Hagemann S, Harmrolfs K, Sandouk A, Hesse C, Castro CN, Bahre H, et al. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat Med. 2014;20:1327–1333. doi: 10.1038/nm.3704. [DOI] [PubMed] [Google Scholar]
- Blagih J, Coulombe F, Vincent EE, Dupuy F, Galicia-Vazquez G, Yurchenko E, Raissi TC, van der Windt GJ, Viollet B, Pearce EL, et al. The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity. 2015;42:41–54. doi: 10.1016/j.immuni.2014.12.030. [DOI] [PubMed] [Google Scholar]
- Borsellino G, Kleinewietfeld M, Di Mitri D, Sternjak A, Diamantini A, Giometto R, Hopner S, Centonze D, Bernardi G, Dell’Acqua ML, et al. Expression of ectonucleotidase CD39 by Foxp3(+) Treg cells: hydrolysis of extracellular ATP and immune suppression. Blood. 2007;110:1225–1232. doi: 10.1182/blood-2006-12-064527. [DOI] [PubMed] [Google Scholar]
- Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–2465. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck MD, O’Sullivan D, Klein Geltink RI, Curtis JD, Chang CH, Sanin DE, Qiu J, Kretz O, Braas D, van der Windt GJ, et al. Mitochondrial Dynamics Controls T Cell Fate through Metabolic Programming. Cell. 2016;166:63–76. doi: 10.1016/j.cell.2016.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck MD, O’Sullivan D, Pearce EL. T cell metabolism drives immunity. J Exp Med. 2015;212:1345–1360. doi: 10.1084/jem.20151159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, Ly A, Lie WR, Hildebrand WH, Mardis ER, et al. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science. 2015;348:803–808. doi: 10.1126/science.aaa3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi R, Foerster H, Fulcher CA, Hopkinson R, Ingraham J, Kaipa P, Krummenacker M, Paley S, Pick J, Rhee SY, et al. MetaCyc: a multiorganism database of metabolic pathways and enzymes. Nucleic acids research. 2006;34:D511–516. doi: 10.1093/nar/gkj128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamoto K, Chowdhury PS, Kumar A, Sonomura K, Matsuda F, Fagarasan S, Honjo T. Mitochondrial activation chemicals synergize with surface receptor PD-1 blockade for T cell-dependent antitumor activity. Proc Natl Acad Sci U S A. 2017;114:E761–E770. doi: 10.1073/pnas.1620433114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CH, Curtis JD, Maggi LB, Jr, Faubert B, Villarino AV, O’Sullivan D, Huang SC, van der Windt GJ, Blagih J, Qiu J, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. 2013;153:1239–1251. doi: 10.1016/j.cell.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CH, Pearce EL. Emerging concepts of T cell metabolism as a target of immunotherapy. Nat Immunol. 2016;17:364–368. doi: 10.1038/ni.3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, Chen Q, Gindin M, Gubin MM, van der Windt GJ, et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell. 2015;162:1229–1241. doi: 10.1016/j.cell.2015.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clever D, Roychoudhuri R, Constantinides MG, Askenase MH, Sukumar M, Klebanoff CA, Eil RL, Hickman HD, Yu Z, Pan JH, et al. Oxygen Sensing by T Cells Establishes an Immunologically Tolerant Metastatic Niche. Cell. 2016;166:1117–1131 e1114. doi: 10.1016/j.cell.2016.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, Cyrus N, Brokowski CE, Eisenbarth SC, Phillips GM, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. 2014;513:559–563. doi: 10.1038/nature13490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossarizza A, Ortolani C, Paganelli R, Barbieri D, Monti D, Sansoni P, Fagiolo U, Castellani G, Bersani F, Londei M, et al. CD45 isoforms expression on CD4+ and CD8+ T cells throughout life, from newborns to centenarians: implications for T cell memory. Mechanisms of ageing and development. 1996;86:173–195. doi: 10.1016/0047-6374(95)01691-0. [DOI] [PubMed] [Google Scholar]
- Crompton JG, Narayanan M, Cuddapah S, Roychoudhuri R, Ji Y, Yang W, Patel SJ, Sukumar M, Palmer DC, Peng W, et al. Lineage relationship of CD8(+) T cell subsets is revealed by progressive changes in the epigenetic landscape. Cell Mol Immunol. 2016;13:502–513. doi: 10.1038/cmi.2015.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crompton JG, Sukumar M, Roychoudhuri R, Clever D, Gros A, Eil RL, Tran E, Hanada K, Yu Z, Palmer DC, et al. Akt inhibition enhances expansion of potent tumor-specific lymphocytes with memory cell characteristics. Cancer Res. 2015;75:296–305. doi: 10.1158/0008-5472.CAN-14-2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson SM, Papagiannakopoulos T, Olenchock BA, Heyman JE, Keibler MA, Luengo A, Bauer MR, Jha AK, O’Brien JP, Pierce KA, et al. Environment Impacts the Metabolic Dependencies of Ras-Driven Non-Small Cell Lung Cancer. Cell Metab. 2016;23:517–528. doi: 10.1016/j.cmet.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVita VT, Jr, Canellos GP, Chabner B, Schein P, Hubbard SP, Young RC. Advanced diffuse histiocytic lymphoma, a potentially curable disease. Lancet. 1975;1:248–250. doi: 10.1016/s0140-6736(75)91142-3. [DOI] [PubMed] [Google Scholar]
