

Abstract
Background
Hypoxia inducible factor 1 (HIF-1) activates protective pathways to counteract hypoxia and prevent tissue damage in conjunction with renal injury. The aim of this study was to evaluate a role of HIF-1 in diabetes-induced kidney damage.
Methods
We used a streptozotocin-induced diabetes mouse model and compared biochemical, histological and molecular parameters associated with kidney damage in Hif1α deficient (Hif1α +/-) and wild-type mice.
Results
We showed that Hif1α deficiency accelerated pathological changes in the early stage of DN. Six weeks after diabetes-induction, Hif1α deficient mice showed more prominent changes in biochemical serum parameters associated with glomerular injury, increased expression of podocyte damage markers, and loss of podocytes compared to wild-type mice. These results indicate that Hif1α deficiency specifically affects podocyte survival in the early phase of DN, resulting in diabetic glomerular injury. In contrast, renal fibrosis was not affected by the global reduction of Hif1α, at least not in the early phase of diabetic exposure.
Conclusions
Together our data reveal that HIF-1 has an essential role in the early response to prevent diabetes-induced tissue damage and that impaired HIF-1 signaling results in a faster progression of DN. Although the modulation of HIF-1 activity is a high-priority target for clinical treatments, further study is required to investigate HIF-1 as a potential therapeutic target for the treatment of DN.
Electronic supplementary material
The online version of this article (doi:10.1186/s12902-017-0200-8) contains supplementary material, which is available to authorized users.
Keywords: Diabetic complications, Diabetic nephropathy, Hypoxia, Podocyte, Mouse model
Background
Diabetic nephropathy (DN) is an endemic complication of diabetes and the leading cause of end-stage renal failure. Clinical features of DN are progressive albuminuria, proteinuria, and an eventual reduction in the glomerular filtration rate [1]. The complex progressive histopathological changes associated with DN include mesangial matrix expansion, thickening of basement membranes, glomerular and tubular hypertrophy, podocyte loss, and glomerulosclerosis and tubulointerstitial fibrosis [2]. High glucose is a primary initiating factor of multiple molecular, metabolic, and hemodynamic changes resulting in kidney damage, including intrarenal tissue hypoxia [3]. Tissue hypoxia activates multiple pathways, such as profibrotic growth factors, hemodynamic cytokines (angiotensin II), advanced glycation end products (AGE), and reactive oxygen species (ROS). Thus, both hyperglycemia and hypoxia are major determinators of the chronic complications associated with diabetes.
A master regulator of transcriptional responses to hypoxia is hypoxia inducible factor 1 (HIF-1). HIF-1 has been recently associated with the progression of chronic renal injuries including DN [4–6]. HIF-1 consists of two subunits, HIF-1α, an O2-labile subunit, and constitutively expressed HIF-1β [7]. Hif1α +/− heterozygote mutants demonstrate impaired responses when challenged with hypoxic conditions after birth [8, 9]. HIF-1 directly regulates the expression of more than 1000 human genes (for review, see [10]). Although the expression of a subset of HIF-1 target genes is induced by hypoxia in most or all cell types, the majority of these genes are induced by hypoxia in a cell type–specific manner. In addition to hypoxia, the HIF-1α subunit activity is regulated by numerous other factors, including growth factors, cytokines, sirtuins, ROS, and intracellular metabolites, even under normoxic conditions [11]. However, the mechanism of HIF-1α stabilization in a hyperglycemic environment is controversial. Hyperglycemia upregulates HIF-1α in the glomeruli of diabetic model mice regardless of the etiology of the diabetes [5, 12]. The activation of HIF-1 in the diabetic kidney may be suboptimal despite profound renal hypoxia, as suggested by a large body of evidence showing that the diabetic milieu deregulates the HIF-1α pathway [13–15]. It remains controversial whether the activation of HIF-1 signaling exerts a beneficial or harmful role in the progression of renal diseases, particularly DN. An indirect approach using YC-1 [3-(5′-hydroxymethyl-2′-furyl)-1-benzyl indazole], a HIF-1 inhibitor, reduced glomerular hypertrophy and AGE-tissue modifications in the type 1 diabetes mouse model [6]. In contrast, an activation of HIF-1α by CoCl2 reduced proteinuria and histological markers of kidney injury in an obese type 2 diabetes model [16] and in STZ-induced DN in rats [3].
To provide more insight into the functional role of HIF-1α pathways, we examine the relationship between diabetes-induced kidney injury and the partial deficiency of HIF-1α caused by the global deletion of the Hif1α functional allele with a specific focus on the early phase of diabetes-exposure. Together, our data suggest the potential roles of HIF-1α and Hif1α genetic variations in the manifestation of DN. Furthermore, our data point out the necessity of optimizing any possible pharmacological inhibition of HIF-1 in therapeutic applications of DN and diabetes-associated pathologies.
Methods
Experimental animals
This study was conducted in accordance with the Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, revised 1996). The experimental protocol was approved by the Animal Care and Use Committee of the Institute of Molecular Genetics, CAS. Diabetes was induced in male inbred FVB (Wt, strain code 207, Charles River) and Hif1α +/− strain on the FVB background, aged 7–9 weeks, by 2 intraperitoneal injections of 100 mg/kg body weight of streptozotocin (STZ; Sigma, St. Louis, MO), as described [17, 18]. Mice were sacrificed after 6 weeks of diabetes at age 15-17 weeks. The Hif1α mutants with the Hif1 atm1Jhu mutant allele [19] were obtained from Prof. Gregg L. Semenza. Hif1α +/− mice showed a partial loss of HIF-1α protein expression levels [20, 21]. The Hif1α +/− mouse colony was bred and maintained in our laboratory. Offspring of Wt x Hif1α +/− matings were genotyped by PCR [22], using DNA isolated from tails and amplifying neomycin (Neo) and Hif1α exon 2 sequences [19]; Neo (463-bp) and Hif1α (317-bp).
Biochemical parameters
Blood serum was collected following a 6-h fast (from 7 a.m. to 1 p.m. as recommended by the NIH for mouse metabolic models [23]) and was analyzed using a Beckman Coulter AU480 Chemistry Analyzer (Beckman) according to the manufacturer’s protocol in the Core Facility of Czech Centre for Phenogenomics in Biocev.
Real-time reverse-transcription PCR (RT-qPCR)
Total RNA was isolated from the renal cortex of diabetic Wt, non-diabetic and diabetic Hif1α +/−(EXP), and from non-diabetic Wt (control); the renal medulla was discarded. Following RT, quantitative real-time PCR (qPCR) was performed as described [9]. The relative expression of a target gene was calculated, based on qPCR efficiencies (E) and the quantification cycle (Cq) difference (Δ) of an experimental sample versus control (ratio = (E target)ΔCq Hif1a(Mean control – Mean EXP)/(E Hprt1)ΔCq Hprt1(Mean control – Mean EXP). RT-qPCR data were analyzed using the GenEX5 program (www.multid.se/genex/genex.html). Primer sequences are presented in Additional file 1: Table S1.
