

Abstract
Introduction
Numerous epidemiological studies indicate an inverse association between birth weight and the risk for chronic kidney disease.
Areas covered
Historically, the first studies to address the developmental origins of chronic disease focused on the inverse relationship between birth weight and blood pressure. A reduction in nephron number was a consistent finding in low birth weight individuals and experimental models of developmental insult. Recent studies indicate that a congenital reduction in renal reserve in conjunction with an increase in blood pressure that has its origins in fetal life increases vulnerability to renal injury and disease.
Expert commentary
Limited experimental studies have investigated the mechanisms that contribute to the developmental origins of kidney disease. Several studies suggest that enhanced susceptibility to renal injury following a developmental insult is altered by sex and age. More in-depth studies are needed to clarify how low birth weight contributes to enhanced renal risk, and how sex and age influence this adverse relationship.
Keywords: Developmental Origins, Low Birth Weight, IUGR, Hypertension, Chronic Kidney Disease, Nephron Number
1. Introduction
1.1. Overview
Kidney disease, one of the leading causes of morbidity and mortality worldwide, contributes to increased risk for cardiovascular disease, recurrent hospitalizations, and billions of dollars in health-care expenditures each year (1). The National Kidney Foundation defines chronic kidney disease (CKD) as “abnormalities of kidney structure or function, present for >3 months, with implications for health (2).” The criteria for CKD includes a reduced glomerular filtration rate (GFR) and markers of kidney damage. Although this primarily refers to albuminuria, other molecular markers of kidney injury, electrolyte imbalances, histological changes, structural abnormalities, or a history of kidney transplantation may also be used for diagnosis. CKD is classified by degree of renal function from normal functioning with secondary evidence of kidney injury to kidney failure. In contrast to CKD, an acute kidney injury (AKI) is marked by a rapid increase in serum creatinine above the patient’s baseline within one week and a reduction in urine output for greater than 6 hours (3). AKI increases the risk for later development of CKD (3), and superimposed acute renal injury in a patient with CKD increases the risk for end-stage renal disease (ESRD) requiring dialysis or renal transplant (4).
Numerous studies indicate that CKD exhibits sex-specific differences (1, 5, 6, 7, 8). Although the overall prevalence of kidney disease is greater in females compared to males (1), perhaps due to the greater life expectancy of females, the decline in renal function and incidence of ESRD is greater in males with CKD compared to female counterparts. In a meta-analysis performed by Neugarten and colleagues (8) involving patients with nondiabetic chronic renal disease (criteria included CKD of mixed etiology, IgA nephropathy, membranous nephropathy, and autosomal dominant polycystic kidney disease), males had a more rapid progression to ESRD than females. Data gathered from the Modification of Diet in Renal Disease clinical trial also showed a greater rate of GFR decline in males versus females (7). In this study, the sex difference in renal susceptibility was abolished when controlled for higher baseline proteinuria and arterial pressure, and lower baseline HDL cholesterol in males (7). These findings suggest that sex differences in renal decline are related to variations in the maintenance of glomerular membrane integrity, hypertensive nephrosclerosis, and lipid metabolism between men and women. Yet, the exact mechanisms that contribute to the sexual dimorphism of renal susceptibility remain unknown.
Diabetes and hypertension are the most common causes of CKD (9). Although not completely understood, reductions in kidney function can be broadly linked to loss of functional nephrons via glomerular injury, tubular injury, interstitial fibrosis, and a combination of these factors. Studies in the past 20 years indicate that influences during fetal life that limit growth may also contribute to greater renal susceptibility. A congenital reduction in nephron number may be a major contributor to the developmental origins of CKD (10). Thus, this review will focus on the developmental origins of enhanced susceptibility to renal injury and increased risk for CKD.
1.2. Developmental origins of chronic health and disease
Dr. David Barker, an English epidemiologist, is credited as the first to propose the hypothesis of fetal or developmental origins of chronic health and disease. In a paper published in 1986, Dr. Barker (11) reported that rates of coronary heart disease were higher in poorer regions of England and Wales in association with higher rates of infant mortality 50–60 years earlier. Based on this geographical association that existed 50 to 60 years apart, Barker hypothesized that similar to the infants who died during the earlier period, those who survived unfavorable conditions during perinatal life underwent physiological adaptations that predisposed them to increased risk for cardiovascular disease in later life (11). Since the reporting of Dr. Barker’s fetal origins hypothesis (12, 13), numerous epidemiological studies have expanded this concept to encompass a role for the development origins of CKD in addition to a host of other chronic diseases.