- DeVita VT, Jr, Canellos GP, Moxley JH., 3rd A decade of combination chemotherapy of advanced Hodgkin’s disease. Cancer. 1972;30:1495–1504. doi: 10.1002/1097-0142(197212)30:6<1495::aid-cncr2820300613>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- Devita VT, Jr, Serpick AA, Carbone PP. Combination chemotherapy in the treatment of advanced Hodgkin’s disease. Annals of internal medicine. 1970;73:881–895. doi: 10.7326/0003-4819-73-6-881. [DOI] [PubMed] [Google Scholar]
- Eil R, Vodnala SK, Clever D, Klebanoff CA, Sukumar M, Pan JH, Palmer DC, Gros A, Yamamoto TN, Patel SJ, et al. Ionic immune suppression within the tumour microenvironment limits T cell effector function. Nature. 2016;537:539–543. doi: 10.1038/nature19364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallarino F, Grohmann U, Hwang KW, Orabona C, Vacca C, Bianchi R, Belladonna ML, Fioretti MC, Alegre ML, Puccetti P. Modulation of tryptophan catabolism by regulatory T cells. Nature Immunology. 2003;4:1206–1212. doi: 10.1038/ni1003. [DOI] [PubMed] [Google Scholar]
- Farber DL, Yudanin NA, Restifo NP. Human memory T cells: generation, compartmentalization and homeostasis. Nat Rev Immunol. 2014;14:24–35. doi: 10.1038/nri3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferris RL, Blumenschein G, Fayette J, Guigay J, Colevas AD, Licitra L, Harrington K, Kasper S, Vokes EE, Even C, et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. New Engl J Med. 2016;375:1856–1867. doi: 10.1056/NEJMoa1602252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franchi L, Monteleone I, Hao LY, Spahr MA, Zhao W, Liu X, Demock K, Kulkarni A, Lesch CA, Sanchez B, et al. Inhibiting Oxidative Phosphorylation In Vivo Restrains Th17 Effector Responses and Ameliorates Murine Colitis. J Immunol. 2017;198:2735–2746. doi: 10.4049/jimmunol.1600810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Pietrocola F, Levine B, Kroemer G. Metabolic control of autophagy. Cell. 2014;159:1263–1276. doi: 10.1016/j.cell.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gattinoni L, Klebanoff CA, Palmer DC, Wrzesinski C, Kerstann K, Yu Z, Finkelstein SE, Theoret MR, Rosenberg SA, Restifo NP. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J Clin Invest. 2005;115:1616–1626. doi: 10.1172/JCI24480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gattinoni L, Klebanoff CA, Restifo NP. Paths to stemness: building the ultimate antitumour T cell. Nat Rev Cancer. 2012;12:671–684. doi: 10.1038/nrc3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, Almeida JR, Gostick E, Yu Z, Carpenito C, et al. A human memory T cell subset with stem cell-like properties. Nat Med. 2011;17:1290–1297. doi: 10.1038/nm.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gattinoni L, Powell DJ, Jr, Rosenberg SA, Restifo NP. Adoptive immunotherapy for cancer: building on success. Nat Rev Immunol. 2006;6:383–393. doi: 10.1038/nri1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gattinoni L, Zhong XS, Palmer DC, Ji Y, Hinrichs CS, Yu Z, Wrzesinski C, Boni A, Cassard L, Garvin LM, et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat Med. 2009;15:808–813. doi: 10.1038/nm.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, Kogadeeva M, Picotti P, Meissner F, Mann M, et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell. 2016;167:829–842 e813. doi: 10.1016/j.cell.2016.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germain RN. T-cell development and the CD4-CD8 lineage decision. Nat Rev Immunol. 2002;2:309–322. doi: 10.1038/nri798. [DOI] [PubMed] [Google Scholar]
- Gerriets VA, Kishton RJ, Johnson MO, Cohen S, Siska PJ, Nichols AG, Warmoes MO, de Cubas AA, MacIver NJ, Locasale JW, et al. Foxp3 and Toll-like receptor signaling balance Treg cell anabolic metabolism for suppression. Nat Immunol. 2016;17:1459–1466. doi: 10.1038/ni.3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerriets VA, Kishton RJ, Nichols AG, Macintyre AN, Inoue M, Ilkayeva O, Winter PS, Liu X, Priyadharshini B, Slawinska ME, et al. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J Clin Invest. 2015;125:194–207. doi: 10.1172/JCI76012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerriets VA, MacIver NJ. Role of T cells in malnutrition and obesity. Front Immunol. 2014;5:379. doi: 10.3389/fimmu.2014.00379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowans JL, Knight EJ. The Route of Re-Circulation of Lymphocytes in the Rat. Proceedings of the Royal Society of London. Series B, Biological sciences. 1964;159:257–282. doi: 10.1098/rspb.1964.0001. [DOI] [PubMed] [Google Scholar]
- Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, Ivanova Y, Hundal J, Arthur CD, Krebber WJ, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014;515:577–581. doi: 10.1038/nature13988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillerey C, Huntington ND, Smyth MJ. Targeting natural killer cells in cancer immunotherapy. Nat Immunol. 2016;17:1025–1036. doi: 10.1038/ni.3518. [DOI] [PubMed] [Google Scholar]
- Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–395. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinrichs CS, Spolski R, Paulos CM, Gattinoni L, Kerstann KW, Palmer DC, Klebanoff CA, Rosenberg SA, Leonard WJ, Restifo NP. IL-2 and IL-21 confer opposing differentiation programs to CD8+ T cells for adoptive immunotherapy. Blood. 2008;111:5326–5333. doi: 10.1182/blood-2007-09-113050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu XJ, Amezquita R, Tsui YC, Cui GL, Micevic G, Perales JC, et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell. 2015;162:1217–1228. doi: 10.1016/j.cell.2015.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho PC, Meeth KM, Tsui YC, Srivastava B, Bosenberg MW, Kaech SM. Immune-based antitumor effects of BRAF inhibitors rely on signaling by CD40L and IFNgamma. Cancer Res. 2014;74:3205–3217. doi: 10.1158/0008-5472.CAN-13-3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosios AM, Hecht VC, Danai LV, Johnson MO, Rathmell JC, Steinhauser ML, Manalis SR, Vander Heiden MG. Amino Acids Rather than Glucose Account for the Majority of Cell Mass in Proliferating Mammalian Cells. Dev Cell. 2016;36:540–549. doi: 10.1016/j.devcel.2016.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Kerstann KW, Ahmadzadeh M, Li YF, El-Gamil M, Rosenberg SA, Robbins PF. Modulation by IL-2 of CD70 and CD27 expression on CD8+ T cells: importance for the therapeutic effectiveness of cell transfer immunotherapy. J Immunol. 2006;176:7726–7735. doi: 10.4049/jimmunol.176.12.7726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Khong HT, Dudley ME, El-Gamil M, Li YF, Rosenberg SA, Robbins PF. Survival, persistence, and progressive differentiation of adoptively transferred tumor-reactive T cells associated with tumor regression. Journal of immunotherapy. 2005;28:258–267. doi: 10.1097/01.cji.0000158855.92792.7a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber M, Heink S, Grothe H, Guralnik A, Reinhard K, Elflein K, Hunig T, Mittrucker HW, Brustle A, Kamradt T, et al. A Th17-like developmental process leads to CD8(+) Tc17 cells with reduced cytotoxic activity. Eur J Immunol. 2009;39:1716–1725. doi: 10.1002/eji.200939412. [DOI] [PubMed] [Google Scholar]
- Huynh A, DuPage M, Priyadharshini B, Sage PT, Quiros J, Borges CM, Townamchai N, Gerriets VA, Rathmell JC, Sharpe AH, et al. Control of PI(3) kinase in Treg cells maintains homeostasis and lineage stability. Nat Immunol. 2015;16:188–196. doi: 10.1038/ni.3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs SR, Herman CE, Maciver NJ, Wofford JA, Wieman HL, Hammen JJ, Rathmell JC. Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J Immunol. 2008;180:4476–4486. doi: 10.4049/jimmunol.180.7.4476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SC, Rabinovitch PS, Kaeberlein M. mTOR is a key modulator of ageing and age-related disease. Nature. 2013;493:338–345. doi: 10.1038/nature11861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagoya Y, Nakatsugawa M, Yamashita Y, Ochi T, Guo TX, Anczurowski M, Saso K, Butler MO, Arrowsmith CH, Hirano N. BET bromodomain inhibition enhances T cell persistence and function in adoptive immunotherapy models. Journal of Clinical Investigation. 2016;126:3479–3494. doi: 10.1172/JCI86437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakarla S, Gottschalk S. CAR T cells for solid tumors: armed and ready to go? Cancer journal. 2014;20:151–155. doi: 10.1097/PPO.0000000000000032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantarjian H, O’Brien S, Jabbour E, Garcia-Manero G, Quintas-Cardama A, Shan J, Rios MB, Ravandi F, Faderl S, Kadia T, et al. Improved survival in chronic myeloid leukemia since the introduction of imatinib therapy: a single-institution historical experience. Blood. 2012;119:1981–1987. doi: 10.1182/blood-2011-08-358135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawalekar OU, O’Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD, Jr, Patel PR, Guedan S, Scholler J, Keith B, et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity. 2016;44:380–390. doi: 10.1016/j.immuni.2016.01.021. [DOI] [PubMed] [Google Scholar]
- Kishton RJ, Barnes CE, Nichols AG, Cohen S, Gerriets VA, Siska PJ, Macintyre AN, Goraksha-Hicks P, de Cubas AA, Liu T, et al. AMPK Is Essential to Balance Glycolysis and Mitochondrial Metabolism to Control T-ALL Cell Stress and Survival. Cell Metab. 2016;23:649–662. doi: 10.1016/j.cmet.2016.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishton RJ, Rathmell JC. Novel therapeutic targets of tumor metabolism. Cancer journal. 2015;21:62–69. doi: 10.1097/PPO.0000000000000099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klapper JA, Downey SG, Smith FO, Yang JC, Hughes MS, Kammula US, Sherry RM, Royal RE, Steinberg SM, Rosenberg S. High-dose interleukin-2 for the treatment of metastatic renal cell carcinoma: a retrospective analysis of response and survival in patients treated in the surgery branch at the National Cancer Institute between 1986 and 2006. Cancer. 2008;113:293–301. doi: 10.1002/cncr.23552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klebanoff CA, Finkelstein SE, Surman DR, Lichtman MK, Gattinoni L, Theoret MR, Grewal N, Spiess PJ, Antony PA, Palmer DC, et al. IL-15 enhances the in vivo antitumor activity of tumor-reactive CD8+ T cells. Proc Natl Acad Sci U S A. 2004;101:1969–1974. doi: 10.1073/pnas.0307298101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klebanoff CA, Gattinoni L, Restifo NP. Sorting through subsets: which T-cell populations mediate highly effective adoptive immunotherapy? Journal of immunotherapy. 