Western blot
The renal cortexices from the diabetic and non-diabetic kidneys were lysed with protease and phosphatase inhibitors to prevent protein degradation and stored at −80 °C until analysis. Fifty microgram of total protein lysates were denatured, resolved using 10% SDS-PAGE, and transferred to a nitrocellulose membrane, as described in detail previously [18]. The membrane was blocked with 5% dry milk and incubated overnight with rabbit anti-CX43 antibody at 1:6000 (#C6219, Sigma), or anti-VEGFA at 1:200 (#sc-7269; Santa Cruz Biotechnology, TX, USA). After incubation with a horseradish peroxidase–conjugated secondary IgG (Sigma), the blots were developed using the SuperSignal™ West Femto Maximum Sensitivity Substrate (#34095; Thermo Scientific, MI, USA). Chemiluminescent signals were captured using an ImageQuant LAS 4000 Imager (GE Healthcare Bio-Sciences AB, Sweden) and analyzed by ImageJ software (http://imagej.nih.gov/ij/download.html). Ponceau S staining was used as the loading control.
Histology and immunohistochemistry
To detect tissue modifications and tissue remodeling we used the Periodic acid–Schiff (PAS) staining system (#395B, Sigma, St. Louis, MO) and Trichrome Stain (Masson) Kit (#HT15-1KT, Sigma), respectively. Paraffin sections (8 μm) were dehydrated and used for the both methods. PAS+ area was delineated using the Adobe Photoshop CS5.1 program. Quantification of the PAS and collagen positive areas was performed using the threshold tool in the ImageJ program ( http://imagej.nih.gov/ij/download.html), separating pixels which fall within a desired range of intensity values from those which do not.
Sections (8 μm) for immunohistochemistry were heated in citrate buffer (0.07 M, pH 6.0) for antigen retrieval and blocked with PBS (pH 7.4) with 0.1% Tween®20 (#P9416, Sigma) and 10% normal goat serum (#005-000-121, Jackson Immuno Research Labs). Primary antibodies used: mouse anti-VEGFA 1:50 (#sc-7269, Santa Cruz Biotechnology), rabbit anti-pHH3 1:100 (#06-570, Merck Millipore), rabbit anti-WT1 1:200 (#CA1026, Merck Millipore) and mouse anti-alpha smooth muscle actin (α-SMA) 1:400 (#A2547, Sigma). Secondary antibodies used: Alexa Fluor® 488 and 594 1:400 (#115-545-146 and #111-585-144, resp., Jackson Immuno Research Labs). The sections were counterstained with Hoechst 33,342 (#14533 Sigma) and imaging with confocal microscope (ZEISS LSM 880 NLO). The areas of VEGFA and α-SMA expression, and a number of WT1+ podocytes and pHH3+ nuclei in the renal cortex were quantified using the ImageJ.
Statistics
All values are means ± SEM. We used two-way ANOVA to compare differences among experimental groups with genotype and experimental condition (diabetes or no diabetes) as categories. When a significant interaction was detected, the differences between subgroups were further analyzed by post hoc Tukey’s multiple comparison tests; significance assigned at the P < 0.05 level (Graph Pad, 2005; Graph Pad, San Diego, CA).
Results
Changes in physiological and biochemical parameters after 6 weeks of diabetes
For this study we used the well-established STZ-induced diabetes mouse model on the FVB genetic background [17, 18]. Age-matched wild-type Hif1α +/+ (Wt) and Hif1α +/− mice were compared. Body weight gain after 6 weeks was significantly decreased in diabetic mice of both genotypes (Fig. 1a; non-diabetic Wt 6.3 ± 0.6 g (n = 9), diabetic Wt 0.6 ± 0.6 g (n = 11), non-diabetic Hif1α +/− 5.9 ± 0.9 g (n = 6) and diabetic Hif1α +/− 1.3 ± 0.7 g (n = 12)). In contrast, the kidney weight-to-body weight ratios were increased in diabetics compared to controls (Fig. 1b; non-diabetic Wt 0.009 ± 0.0004 g (n = 17), diabetic Wt 0.014 ± 0.001 g (n = 8), non-diabetic Hif1α +/− 0.010 ± 0.001 g (n = 6) and diabetic Hif1α +/− 0.017 ± 0.004 g (n = 11)), consistent with diabetic renal hypertrophy phenotype [4, 24].
Fig. 1.
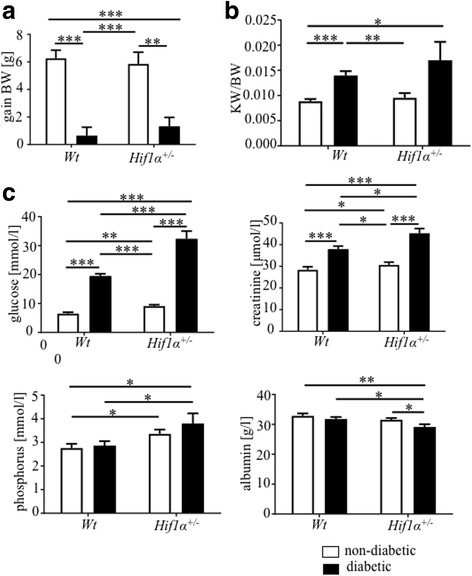
Physiological and biochemical parameters. a Body weight gain after 6 weeks from the induction of diabetes. b Changes in kidney/body weight ratio (KW/BW) after 6 weeks of diabetes exposure. The values represent means ± SEM (non-diabetic Wt (n = 9), diabetic Wt (n = 11), non-diabetic Hif1α +/− (n = 6) and diabetic Hif1α +/− (n = 12)). Two-way ANOVA showed significant effect of diabetes in the body weight gain (P < 0.0001) and in the KW/BW (P = 0.0005). c The levels of glucose, creatinine, phosphorus, and albumin in the blood serum of Wt and Hif1α +/− mice after 6 h fasting and collected at the end of experiment (6 weeks from the induction of diabetes). The values represent means ± SEM (n = 8 mice in each group). Statistical significance assessed by two-way ANOVA: genotype effect (creatinine, P < 0.001; phosphorus, P = 0.0037; albumin, P = 0.027); diabetes effect (creatinine, P < 0.0001; albumin, P = 0.027); and interaction between genotype and diabetes: glucose P < 0.0001. *Significant differences by post hoc pairwise comparison tests, *P < 0.05, **P < 0.01, ***P < 0.001
Increased serum levels of creatinine and phosphorus, and decreased serum levels of albumin are the first markers of kidney damage due to high glucose concentrations [4]. Blood serum was collected from non-diabetic and diabetic Wt and Hif1α +/− mice after 6 h-fasting. Both Wt and Hif1α +/− mice developed high levels of hyperglycemia after STZ injections over the 6-week study (Fig. 1c). Interestingly, serum glucose levels were significantly higher in diabetic Hif1α +/− compare to diabetic Wt mice. Serum phosphorus levels were also slightly higher in non-diabetic Hif1α +/− compared to non-diabetic Wt mice (Fig. 1c). As serum phosphorus is a cardiovascular risk factor [25], these data correspond with a predisposition of Hif1α +/− mutation for endothelial dysfunction and cardiovascular disease [8, 9, 18]. Phosphorus and creatinine levels were significantly increased, whereas the levels of albumin were significantly reduced in diabetic Hif1α +/− compared to diabetic Wt mice (Fig. 1c).
Tissue modification and remodeling in the renal cortex
Periodic acid–Schiff (PAS) staining is a method for the detection of AGE, non-enzymatic tissue modifications [26]. A weak positive staining was detected in the tubular part of the renal cortex of non-diabetic Hif1α +/− mice (Fig. 2a, b), suggesting AGE modifications due to Hif1α deficiency even under normal conditions. A significantly higher production of AGE products was detected in both the diabetic Wt and Hif1α +/− renal cortex (Fig. 2e). Using Masson’s trichrome staining, we analyzed interstitial collagen deposition in the renal cortex as an index of interstitial fibrosis and overall tissue remodeling (Fig. 2c, d). We did not detect any differences in collagen deposition between non-diabetic Wt and Hif1α +/−. Collagen accumulation was significantly increased in both Wt and Hif1α +/− diabetic groups, although the trend of more abundant tissue remodeling was evident in the diabetic Hif1α +/− renal cortex (Fig. 2f). Based on light microscopy evaluation, we detected only mild mesangial expansion without nodular sclerosis (Fig. 2c, arrow), classified as class I/II DN, which is a characteristic early stage of DN [27].
