

Summary
The Toll‐like receptor (TLR) adaptor proteins myeloid differentiating factor 88 (MyD88) and Toll, interleukin‐1 receptor and resistance protein (TIR) domain‐containing adaptor inducing interferon‐β (TRIF) comprise the two principal limbs of the TLR signalling network. We studied the role of these adaptors in the TLR4‐dependent inhibition of allergic airway disease and induction of CD4+ ICOS + T cells by nasal application of Protollin™, a mucosal adjuvant composed of TLR2 and TLR4 agonists. Wild‐type (WT), Trif −/− or Myd88 −/− mice were sensitized to birch pollen extract (BPEx), then received intranasal Protollin followed by consecutive BPEx challenges. Protollin's protection against allergic airway disease was TRIF‐dependent and MyD88‐independent. TRIF deficiency diminished the CD4+ ICOS + T‐cell subsets in the lymph nodes draining the nasal mucosa, as well as their recruitment to the lungs. Overall, TRIF deficiency reduced the proportion of cervical lymph node and lung CD4+ ICOS + Foxp3− cells, in particular. Adoptive transfer of cervical lymph node cells supported a role for Protollin‐induced CD4+ ICOS + cells in the TRIF‐dependent inhibition of airway hyper‐responsiveness. Hence, our data demonstrate that stimulation of the TLR4‐TRIF pathway can protect against the development of allergic airway disease and that a TRIF‐dependent adjuvant effect on CD4+ ICOS + T‐cell responses may be a contributing mechanism.
Keywords: Asthma; inducible co‐stimulator; T helper type 2; Toll‐like receptor 4; Toll, interleukin‐1R and resistance protein (TIR) domain‐containing adaptor inducing interferon‐β
Abbreviations
- AHR
airway hyperresponsiveness
- BAL
bronchoalveolar lavage
- BPEx
birch pollen allergen extract
- Foxp3
Forkhead box p3
- ICOS
inducible co‐stimulator
- ICOS‐L
ICOSy molecule ligand
- IFN
interferon
- i.p.
intraperitoneal
- LPS
lipopolysaccharide
- MCh
acetyl‐β‐methylcholine
- MyD88
myeloid differentiating factor 88
- poly(I:C)
polyinosinic‐polycytidylic acid
- Th2
T helper type 2
- TLR
Toll‐like receptor
- TRIF
Toll, interleukin‐1R and resistance protein (TIR) domain‐containing adaptor inducing interferon‐β
- WT
wild‐type
Introduction
Epidemiological, genetic, clinical and experimental data indicate a potential for the Toll‐like receptor 4 (TLR4) to initiate, exacerbate or conversely prevent allergic airway disease.1, 2 These apparently contradictory findings are probably due to several factors, such as the capacity of an allergen and immunomodulatory microbial exposures to activate multiple TLRs or interacting pattern recognition receptors simultaneously, the effect of gene polymorphisms upon the host response to the microbial stimuli, the structure of the specific TLR ligand(s), as well as the dose and the timing of exposure relative to the development of the immune system, disease onset or exacerbation. Elucidating the role of specific TLR signalling pathways in regulating the predominantly type 2 inflammatory response of the airways to aeroallergens that is characteristic of allergic asthma may also be an important step towards defining the specific conditions and mechanisms by which TLRs influence allergic disease. We have previously demonstrated that selective intranasal application of Protollin™, a mucosal adjuvant composed of purified bacterial TLR2 and TLR4 ligands, vesicles of hydrophobic outer‐membrane proteins from Neisseria meningitidis and lipopolysaccharide (LPS) from Shigella flexneri, to sensitized animals before the allergen challenge prevented the development of allergen‐induced airway hyper‐responsiveness (AHR) and inflammation.3 This effect was associated with an expansion of CD4+ T cells in the cervical lymph nodes draining the nasal mucosa expressing the inducible co‐stimulatory molecule (ICOS), as well as an increase in lung CD4+ ICOS+ cells. However, the roles of myeloid differentiating factor 88 (MyD88) and Toll, interleukin‐1R and resistance protein (TIR) domain‐containing adaptor inducing interferon‐β (TRIF) in mediating the TLR4‐dependent inhibition of allergic airway disease was not explored.
The adaptor proteins MyD88 and TRIF mediate distinct, but interacting, signalling cascades downstream of TLR ligation, ultimately leading to the production of pro‐inflammatory mediators and type I interferons (IFNs).4, 5 Mice that are deficient in both proteins cannot signal via any of the 13 TLRs discovered to date.6 All TLRs are thought to signal via MyD88, with the exception of TLR3, which relies solely on TRIF.7 TLR4 was thought to be the only TLR that signals through both adaptors but TLRs 2 and 5 can also do so in certain conditions.8, 9 To date, the TLR4–MyD88 pathway has been frequently implicated in allergic airway disease, particularly in allergic sensitization by way of the respiratory mucosa or induction of type 2 immunity by inhaled antigens10, 11, 12, 13, 14, 15 and the oxidizing pollutant ozone.16 TLR4–MyD88 signalling augments T helper type 2 (Th2) ‐promoting molecules on dendritic cells17, 18 and epithelial and inflammatory cell production of a range of cytokines and growth factors.19, 20, 21, 22 MyD88 is also involved in the signal transduction of mediators associated with severe asthma and/or corticosteroid resistance such as interleukin‐33 (IL‐33) and S100A8,23, 24 whereas LPS inhalation with ovalbumin can promote TLR4–MyD88‐dependent glucocorticoid‐resistant AHR in mice.25 Conversely, TLR4–MyD88 signalling has also been documented to inhibit the development of AHR and/or type 2 airway inflammation by LPS administered systemically26 or by repeated inhalational exposure.27 Oral or respiratory exposure to a non‐pathogenic cowshed bacterium,28, 29 commercial bacterial extracts,30 or probiotic strains31 also offers MyD88‐dependent protection against the development of allergic airway disease.
The role of TRIF signalling in relation to allergic asthma has been examined to a lesser extent. Activation of TLR3 by the synthetic double‐stranded RNA, polyinosinic‐polycytidylic acid [poly(I:C)], was confirmed to elicit32 as well as exacerbate33 type 2 airway disease in animals TRIF‐dependently. However, poly(I:C) was also reported to inhibit experimental allergic asthma in mice,34 but it was not confirmed whether this was TRIF‐dependent and poly(I:C) can also activate the TRIF‐independent RNA‐sensing protein kinase R, retinoic acid‐inducible gene I and melanoma differentiation‐associated gene 5. Moreover, TLR4–TRIF signalling is important in the development of lung Th17 and neutrophilic inflammation following house dust extract‐induced allergic sensitization to ovalbumin.35 However, there are no reports to date confirming an anti‐inflammatory role of the TRIF pathway in allergic asthma. In the current study, we sought to elucidate the roles of MyD88 and TRIF in mediating the TLR4‐dependent inhibition of allergic airway disease development by intranasal Protollin and the induction of CD4+ ICOS+ cells. Here, we show that activation of TLR4 signalling through the TRIF pathway prevents the development of allergic airway disease in mice and that the recruitment of CD4+ ICOS+ cells to the lungs may be one contributing TRIF‐dependent mechanism.
Materials and methods
Animal treatments
Six‐ to nine‐week‐old, female MyD88 knockout mice on a BALB/c background (supplied by S. Qureshi) and breeding pairs of C57BL/6J Ticam1/Lps2 (Trif knockout) mice (Jackson Laboratories, Bar Harbor, ME) were bred in the Animal Care Facilities of the McGill University Health Centre. Wild‐type (WT) C57BL/6J mice were also purchased from Jackson Laboratories. All animals were housed in a specific pathogen‐free animal facility under a 12 hr light/dark cycle with free access to food and water. All animals were sensitized on day 0 with a single 0·15 ml intraperitoneal (i.p.) injection of 20 protein nitrogen units of birch pollen allergen extract (BPEx; Greer Laboratories, Lenoir, NC) and 3 mg aluminium hydroxide (Alum hydrogel 2%; Brenntag Biosector, Frederiksund, Denmark). This BPEx extract is used for clinical purposes in intradermal desensitization and so is low in endotoxin (< 5 EU/ml; equivalent to < 0·05 EU/kg body weight). Experimental procedures were approved by the McGill University Animal Care Committee.