2012;35:651–660. doi: 10.1097/CJI.0b013e31827806e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klebanoff CA, Gattinoni L, Torabi-Parizi P, Kerstann K, Cardones AR, Finkelstein SE, Palmer DC, Antony PA, Hwang ST, Rosenberg SA, et al. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc Natl Acad Sci U S A. 2005;102:9571–9576. doi: 10.1073/pnas.0503726102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klebanoff CA, Rosenberg SA, Restifo NP. Prospects for gene-engineered T cell immunotherapy for solid cancers. Nat Med. 2016;22:26–36. doi: 10.1038/nm.4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, Maric I, Raffeld M, Nathan DA, Lanier BJ, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116:4099–4102. doi: 10.1182/blood-2010-04-281931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohanbash G, Carrera DA, Shrivastav S, Ahn BJ, Jahan N, Mazor T, Chheda ZS, Downey KM, Watchmaker PB, Beppler C, et al. Isocitrate dehydrogenase mutations suppress STAT1 and CD8+ T cell accumulation in gliomas. J Clin Invest. 2017;127:1425–1437. doi: 10.1172/JCI90644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le DT, Uram JN, Wang H, Kemberling H, Eyring A, Bartlett B, Goldberg RM, Crocenzi TS, Fisher GA, Lee JJ, et al. PD-1 blockade in mismatch repair deficient non-colorectal gastrointestinal cancers. Journal of Clinical Oncology. 2016;34 [Google Scholar]
- Lee SH, Lee SK, Paik D, Min KJ. Overexpression of Fatty-Acid-beta-Oxidation-Related Genes Extends the Lifespan of Drosophila melanogaster. Oxid Med Cell Longev. 2012 doi: 10.1155/2012/854502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim WA, June CH. The Principles of Engineering Immune Cells to Treat Cancer. Cell. 2017;168:724–740. doi: 10.1016/j.cell.2017.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SJ, Kaeberlein M, Andalis AA, Sturtz LA, Defossez PA, Culotta VC, Fink GR, Guarente L. Calorie restriction extends Saccharomyces cerevisiae lifespan by increasing respiration. Nature. 2002;418:344–348. doi: 10.1038/nature00829. [DOI] [PubMed] [Google Scholar]
- Liu C, Chapman NM, Karmaus PW, Zeng H, Chi H. mTOR and metabolic regulation of conventional and regulatory T cells. J Leukoc Biol. 2015;97:837–847. doi: 10.1189/jlb.2RI0814-408R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locke FL, Neelapu SS, Bartlett NL, Siddiqi T, Chavez JC, Hosing CM, Ghobadi A, Budde LE, Bot A, Rossi JM, et al. Phase 1 Results of ZUMA-1: A Multicenter Study of KTE-C19 Anti-CD19 CAR T Cell Therapy in Refractory Aggressive Lymphoma. Molecular therapy: the journal of the American Society of Gene Therapy. 2017;25:285–295. doi: 10.1016/j.ymthe.2016.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Otin C, Galluzzi L, Freije JM, Madeo F, Kroemer G. Metabolic Control of Longevity. Cell. 2016;166:802–821. doi: 10.1016/j.cell.2016.07.031. [DOI] [PubMed] [Google Scholar]
- Lord GM, Matarese G, Howard JK, Baker RJ, Bloom SR, Lechler RI. Leptin modulates the T-cell immune response and reverses starvation-induced immunosuppression. Nature. 1998;394:897–901. doi: 10.1038/29795. [DOI] [PubMed] [Google Scholar]
- Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, Rossig C, Russell HV, Diouf O, Liu E, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood. 2011;118:6050–6056. doi: 10.1182/blood-2011-05-354449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, Anderson SM, Abel ED, Chen BJ, Hale LP, et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. 2014;20:61–72. doi: 10.1016/j.cmet.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macintyre AN, Rathmell JC. Activated lymphocytes as a metabolic model for carcinogenesis. Cancer Metab. 2013;1:5. doi: 10.1186/2049-3002-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacIver NJ, Blagih J, Saucillo DC, Tonelli L, Griss T, Rathmell JC, Jones RG. The liver kinase B1 is a central regulator of T cell development, activation, and metabolism. J Immunol. 2011;187:4187–4198. doi: 10.4049/jimmunol.1100367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annual review of immunology. 2013;31:259–283. doi: 10.1146/annurev-immunol-032712-095956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino G, Pietrocola F, Eisenberg T, Kong Y, Malik SA, Andryushkova A, Schroeder S, Pendl T, Harger A, Niso-Santano M, et al. Regulation of autophagy by cytosolic acetyl-coenzyme A. Molecular cell. 2014;53:710–725. doi: 10.1016/j.molcel.2014.01.016. [DOI] [PubMed] [Google Scholar]
- Martin-Montalvo A, Mercken EM, Mitchell SJ, Palacios HH, Mote PL, Scheibye-Knudsen M, Gomes AP, Ward TM, Minor RK, Blouin MJ, et al. Metformin improves healthspan and lifespan in mice. Nat Commun. 2013;4:2192. doi: 10.1038/ncomms3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masopust D, Schenkel JM. The integration of T cell migration, differentiation and function. Nat Rev Immunol. 2013;13:309–320. doi: 10.1038/nri3442. [DOI] [PubMed] [Google Scholar]
- Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371:1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maude SL, Teachey DT, Porter DL, Grupp SA. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood. 2015;125:4017–4023. doi: 10.1182/blood-2014-12-580068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, Sullivan SA, Nichols AG, Rathmell JC. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol. 2011;186:3299–3303. doi: 10.4049/jimmunol.1003613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munn DH, Shafizadeh E, Attwood JT, Bondarev I, Pashine A, Mellor AL. Inhibition of T cell proliferation by macrophage tryptophan catabolism. Journal of Experimental Medicine. 1999;189:1363–1372. doi: 10.1084/jem.189.9.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muranski P, Boni A, Antony PA, Cassard L, Irvine KR, Kaiser A, Paulos CM, Palmer DC, Touloukian CE, Ptak K, et al. Tumor-specific Th17-polarized cells eradicate large established melanoma. Blood. 2008;112:362–373. doi: 10.1182/blood-2007-11-120998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muranski P, Borman ZA, Kerkar SP, Klebanoff CA, Ji Y, Sanchez-Perez L, Sukumar M, Reger RN, Yu Z, Kern SJ, et al. Th17 cells are long lived and retain a stem cell-like molecular signature. Immunity. 2011;35:972–985. doi: 10.1016/j.immuni.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muul LM, Spiess PJ, Director EP, Rosenberg SA. Identification of specific cytolytic immune responses against autologous tumor in humans bearing malignant melanoma. J Immunol. 1987;138:989–995. [PubMed] [Google Scholar]
- Nakaya M, Xiao Y, Zhou X, Chang JH, Chang M, Cheng X, Blonska M, Lin X, Sun SC. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity. 2014;40:692–705. doi: 10.1016/j.immuni.2014.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolich-Zugich J, Slifka MK, Messaoudi I. The many important facets of T-cell repertoire diversity. Nat Rev Immunol. 2004;4:123–132. doi: 10.1038/nri1292. [DOI] [PubMed] [Google Scholar]
- Nitenberg G, Raynard B. Nutritional support of the cancer patient: issues and dilemmas. Crit Rev Oncol Hematol. 2000;34:137–168. doi: 10.1016/s1040-8428(00)00048-2. [DOI] [PubMed] [Google Scholar]
- O’Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. 2016;16:553–565. doi: 10.1038/nri.2016.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill LA, Pearce EJ. Immunometabolism governs dendritic cell and macrophage function. J Exp Med. 2016;213:15–23. doi: 10.1084/jem.20151570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Sullivan D, Pearce EL. Targeting T cell metabolism for therapy. Trends in immunology. 2015;36:71–80. doi: 10.1016/j.it.2014.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Sullivan D, van der Windt GJ, Huang SC, Curtis JD, Chang CH, Buck MD, Qiu J, Smith AM, Lam WY, Di Plato LM, et al. Memory CD8(+) T cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity. 2014;41:75–88. doi: 10.1016/j.immuni.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oriol A, Vives S, Hernandez-Rivas JM, Tormo M, Heras I, Rivas C, Bethencourt C, Moscardo F, Bueno J, Grande C, et al. Outcome after relapse of acute lymphoblastic leukemia in adult patients included in four consecutive risk-adapted trials by the PETHEMA Study Group. Haematologica. 2010;95:589–596. doi: 10.3324/haematol.2009.014274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paik D, Jang YG, Lee YE, Lee YN, Yamamoto R, Gee HY, Yoo S, Bae E, Min KJ, Tatar M, et al. Misexpression screen delineates novel genes controlling Drosophila lifespan. Mechanisms of ageing and development. 2012;133:234–245. doi: 10.1016/j.mad.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JH, Geyer MB, Brentjens RJ. CD19-targeted CAR T-cell therapeutics for hematologic malignancies: interpreting clinical outcomes to date. Blood. 2016;127:3312–3320. doi: 10.1182/blood-2016-02-629063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, Karoly ED, Freeman GJ, Petkova V, Seth P, et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nature communications. 2015;6:6692. doi: 10.1038/ncomms7692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, Jones RG, Choi Y. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature. 2009;460:103–107. doi: 10.1038/nature08097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng M, Yin N, Chhangawala S, Xu K, Leslie CS, Li MO. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science. 2016;354:481–484. doi: 10.1126/science.aaf6284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfleger J, He M, Abdellatif M. Mitochondrial complex II is a source of the reserve respiratory capacity that is regulated by metabolic sensors and promotes cell survival. Cell death & disease. 2015;6:e1835. doi: 10.1038/cddis.2015.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan AT, Doedens AL, Palazon A, Tyrakis PA, Cheung KP, Johnson RS, Goldrath AW. Constitutive Glycolytic Metabolism Supports CD8+ T Cell Effector Memory Differentiation during Viral Infection. Immunity. 2016;45:1024–1037. doi: 10.1016/j.immuni.2016.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollizzi KN, Patel CH, Sun IH, Oh MH, Waickman AT, Wen J, Delgoffe GM, Powell JD. mTORC1 and mTORC2 selectively regulate CD8(+) T cell differentiation. J Clin Invest. 2015;125:2090–2108. doi: 10.1172/JCI77746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollizzi KN, Sun IH, Patel CH, Lo YC, Oh MH, Waickman AT, Tam AJ, Blosser RL, Wen J, Delgoffe GM, et al. Asymmetric inheritance of mTORC1 kinase activity during division dictates CD8(+) T cell differentiation. Nature immunology. 2016;17:704–711. doi: 10.1038/ni.3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powles T, Eder JP, Fine GD, Braiteh FS, Loriot Y, Cruz C, Bellmunt J, Burris HA, Petrylak DP, Teng SL, et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature. 2014;515:558–+. doi: 10.1038/nature13904. [DOI] [PubMed] [Google Scholar]