Fig. 2.
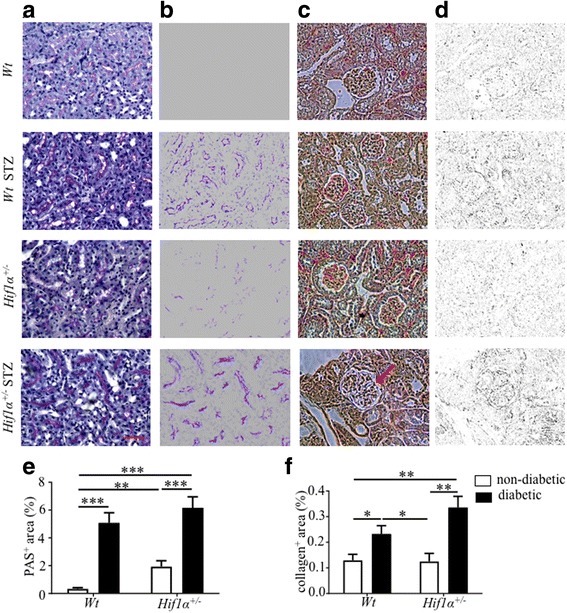
PAS and trichrome-staining of the renal cortex in non-diabetic and diabetic Wt and Hif1α +/− mice. a Representative PAS staining of 8 μm kidney sections showed advanced glycation end products in diabetic Wt (Wt STZ) and diabetic Hif1α +/− (Hif1α +/− STZ). The strongest positive staining was detected in the tubular part of the kidney section. b Delineated PAS+ area in the kidney section using Adobe Photoshop. c Representative Masson’s trichrome-staining of 8 μm kidney sections showed increased fibrosis with increased collagen fibers in the renal cortex of diabetic kidneys. d Delineated collagen+ area in the kidney section by ImageJ. e-f A relative quantification of staining was determined as a percentage of positive area in the field of view by ImageJ. Scale bar 100 μm. The values represent means ± SEM (n = 3 sections/3 samples/group). Statistical significance differences were tested by two-way ANOVA (diabetes effect: PAS and trichrome (P < 0.0001); effect of genotype in PAS staining (P = 0.04)). *Significant differences by post hoc pairwise comparison, *P < 0.05, **P < 0.01, ***P < 0.0001
Molecular changes in the renal cortex of diabetic mice with Hif1α+/− deficiency
This study assesses the effects of partial Hif1α +/− deficiency on the progression of DN in early stages of diabetes. We analyzed the expression of genes associated with extracellular matrix expansion, podocyte dysfunction, and profibrotic responses with a specific focus on HIF-1α direct target genes (Fig. 3a). Hif1α partial deficiency was demonstrated by a reduced expression of HIF-1-targeted genes (Pdk1, Ntn1, Ctgf, and Fn1) in the renal cortex under non-diabetic conditions. The mRNA level of adrenomedullin (Adm), a potent vasodilatory peptide hormone, was increased in the diabetic Hif1α +/− kidney cortex compared to other experimental groups. The relative gene expression of podocin (Nphs2), a marker for podocyte damage, was significantly elevated only in the diabetes-exposed Hif1α +/− renal cortex. Collagen accumulation has been associated with the up-regulation of the transcription factor Sox9, a direct HIF-1α target [28]. A significant increase in the mRNA level of the Sox9 gene was detected in the diabetic Hif1α +/− kidney cortex. A pivotal cytokine in the profibrotic responses [29], transforming growth factor beta 1 (Tgfβ1), was significantly increased in both diabetic groups (the effect of diabetes, P < 0.05 by two-way ANOVA). Fibronectin (Fn1) and connective tissue growth factor (Ctgf), classical markers of fibrosis and indicators of extracellular matrix accumulation, were increased in diabetic mice (significant effect of diabetes P < 0.008 and effect of genotype P < 0.01 by two-way ANOVA). As HIF-1α direct target genes, both Fn1 and Ctgf were significantly decreased in non-diabetic Hif1α +/− mice, suggesting impaired HIF-1α regulation. The partial deficiency of netrin-1 (Ntn1) results in kidney microvascular dysfunction and accelerated DN [30]. Correspondingly, the expression of Ntn1 was reduced in the diabetic Wt and Hif1α +/− renal cortex compared with non-diabetic Wt. We also found a significant reduction of Ntn1, a direct HIF-1α target, in the non-diabetic Hif1α +/− compared to Wt mice, indicating altered HIF-1α regulation. We found that the expression of Cx43 [31] in the renal cortex was significantly attenuated in non-diabetic Hif1α +/−. Cx43 participates in intercellular communication and is down-regulated by diabetes [32]. Accordingly, in our experimental diabetic model, both diabetic groups Wt and Hif1α +/− showed decreased Cx43 expression compared to non-diabetic Wt. Consistently, decreased protein levels of CX43 were detected in the renal cortex of diabetic Hif1α +/− mice, indicating impaired intercellular communication that may cause endothelial cell dysfunction and glomerular injury (Fig. 3b, c).
Fig. 3.
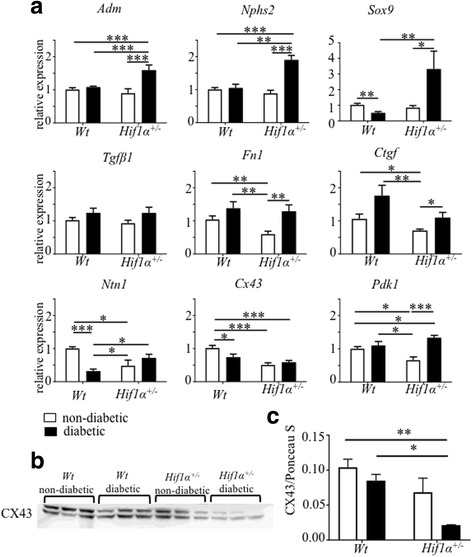
Gene expression changes in the renal cortex of diabetic and non-diabetic Wt and Hif1α +/− mice. a The relative gene expression changes were analyzed using RT-qPCR and quantified with ΔΔCT method. The values represent means ± SEM (n = 8 samples/group). Statistical significance differences in normalized Ct values were tested by two-way ANOVA followed by post hoc pairwise comparison tests *P < 0.05, **P < 0.01, ***P < 0.001. b The renal cortex extracts were prepared and probed on Western blots with antibodies directed against connexin 43 (CX43; ~43 kDa). Representative immunoblot for CX43 is shown. c Combined results obtained by densitometric evaluation of the Western blots of CX43 using cortical extracts from three mice per group. Ponceau S staining was used as the loading control. Data were analyzed by ImageJ software. Statistical significance differences were tested by two-way ANOVA followed by post hoc pairwise comparison tests *P < 0.05, **P < 0.01. Abbreviations: adrenomedullin (Adm), podocin (Nphs2), SRY (Sex Determining Region Y)-Box 9 (Sox9)), transforming growth factor beta 1 (Tgfβ1), fibronectin 1 (Fn1), connective tissue growth factor (Ctgf), netrin (Ntn1), connexin 43 (Cx43), and pyruvatdehydrogenase kinase1 (Pdk1)
Podocyte dysfunction in diabetic Hif1α+/− mice
Podocytes highly express vascular endothelial growth factor (VEGFA) and any small changes in VEGFA levels cause significant aberrations in glomerular structure [33]. We detected significantly higher VEGFA expression in the glomerulus of diabetic Hif1α +/− mice compared to diabetic Wt (Fig. 4a–d), indicating early deleterious changes in diabetic disease. Podocyte loss was also demonstrated by staining with WT1 (Fig. 4c), a nuclear marker of mature and fully functional podocytes [34]. The number of WT1 positive podocytes per glomerulus area was significantly decreased in diabetic Hif1α +/− compared to diabetic Wt (Fig. 4e). Together with the increased expression Nphs2 (Fig. 3a), a marker for podocyte damage, our data thus indicate that Hif1α partial deficiency combined with diabetes accelerates podocyte loss and the inability to sustain the glomerular filtration barrier.