Prior to discussing the developmental origins of renal disease, it is important to define the terminologies. The terminology most commonly used to indicate an adverse fetal environment, regardless of the etiology, is “intrauterine growth restriction” or IUGR. IUGR indicates an inability of the fetus to reach its growth potential when compared to the expected trajectory at any point during gestation (14, 15). Low birth weight (LBW) has been used as a non-specific marker of an adverse fetal environment, and is defined by the World Health Organization as a birth weight of less than 2500 grams (16). By this definition, LBW may refer to both full term and preterm infants or those born before the 37th week of gestation. “Small for gestational age” (SGA) is a narrower term that refers to infants with birth weights below the 10th percentile for their gestational age (17, 18). Therefore, SGA may be a better indicator of IUGR (19). However, weight below 2500g or LBW remains the standard utilized in studies related to later increased risk of chronic health and disease that follows slow or impaired growth during fetal life. In practice, the term LBW is used more commonly than SGA, and it is often used interchangeably with IUGR.
The field of developmental origins of chronic health and disease has broadened since Barker’s first study that implicated a relationship between infant mortality and death from coronary heart disease (11). Although early investigators focused on maternal undernutrition as the primary cause of IUGR, the etiology of IUGR is multi-faceted (20). Preeclampsia is one of the most common causes of fetal growth restriction in the United States, and its incidence has risen continually over the last three decades (21). However, other placental disorders, perinatal exposures to nicotine, other synthetic chemicals or environmental pollutants, chronic maternal illness or stress, and maternal infections are also associated with IUGR (18, 22, 23). Numerous epidemiological and experimental studies focus on the link between IUGR of different origins and the chronic health outcomes of offspring in later life. Brenner (10) and others (24, 25, 26) have applied this hypothesis to the etiology of kidney disease. Like Barker, Brenner postulated that IUGR leads to a redistribution of nutrient delivery to supply essential tissues during development at the expense of organs like the kidney which typically have a high functional reserve (10). Brenner hypothesized that redirection of blood flow away from the kidneys results in a congenital lack of nephrons (10). A reduction in nephron number leading to chronic hyper-filtration and hypertension would contribute to increased susceptibility to a secondary renal insult, particularly with aging, due to enhanced loss of renal reserve (10). Normal congenital nephron number is far in excess of what is needed for normal function, but a loss of the renal reserve which usually compensates for renal damage that occurs throughout the lifespan puts individuals with low renal reserve at greater risk. Thus, a loss of excess nephron stores in response to a developmental insult may lead to the development of kidney disease over time, in particular following a secondary insult (Figure).
Figure.
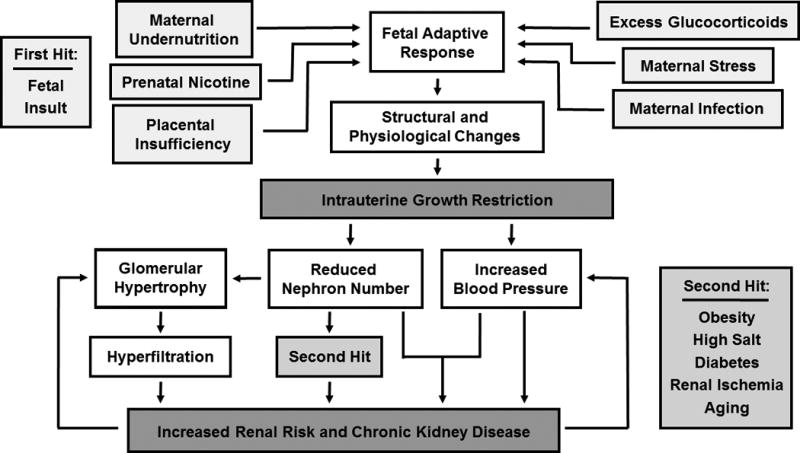
A schematic representing the pathway by which numerous adverse exposures during fetal life program an increased risk for renal susceptibility and chronic renal disease.