Experimental asthma protocol and nasal immunomodulation
Allergic airway disease was induced in mice as described previously (Fig. 1a).3 Following sensitization, on each of days 7, 10 and 13, awake mice received nasal applications, without prior anaesthesia, of 15 μl of either PBS or Protollin (GlaxoSmithKline Biologicals North America, Laval, QC, Canada). Previously, it has been confirmed that > 90% of fluid administered in this manner deposits in the nose/upper airways and the remaining portion is found largely in the gastrointestinal tract with very little in the lungs.36 Protollin consisted of a 1 : 1·1 ratio of Neisseria proteins to Shigella LPS at a concentration of 1 μg/μl LPS, resulting in an intranasal dose of approximately 15 μg of LPS and Neisseria proteosomes on each of the indicated days. On days 15, 16 and 17, mice were allergen challenged intranasally under light isoflurane anaesthesia (4%) with a dose of 25 protein nitrogen units of BPEx in a volume of 36 μl sterile PBS. Control mice were sham‐challenged with sterile PBS only.
Figure 1.
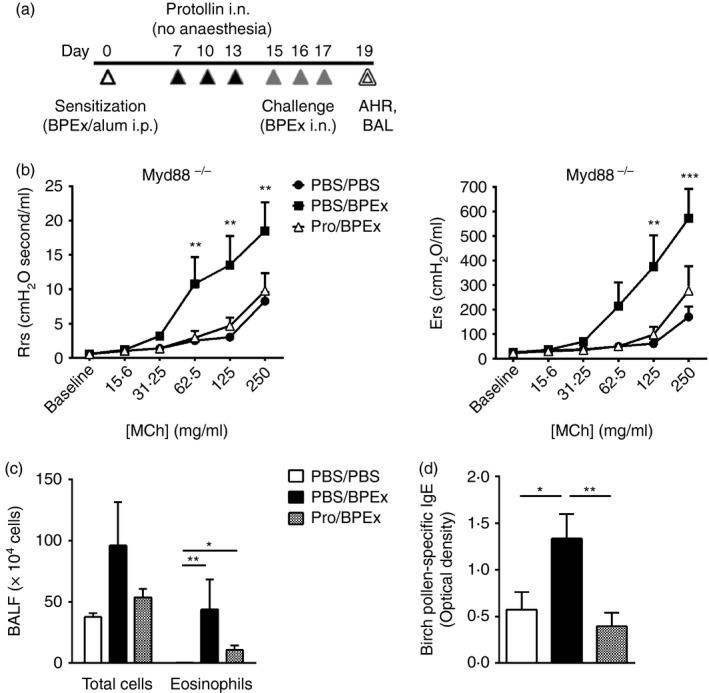
The inhibition of allergic airway disease by intranasal Protollin is MyD88‐independent. Airway responses (respiratory system resistance and elastance) to aerosolized acetyl‐β‐methylcholine (MCh) (b), bronchoalveolar lavage (BAL) fluid total inflammatory cells and eosinophils (c) and serum birch pollen allergen extract (BPEx) ‐specific IgE (d) were measured in MyD88−/− mice following nasal application to conscious mice of either PBS or Protollin (‘Pro’) and then intranasal sham (‘PBS’) or allergen (‘BPEx’) challenge with light anaesthesia (a) (n = 6 to n = 9 animals/group pooled from greater than three independent experiments; *P < 0·05, **P < 0·01, ***P < 0·001).
Assessment of allergen‐induced AHR
Airway responses to methacholine (MCh; acetyl‐β‐methylcholine; Sigma‐Aldrich Canada Ltd., Oakville, ON, Canada) were assessed on day 19, 48 hr after the final allergen challenge. Mice were anaesthetized with an injection of xylazine hydrochloride (10 mg/kg, i.p.) followed by i.p. administration of sodium pentobarbital (32 mg/kg). Mice were tracheotomized using a 19G metal cannula and connected via the endotracheal cannula to a commercial small animal ventilator (FlexiVent, SCIREQ Inc., Montreal, QC, Canada). The animal was ventilated at a respiratory rate of 150 breaths/min and tidal volume of 10 ml/kg against a positive end‐expiratory pressure of 3 cm H2O. Paralysis was induced with a 1 mg/kg pancuronium bromide i.p. injection before the measurement of baseline respiratory mechanics. A 1·2‐second, 2·5‐Hz single‐frequency forced oscillation manoeuvre was performed at 10‐second intervals and respiratory system resistance and elastance were calculated with commercial software. Doubling concentrations of MCh from 15·6–250 mg/kg were delivered to the mouse as an aerosol using a 4‐second nebulization period synchronized with inspiration. Allergen‐induced AHR was assessed by recording the peak respiratory system resistance and elastance following each dose of MCh administered.
Assessment of airway inflammation
On day 19, bronchoalveolar lavage (BAL) was performed using saline containing 10% fetal bovine serum. The recovered cell pellet was used to measure the total number of cells in the BAL and cytospins were prepared and stained with Diff‐Quik stain (Diff‐Quik® method; Medical Diagnostics, Düdingen, Germany) for differential cell counts.
Measurement of BPEx‐specific serum IgE
On day 19, blood was collected by exsanguination into serum separator tubes and left at room temperature to clot. Samples were centrifuged at 4000 g for 5 min and the serum was collected and stored at −20°. BPEx‐specific serum IgE was measured by ELISA, according to the manufacturer's instructions (BioLegend, San Diego, CA).
Flow cytometric analysis of cervical lymph node and lung CD4 T‐cell ICOS expression
To characterize Protollin's adjuvant effect on the CD4+ T helper cell and regulatory T‐cell responses, cervical lymph nodes and lungs were harvested from BPEx‐sensitized mice on days 16 and 17, respectively, 24 hr after one or two BPEx allergen challenges, following three administrations (days 7, 10 and 13) of either PBS or Protollin alone. Superficial cervical lymph nodes were isolated and placed in RPMI‐1640 medium, containing 8% heat‐inactivated FBS, 2 mm l‐glutamine, 50 μg/ml gentamycin and 10 mm HEPES. A single‐cell suspension was obtained by mincing and crushing the tissue on a 70‐μm BD Falcon cell strainer. Lungs were injected and minced ex vivo in a solution of RPMI‐1640 (Invitrogen, Life Technologies Inc., Burlington, ON, Canada), supplemented with 0·2 Wunsch units/ml Collagenase from Clostridium histolyticum (Type XI‐S), 1000 Dornase units/ml DNAse I (Type II‐S) (Sigma‐Aldrich Canada Ltd., Oakville, ON, Canada) and 0·5 mm Ca2+. The tissue was then incubated on an orbital shaker at 37° for 1 hr after which digestion was inhibited by the addition of cold complete RPMI medium with 2 mm EDTA and 50 μM β‐mercaptoethanol. Red blood cells were lysed with ammonium chloride solution. Isolated lymph node or lung cells were stained as described previously. Briefly, cells were stained with FITC‐conjugated anti‐CD4, phycoerythrin‐conjugated anti‐ICOS, and allophycocyanin‐conjugated anti‐Forkhead box p3 (Foxp3) monoclonal antibodies or the appropriate isotype controls. Cell acquisition was performed using either the BD FacsCalibur or LSRII and the percentage of ICOS‐expressing CD4+ Foxp3+ or CD4+ Foxp3− cells was analysed with cell quest pro or flowjo software, respectively (BD Biosciences, Mississauga, ON, Canada).