- Priebe S, Menzel U, Zarse K, Groth M, Platzer M, Ristow M, Guthke R. Extension of Life Span by Impaired Glucose Metabolism in Caenorhabditis elegans Is Accompanied by Structural Rearrangements of the Transcriptomic Network. Plos One. 2013;8 doi: 10.1371/journal.pone.0077776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Procaccini C, Carbone F, Di Silvestre D, Brambilla F, De Rosa V, Galgani M, Faicchia D, Marone G, Tramontano D, Corona M, et al. The Proteomic Landscape of Human Ex Vivo Regulatory and Conventional T Cells Reveals Specific Metabolic Requirements. Immunity. 2016;44:406–421. doi: 10.1016/j.immuni.2016.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quezada SA, Simpson TR, Peggs KS, Merghoub T, Vider J, Fan X, Blasberg R, Yagita H, Muranski P, Antony PA, et al. Tumor-reactive CD4(+) T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J Exp Med. 2010;207:637–650. doi: 10.1084/jem.20091918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reck M, Rodriguez-Abreu D, Robinson AG, Hui R, Csoszi T, Fulop A, Gottfried M, Peled N, Tafreshi A, Cuffe S, et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N Engl J Med. 2016;375:1823–1833. doi: 10.1056/NEJMoa1606774. [DOI] [PubMed] [Google Scholar]
- Restifo NP, Gattinoni L. Lineage relationship of effector and memory T cells. Current opinion in immunology. 2013;25:556–563. doi: 10.1016/j.coi.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins PF, Dudley ME, Wunderlich J, El-Gamil M, Li YF, Zhou J, Huang J, Powell DJ, Jr, Rosenberg SA. Cutting edge: persistence of transferred lymphocyte clonotypes correlates with cancer regression in patients receiving cell transfer therapy. J Immunol. 2004;173:7125–7130. doi: 10.4049/jimmunol.173.12.7125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins PF, Lu YC, El-Gamil M, Li YF, Gross C, Gartner J, Lin JC, Teer JK, Cliften P, Tycksen E, et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med. 2013;19:747–752. doi: 10.1038/nm.3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, Wunderlich JR, Nahvi AV, Helman LJ, Mackall CL, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2011;29:917–924. doi: 10.1200/JCO.2010.32.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, Delgado A, Correa P, Brayer J, Sotomayor EM, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. 2004;64:5839–5849. doi: 10.1158/0008-5472.CAN-04-0465. [DOI] [PubMed] [Google Scholar]
- Rolf J, Zarrouk M, Finlay DK, Foretz M, Viollet B, Cantrell DA. AMPK alpha 1: A glucose sensor that controls CD8 T-cell memory. European Journal of Immunology. 2013;43:889–896. doi: 10.1002/eji.201243008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg SA. Raising the bar: the curative potential of human cancer immunotherapy. Science translational medicine. 2012;4:127ps128. doi: 10.1126/scitranslmed.3003634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg SA, Packard BS, Aebersold PM, Solomon D, Topalian SL, Toy ST, Simon P, Lotze MT, Yang JC, Seipp CA, et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N Engl J Med. 1988;319:1676–1680. doi: 10.1056/NEJM198812223192527. [DOI] [PubMed] [Google Scholar]
- Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 2015;348:62–68. doi: 10.1126/science.aaa4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg SA, Spiess P, Lafreniere R. A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science. 1986;233:1318–1321. doi: 10.1126/science.3489291. [DOI] [PubMed] [Google Scholar]
- Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, Citrin DE, Restifo NP, Robbins PF, Wunderlich JR, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clinical cancer research: an official journal of the American Association for Cancer Research. 2011;17:4550–4557. doi: 10.1158/1078-0432.CCR-11-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sad S, Marcotte R, Mosmann TR. Cytokine-induced differentiation of precursor mouse CD8+ T cells into cytotoxic CD8+ T cells secreting Th1 or Th2 cytokines. Immunity. 1995;2:271–279. doi: 10.1016/1074-7613(95)90051-9. [DOI] [PubMed] [Google Scholar]
- Saucillo DC, Gerriets VA, Sheng J, Rathmell JC, Maciver NJ. Leptin metabolically licenses T cells for activation to link nutrition and immunity. J Immunol. 2014;192:136–144. doi: 10.4049/jimmunol.1301158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanlan MJ, Gure AO, Jungbluth AA, Old LJ, Chen YT. Cancer/testis antigens: an expanding family of targets for cancer immunotherapy. Immunol Rev. 2002;188:22–32. doi: 10.1034/j.1600-065x.2002.18803.x. [DOI] [PubMed] [Google Scholar]