Fig. 4.
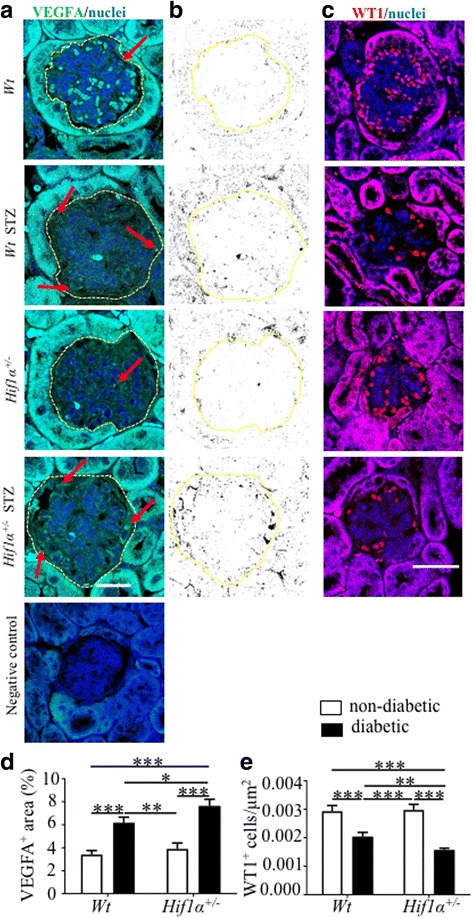
Molecular changes in the glomerulus of Wt and Hif1α +/− mice. a Representative confocal microscopy images of 8 μm sections of the glomerulus stained with anti-VEGFA antibody (green) show the highest expression of VEGFA in diabetic Hif1α +/− kidneys. The area of the glomerulus is outlined by the yellow dashed line. Arrows indicate VEGFA expression (sharp green) in the glomerulus. Negative control is without primary antibody. Images are stacked Z-plane sections; nuclei are counterstained with Hoechst 33,342 (blue); scale bar 20 μm. b Delineated VEGFA+ area by ImageJ. c Staining of mature and fully functional podocytes with anti-WT1 antibody (red) shows the largest loss of podocytes in the diabetic Hif1α +/− renal cortex; nuclei are counterstained with Hoechst 33,342 (blue); scale bar 50 μm. d Relative quantification of VEGFA expression was determined as a percentage of VEGFA+ area per glomerular area. e Quantification of podocyte density as a number of WT1+ cells per glomerular area. The values represent means ± SEM (n = 10 glomerulus/3 sections/3 mice/group). Two-way ANOVA detected statistical significance of diabetes effect (P < 0.0001) followed by post hoc pairwise comparison tests *P < 0.05, **P < 0.01, ***P < 0.001. Abbreviations: vascular endothelial growth factor (VEGFA), Wilms tumor 1 (WT1)
Diabetes-induced changes associated with profibrotic processes and accumulation of extracellular matrix
Our gene expression profiling analyses showed significant changes in the expression of profibrotic markers induced by the diabetic milieu. We analyzed the expression of α-SMA, an excellent prognostic indicator of renal fibrosis progression and marker of extracellular matrix accumulation [35]. The expression of α-SMA was significantly increased in both diabetic Wt and Hif1α +/− mice (Fig. 5a, b). The diabetic milieu triggers early tubular cell proliferation. Proximal tubule growth involves an early period of hyperplasia followed by a shift to hypertrophy [24]. Consistent with a hyperplasia phenotype in an early stage of DN, the number of mitotic cells found in tubular cells was increased in both diabetic Wt and Hif1α +/− kidneys in comparison with non-diabetics (Fig. 5a, b). We did not detect any significant differences associated with the Hif1α +/− phenotype.
Fig. 5.
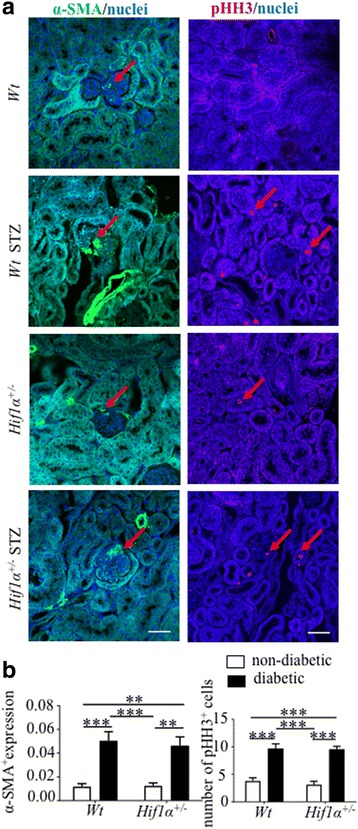
Diabetes-induced changes in α-SMA and pHH3 expression in the kidney of Wt and Hif1α +/− mice. a Confocal imaging of 8 μm sections of the kidney stained with anti-α-SMA and anti-pHH3 antibodies. Images are stacked Z-plane sections; nuclei are counterstained with Hoechst 33,342 (blue); scale bars 50 μm. b Quantification of α-SMA+ area was done per glomerular area (10 glomerulus/each section) and the positive pHH3+ staining was determined as a number of the positive nuclei in the field of view of the renal cortex. The values represent means ± SEM (n = 3 sections/3mice/group). Statistical significance assessed by two-way ANOVA: diabetes effect in the expression of pHH3 and α-SMA (P < 0.0001). Post hoc pairwise comparison **P < 0.01, ***P < 0.001. Abbreviations: alpha smooth muscle actin (α-SMA), phospho-histone H3 (pHH3)
Discussion
Our partial deficiency Hif1α model provides the first model that tests in vivo the function of HIF-1α in the development and progression of diabetes-induced renal damage. Previous work has only provided indirect evidence for the role of HIF-1α, using a HIF-1 inhibitor [6] or HIF-1 activator [16]. Our data extend previous findings that HIF-1α signaling is activated in the kidneys of experimental models with type I and type II diabetes and that it may be relevant to the development of DN [4, 5, 12]. We examined the role of HIF-1α in the early stage of disease using the STZ-induced diabetic mouse model characterized by hyperglycemia (blood glucose levels > 13.9 mmol/L) and insulinopenia. We found that Hif1α partial deficiency significantly accelerated the manifestation of pathological changes associated with the progression of DN. Changes in serum biochemical parameters associated with diabetic glomerular injury and progression of chronic kidney disease were more significant in diabetic Hif1α +/− compared to diabetic Wt mice. The combination of Hif1α deficiency and diabetes resulted in an altered transcriptional expression profile of the renal cortex and decreased survival of podocytes.