Adoptive transfer of FACS‐sorted WT CD4+ ICOS+ cells to Trif knockout mice
An adoptive transfer of 0·3 million cervical lymph node CD4+ ICOS+ cells was previously shown to be effective in inhibiting the development of AHR and airway inflammation in WT BALB/c mice. In the current study, cervical lymph node CD4+ ICOS+ cells were sorted and adoptively transferred to demonstrate an equivalent effect of these cells in C57BL/6J mice. On day 14, cervical lymph nodes were harvested and pooled from WT C57BL/6J BPEx‐sensitized mice that received nasal applications of Protollin on days 7, 10 and 13, and isolated cells were stained with FITC‐conjugated anti‐CD4 and phycoerythrin‐conjugated anti‐ICOS monoclonal antibodies. CD4+ ICOS+ cells were sorted by flow cytometry using a Beckman Coulter MoFlo cell sorter and either 0·1 or 0·3 million cells were adoptively transferred (i.p.) in 0·3 ml sterile PBS to Trif−/− mice that had been sensitized with BPEx in parallel with the WT animals, but that were otherwise untreated. Mice were then challenged on days 15–17 and underwent testing of airway responses to MCh, as well as BAL collection on day 19 (Fig. 5a).
Figure 5.
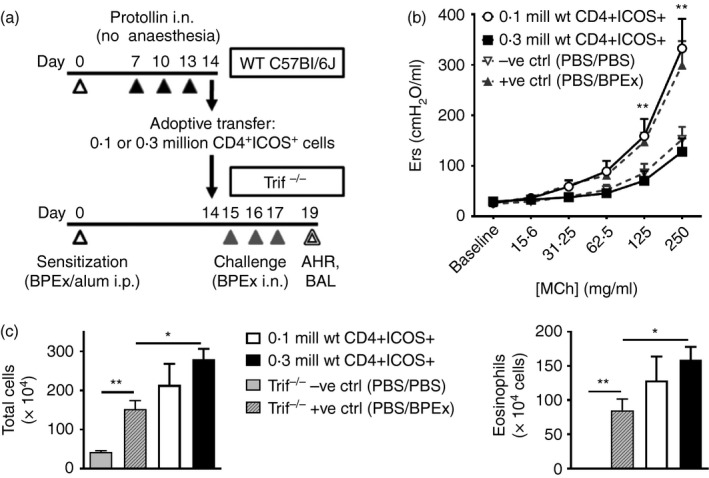
CD4+ ICOS + cells from the cervical lymph nodes of Protollin‐treated wild‐type (WT) mice have the capacity to inhibit airway hyper‐responsiveness when adoptively transferred to Trif−/− mice. CD4+ ICOS + cells sorted by FACS from lymph nodes of WT Protollin‐treated mice on day 14 were adoptively transferred (0·1 or 0·3 million cells, i.p.) to Trif−/− mice that had been sensitized in parallel but that did not receive Protollin, after which the recipient mice were challenged with birch pollen allergen extract (BPEx), as described in (a). Airway responses to aerosolized acetyl‐β‐methylcholine (MCh) (b) and bronchoalveolar lavage (BAL) fluid total inflammatory cells and eosinophils (c) were quantified on day 19 as in previous experiments. Historical −ve and +ve controls are shown in dashed lines (b) or grey bars (c) (n ≥ 5 animals/group from three independent experiments; *P < 0·05, **P < 0·01).
Adoptive transfer of MACS‐sorted total CD4+ cells to WT mice
Total cervical lymph node CD4+ cells were sorted from Protollin‐treated WT or Trif−/− mice to confirm the importance of TRIF‐induced CD4+ ICOS+ cells in the inhibition of allergic airway disease. On day 14, cervical lymph nodes were harvested and pooled from WT or Trif−/− BPEx‐sensitized mice that received nasal applications of either PBS or Protollin on days 7, 10 and 13. In these experiments, CD4+ cells were isolated by positive selection using mouse CD4 (L3T4) MicroBeads and magnetic cell sorting (Miltenyi Biotec Inc., Auburn, CA). Three million sorted cells from WT PBS‐treated mice or Protollin‐treated WT or Trif−/− mice were then adoptively transferred (i.p.) in 0·3 ml sterile PBS to WT mice that had been sensitized in parallel but that were otherwise untreated. Mice were then challenged on days 15–17 and underwent lung function testing in response to MCh, as well as BAL collection on day 19 (Fig. 6a).
Figure 6.
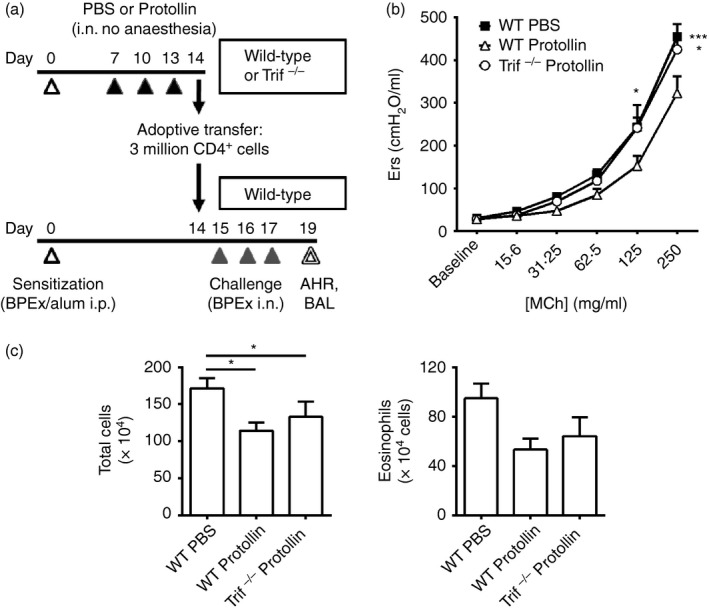
CD4+ cells from lymph nodes of Protollin‐treated wild‐type (WT) mice are capable of significantly inhibiting airway hyper‐responsiveness upon adoptive transfer whereas CD4+ cells from Protollin‐treated Trif−/− mice are not. Total CD4+ cells were sorted by MACS from lymph nodes of WT Protollin‐treated, PBS‐treated, or Trif−/− Protollin‐treated mice on day 14 and were adoptively transferred (3 million cells, i.p.) to WT C57BL/6J mice that had been sensitized in parallel but that were otherwise untreated, after which the recipient mice were challenged with birch pollen allergen extract (BPEx), as described in (a). Airway responses to aerosolized acetyl‐β‐methylcholine (MCh) (b; n = 6 or n = 7 animals/group from three independent experiments) and bronchoalveolar lavage (BAL) fluid total inflammatory cells and eosinophils (c; n = 12 to n = 14 animals/group from greater than three independent experiments) were quantified on day 19 as in previous experiments (*P < 0·05, ***P < 0·001).
Statistical analysis
Airway responses to MCh bronchoprovocation were analysed in graphpad prism Version 5 (GraphPad software, San Diego, CA) by two‐way analysis of variance (anova) followed by Bonferroni post‐tests comparing all experimental groups to each other. One‐way anova and post‐hoc Newman‐Keuls’ tests were used for all other analyses involving three or more groups, or unpaired Student's t‐test was used in the case where only two experimental groups were compared. Data were log‐transformed before statistical analysis when not normally distributed.
Results
TRIF signalling can prevent the development of experimental allergic airway disease
We have previously reported that the inhibition of allergen‐induced AHR and BAL eosinophilia in BALB/c mice by nasal Protollin administration before allergen challenge was TLR4‐ and not TLR2‐dependent.3 In the current study, we investigated the effects of Protollin in allergen‐challenged MyD88−/− mice. Mice that received Protollin exhibited significantly lower respiratory system resistance and elastance values in response to increasing doses of MCh (Fig. 1b) and serum BPEx‐specific IgE (Fig. 1d) compared with mice that were BPEx‐challenged but that had not received Protollin. Total inflammatory cells and eosinophils were not significantly lower in the BAL fluid of Protollin‐treated mice (Fig. 1c), suggesting a contribution of the MyD88 pathway to the inhibition of airway inflammation but not AHR. Trif−/− mice were only available on the C57BL/6J background but intranasal Protollin also potently inhibited AHR in WT mice of this strain (Fig. 2a) and significantly reduced total BAL cell counts and eosinophils (Fig. 2b). In contrast, Protollin failed to inhibit AHR (Fig. 2c) and airway inflammation (Fig. 2d) in Trif−/− mice. Serum BPEx‐specific IgE levels in either strain of C57BL/6J mice did not exceed the lower limit of detection and allergen‐associated increases could not be detected to allow confirmation of whether Protollin's inhibition of IgE synthesis was also TRIF‐dependent.