2. Epidemiological studies linking birth weight and renal disease
2.1. Methodology: nephron number
Despite the high degree of variability in nephron number among the general population that ranges from 200,000 to 2.5 million per kidney with an average of 900,000 to one million (26), human studies consistently show a significant inverse relationship between birth weight and nephron endowment in accordance with Brenner’s hypothesis (27, 28, 29). However, evidence directly linking nephron number to kidney disease is inconclusive because until recently, nephron number was always determined post-mortem with kidney injury determined by histological assessment of the ex vivo kidney rather than measurement of renal hemodynamics, proteinuria or other functional studies. Another caveat related to measurement of nephron number at autopsy involves the ability to clarify whether an individual had a congenital reduction in nephron number that predated the development of kidney disease, or if a greater destruction of nephrons developed after injury or disease. Even independent of kidney disease, there is a normal loss of nephrons with aging; this further limits the ability to make conclusions from currently available findings. Loss of functional nephrons occurs at a rate of approximately 10% per decade after 40 years of age. Denic and colleagues at the Mayo and Cleveland Clinics used biopsy samples from living kidney donors to estimate functional nephron number (30). Their results published in 2016 indicate that loss of nephrons with aging may be underestimated when using kidneys obtained at autopsy (30). This underestimate of nephron loss determined at autopsy results from the practice of using glomerulosclerosis as a marker of nephron loss, despite the occurrence of atrophy and resorption of damaged tissue that could introduce another source of variability in estimated values. In addition, the various methods by which nephron complement is determined are semi-quantitative and prone to error. Due to these confounding factors, it is difficult to determine if nephron loss results from a congenital, early-life nephron deficit, an accelerated nephron loss with aging, or an additional decrease in nephrons due to a secondary renal insult resulting from kidney injury or disease. These biases complicate findings related to nephron number and renal disease.
2.2. Birth weight and renal function and disease
With the popularization and acceptance of the hypothesis of developmental origins of chronic disease and health, a host of observational studies were performed to determine whether an inverse association exists between birth weight and kidney disease. White and colleagues performed a systematic review and meta-analysis of the literature published prior to 2008 with birth weight as a study factor (26). Studies with valid measurement of kidney function were selected. Analysis of the collected findings indicated that very LBW (<1500g) individuals had a greater subsequent risk for development of CKD, determined by albumin/protein excretion, serum creatinine level, creatinine clearance, GFR or presence of ESRD, compared to normal birth weight counterparts (25). Das et al. noted similar findings in a study published in 2016 (24). In this systemic review and meta-analysis of studies published prior to September 2015, LBW was associated with lower GFR (24). In addition, over one third of those with diabetes had LBW and CKD, defined by GFR and the microalbuminuria/albumin excretion rate (AER) /urinary albumin creatinine ratio (ACR) (23). Although these studies used different exclusion criteria and outcome measurements, these studies provided strong support for an association between birth weight and renal disease. A review by Luyckx and Brenner further reported that in addition to CKD as a broad category, LBW was also associated with a more rapid progression and greater severity of other renal diseases such as IgA nephropathy, membranous nephropathy, minimal change disease, nephrotic syndrome, and chronic pyelonephritis (31). Although impairment in nephron mass may be a contributory component, heightened risk for hypertension and diabetes in LBW individuals may also be contributory factors (31). Studies examining the relationship between birth weight and renal disease in childhood or prior to confounding variables such as obesity, smoking, or other chronic disease also demonstrate that the effect of LBW on renal function initiates in early life. Cassidy-Bushrow and colleagues reported that GFR was reduced as early as one year of age in LBW children (32); in a study by Frankfort et al., GFR decreased between one and three years of age in children with the lowest birth weights (33). Khasla et al. showed that by age 12, LBW adolescents had a greater risk for decreased eGFR in association with increased systolic blood pressure (34). CKD is common in the elderly. Yet, a study by Ruggjo et al. demonstrated that LBW and SGA were associated with a higher risk for development of ESRD by age 40 with onset defined as the date of initiating dialysis treatment or undergoing kidney transplantation (35). Taken together, these studies suggest that LBW programs the development of impaired renal function and renal injury in early life, enhancing the risk for CKD by middle age. Collectively, these findings substantiate the need for additional studies that incorporate serial measurements for renal function and markers of renal injury across the lifespan to clarify the etiology for the risk for CKD that has its origins in fetal life.