Hypoxia represents an early and potentially initiating factor in the development and progression of chronic kidney diseases including DN [4, 36]. HIF-1 mediates hypoxia-induced cellular responses through the regulation of genes involved in cell metabolism, glucose utilization, angiogenesis, oxidative stress, apoptosis, and proliferation. However, the activation of HIF-1 in the diabetic kidney may be suboptimal despite profound renal hypoxia, as suggested by a large body of evidence showing that the diabetic milieu deregulates the HIF-1α pathway [13–15]. In recent years, HIF-1α genetic polymorphisms have emerged as potentially important determinants of disease severity and adverse outcomes [37, 38]. Nonetheless, given the diversity of HIF-1 signaling, it remains controversial whether the activation of HIF-1 signaling exerts a beneficial or harmful role in the progression of renal diseases, particularly DN.
Persistent, chronic exposure to hypoxia is associated with structural tissue remodeling, such as renal fibrosis, inflammation, apoptosis and loss of microvasculature. HIF-1 signaling is an important protective physiological mechanism activated to counteract hypoxia and prevent renal damage (for review, see [39]). For example, the global inactivation of the Vhlh gene by the Cre-loxP system resulted in HIF-1α and HIF-2α stabilization and suppressed fibrogenesis in mice subjected to unilateral ureteral obstruction [40]. Other studies using pharmacological approaches for systemic HIF-1 activation demonstrated improved proteinuria and histological parameters in experimental chronic kidney disease models [41, 42]. In contrast, other studies have shown that sustained HIF-1 activation may have unfavorable effects. Genetic inactivation of the Vhlh gene in tubular epithelial cells resulted in constitutive HIF-1α stabilization and accelerated renal fibrosis [43]. Similarly, the genetic ablation of Hif1α in the renal proximal tubule inhibited tubulointerstitial fibrosis in the in vivo model of unilateral ureteral obstruction [44]. These data suggest that HIF-1α may play different roles in the progression of chronic kidney diseases depending on the mode of activation, cell-type specific action, and local versus global HIF-1α stabilization. Thus, these conflicting results reflect the complexity of the adaptive responses mediated by HIF-1.
Similar discrepancies have been reported regarding the role of HIF-1 in DN. An indirect approach using YC-1 [3-(5′-hydroxymethyl-2′-furyl)-1-benzyl indazole], a HIF-1 inhibitor, reduced glomerular hypertrophy and AGE in the type 1 diabetes mouse model [6]. In contrast, an induction of HIF-1α by CoCl2 reduced proteinuria and histological markers of kidney injury in an obese type 2 diabetes model [16] and in STZ-induced DN in rats [3]. In conjunction with these studies, our data demonstrate that a partial Hif1α deficiency promotes the diabetes-induced kidney injury. Hif1α partial deficiency was associated with a reduced expression of HIF-1-targeted genes Pdk1, Ntn1, Ctgf, and Fn1. Serum glucose levels were significantly increased in Hif1α +/− mice compared to Wt, implying systemic changes in glucose metabolism in association with Hif1α partial deletion, which may contribute to the enhanced pathogenesis. HIF-1, by regulating the expression of glucose transporter GLUT1 and glycolytic enzymes, affects glucose homeostasis, including the regulation of glucose-stimulated insulin secretion (GSIS) from the pancreatic beta-cells [45]. Targeted disruption of Hif1α in pancreatic beta-cells resulted in glucose intolerance, impaired GSIS, and beta-cell dysfunction [46]. Thus, the increased serum glucose levels in our diabetic Hif1α +/− mice were in accordance with the changes in beta-cell function and impaired glucose homeostasis.
These changes were accompanied by glomerular damage, as indicated by a significant loss of podocytes and increased expression of podocin, a marker for podocyte damage, in the diabetic Hif1α +/− renal cortex. These results suggest that HIF-1α functional impairment affected the survival of podocytes in the diabetes-exposed kidney. It is important to notice that systemic pharmacological approaches used in previous studies of DN [3, 6, 16] may produce HIF-1-independent effects and may also affect other tissues resulting in different responses in diabetes-exposed kidneys.
In response to injury, mesangial cells transdifferentiate and synthesize different extracellular matrix proteins, which is an important pathological event during glomerulosclerosis and the progression of DN. The increased expression of transcription factor SOX9 has been associated with changes in mesangial cells and expansion of the mesangial area in the progression of DN [28]. Additionally, the activation of SOX9 is critical for the early damage and repair response of injured renal tubule cells [47]. This repair response in the chronically active form may represent an additional mechanism triggering long-term pathological responses resulting in kidney damage. Not only HIF-1 mediates Sox9 expression, ERK1/2 signaling [48] or BMP4 [28] may also induce Sox9 expression. Furthermore, advanced glycation end products (AGEs) have been shown to induce Sox9 expression [28]. Thus, we can postulate that increased Sox9 expression in the diabetic Hif1α +/− renal cortex may indicate a) an early transcriptional response to renal injury or/and b) regulatory compensatory response to Hif1α deficiency and diabetic environment.
We found increased collagen accumulation in both diabetic Hif1α +/− and Wt mice. Correspondingly, the expression of markers of fibrosis and extracellular matrix accumulation, Tgfβ1, fibronectin, Ctgf, and α-SMA were increased in both diabetic Hif1α +/− and Wt mice. These results indicate that fibrosis in the diabetic kidney was not affected by the global reduction of Hif1α, at least not in the early phase of diabetic exposure. In line with our observations are studies where the global Hif1α deletion using the Ubc-cre/ERT2 system did not affect collagen accumulation, although inflammation and renal injury were enhanced by Hif1α deletion in the model of unilateral ureteral obstruction [49].
VEGFA stimulates endothelial cell proliferation and has a key role in physiologic and pathologic angiogenesis in different tissues. In the kidney, VEGFA regulates glomerular permeability and maintenance of the glomerular tuft, and overall maintenance of kidney integrity [50]. VEGFA is tightly regulated as shown by glomerular-selective overexpression or deletion of VEGFA resulting in severe and early renal pathologies [33]. Renal diseases are frequently associated with impaired angiogenesis, capillary loss, and a reduction of VEGFA expression. In contrast, in diabetic nephropathy, renal VEGFA levels are elevated in experimental models as well as in diabetic patients [51–53] The upregulation of VEGFA has been proposed as a contributing mechanism to renal dysfunction during the early phase of diabetes [53, 54]. Inhibition of VEGFA at the onset of diabetes abolished the associated diabetes-glomerular hyperfiltration, glomerular hypertrophy, and urinary albumin excretion in the type I diabetes model [53]. In our study, consistent with previously published data, VEGFA expression was significantly increased in the glomerulus of diabetic Hif1α +/− compared to the diabetic Wt, indicating a faster progression of renal dysfunction in diabetes (Fig. 4). The cause of the upregulation of VEGFA in the diabetic kidney remains speculative; however, multiple factors may be implicated [53]. Renal dysfunction of diabetic Hif1α +/− mice was further supported by the increased expression of Adm in the diabetic Hif1α +/− renal cortex. The upregulation of Adm, which encodes a potent vasorelaxant peptide, is associated with glomerular hyperfiltration and dilatation of the glomerular capillaries in the acute phase of type 1 diabetes [55]. Notably, serum albumin levels were significantly decreased in diabetic Hif1α +/− mice (Fig.1).