- Scharping NE, Menk AV, Moreci RS, Whetstone RD, Dadey RE, Watkins SC, Ferris RL, Delgoffe GM. The Tumor Microenvironment Represses T Cell Mitochondrial Biogenesis to Drive Intratumoral T Cell Metabolic Insufficiency and Dysfunction. Immunity. 2016;45:374–388. doi: 10.1016/j.immuni.2016.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz G, Jandus C, Zhang L, Grandclement C, Lopez-Mejia IC, Soneson C, Delorenzi M, Fajas L, Held W, Dormond O, et al. Modulation of mTOR Signalling Triggers the Formation of Stem Cell-like Memory T Cells. EBioMedicine. 2016;4:50–61. doi: 10.1016/j.ebiom.2016.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz TJ, Zarse K, Voigt A, Urban N, Birringer M, Ristow M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 2007;6:280–293. doi: 10.1016/j.cmet.2007.08.011. [DOI] [PubMed] [Google Scholar]
- Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, Wang CR, Schumacker PT, Licht JD, Perlman H, et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity. 2013;38:225–236. doi: 10.1016/j.immuni.2012.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma U, Rando OJ. Metabolic Inputs into the Epigenome. Cell Metabolism. 2017;25:544–558. doi: 10.1016/j.cmet.2017.02.003. [DOI] [PubMed] [Google Scholar]
- Shrestha S, Yang K, Guy C, Vogel P, Neale G, Chi H. Treg cells require the phosphatase PTEN to restrain TH1 and TFH cell responses. Nat Immunol. 2015;16:178–187. doi: 10.1038/ni.3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrestha S, Yang K, Wei J, Karmaus PW, Neale G, Chi H. Tsc1 promotes the differentiation of memory CD8+ T cells via orchestrating the transcriptional and metabolic programs. Proc Natl Acad Sci U S A. 2014;111:14858–14863. doi: 10.1073/pnas.1404264111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair LV, Rolf J, Emslie E, Shi YB, Taylor PM, Cantrell DA. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat Immunol. 2013;14:500–508. doi: 10.1038/ni.2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siska PJ, Rathmell JC. T cell metabolic fitness in antitumor immunity. Trends in immunology. 2015;36:257–264. doi: 10.1016/j.it.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siska PJ, van der Windt GJ, Kishton RJ, Cohen S, Eisner W, MacIver NJ, Kater AP, Weinberg JB, Rathmell JC. Suppression of Glut1 and Glucose Metabolism by Decreased Akt/mTORC1 Signaling Drives T Cell Impairment in B Cell Leukemia. Journal of immunology. 2016;197:2532–2540. doi: 10.4049/jimmunol.1502464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TR, Kumar V. Revival of CD8+ Treg-mediated suppression. Trends in immunology. 2008;29:337–342. doi: 10.1016/j.it.2008.04.002. [DOI] [PubMed] [Google Scholar]
- Smith-Garvin JE, Koretzky GA, Jordan MS. T cell activation. Annual review of immunology. 2009;27:591–619. doi: 10.1146/annurev.immunol.021908.132706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sridhara R, Johnson JR, Justice R, Keegan P, Chakravarty A, Pazdur R. Review of oncology and hematology drug product approvals at the US Food and Drug Administration between July 2005 and December 2007. Journal of the National Cancer Institute. 2010;102:230–243. doi: 10.1093/jnci/djp515. [DOI] [PubMed] [Google Scholar]
- Su S, Hu B, Shao J, Shen B, Du J, Du Y, Zhou J, Yu L, Zhang L, Chen F, et al. CRISPR-Cas9 mediated efficient PD-1 disruption on human primary T cells from cancer patients. Sci Rep. 2016;6:20070. doi: 10.1038/srep20070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukumar M, Kishton RJ, Restifo NP. Metabolic reprograming of anti-tumor immunity. Current opinion in immunology. 2017;46:14–22. doi: 10.1016/j.coi.2017.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z, Roychoudhuri R, Palmer DC, Muranski P, Karoly ED, et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Invest. 2013;123:4479–4488. doi: 10.1172/JCI69589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukumar M, Liu J, Mehta GU, Patel SJ, Roychoudhuri R, Crompton JG, Klebanoff CA, Ji Y, Li P, Yu Z, et al. Mitochondrial Membrane Potential Identifies Cells with Enhanced Stemness for Cellular Therapy. Cell Metab. 2016;23:63–76. doi: 10.1016/j.cmet.2015.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun D, Luo M, Jeong M, Rodriguez B, Xia Z, Hannah R, Wang H, Le T, Faull KF, Chen R, et al. Epigenomic profiling of young and aged HSCs reveals concerted changes during aging that reinforce self-renewal. Cell stem cell. 2014;14:673–688. doi: 10.1016/j.stem.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran E, Robbins PF, Lu YC, Prickett TD, Gartner JJ, Jia L, Pasetto A, Zheng Z, Ray S, Groh EM, et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N Engl J Med. 2016;375:2255–2262. doi: 10.1056/NEJMoa1609279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran E, Robbins PF, Rosenberg SA. ‘Final common pathway’ of human cancer immunotherapy: targeting random somatic mutations. Nature immunology. 2017;18:255–262. doi: 10.1038/ni.3682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, Wunderlich JR, Somerville RP, Hogan K, Hinrichs CS, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science. 2014;344:641–645. doi: 10.1126/science.1251102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyrakis PA, Palazon A, Macias D, Lee KL, Phan AT, Velica P, You J, Chia GS, Sim J, Doedens A, et al. S-2-hydroxyglutarate regulates CD8+ T-lymphocyte fate. Nature. 