Gestational age in addition to IUGR may be a contributory factor in the developmental origins of increased kidney disease. Hirano and colleagues demonstrated a strong association between birth weight and gestational age with childhood-onset CKD (36). In babies born at term, the majority of nephron formation occurs during the last trimester of gestation with nephrogenesis completed by week 36 of gestation (37). In infants born preterm, nephrogenesis continues after birth (38) with the final nephron count determined during the postnatal developmental period (39). The addition of preventative treatments in infants born preterm may tax continued renal development during the postnatal period (39). Regardless, abnormal glomerular morphology is observed in kidneys of infants born preterm (40), making assessment of renal function problematic in preterm infants due to immaturity of the kidneys (39). The incidence of preterm birth has increased over the last several decades. Thus, the relative importance of preterm birth versus IUGR on renal disease is another area that requires additional clarification, and advances in accelerating or prolonging nephrogenesis in preterm infants are required to improve renal health and prevent kidney disease.
2.3. Sex Differences in the developmental origins of renal disease
Within the general population the prevalence of CKD is greater in women; however, disease severity is greater in men (5). Therefore, another observation that warrants further investigation involves the effect of sex on the developmental origins of enhanced susceptibility to renal injury and disease. Studies investigating sex differences in renal risk that has its origin in early life are very limited. The Kidney Early Evaluation Program (KEEP) published by the National Kidney Foundation compared 12,364 patients at 18 years and older and reported that an association between CKD was observed only in males; criteria for CKD included serum creatinine, GFR and prevalence of albumin/creatinine ratio and LBW (41). In a prospective cohort study involving young adults between 20 to 30 years of age, Hallen et al. demonstrated that an association between IUGR and low-normal kidney function was present, but was more pronounced in males that females (42). Yet, Lackland et al. demonstrated that early-onset chronic renal failure classified based on Medicare criteria was associated with LBW in both men and women (25). Differences in severity or stage of disease progression evaluated in these studies may contribute to these sex-specific differential outcomes. Regardless, how sex alters susceptibility to renal injury and disease that has its origins in fetal life adds another layer of complexity that is yet to be revealed.
Observational studies within the human population provide correlative support for an inverse association between birth weight and CKD. To provide proof of principle and investigate the mechanisms by which low birth weight increases renal risk in later life, experimental models of LBW are needed to determine how exposure to an adverse fetal environment programs increased susceptibility to kidney injury and disease.
3. Experimental studies linking birth weight and renal risk
3.1. Methodology: nephron number and glomerular filtration rate
In order to evaluate the validity of studies that investigate how an insult during development alters renal structure and function to increase renal risk, a broad knowledge of methods for determining nephron number and evaluating renal function is needed. As in human studies, measurement of nephron number within animal models is challenging, and often lacks reproducibility. Additionally, methods for measurement of renal function vary in regards to reliability and method.
A histological method for estimating nephron number involves counting of glomeruli (the dense capillary beds that are the filtration component of each nephron) within selected kidney sections. This method is limited by the assumption that glomeruli are equally distributed throughout the kidney and also by the inability to detect glomeruli that lie longitudinal to the axis (43). The gold standard method for assessment of glomerular number that uses the three-dimensional structure of the kidney is the physical dissector/fractionation method (44, 45). This method, based on the principle of Sterio (46), is an unbiased approach that uses the whole kidney to provide the most accurate and precise method for estimation of nephron number. Despite this, stereology is not often used. Caveats include substantial histological sample preparation, specialized equipment to allow for simultaneous projection of identical fields, and expense due to labor-intensive counting of glomeruli (44, 45). A simpler, inexpensive alternative to stereology called acid maceration was described by Kaufman et al. in 1975 (47). Slight variations on this technique are widely used by many researchers and this approach estimates glomerular number based on aliquots from whole kidney incubated in a weak acid (44, 45). Although more rapid and less costly, acid maceration is less accurate than the stereology approach (44). If the acid incubation period is too brief or too long, it may result in glomeruli remaining trapped within surrounding tissue or destroyed. Tubular segments that coil during the preparation process take on the appearance of the rounded glomeruli. These variables make structures difficult to identify resulting in some glomeruli being ignored or counted multiple times. Non-uniform suspension of sample aliquot can also lead to counting error (44). Despite these concerns, this process yields reproducible results and can serve as a reasonable estimate for determining nephron numbers. Magnetic resonance imaging (MRI) is an emerging field in the estimation of nephron number. MRI is the most rapid method for nephron quantitation, and it is the only technique that can be utilized in vivo. This method labels glomeruli with ferritin, a protein-based nanoparticle that accumulates within the glomerular basement membrane, to provide a direct measurement of every glomerulus in the kidney (44, 45). This method also allows determination of intra-renal glomerular distribution (45). Although this methodology may become a useful clinical tool, glomeruli can go undetected in the presence of pre-existing renal pathology due to loss of the contrast agent in kidneys with extensive glomerulosclerosis (44, 45). MRI also requires a substantial investment in equipment making the cost prohibitive for most laboratories (44, 48).