A limitation of our study is the global nature of the Hif1α deletion. We are unable to determine which cell type or which combinations of cell types are contributing to the increased susceptibility of Hif1α +/− mice to DN. The global deletion of Hif1α may affect other tissues and it may indirectly escalate pathological functional and structural changes in the kidney of Hif1α +/− mutants. At the same time, our model reproduces the conditions of a global inhibition of HIF-1 signaling, such as in pharmacological targeted-HIF-1 inhibition.
Conclusions
Taken together, our studies point to a protective role of HIF-1 signaling in the early phase of adaptive responses to diabetic environment and that impaired HIF-1 signaling results in a faster progression of DN. Furthermore, our data suggest a potential role of Hif1α genetic variations in the manifestation of DN. Although the modulation of HIF-1 activity is a high-priority target for clinical therapies, our data accentuate the necessity of optimizing possible pharmacological inhibition of HIF-1 in therapeutic applications for the treatment of DN.
Acknowledgements
We thank A. Pavlinek for editing the manuscript. Serum analysis were done by the Czech Centre for Phenogenomics supported by LM2015040, LQ1604 (NPU II), by MEYS and CAS (RVO 68378050). We thank the Imaging Methods Core Facility at BIOCEV supported by the MEYS CR (LM2015062 Czech-BioImaging).
Funding
This work was supported by the Czech Science Foundation (Grant Agreement No. 16-06825S to GP); by BIOCEV CZ.1.05/1.1.00/02.0109 from the ERDF; and AVOZ50520701 from the MEYS; by the Charles University in Prague (GA UK No. 228416 to RC).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- Adm
Adrenomedullin
- AGE
Advanced glycation end products
- Ctgf
Connective tissue growth factor
- Cx43
Connexin 43
- DN
Diabetic nephropathy
- Fn1
Fibronectin 1
- GSIS
Glucose-stimulated insulin secretion
- HIF-1
Hypoxia inducible factor 1
- Nphs2
Podocin
- Ntn1
Netrin
- PAS
Periodic acid–Schiff staining
- Pdk1
Pyruvatdehydrogenase kinase1
- ROS
Reactive oxygen species
- Sox9
SRY (Sex Determining Region Y)-Box 9
- STZ
Streptozotocin
- Tgfβ1
Transforming growth factor beta 1
- Vegfa
Vascular endothelial growth factor
- Wt
Wild-type
- Wt1
Wilms tumor 1 homolog
- α-SMA
Alpha 2 smooth muscle actin
Additional file
{kind=link}
The additional file lists primer sequences for genes analyzed by qPCR. (JPEG 122 kb)
Authors’ contributions
All authors have read and approved the manuscript. G.P. conceived the study and takes responsibility for the integrity of the data. GP and RB co-wrote the manuscript. RC provided critical reading of the manuscript. RB and KN conducted animal study. RB and KN designed and performed qPCR analyses. RB performed Western blotting and confocal immunohistochemical analyses. RC designed and conducted histological and morphological analyses, and interpreted acquired data.
Ethics approval and consent to participate
This study was conducted in accordance with the Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, revised 1996). The experimental protocol was approved by the Animal Care and Use Committee of the Institute of Molecular Genetics, CAS. All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. All procedures performed in studies involving animals were in accordance with the ethical standards of the Institute of Molecular Genetics CAS at which the studies were conducted.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s12902-017-0200-8) contains supplementary material, which is available to authorized users.
Contributor Information
Romana Bohuslavova, Email: romana.bohuslavova@ibt.cas.cz.
Radka Cerychova, Email: radka.cerychova@ibt.cas.cz.
Katerina Nepomucka, Email: katerina.nepomucka@img.cas.cz.
Gabriela Pavlinkova, Phone: (+420) 32587-3794, Email: gpavlinkova@ibt.cas.cz.
References
- 1.Parving HH. Blockade of the renin-angiotensin-aldosterone system and renal protection in diabetes mellitus. J Renin-Angiotensin-Aldosterone Syst. 2000;1(1):30–31. doi: 10.3317/jraas.2000.006. [DOI] [PubMed] [Google Scholar]
- 2.Taft JL, Nolan CJ, Yeung SP, Hewitson TD, Martin FI. Clinical and histological correlations of decline in renal function in diabetic patients with proteinuria. Diabetes. 1994;43(8):1046–1051. doi: 10.2337/diab.43.8.1046. [DOI] [PubMed] [Google Scholar]
- 3.Nordquist L, Friederich-Persson M, Fasching A, Liss P, Shoji K, Nangaku M, Hansell P, Palm F. Activation of hypoxia-inducible factors prevents diabetic nephropathy. J Am Soc Nephrol. 2015;26(2):328–338. doi: 10.1681/ASN.2013090990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rosenberger C, Khamaisi M, Abassi Z, Shilo V, Weksler-Zangen S, Goldfarb M, Shina A, Zibertrest F, Eckardt KU, Rosen S, et al. Adaptation to hypoxia in the diabetic rat kidney. Kidney Int. 2008;73(1):34–42. doi: 10.1038/sj.ki.5002567. [DOI] [PubMed] [Google Scholar]
- 5.Isoe T, Makino Y, Mizumoto K, Sakagami H, Fujita Y, Honjo J, Takiyama Y, Itoh H, Haneda M. High glucose activates HIF-1-mediated signal transduction in glomerular mesangial cells through a carbohydrate response element binding protein. Kidney Int. 2010;78(1):48–59. doi: 10.1038/ki.2010.99. [DOI] [PubMed] [Google Scholar]
- 6.Nayak BK, Shanmugasundaram K, Friedrichs WE, Cavaglierii RC, Patel M, Barnes J, Block K. HIF-1 mediates renal fibrosis in OVE26 type 1 diabetic mice. Diabetes. 2016;65(5):1387–1397. doi: 10.2337/db15-0519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Semenza GL. Oxygen sensing, homeostasis, and disease. N Engl J Med. 2011;365(6):537–547. doi: 10.1056/NEJMra1011165. [DOI] [PubMed] [Google Scholar]
- 8.Li J, Bosch-Marce M, Nanayakkara A, Savransky V, Fried SK, Semenza GL, Polotsky VY. Altered metabolic responses to intermitternt hypoxia in mice with partial deficiency of hypoxia-inducible factor 1 a. Physiol Genomics. 2006;25:450–457. doi: 10.1152/physiolgenomics.00293.2005. [DOI] [PubMed] [Google Scholar]
- 9.Bohuslavova R, Kolar F, Kuthanova L, Neckar J, Tichopad A, Pavlinkova G. Gene expression profiling of sex differences in HIF1-dependent adaptive cardiac responses to chronic hypoxia. J Appl Physiol. 2010;109(4):1195–1202. doi: 10.1152/japplphysiol.00366.2010. [DOI] [PubMed] [Google Scholar]