Figure 2.
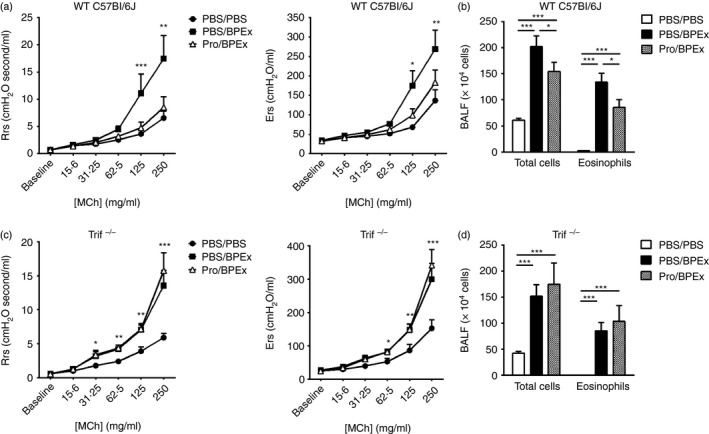
The inhibition of allergic airway disease by intranasal Protollin is TRIF‐dependent. Airway responses (respiratory system resistance and elastance) to aerosolized acetyl‐β‐methylcholine (MCh) and bronchoalveolar lavage (BAL) fluid total inflammatory cells and eosinophils were quantified in C57BL/6J wild‐type (WT) mice (a and b; n = 10 to n = 14 animals/group from more than three independent experiments) or Trif−/− mice (c and d; n = 7 or n = 8 animals/group from more than three independent experiments) following nasal PBS or Protollin applications and intranasal PBS or birch pollen allergen extract (BPEx) challenges (*P < 0·05, **P < 0·01, ***P < 0·001).
Induction of ICOS in CD4+ cells and expansion of CD4+ ICOS+ T cells is mediated by TRIF
Nasal application of Protollin to conscious animals increases ICOS mRNA expression in the nasal‐associated lymphoid tissues, as well as protein expression in both the CD4+ Foxp3+ regulatory cells and CD4+ ICOS− T‐cell populations in the cervical lymph nodes draining the nasal mucosa.3 Here, we assessed the effects of Protollin upon the cervical lymph nodes at the same time‐point as previously examined, following intranasal PBS or Protollin administration and a single intrapulmonary allergen challenge. Total lymph node cell numbers were similarly augmented by Protollin in WT and MyD88−/− BALB/c mice, but were unaltered in Tlr4−/− mice, suggesting that the TLR4‐dependent lymphoproliferation was intact even in the absence of MyD88 (see Supplementary material, Fig. S1b). Total lymph node cell numbers were also significantly augmented in WT C57BL/6J mice but not in the Trif−/− strain (see Supplementary material, Fig. S1b).
The absolute number of CD4+ ICOS+ Foxp3+ cells was significantly increased by Protollin in WT mice, as well as in MyD88−/− mice, albeit to a lesser degree (Fig. 3c). These cells were not significantly augmented by Protollin in Trif−/− mice. Also, the relative proportion of CD4+ ICOS+ Foxp3+ to CD4+ ICOS− Foxp3− cells was not significantly increased in Trif−/− mice (see Supplementary material, Fig. S2c), suggesting that the expansion of these cells is primarily TRIF‐dependent.
Figure 3.
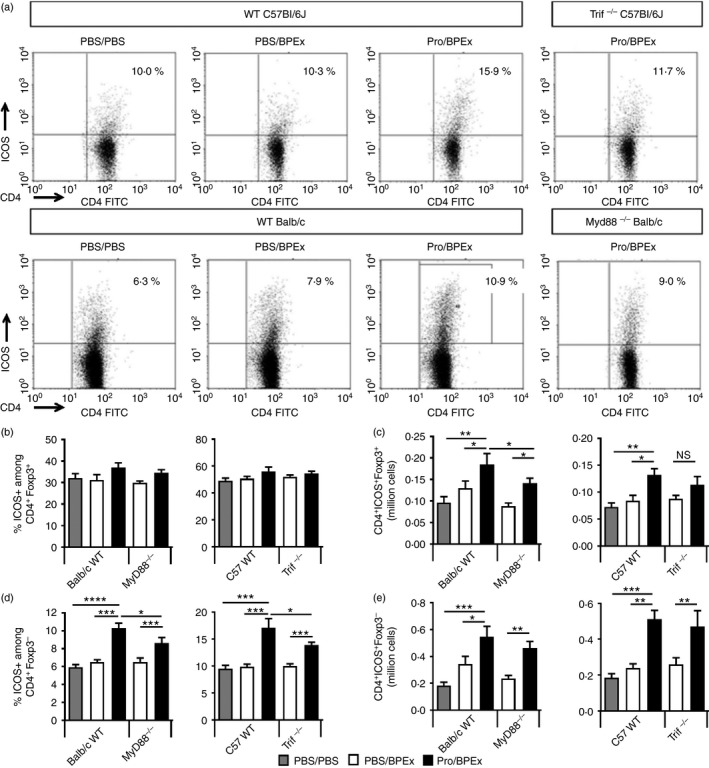
The induction and expansion of ICOS molecule ‐expressing CD4+ T cells in the cervical lymph nodes is significantly dependent on TRIF. Lymph node cells harvested from C57BL/6J wild‐type (WT) or Trif−/− mice or BALB/c WT or MyD88−/− mice on day 16, 24 hr after a single PBS or birch pollen allergen extract (BPEx) challenge following nasal applications of either PBS or Protollin were stained for CD4, ICOS and Foxp3. Representative dot plots show ICOS expression among gated CD4+ Foxp3− cells (a). Percentage of ICOS‐expressing CD4+ Foxp3+ (b) or CD4+ Foxp3− cells (d). Absolute number of lymph node CD4+ ICOS + Foxp3+ (c) and CD4+ ICOS + Foxp3− cells (e). (BALB/c MyD88−/−, n = 9 or n = 10 animals/group from greater than three independent experiments; C57BL/6J WT or Trif−/−, n = 7 animals/group from at least two independent experiments per strain; *P < 0·05, **P < 0·01, ***P < 0·001).
Protollin significantly increased the percentage of ICOS+ cells among the CD4+ Foxp3− population even in the absence of MyD88 or TRIF signalling. However, the degree of induction of ICOS in this population in either MyD88−/− or Trif−/− mice was significantly lower than in their WT counterparts (Fig. 3a,d). Finally, Protollin increased the absolute number of CD4+ ICOS+ Foxp3− cells in the cervical lymph nodes of all strains (Fig. 3e), indicating redundancy between the MyD88 and TRIF pathways and a contribution from both pathways to the induction of ICOS+ CD4 T cells. Overall, TRIF deficiency resulted in a reduced proportion of CD4+ Foxp3− and CD4+ Foxp3+ cells expressing ICOS in the cervical lymph nodes of Protollin‐treated mice (see Supplementary material, Fig. S2a,c).