Abnormal urinary excretion of albuminuria is a marker of renal damage (49). 24-hour collection for analysis albuminuria is the gold standard; however, an acute measurement when adjusted to urinary creatinine can correct for variations in urinary flow rate and concentration (49). As in clinical practice, the primary indicator of kidney function in experimental models is measurement of GFR (50). Assessment of GFR may involve indirect or direct methods. Creatinine, an endogenous waste production of muscle metabolism that is freely filtered by the kidney, provides a reliable estimate of GFR. This indirect approach requires only a single blood sample. However, it is a less accurate estimate of GFR at extremes of renal function and it may vary between individuals due to differences in muscle mass, age, and sex (51). A more direct method for determination of GFR involves measurement of the clearance rate of an exogenously administered substance that is also freely filtered (50). Historically, this approach was cumbersome because it required catheterization of the animal to allow for venous administration of the exogenous substance, and arterial and bladder catheterization for collection of plasma and urine for determination of the plasma clearance rate of the exogenous substance. Inulin and iothalamate fit the proper criteria for clearance by the kidneys (50), but because of the difficulty of these surgeries, GFR was generally measured only at the endpoint of an experiment. New technologies use a similar principle, but provide a refinement in the approach for direct evaluation of renal function. Transcutaneous measurement of GFR can detect the clearance rate of a fluorescent inulin-like substance which can be injected through a venous catheter or via tail vein to avoid catheterization thus allowing serial measurements (52, 53). In a study by Carrara et al., injection of non-radiolabeled ioxhexol into conscious restrained rats followed by four small blood draws from a tail vein was sufficient to calculate GFR (54). This procedure, which requires analysis of iohexol by HPLC, was validated for male and female rats under healthy conditions or following renal injury (54). MRI with measurement of clearance by a contrast agent can also be used for estimation of GFR (55). MRI allows for serial determination, but a caveat involves the association of larger cost (55). Use of these approaches to determine nephron number and assess renal function provide the basis for mechanistic investigations into how adverse events during development programs increased susceptibility to kidney injury.
Accurate measurement of renal function is also of great importance in the clinical setting. Traynor et al. provide an excellent overview of current approaches for assessment of GFR and highlight that serum creatinine predictions methods are the most widely reported approaches (56). However, Delanghe suggests that inter-laboratory variability makes this method unacceptable for use in infants and young children (57). A need for repeated measurements limits use of radiolabeled substrates for detection of GFR in children and adolescents (58). Thus, Schwartz and Work propose use of ioxhexol for measurement of GFR in children (58). Further discussion of methods for detection of GFR in humans is beyond the scope of this review. However, interpretation of GFR in studies investigating the relationship between birth weight and renal disease require knowledge of best practices regardless of species.
3.2. Intrauterine growth restriction, nephron number and glomerular filtration rate
Early experimental models of developmental programing were designed to replicate clinical findings linking LBW and later cardiovascular risk. The first studies to provide proof of principle involved protein or caloric restriction in pregnant animals (59, 60). Although maternal undernutrition may be a causative contributor to LBW in developing countries, placental insufficiency is the most common cause of LBW in well-nourished populations (61). Although not fully understood, the pathophysiology of preeclampsia is thought to be due to a failure of spiral artery remodeling in the uterus, leading to placental ischemia and release of angiogenic factors into the systemic circulation that contribute to pathology within the mother (62). Numerous studies indicate that offspring of pregnancies complicated by preeclampsia have an increase in blood pressure (63). Thus, models of placental insufficiency induced via bilateral uterine ligation (64) or reduced uterine perfusion pressure (65) were developed to study the etiology of hypertension programmed by fetal exposure to placental ischemia. Additional models of perinatal insult used for investigation into the developmental origins of chronic disease include prenatal exposure to nicotine (66), glucocorticoid excess (67), vitamin (68) and mineral deficiencies (69). Studies utilizing these numerous experimental models of developmental insult have focused extensively on the developmental programming of increased blood pressure (59, 60, 64, 65, 66, 67, 68, 69). Numerous studies have also focused on metabolic disease and impaired glucose homeostasis (70, 71, 72). However, studies indicating that LBW is linked with an increase in blood pressure in addition to a decrease in GFR, risk factors for CKD (34), have led to expanded investigation into the developmental origins of kidney disease.