2016;540:236–241. doi: 10.1038/nature20165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Windt GJ, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, Pearce EJ, Pearce EL. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity. 2012;36:68–78. doi: 10.1016/j.immuni.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rooij N, van Buuren MM, Philips D, Velds A, Toebes M, Heemskerk B, van Dijk LJ, Behjati S, Hilkmann H, El Atmioui D, et al. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2013;31:e439–442. doi: 10.1200/JCO.2012.47.7521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannini N, Girotra M, Naveiras O, Nikitin G, Campos V, Giger S, Roch A, Auwerx J, Lutolf MP. Specification of haematopoietic stem cell fate via modulation of mitochondrial activity. Nat Commun. 2016;7:13125. doi: 10.1038/ncomms13125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbist KC, Guy CS, Milasta S, Liedmann S, Kaminski MM, Wang R, Green DR. Metabolic maintenance of cell asymmetry following division in activated T lymphocytes. Nature. 2016;532:389–393. doi: 10.1038/nature17442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Holst J. L-type amino acid transport and cancer: targeting the mTORC1 pathway to inhibit neoplasia. American journal of cancer research. 2015;5:1281–1294. [PMC free article] [PubMed] [Google Scholar]
- Wang QJ, Yu Z, Griffith K, Hanada K, Restifo NP, Yang JC. Identification of T-cell Receptors Targeting KRAS-Mutated Human Tumors. Cancer immunology research. 2016;4:204–214. doi: 10.1158/2326-6066.CIR-15-0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, McCormick LL, Fitzgerald P, Chi H, Munger J, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. 2011;35:871–882. doi: 10.1016/j.immuni.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Long L, Yang K, Guy C, Shrestha S, Chen Z, Wu C, Vogel P, Neale G, Green DR, et al. Autophagy enforces functional integrity of regulatory T cells by coupling environmental cues and metabolic homeostasis. Nat Immunol. 2016;17:277–285. doi: 10.1038/ni.3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weindruch R, Walford RL. Dietary restriction in mice beginning at 1 year of age: effect on life-span and spontaneous cancer incidence. Science. 1982;215:1415–1418. doi: 10.1126/science.7063854. [DOI] [PubMed] [Google Scholar]
- Weng NP, Levine BL, June CH, Hodes RJ. Human naive and memory T lymphocytes differ in telomeric length and replicative potential. Proc Natl Acad Sci U S A. 1995;92:11091–11094. doi: 10.1073/pnas.92.24.11091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu XJ, Araki K, Li SZ, Han JH, Ye LL, Tan WG, Konieczny BT, Bruinsma MW, Martinez J, Pearce EL, et al. Autophagy is essential for effector CD8(+) T cell survival and memory formation. Nature Immunology. 2014;15:1152–1161. doi: 10.1038/ni.3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Youm YH, Vandanmagsar B, Ravussin A, Gimble JM, Greenway F, Stephens JM, Mynatt RL, Dixit VD. Obesity increases the production of proinflammatory mediators from adipose tissue T cells and compromises TCR repertoire diversity: implications for systemic inflammation and insulin resistance. J Immunol. 2010;185:1836–1845. doi: 10.4049/jimmunol.1000021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Bai Y, Xiong Y, Zhang J, Chen S, Zheng X, Meng X, Li L, Wang J, Xu C, et al. Potentiating the antitumour response of CD8(+) T cells by modulating cholesterol metabolism. Nature. 2016;531:651–655. doi: 10.1038/nature17412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong CS, Dardalhon V, Devaud C, Taylor N, Darcy PK, Kershaw MH. CAR T-cell therapy of solid tumors. Immunology and cell biology. 2017 doi: 10.1038/icb.2016.128. [DOI] [PubMed] [Google Scholar]
- Yu J, Du W, Yan F, Wang Y, Li H, Cao S, Yu W, Shen C, Liu J, Ren X. Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J Immunol. 2013;190:3783–3797. doi: 10.4049/jimmunol.1201449. [DOI] [PubMed] [Google Scholar]
- Zhang N, Bevan MJ. CD8(+) T cells: foot soldiers of the immune system. Immunity. 2011;35:161–168. doi: 10.1016/j.immuni.2011.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Zheng Z, Robbins PF, Khong HT, Rosenberg SA, Morgan RA. Primary human lymphocytes transduced with NY-ESO-1 antigen-specific TCR genes recognize and kill diverse human tumor cell lines. J Immunol. 2005;174:4415–4423. doi: 10.4049/jimmunol.174.7.4415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Shen X, Huang J, Hodes RJ, Rosenberg SA, Robbins PF. Telomere length of transferred lymphocytes correlates with in vivo persistence and tumor regression in melanoma patients receiving cell transfer therapy. J Immunol. 2005;175:7046–7052. doi: 10.4049/jimmunol.175.10.7046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*) Annual review of immunology. 2010;28:445–489. doi: 10.1146/annurev-immunol-030409-101212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler SF. Division of labour by CD4(+) T helper cells. Nat Rev Immunol. 2016;16:403. doi: 10.1038/nri.2016.53. [DOI] [PubMed] [Google Scholar]