TRIF signalling is necessary for the recruitment of CD4+ ICOS+ cells to the lungs
We have previously shown that CD4+ ICOS+ Foxp3+ and CD4+ ICOS+ Foxp3− cell numbers are elevated in the lungs of Protollin‐treated WT BALB/c mice after successive allergen challenges,3 implying a recruitment of these cells to the lungs. However, in WT C57BL/6J mice, Protollin exposure increased only the percentage of CD4+ Foxp3− cells expressing ICOS (Fig. 4b) and not CD4+ Foxp3+ cells (Fig. 4a). Moreover, only CD4+ ICOS+ Foxp3− cells (Fig. 4d), and not CD4+ ICOS+ Foxp3+ cells (Fig. 4c), were increased in absolute numbers by Protollin. Significantly, the proportion of CD4+ Foxp3− cells expressing ICOS (Figs 4b and 5b), as well as the absolute number of CD4+ ICOS+ Foxp3− cells in the lungs (Fig. 4d), was not increased by Protollin in Trif−/− mice, despite the expansion of these cells in the lymph nodes, supporting a role for TRIF signalling in mediating the recruitment of the CD4+ ICOS+ cells to the lungs.
Figure 4.
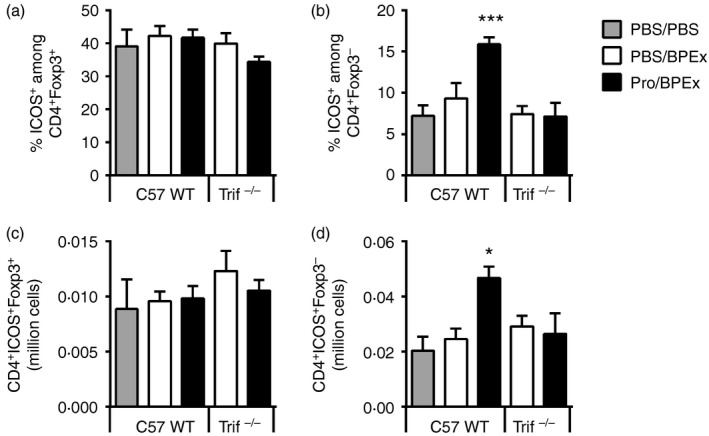
Protollin increases the percentage and absolute number of ICOS molecule ‐expressing CD4+ Foxp3− T cells but not CD4+ Foxp3+ regulatory T cells in the lungs, which occurs TRIF dependently. Percentage of CD4+ Foxp3+ and CD4+ Foxp3− cells expressing ICOS (a and b, respectively) and absolute numbers of CD4+ ICOS + Foxp3+ and CD4+ ICOS + Foxp3− cells (c and d, respectively) in lungs harvested from C57BL/6J WT or Trif−/− mice on day 17, 24 hr after two PBS or birch pollen allergen extract (BPEx) challenges following nasal applications of either PBS or Protollin (n = 6 to n = 8 animals/group from at least two independent experiments per strain; *P < 0·05, ***P < 0·001).
CD4+ ICOS+ cells contribute to the TRIF‐dependent inhibition of allergen‐induced AHR
To confirm the immunoregulatory potential of CD4+ ICOS+ cells in C57BL/6J mice, FACS‐sorted CD4+ ICOS+ cells isolated from the cervical lymph nodes of Protollin‐treated WT C57BL/6J mice were adoptively transferred to Trif−/− recipients (Fig. 5a). An adoptive transfer of 0·3 million CD4+ ICOS+ cells, but not 0·1 million cells, potently inhibited AHR (Fig. 5b). This is consistent with our previously published findings that even CD4+ ICOS+ cells isolated from non‐Protollin‐treated mice are capable of suppressing AHR when transferred in sufficient numbers3 and therefore, that TLR4 stimulation of the nasal mucosa by Protollin elicits a numerical increase in CD4+ ICOS+ cells that is protective against AHR. Surprisingly, the adoptive transfer of CD4+ ICOS+ cells was ineffective in reducing airway inflammatory cell numbers and, if anything, appeared to augment airway inflammation relative to historical controls that did not receive an adoptive transfer (Fig. 5c). These data indicate that WT CD4+ ICOS+ cells adoptively transferred via the systemic route can bypass the requirement for TRIF on endogenous immune effector cells to inhibit AHR but not airway inflammation in C57BL/6J mice.
Given that the proportion of CD4+ cells expressing ICOS was lower in the cervical lymph nodes of sensitized WT PBS‐treated mice and Trif−/− Protollin‐treated mice compared with WT Protollin‐treated mice (Fig. 3d and see Supplementary material, Fig. S2a,c), we proceeded to adoptively transfer an equivalent number of lymph node CD4+ cells from each of these groups of mice to WT C57BL/6J recipients before their allergen challenge to determine the capacity of these cells to inhibit allergen‐induced airway disease (Fig. 6a). Notably, only the CD4+ cells from Protollin‐treated WT C57BL/6J and not from Trif−/− donors attenuated AHR (Fig. 6b), consistent with the notion that CD4+ ICOS+ cells contribute to the TRIF‐dependent inhibition of AHR. Surprisingly, the CD4+ cells from both WT and Trif−/− Protollin‐treated donors reduced airway inflammation, namely BAL total inflammatory cells and eosinophils (Fig. 6c), as compared to the effects of CD4+ cells adoptively transferred from WT PBS‐treated mice. Collectively, this suggests that Protollin is also evoking MyD88‐dependent modulation of CD4+ ICOS− cells in the cervical lymph nodes which are capable of inhibiting allergic airway inflammation when adoptively transferred via the systemic route to WT C57BL/6J mice.
Discussion
Our understanding of the preconditions and mechanisms, including the role of specific signalling pathways, through which TLR4 influences allergen‐induced airway disease and adaptive immune responses is incomplete. In the current study, we sought to identify the role of the primary TLR adaptor proteins MyD88 and TRIF in mediating the previously described TLR4‐dependent inhibition of allergic airway disease development and induction of CD4+ ICOS+ T cells.3 We report that TRIF signalling via TLR4 can prevent the development of experimental allergic airway disease in mice in the context of nasal application of a TLR4‐stimulating mucosal adjuvant, Protollin. Our data also support a role for CD4+ ICOS+ cells in the TLR4–TRIF‐dependent inhibition of AHR in this model.