Numerous studies using animal models of developmental insult demonstrate a link between adverse conditions during development and various degrees of kidney dysfunction in later life (Figure). A reduction in nephron number is a common finding in models of prenatally programmed hypertension including rat offspring exposed to placental insufficiency (64), rat offspring exposed to excess glucocorticoids during fetal life (73), and prenatal exposure to protein restriction in the rat (74, 75) and sheep (76). Prenatal exposure to protein restriction in the rat programs a reduction in creatinine clearance in low protein offspring compared to control offspring by 2 months of age (75). In this study by Xie and colleagues, blood urea nitrogen and proteinuria, markers of renal injury, were elevated in the low protein offspring as early as 3 months of age (75). Glomerular number, measured by the method of acid maceration, was significantly lower, and an inverse relationship between glomerular number and blood pressure was reported for all animals in this study (75). This study did not involve a direct measurement of GFR (75). However, despite the finding that creatinine clearance was elevated in the study by Xie, a reduction in absolute GFR is not always observed in this and other experimental models of developmental insult. In a study by Woods et al., absolute GFR was not reduced in rat offspring exposed to prenatal protein restriction (74). Subtle differences in the degree of prenatal protein restriction may account for potential differential findings; 8.5% low-protein in the study by Woods et al. (74) versus 10% low-protein in the study by Xie (75). However, no change in absolute GFR is also reported in rat offspring exposed to placental insufficiency (65) or prenatal dexamethasone exposure (73). Thus, despite the presence of hypertension (65, 73, 74) and reduced glomerular number (73, 74), absolute GFR is not altered under baseline conditions in many experimental models of developmental insult. In the study by Woods et al., the gold-standard method of stereology was used for estimation of glomerular number (74). Woods and colleagues also reported that GFR, when adjusted per kidney weight, was reduced in the offspring exposed to prenatally protein restriction; however, glomerular volume was increased (74). Thus, collectively findings from the study by Woods et al. suggest that GFR at the level of the nephron may be impaired following a developmental insult. However, glomerular hypertrophy in conjunction with reduced nephron number serves as a compensatory response that contributes to maintenance of absolute GFR. A reduction in nephron number associated with a compensatory increase in glomerular volume is observed in IUGR offspring exposed to placental insufficiency (64). Proteinuria is also observed in IUGR offspring (77) suggesting that maintenance of global GFR by glomerular hypertrophy may occur at the expense of enhanced risk for renal injury (Figure).
Zimanyi and colleagues demonstrated that a reduction in nephron number in low protein offspring is associated with greater vulnerability to a secondary renal insult (78). GFR did not differ under baseline conditions, but GFR was reduced in low protein offspring exposed to a chronic infusion of advanced glycation end-products, a by-product linked to renal disease (78). Susceptibility to a second renal insult is also increased in the model of placental insufficiency as demonstrated in a study by Ojeda (79). Ojeda reported that absolute GFR did not differ in IUGR relative to control rats under baseline conditions. But following exposure to a secondary renal insult of mild ischemia-reperfusion (I/R) (15 minutes of ischemia followed by 2 hours of reperfusion), male IUGR offspring exhibited a significant decrease in absolute GFR that was not observed in control offspring exposed to mild I/R (79). Histological evidence of tubular injury was also observed in I/R exposed IUGR offspring whereas I/R exposed control offspring were unaffected (79). A congenital reduction in nephron number also enhances susceptibility to renal disease in the presence of obesity or overfeeding during post-natal life (80, 81). Taken together, these studies suggest that exposure to an insult during development programs an increased susceptibility to renal injury in response to a secondary insult in later life (Figure). The congenital reduction in nephron number that reduces functional nephron reserve may act to lower the threshold of injury and serve as one mechanism by which enhanced susceptibility to renal injury and disease have their origin in early life. Thus, these studies demonstrate that findings of increased renal risk in animal models of developmental programming share homology with LBW humans, and that use of animal models, despite differences in their method of developmental insult, can provide valuable insight to our understanding of the mechanisms linking an adverse environment during development with increased susceptibility to kidney disease in later life.