- 10.Semenza GL. Hypoxia-inducible factor 1 and cardiovascular disease. Annu Rev Physiol. 2014;76:39–56. doi: 10.1146/annurev-physiol-021113-170322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell. 2010;40(2):294–309. doi: 10.1016/j.molcel.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Makino H, Miyamoto Y, Sawai K, Mori K, Mukoyama M, Nakao K, Yoshimasa Y, Suga S. Altered gene expression related to glomerulogenesis and podocyte structure in early diabetic nephropathy of db/db mice and its restoration by pioglitazone. Diabetes. 2006;55(10):2747–2756. doi: 10.2337/db05-1683. [DOI] [PubMed] [Google Scholar]
- 13.Catrina SB, Okamoto K, Pereira T, Brismar K, Poellinger L. Hyperglycemia regulates hypoxia-inducible factor-1alpha protein stability and function. Diabetes. 2004;53(12):3226–3232. doi: 10.2337/diabetes.53.12.3226. [DOI] [PubMed] [Google Scholar]
- 14.Thangarajah H, Yao D, Chang EI, Shi Y, Jazayeri L, Vial IN, Galiano RD, Du XL, Grogan R, Galvez MG, et al. The molecular basis for impaired hypoxia-induced VEGF expression in diabetic tissues. Proc Natl Acad Sci U S A. 2009;106(32):13505–13510. doi: 10.1073/pnas.0906670106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bento CF, Pereira P. Regulation of hypoxia-inducible factor 1 and the loss of the cellular response to hypoxia in diabetes. Diabetologia. 2011;54(8):1946–1956. doi: 10.1007/s00125-011-2191-8. [DOI] [PubMed] [Google Scholar]
- 16.Ohtomo S, Nangaku M, Izuhara Y, Takizawa S, Strihou C, Miyata T. Cobalt ameliorates renal injury in an obese, hypertensive type 2 diabetes rat model. Nephrol Dial Transplant. 2008;23(4):1166–1172. doi: 10.1093/ndt/gfm715. [DOI] [PubMed] [Google Scholar]
- 17.Salbaum JM, Kruger C, Zhang X, Delahaye NA, Pavlinkova G, Burk DH, Kappen C. Altered gene expression and spongiotrophoblast differentiation in placenta from a mouse model of diabetes in pregnancy. Diabetologia. 2011;54(7):1909–1920. doi: 10.1007/s00125-011-2132-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bohuslavova R, Kolar F, Sedmera D, Skvorova L, Papousek F, Neckar J, Pavlinkova G. Partial deficiency of HIF-1alpha stimulates pathological cardiac changes in streptozotocin-induced diabetic mice. BMC Endocr Disord. 2014;14(1):11. doi: 10.1186/1472-6823-14-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, Gassmann M, Gearhart JD, Lawler AM, Yu AY, et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 1998;12(2):149–162. doi: 10.1101/gad.12.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peng YJ, Yuan G, Ramakrishnan D, Sharma SD, Bosch-Marce M, Kumar GK, Semenza GL, Prabhakar NR. Heterozygous HIF-1alpha deficiency impairs carotid body-mediated systemic responses and reactive oxygen species generation in mice exposed to intermittent hypoxia. J Physiol. 2006;577(Pt 2):705–716. doi: 10.1113/jphysiol.2006.114033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bosch-Marce M, Okuyama H, Wesley JB, Sarkar K, Kimura H, Liu YV, Zhang H, Strazza M, Rey S, Savino L, et al. Effects of aging and hypoxia-inducible factor-1 activity on angiogenic cell mobilization and recovery of perfusion after limb ischemia. Circ Res. 2007;101(12):1310–1318. doi: 10.1161/CIRCRESAHA.107.153346. [DOI] [PubMed] [Google Scholar]
- 22.Bohuslavova R, Skvorova L, Sedmera D, Semenza GL, Pavlinkova G. Increased susceptibility of HIF-1alpha heterozygous-null mice to cardiovascular malformations associated with maternal diabetes. J Mol Cell Cardiol. 2013;60:129–141. doi: 10.1016/j.yjmcc.2013.04.015. [DOI] [PubMed] [Google Scholar]
- 23.Breyer MD, Bottinger E, Brosius FC, 3rd, Coffman TM, Harris RC, Heilig CW, Sharma K, Amdcc Mouse models of diabetic nephropathy. J Am Soc Nephrol. 2005;16(1):27–45. doi: 10.1681/ASN.2004080648. [DOI] [PubMed] [Google Scholar]
- 24.Huang HC, Preisig PA. G1 kinases and transforming growth factor-beta signaling are associated with a growth pattern switch in diabetes-induced renal growth. Kidney Int. 2000;58(1):162–172. doi: 10.1046/j.1523-1755.2000.00151.x. [DOI] [PubMed] [Google Scholar]
- 25.Perticone M, Maio R, Sciacqua A, Cimellaro A, Andreucci M, Tripepi G, Zoccali C, Sesti G, Perticone F. Serum phosphorus levels are associated with endothelial dysfunction in hypertensive patients. Nutr Metab Cardiovasc Dis. 2016;26(8):683–688. doi: 10.1016/j.numecd.2016.02.003. [DOI] [PubMed] [Google Scholar]
- 26.Soulis T, Thallas V, Youssef S, Gilbert RE, McWilliam BG, Murray-McIntosh RP, Cooper ME. Advanced glycation end products and their receptors co-localise in rat organs susceptible to diabetic microvascular injury. Diabetologia. 1997;40(6):619–628. doi: 10.1007/s001250050725. [DOI] [PubMed] [Google Scholar]
- 27.Tervaert TW, Mooyaart AL, Amann K, Cohen AH, Cook HT, Drachenberg CB, Ferrario F, Fogo AB, Haas M, de Heer E, et al. Pathologic classification of diabetic nephropathy. J Am Soc Nephrol. 2010;21(4):556–563. doi: 10.1681/ASN.2010010010. [DOI] [PubMed] [Google Scholar]
- 28.Kishi S, Abe H, Akiyama H, Tominaga T, Murakami T, Mima A, Nagai K, Kishi F, Matsuura M, Matsubara T, et al. SOX9 protein induces a chondrogenic phenotype of mesangial cells and contributes to advanced diabetic nephropathy. J Biol Chem. 2011;286(37):32162–32169. doi: 10.1074/jbc.M111.244541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bottinger EP. TGF-beta in renal injury and disease. Semin Nephrol. 2007;27(3):309–320. doi: 10.1016/j.semnephrol.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 30.Rosenberger P, Schwab JM, Mirakaj V, Masekowsky E, Mager A, Morote-Garcia JC, Unertl K, Eltzschig HK. Hypoxia-inducible factor-dependent induction of netrin-1 dampens inflammation caused by hypoxia. Nat Immunol. 2009;10(2):195–202. doi: 10.1038/ni.1683. [DOI] [PubMed] [Google Scholar]
- 31.Tittarelli A, Janji B, Van Moer K, Noman MZ, Chouaib S. The selective degradation of synaptic connexin 43 protein by hypoxia-induced autophagy impairs natural killer cell-mediated tumor cell killing. J Biol Chem. 2015;290(39):23670–23679. doi: 10.1074/jbc.M115.651547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sato T, Haimovici R, Kao R, Li AF, Roy S. Downregulation of connexin 43 expression by high glucose reduces gap junction activity in microvascular endothelial cells. Diabetes. 2002;51(5):1565–1571. doi: 10.2337/diabetes.51.5.1565. [DOI] [PubMed] [Google Scholar]
- 33.Eremina V, Sood M, Haigh J, Nagy A, Lajoie G, Ferrara N, Gerber HP, Kikkawa Y, Miner JH, Quaggin SE. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J Clin Invest. 2003;111(5):707–716. doi: 10.1172/JCI17423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guo JK, Menke AL, Gubler MC, Clarke AR, Harrison D, Hammes A, Hastie ND, Schedl A. WT1 is a key regulator of podocyte function: reduced expression levels cause crescentic glomerulonephritis and mesangial sclerosis. Hum Mol Genet. 2002;11(6):651–659. doi: 10.1093/hmg/11.6.651. [DOI] [PubMed] [Google Scholar]