Using a model of birch pollen allergen extract (BPEx) ‐induced experimental asthma, we have shown previously that the application of Protollin to the nares alone prevented AHR, eosinophilic airway inflammation and production of BPEx‐specific serum IgE independently of TLR2 but required TLR4.3 In the current study, we have demonstrated that Protollin's inhibitory effects did not require signalling through MYD88 but were absent in Trif−/− mice, indicating that stimulation of the TLR4–TRIF pathway via the nasal mucosa can offer protection against allergic lower airway disease and may be a promising avenue for adjuvant‐based immunotherapy. We have previously also examined the role of TRIF in the context of inhalational exposures to birch pollen allergen alone (and so minimal LPS and TLR4 stimulation) and reported that whereas TRIF signalling was not necessary for the induction of allergic airway disease and did not influence AHR in this context, it was important in restraining eosinophilic airway inflammation.36 Our current data indicate that when TLR4 is further stimulated by an agonist, and selectively in the nasal mucosa, TRIF signalling is additionally capable of inhibiting AHR. Whether this pathway is also relevant to the effects of TRIF‐biased adjuvants such as Monophosphoryl Lipid A in allergy therapy remains to be investigated.37
The dose of LPS has a substantial influence on the development of allergic airway disease1, 2, 38 and there is evidence linking atopy and allergic asthma with gene–environment interactions between polymorphisms of the TLR4 accessory molecule CD14 gene and varying levels of LPS exposure.1, 39 Hence, in the broader context, given that we have administered a moderate–high dose of agonist to the nares, our finding suggests that the TLR4–TRIF pathway may perhaps be important in the protective effects of higher ambient levels of endotoxin upon allergic disease, such as those associated with living in a farming environment. CD14 has been identified to be of paramount importance to LPS‐induced MyD88‐independent TLR4 signalling through the TRIF pathway.40, 41 Whether a CD14 genotype that offers protection against the development of allergic asthma might do so by conferring enhanced TRIF‐dependent signalling is an interesting possibility. Notably, a CD14 genotype and LPS burden associated with protection against allergy in infants was also associated with higher numbers of peripheral blood CD4+ Foxp3− cells.42
The role of MyD88 and TRIF in mediating the adjuvant effect of TLR4 ligands upon T‐cell responses is an area of active investigation. Priming of T‐cell responses by TLR4 agonists can occur MyD88‐independently,43, 44, 45 whereas in vivo clonal expansion of CD4+ and CD8+ T cells by Monophosphoryl Lipid A,43 as well as up‐regulation of co‐stimulatory molecules, such as CD40, CD80 and CD86 on antigen‐presenting cells by LPS has been shown to be TRIF‐dependent and MyD88‐independent.46 Interestingly, the immunostimulatory effects of the N. meningitidis outer membrane vesicle vaccine that is relatively similar in structure to Protollin and the whole‐cell Pertussis vaccine, both of which contain LPS and lipoproteins that can activate TLR4 and TLR2, respectively, were shown to be TLR2‐independent but TLR4‐ and TRIF‐dependent, supporting the TLR4–TRIF pathway as an attractive target for vaccine adjuvants.47 We extended our previous observation of TLR4‐mediated induction of the T‐cell‐expressed co‐stimulatory molecule belonging to the CD28 family, ICOS, and expansion of CD4+ ICOS+ cells. ICOS is differentially inducible among T regulatory cells and effector T cells and its expression in CD4+ Foxp3+ cells has been demonstrated to be important in limiting airway inflammation in a model of intranasal allergen‐induced tolerance.48 Moreover, sensitization to ovalbumin via the airways with higher doses of LPS leads to the accumulation of lung CD4+ ICOS+ Foxp3+ cells and resultant tolerance.49 Our data demonstrate that Trif‐deficient mice exhibit impaired expansion of CD4+ ICOS+ Foxp3+ cells in the cervical lymph nodes draining the nasal mucosa and a reduced proportion of ICOS‐expressing CD4+ Foxp3− and CD4+ Foxp3+ cells. MyD88‐deficient mice demonstrated significant induction and expansion of ICOS+ cells within both of these populations, albeit lower than WT mice. Consistent with our observation of predominantly CD4+ ICOS+ Foxp3− cell expansion, the priming of antigen‐specific T‐cell responses by systemic LPS has been associated with selective induction of ICOS, resulting in preferential amplification of effector rather than regulatory T cells.50 Hence, the induction of ICOS on CD4+ cells has been described as a distinguishing marker of a pro‐inflammatory LPS‐driven immune response,50 whereas our data indicate that similar processes elicited by LPS via the nasal mucosa before pulmonary allergen challenge could perhaps be anti‐inflammatory in the context of allergen‐induced type 2 airway disease.
TLR4–TRIF signalling during allergic sensitization through the airways has been reported to be critical for the development of lung Th17 cells, which are known to express ICOS, and neutrophilic inflammation.35 ICOS is similarly critical for T follicular helper cell development and function;51, 52 however, our analysis of these cells did not indicate that they preferentially expressed Th17 or T follicular helper‐associated genes at the transcriptional level (ref. 3 and data not shown). We have previously demonstrated that CD4+ ICOS+ Foxp3+ and CD4+ ICOS+ Foxp3− cells are detected in higher numbers in the lungs of WT BALB/c mice at a later time‐point than in the cervical lymph nodes, suggesting that these cells migrate towards the lungs.3 In C57BL/6J mice, we were only able to detect CD4+ ICOS+ Foxp3− cells in higher numbers in the lungs of WT Protollin‐treated mice and additionally found this to be TRIF‐dependent. That Trif−/− mice displayed similar absolute numbers of CD4+ ICOS+ Foxp3− cells in the cervical lymph nodes compared with WT mice but lower numbers in the lungs, suggests that these cells probably traffic towards the lungs and that this process is impaired in the absence of functional TRIF. This is consistent with a report by McAleer et al.53 demonstrating that TRIF potentiated effector T‐cell migration to non‐lymphoid tissues, including the lungs, following intraperitoneal LPS injection, whereas T‐cell accumulation in lymphoid tissues was normal. Moreover, ICOS‐L−/− mice displayed reduced accumulation of CD4+ T cells to the lungs following systemic LPS boosting of the T‐cell response to ovalbumin and subsequent intranasal ovalbumin/LPS challenge.50 We determined that adoptive transfer of lymph node CD4+ ICOS+ cells from WT Protollin‐treated mice was capable of rescuing the inhibition of AHR in Trif−/− mice. Also, CD4+ cells from the lymph nodes of WT Protollin‐treated mice but not from WT PBS− or Trif−/− Protollin‐treated mice, which contained a lower proportion of ICOS‐expressing cells compared with the preceding group, could inhibit AHR to a significant degree. Taken together, these results indicate a capacity for lymph node CD4+ ICOS+ cells and accumulation of CD4+ ICOS+ Foxp3− cells in the lungs to contribute to the TLR4–TRIF‐dependent inhibition of AHR.
The mechanism for the strain‐specific difference in the accumulation of CD4+ ICOS+ Foxp3+ cells in the lungs is unclear. Moreover, C57BL/6J mice exhibited greater BAL eosinophilia than BALB/c mice and Protollin appeared to inhibit eosinophilic inflammation more robustly in BALB/c compared with C57BL/6 mice, suggesting that the accumulation of CD4+ ICOS+ Foxp3+ cells in the lungs may play a role in the suppression of eosinophilic inflammation. Notably, a recent study demonstrated that inducible regulatory T cells, but not natural regulatory T cells, were capable of inhibiting type 2 innate lymphoid cell (ILC2) ‐mediated AHR and airway inflammation upon adoptive transfer of these cells, and that the suppression depended on regulatory T cell ICOS interaction with ICOS‐L on ILC2s.54 However, our adoptive transfer experiments indicated that WT CD4+ ICOS+ cells failed to suppress BPEx‐induced airway inflammation in Trif−/− mice whereas MACS‐sorted CD4+ cells from both WT and Trif−/− mice inhibited airway inflammation when transferred to WT mice. These results indicate a dissociation between the regulation of these inflammatory outcomes and AHR and imply that the ICOS‐negative constituency of CD4+ cells possesses the relevant anti‐inflammatory capacity when adoptively transferred. Moreover, TRIF can induce type I IFNs, particularly IFN‐β, through its association with the tumour necrosis factor receptor‐associated factor 3 and the interferon regulatory factors IRF‐3 and IRF‐7.55 Type I IFNs have also recently been reported to directly inhibit ILC2 function, proliferation and survival, which may be an alternative mechanism, independent of ICOS, through which TLR4–TRIF signalling could attenuate pulmonary type 2 inflammation.56 Therefore, it is likely that multiple mechanisms underlie the effects of TLR4–TRIF signalling in this model. Overall, our data support that TLR4–TRIF‐dependent signals possess the capacity to inhibit allergic airway disease, which is likely mediated in part by the induction of CD4+ ICOS+ cells.
Disclosures
The authors have no financial or commercial conflicts of interest to declare.
Supporting information
Figure S1. Protollin induces significant lymphoproliferation (based on total cell numbers) within the cervical lymph nodes draining the nasal mucosa.
Figure S2. Protollin increases the relative proportion of CD4+ ICOS+ cells in the lymph nodes and lungs of C57BL/6J mice in TRIF‐dependent fashion.
Acknowledgements
We kindly thank Dr Clément Rioux and GlaxoSmithKline Biologicals North America (Laval, QC, Canada) for generously providing us with Protollin. We also thank Ms Jamilah Saeed for outstanding technical assistance, as well as Ms Julie Lord and Mr Eric Massicotte from the Institut de Recherches Cliniques de Montreal for cell‐sorting services. This work was supported by the Canadian Institutes of Health Research (CIHR) Grant MOP‐93747 awarded to JGM, and the J.T. Costello Memorial Fund, as well as CIHR MOP‐102494, awarded to STQ. KHS received fellowships from the Canadian Lung Association and Quebec Respiratory Health Training Program.