3.3. Nephron number, blood pressure and renal injury
A significant increase in blood pressure associated with a significant reduction in nephron number are common findings in different models of developmental insult (Figure) (64, 73, 74, 75). Hypertension is a common cause of CKD; a reduction in congenital renal reserve is strongly associated with increased renal risk. Yet, the extent of nephron loss, or the relative importance of a congenital reduction in nephron number versus an associated increase in blood pressure in the etiology of increased renal susceptibility are not clear. To address whether the degree of nephron loss affects the progression of renal injury, Boubred and colleagues utilized two different models of developmental insult that program a similar increase in blood pressure associated with a differing degree in the congenital reduction in nephron number (82). Their study demonstrated that a greater loss of nephron number in offspring exposed to prenatal betamethasone was associated with a two-fold greater increase in glomerulosclerosis compared to offspring exposed to prenatal protein restriction (82) suggesting that the magnitude of congenital nephron loss contributes to the extent of renal injury (82). However, caveats to the study included methodology and timing for determination of nephron number. Nephron number was determined by acid maceration; blood pressure was determined by tail-cuff, a method that is prone to stress-induced changes in blood pressure (83). Determination of nephron number was at day 1 during nephrogenesis and 22 months of age (82) after established hypertension, a potential confounding variable.
Blood pressure in adulthood is increased in rat (84) or sheep (85) that undergo a surgical reduction in renal mass during development, suggesting that a congenital reduction in nephron number is sufficient to initiate the development hypertension. However, in order to discern the effect of a congenital reduction in nephron number independent of another in utero insult on renal injury and blood pressure, Ruta and colleagues (86) used a genetic mouse model in which a nephron deficit develops spontaneously in a subset of the offspring. Nephron number was measured via stereology with wild type mice compared to genetic mice that exhibited a 65% or 25% reduction in nephron number (86). Blood pressure was measured by the gold-standard method of radio-telemetry which provides an accurate measurement of blood pressure in the absence of stress (83). Blood pressure did not differ between wild type and reduced nephron number groups (86). 24-hour albuminuria and histological analysis for renal injury also did not differ between wild type control and reduced nephron number groups (86). Total renal volume was increased in both nephron deficient groups relative to wild type controls (86). Thus, this study using the best available methods indicated that a congenital deficit in nephron number alone was not sufficient to induce a chronic increase in blood pressure. Importantly, nephron loss in the absence of an increase in blood pressure was not associated with an increase in renal injury. However, when challenged with a chronic salt load, blood pressure was increased in the nephron deficient groups (86), suggesting that a congenital reduction in nephron number can program a greater susceptibility to salt-sensitive hypertension indicative of impaired renal function.
Thus, these studies suggest that a reduction in nephron number initiated in early life lowers the threshold for renal injury, programming an enhanced susceptibility to renal injury and disease. The presence of increased blood pressure may be required to induce enhanced renal susceptibility in the absence of a secondary insult. However, a congenital reduction in renal reserve per se, is not sufficient to program hypertension (Figure).
4. Sex, Age and Renal Dysfunction
A sex difference in the enhanced risk for increased blood pressure is observed in experimental models of developmental insult. Numerous studies using different experimental models demonstrate that female offspring are protected against programmed increases in blood pressure in young adulthood relative to male littermates (65, 673, 87). However, this protection is lost with aging (88, 89, 90, 91). Anderson et al. report that the prevalence of hypertension, a risk factor for CKD, is greater in low birth weight women relative to normal birth weight counterparts by 60 years of age but not at 50 years of age (92). The etiology for the enhanced prevalence of higher blood pressure with age in low birth weight women is not known. Whether similar findings extend to renal disease is not yet clear. Yet, these studies provide rationale for sex- and age-specific effects on the association between birth weight and renal disease, and indicate a need for additional studies to clarify how sex and age influence this adverse relationship.