- 35.Essawy M, Soylemezoglu O, Muchaneta-Kubara EC, Shortland J, Brown CB, el Nahas AM. Myofibroblasts and the progression of diabetic nephropathy. Nephrol Dial Transplant. 1997;12(1):43–50. doi: 10.1093/ndt/12.1.43. [DOI] [PubMed] [Google Scholar]
- 36.Palm F, Hansell P, Ronquist G, Waldenstrom A, Liss P, Carlsson PO. Polyol-pathway-dependent disturbances in renal medullary metabolism in experimental insulin-deficient diabetes mellitus in rats. Diabetologia. 2004;47(7):1223–1231. doi: 10.1007/s00125-004-1434-3. [DOI] [PubMed] [Google Scholar]
- 37.Kolyada AY, Tighiouart H, Perianayagam MC, Liangos O, Madias NE, Jaber BL. A genetic variant of hypoxia-inducible factor-1alpha is associated with adverse outcomes in acute kidney injury. Kidney Int. 2009;75(12):1322–1329. doi: 10.1038/ki.2009.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gu HF, Zheng X, Abu Seman N, Gu T, Botusan IR, Sunkari VG, Lokman EF, Brismar K, Catrina SB. Impact of the hypoxia-inducible factor-1 alpha (HIF1A) Pro582Ser polymorphism on diabetes nephropathy. Diabetes Care. 2013;36(2):415–421. doi: 10.2337/dc12-1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Haase VH. Hypoxia-inducible factors in the kidney. Am J Physiol Renal Physiol. 2006;291(2):F271–F281. doi: 10.1152/ajprenal.00071.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brukamp K, Jim B, Moeller MJ, Haase VH. Hypoxia and podocyte-specific Vhlh deletion confer risk of glomerular disease. Am J Physiol Renal Physiol. 2007;293(4):F1397–F1407. doi: 10.1152/ajprenal.00133.2007. [DOI] [PubMed] [Google Scholar]
- 41.Song YR, You SJ, Lee YM, Chin HJ, Chae DW, Oh YK, Joo KW, Han JS, Na KY. Activation of hypoxia-inducible factor attenuates renal injury in rat remnant kidney. Nephrol Dial Transplant. 2010;25(1):77–85. doi: 10.1093/ndt/gfp454. [DOI] [PubMed] [Google Scholar]
- 42.Deng A, Arndt MA, Satriano J, Singh P, Rieg T, Thomson S, Tang T, Blantz RC. Renal protection in chronic kidney disease: hypoxia-inducible factor activation vs. angiotensin II blockade. Am J Physiol Renal Physiol. 2010;299(6):F1365–F1373. doi: 10.1152/ajprenal.00153.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kimura K, Iwano M, Higgins DF, Yamaguchi Y, Nakatani K, Harada K, Kubo A, Akai Y, Rankin EB, Neilson EG, et al. Stable expression of HIF-1alpha in tubular epithelial cells promotes interstitial fibrosis. Am J Physiol Renal Physiol. 2008;295(4):F1023–F1029. doi: 10.1152/ajprenal.90209.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Higgins DF, Kimura K, Bernhardt WM, Shrimanker N, Akai Y, Hohenstein B, Saito Y, Johnson RS, Kretzler M, Cohen CD, et al. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J Clin Invest. 2007;117(12):3810–3820. doi: 10.1172/JCI30487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cantley J, Selman C, Shukla D, Abramov AY, Forstreuter F, Esteban MA, Claret M, Lingard SJ, Clements M, Harten SK, et al. Deletion of the von Hippel-Lindau gene in pancreatic beta cells impairs glucose homeostasis in mice. J Clin Invest. 2009;119(1):125–135. doi: 10.1172/JCI26934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cheng K, Ho K, Stokes R, Scott C, Lau SM, Hawthorne WJ, O'Connell PJ, Loudovaris T, Kay TW, Kulkarni RN, et al. Hypoxia-inducible factor-1alpha regulates beta cell function in mouse and human islets. J Clin Invest. 2010;120(6):2171–2183. doi: 10.1172/JCI35846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kumar S, Liu J, Pang P, Krautzberger AM, Reginensi A, Akiyama H, Schedl A, Humphreys BD, McMahon AP. Sox9 activation highlights a cellular pathway of renal repair in the acutely injured mammalian kidney. Cell Rep. 2015;12(8):1325–1338. doi: 10.1016/j.celrep.2015.07.034. [DOI] [PubMed] [Google Scholar]
- 48.Ling S, Chang X, Schultz L, Lee TK, Chaux A, Marchionni L, Netto GJ, Sidransky D, Berman DM. An EGFR-ERK-SOX9 signaling cascade links urothelial development and regeneration to cancer. Cancer Res. 2011;71(11):3812–3821. doi: 10.1158/0008-5472.CAN-10-3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kobayashi H, Gilbert V, Liu Q, Kapitsinou PP, Unger TL, Rha J, Rivella S, Schlondorff D, Haase VH. Myeloid cell-derived hypoxia-inducible factor attenuates inflammation in unilateral ureteral obstruction-induced kidney injury. J Immunol. 2012;188(10):5106–5115. doi: 10.4049/jimmunol.1103377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schrijvers BF, Flyvbjerg A, De Vriese AS. The role of vascular endothelial growth factor (VEGF) in renal pathophysiology. Kidney Int. 2004;65(6):2003–2017. doi: 10.1111/j.1523-1755.2004.00621.x. [DOI] [PubMed] [Google Scholar]
- 51.Cooper ME, Vranes D, Youssef S, Stacker SA, Cox AJ, Rizkalla B, Casley DJ, Bach LA, Kelly DJ, Gilbert RE. Increased renal expression of vascular endothelial growth factor (VEGF) and its receptor VEGFR-2 in experimental diabetes. Diabetes. 1999;48(11):2229–2239. doi: 10.2337/diabetes.48.11.2229. [DOI] [PubMed] [Google Scholar]
- 52.Nakagawa T. Uncoupling of the VEGF-endothelial nitric oxide axis in diabetic nephropathy: an explanation for the paradoxical effects of VEGF in renal disease. Am J Physiol Renal Physiol. 2007;292(6):F1665–F1672. doi: 10.1152/ajprenal.00495.2006. [DOI] [PubMed] [Google Scholar]
- 53.de Vriese AS, Tilton RG, Elger M, Stephan CC, Kriz W, Lameire NH. Antibodies against vascular endothelial growth factor improve early renal dysfunction in experimental diabetes. J Am Soc Nephrol. 2001;12(5):993–1000. doi: 10.1681/ASN.V125993. [DOI] [PubMed] [Google Scholar]
- 54.Flyvbjerg A, Dagnaes-Hansen F, De Vriese AS, Schrijvers BF, Tilton RG, Rasch R. Amelioration of long-term renal changes in obese type 2 diabetic mice by a neutralizing vascular endothelial growth factor antibody. Diabetes. 2002;51(10):3090–3094. doi: 10.2337/diabetes.51.10.3090. [DOI] [PubMed] [Google Scholar]
- 55.Hiragushi K, Wada J, Eguchi J, Matsuoka T, Yasuhara A, Hashimoto I, Yamashita T, Hida K, Nakamura Y, Shikata K, et al. The role of adrenomedullin and receptors in glomerular hyperfiltration in streptozotocin-induced diabetic rats. Kidney Int. 2004;65(2):540–550. doi: 10.1111/j.1523-1755.2004.00407.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.