Senior author: Dr James G. Martin
References
- 1. Simpson A, Martinez FD. The role of lipopolysaccharide in the development of atopy in humans. Clin Exp Allergy 2010; 40:209–23. [DOI] [PubMed] [Google Scholar]
- 2. Zhu Z, Oh SY, Zheng T, Kim YK. Immunomodulating effects of endotoxin in mouse models of allergic asthma. Clin Exp Allergy 2010; 40:536–46. [DOI] [PubMed] [Google Scholar]
- 3. Shalaby KH, Jo T, Nakada E, Allard‐Coutu A, Tsuchiya K, Hirota N et al ICOS‐expressing CD4 T cells induced via TLR4 in the nasal mucosa are capable of inhibiting experimental allergic asthma. J Immunol 2012; 189:2793–804. [DOI] [PubMed] [Google Scholar]
- 4. Muzio M, Natoli G, Saccani S, Levrero M, Mantovani A. The human toll signaling pathway: divergence of nuclear factor κB and JNK/SAPK activation upstream of tumor necrosis factor receptor‐associated factor 6 (TRAF6). J Exp Med 1998; 187:2097–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sato S, Sugiyama M, Yamamoto M, Watanabe Y, Kawai T, Takeda K et al Toll/IL‐1 receptor domain‐containing adaptor inducing IFN‐β (TRIF) associates with TNF receptor‐associated factor 6 and TANK‐binding kinase 1, and activates two distinct transcription factors, NF‐κB and IFN‐regulatory factor‐3, in the Toll‐like receptor signaling. J Immunol 2003; 171:4304–10. [DOI] [PubMed] [Google Scholar]
- 6. Hoebe K, Du X, Georgel P, Janssen E, Tabeta K, Kim SO et al Identification of Lps2 as a key transducer of MyD88‐independent TIR signalling. Nature 2003; 424:743–8. [DOI] [PubMed] [Google Scholar]
- 7. Jiang Z, Mak TW, Sen G, Li X. Toll‐like receptor 3‐mediated activation of NF‐κB and IRF3 diverges at Toll‐IL‐1 receptor domain‐containing adapter inducing IFN‐β . Proc Natl Acad Sci USA 2004; 101:3533–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Aubry C, Corr SC, Wienerroither S, Goulard C, Jones R, Jamieson AM et al Both TLR2 and TRIF contribute to interferon‐β production during Listeria infection. PLoS One 2012; 7:e33299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Choi YJ, Im E, Chung HK, Pothoulakis C, Rhee SH. TRIF mediates Toll‐like receptor 5‐induced signaling in intestinal epithelial cells. J Biol Chem 2010; 285:37570–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hammad H, Plantinga M, Deswarte K, Pouliot P, Willart MA, Kool M et al Inflammatory dendritic cells–not basophils–are necessary and sufficient for induction of Th2 immunity to inhaled house dust mite allergen. J Exp Med 2010; 207:2097–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li DQ, Zhang L, Pflugfelder SC, De Paiva CS, Zhang X, Zhao G et al Short ragweed pollen triggers allergic inflammation through Toll‐like receptor 4‐dependent thymic stromal lymphopoietin/OX40 ligand/OX40 signaling pathways. J Allergy Clin Immunol 2011; 128:1318–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ng N, Lam D, Paulus P, Batzer G, Horner AA. House dust extracts have both TH2 adjuvant and tolerogenic activities. J Allergy Clin Immunol 2006; 117:1074–81. [DOI] [PubMed] [Google Scholar]
- 13. Page K, Zhou P, Ledford JR, Day SB, Lutfi R, Dienger K et al Early immunological response to German cockroach frass exposure induces a Th2/Th17 environment. J Innate Immun 2011; 3:167–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Phipps S, Lam CE, Kaiko GE, Foo SY, Collison A, Mattes J et al Toll/IL‐1 signaling is critical for house dust mite‐specific helper T cell type 2 and type 17 [corrected] responses. Am J Respir Crit Care Med 2009; 179:883–93. [DOI] [PubMed] [Google Scholar]
- 15. Piggott DA, Eisenbarth SC, Xu L, Constant SL, Huleatt JW, Herrick CA et al MyD88‐dependent induction of allergic Th2 responses to intranasal antigen. J Clin Invest 2005; 115:459–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li Z, Potts‐Kant EN, Garantziotis S, Foster WM, Hollingsworth JW. Hyaluronan signaling during ozone‐induced lung injury requires TLR4, MyD88, and TIRAP. PLoS One 2011; 6:e27137. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 17. Duan W, So T, Croft M. Antagonism of airway tolerance by endotoxin/lipopolysaccharide through promoting OX40L and suppressing antigen‐specific Foxp3+ T regulatory cells. J Immunol 2008; 181:8650–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tan AM, Chen HC, Pochard P, Eisenbarth SC, Herrick CA, Bottomly HK. TLR4 signaling in stromal cells is critical for the initiation of allergic Th2 responses to inhaled antigen. J Immunol 2010; 184:3535–44. [DOI] [PubMed] [Google Scholar]
- 19. Hammad H, Chieppa M, Perros F, Willart MA, Germain RN, Lambrecht BN. House dust mite allergen induces asthma via Toll‐like receptor 4 triggering of airway structural cells. Nat Med 2009; 15:410–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ho LH, Ohno T, Oboki K, Kajiwara N, Suto H, Iikura M et al IL‐33 induces IL‐13 production by mouse mast cells independently of IgE‐FcεRI signals. J Leukoc Biol 2007; 82:1481–90. [DOI] [PubMed] [Google Scholar]
- 21. Kim YS, Hong SW, Choi JP, Shin TS, Moon HG, Choi EJ et al Vascular endothelial growth factor is a key mediator in the development of T cell priming and its polarization to type 1 and type 17 T helper cells in the airways. J Immunol 2009; 183:5113–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kroeger KM, Sullivan BM, Locksley RM. IL‐18 and IL‐33 elicit Th2 cytokines from basophils via a MyD88‐ and p38α‐dependent pathway. J Leukoc Biol 2009; 86:769–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Halayko AJ, Ghavami S. S100A8/A9: a mediator of severe asthma pathogenesis and morbidity? Can J Physiol Pharmacol 2009; 87:743–55. [DOI] [PubMed] [Google Scholar]
- 24. Luzina IG, Pickering EM, Kopach P, Kang PH, Lockatell V, Todd NW et al Full‐length IL‐33 promotes inflammation but not Th2 response in vivo in an ST2‐independent fashion. J Immunol 2012; 189:403–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yang M, Kumar RK, Foster PS. Pathogenesis of steroid‐resistant airway hyperresponsiveness: interaction between IFN‐γ and TLR4/MyD88 pathways. J Immunol 2009; 182:5107–15. [DOI] [PubMed] [Google Scholar]
- 26. Bortolatto J, Borducchi E, Rodriguez D, Keller AC, Faquim‐Mauro E, Bortoluci KR et al Toll‐like receptor 4 agonists adsorbed to aluminium hydroxide adjuvant attenuate ovalbumin‐specific allergic airway disease: role of MyD88 adaptor molecule and interleukin‐12/interferon‐γ axis. Clin Exp Allergy 2008; 38:1668–79. [DOI] [PubMed] [Google Scholar]
- 27. Arora M, Poe SL, Oriss TB, Krishnamoorthy N, Yarlagadda M, Wenzel SE et al TLR4/MyD88‐induced CD11b+Gr‐1 int F4/80+ non‐migratory myeloid cells suppress Th2 effector function in the lung. Mucosal Immunol 2010; 3:578–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Conrad ML, Ferstl R, Teich R, Brand S, Blumer N, Yildirim AO et al Maternal TLR signaling is required for prenatal asthma protection by the nonpathogenic microbe Acinetobacter lwoffii F78. J Exp Med 2009; 206:2869–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. DeBarry J, Hanuszkiewicz A, Stein K, Holst O, Heine H. The allergy‐protective properties of Acinetobacter lwoffii F78 are imparted by its lipopolysaccharide. Allergy 2010; 65:690–7. [DOI] [PubMed] [Google Scholar]
- 30. Navarro S, Cossalter G, Chiavaroli C, Kanda A, Fleury S, Lazzari A et al The oral administration of bacterial extracts prevents asthma via the recruitment of regulatory T cells to the airways. Mucosal Immunol 2011; 4:53–65. [DOI] [PubMed] [Google Scholar]
- 31. Aumeunier A, Grela F, Ramadan A, Pham VL, Bardel E, Gomez AA et al Systemic Toll‐like receptor stimulation suppresses experimental allergic asthma and autoimmune diabetes in NOD mice. PLoS One 2010; 5:e11484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Matsumoto K, Kan O, Eguchi‐Tsuda M, Fukuyama S, Asai Y, Matsumoto T et al Essential role of B7‐H1 in double‐stranded RNA‐induced augmentation of an asthma phenotype in mice. Am J Respir Cell Mol Biol 2011; 45:31–9. [DOI] [PubMed] [Google Scholar]
- 33. Torres D, Dieudonne A, Ryffel B, Vilain E, Si‐Tahar M, Pichavant M et al Double‐stranded RNA exacerbates pulmonary allergic reaction through TLR3: implication of airway epithelium and dendritic cells. J Immunol 2010; 185:451–9. [DOI] [PubMed] [Google Scholar]
- 34. Sel S, Wegmann M, Sel S, Bauer S, Garn H, Alber G et al Immunomodulatory effects of viral TLR ligands on experimental asthma depend on the additive effects of IL‐12 and IL‐10. J Immunol 2007; 178:7805–13. [DOI] [PubMed] [Google Scholar]
- 35. Hsia BJ, Whitehead GS, Thomas SY, Nakano K, Gowdy KM, Aloor JJ et al Trif‐dependent induction of Th17 immunity by lung dendritic cells. Mucosal Immunol 2015; 8:186–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shalaby KH, Allard‐Coutu A, O'Sullivan MJ, Nakada E, Qureshi ST, Day BJ et al Inhaled birch pollen extract induces airway hyperresponsiveness via oxidative stress but independently of pollen‐intrinsic NADPH oxidase activity, or the TLR4‐TRIF pathway. J Immunol 2013; 191:922–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pfaar O, Barth C, Jaschke C, Hormann K, Klimek L. Sublingual allergen‐specific immunotherapy adjuvanted with monophosphoryl lipid A: a phase I/IIa study. Int Arch Allergy Immunol 2011; 154:336–44. [DOI] [PubMed] [Google Scholar]
- 38. Wills‐Karp M. Allergen‐specific pattern recognition receptor pathways. Curr Opin Immunol 2010; 22:777–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhao L, Bracken MB. Association of CD14 ‐260 (‐159) C>T and asthma: a systematic review and meta‐analysis. BMC Med Genet 2011; 12:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jiang Z, Georgel P, Du X, Shamel L, Sovath S, Mudd S et al CD14 is required for MyD88‐independent LPS signaling. Nat Immunol 2005; 6:565–70. [DOI] [PubMed] [Google Scholar]
- 41. Zanoni I, Ostuni R, Marek LR, Barresi S, Barbalat R, Barton GM et al CD14 controls the LPS‐induced endocytosis of Toll‐like receptor 4. Cell 2011; 147:868–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Williams LK, Oliver J, Peterson EL, Bobbitt KR, McCabe MJ Jr, Smolarek D et al Gene‐environment interactions between CD14 C‐260T and endotoxin exposure on Foxp3+ and Foxp3− CD4+ lymphocyte numbers and total serum IgE levels in early childhood. Ann Allergy Asthma Immunol 2008; 100:128–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mata‐Haro V, Cekic C, Martin M, Chilton PM, Casella CR, Mitchell TC. The vaccine adjuvant monophosphoryl lipid A as a TRIF‐biased agonist of TLR4. Science 2007; 316:1628–32. [DOI] [PubMed] [Google Scholar]
- 44. Sukhumavasi W, Egan CE, Warren AL, Taylor GA, Fox BA, Bzik DJ et al TLR adaptor MyD88 is essential for pathogen control during oral Toxoplasma gondii infection but not adaptive immunity induced by a vaccine strain of the parasite. J Immunol 2008; 181:3464–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Thompson BS, Chilton PM, Ward JR, Evans JT, Mitchell TC. The low‐toxicity versions of LPS, MPL adjuvant and RC529, are efficient adjuvants for CD4+ T cells. J Leukoc Biol 2005; 78:1273–80. [DOI] [PubMed] [Google Scholar]
- 46. Hoebe K, Janssen EM, Kim SO, Alexopoulou L, Flavell RA, Han J et al Upregulation of costimulatory molecules induced by lipopolysaccharide and double‐stranded RNA occurs by Trif‐dependent and Trif‐independent pathways. Nat Immunol 2003; 4:1223–9. [DOI] [PubMed] [Google Scholar]
- 47. Fransen F, Stenger RM, Poelen MC, van Dijken HH, Kuipers B, Boog CJ et al Differential effect of TLR2 and TLR4 on the immune response after immunization with a vaccine against Neisseria meningitidis or Bordetella pertussis . PLoS One 2010; 5:e15692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Busse M, Krech M, Meyer‐Bahlburg A, Hennig C, Hansen G. ICOS mediates the generation and function of CD4+ CD25+ Foxp3+ regulatory T cells conveying respiratory tolerance. J Immunol 2012; 189:1975–82. [DOI] [PubMed] [Google Scholar]
- 49. Whitehead GS, Thomas SY, Cook DN. Modulation of distinct asthmatic phenotypes in mice by dose‐dependent inhalation of microbial products. Environ Health Perspect 2014; 122:34–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lischke T, Hegemann A, Gurka S, Vu VD, Burmeister Y, Lam KP et al Comprehensive analysis of CD4+ T cells in the decision between tolerance and immunity in vivo reveals a pivotal role for ICOS. J Immunol 2012; 189:234–44. [DOI] [PubMed] [Google Scholar]
- 51. Bauquet AT, Jin H, Paterson AM, Mitsdoerffer M, Ho IC, Sharpe AH et al The costimulatory molecule ICOS regulates the expression of c‐Maf and IL‐21 in the development of follicular T helper cells and TH‐17 cells. Nat Immunol 2009; 10:167–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Choi YS, Kageyama R, Eto D, Escobar TC, Johnston RJ, Monticelli L, et al ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity 2011; 34:932–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. McAleer JP, Rossi RJ, Vella AT. Lipopolysaccharide potentiates effector T cell accumulation into nonlymphoid tissues through TRIF. J Immunol 2009; 182:5322–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Rigas D, Lewis G, Aron JL, Wang B, Banie H, Sankaranarayanan I et al Type 2 innate lymphoid cell suppression by regulatory T cells attenuates airway hyperreactivity and requires inducible T‐cell costimulator‐inducible T‐cell costimulator ligand interaction. J Allergy Clin Immunol 2017; 139:1468–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhu J, Mohan C. Toll‐like receptor signaling pathways – therapeutic opportunities. Mediators Inflamm 2010; 2010:781235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Duerr CU, McCarthy CD, Mindt BC, Rubio M, Meli AP, Pothlichet J et al Type I interferon restricts type 2 immunopathology through the regulation of group 2 innate lymphoid cells. Nat Immunol 2016; 17:65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Protollin induces significant lymphoproliferation (based on total cell numbers) within the cervical lymph nodes draining the nasal mucosa.
Figure S2. Protollin increases the relative proportion of CD4+ ICOS+ cells in the lymph nodes and lungs of C57BL/6J mice in TRIF‐dependent fashion.