5. Clinical Caveats
All-cause mortality including risk of renal disease does not differ upon comparison of kidney donors to the general population (93). Yet, recent studies indicate otherwise for individuals with a congenital deficient in nephron number. Long-term outcome including increased blood pressure, established proteinuria, and reduced GFR are reported for Aborigine kidney donors in Australia relative to non-Aborigine donors (94). Glomerular number is significantly reduced in Aborigines relative to non-Aboriginal individuals (95) suggesting that a loss of renal reserve in later life in those already compromised from birth may increase the risk for kidney disease. A recent study by Berglund et al. reported that birth weight in living kidney donors is associated with albuminuria, a powerful predictor for later renal disease (96). Collectively, these studies indicate that exposure to a sub-optimal environment that slows fetal growth and impairs renal development may adversely affect long-term outcome in living kidney donors. However, the effect of in utero programming of increased renal risk may also extend to the allograph recipient. Mismatch between recipient size and donor renal transplant size can adversely affect transplant outcome (97, 98). Using kidney weight as an indicator of nephron mass, a study by Kim et al. showed that a greater ratio of donor kidney weight to recipient body weight led to better 3-year allograph outcome (98). Using a calculated renal reserve in cadaveric renal transplant donor kidneys, Nicholson and colleagues reported that higher nephron dose was also associated with greater allograph renal function outcome (97). For all studies in living donors, kidney function was normal prior to donation. Thus, awareness to the long-term effect of reduced congenital renal reserve on outcome of in renal transplantation are needed.
6. Expert Commentary
Numerous epidemiological studies indicate that LBW is associated with a higher risk for CKD. Experimental studies provide proof of concept. Numerous experimental studies have investigated the mechanisms involved in the development of risk factors associated with CKD including hypertension and cardiovascular disease. Studies have already demonstrated sex differences in the susceptibility to programmed hypertension, with age abolishing the sexual dimorphism seen in low birth weight populations. Yet, few experimental studies are investigating the mechanisms that contribute directly to the developmental origins of kidney disease. Although the field of developmental origins of cardiovascular-renal disease is expanding, more research is needed to clarify the mechanisms that contribute to the association between LBW and heightened susceptibility for CKD in later life. Understanding the mechanisms that contribute to enhanced renal susceptibility will be critical to developing therapeutic interventions and better treatment options for this subset of the population that is at higher risk. As LBW is increasing in acceptance and awareness as a risk factor for CKD, primary care physicians should include questions about a patient’s birth history in addition to traditional risk factors for chronic disease like smoking and dietary habits. Although current evidence about how this might affect treatment strategies is limited, clinicians should be on alert for earlier signs of kidney disease in this population; nephrologists should monitor progression of renal disease in this subset of patients.
7. Five-year view
Although the risk for hypertension and renal disease is low in living kidney donors, a recent study by Berglund et al. reported that living kidney donors born LBW had a greater risk for development of albuminuria. Albuminuria as a marker of early end organ damage introduces a caveat to numerous studies that suggest kidney donation is relatively safe in terms of chronic disease risk to the donor. Numerous epidemiological studies indicate that LBW and/or preterm birth increases the risk for renal injury and disease in later life. Thus, this study by Berglund and colleagues adds another consideration to the field of developmental origins of chronic disease and health by suggesting that a perinatal history should be included during a medical evaluation for consideration as a candidate for kidney donation. The multitude of studies supporting the developmental origins of health and disease indicate that a perinatal history could also be vital in determination of preventative and therapeutic options for an individual’s health. Studies in human cohorts addressing serial changes in renal function and blood pressure with control for confounding variables are needed to discern the progressive nature by which adverse events during development enhance the risk for renal injury and the development of chronic kidney disease. Retrospective longitudinal studies provide insight into adult outcomes in populations born LBW or preterm (99, 100). However, in the decades ahead, findings from several well-characterized cohorts that are tracking the health of babies born LBW and/or preterm will provide important information related to the rate of progression and degree of severity of renal injury and kidney disease in these populations (101, 102). Research using experimental models is expanding. Using “LBW” or “preterm birth” or “SGA” and “renal injury” or “kidney disease” as search terms, over 9,600 search results are reported for 2016 using the PubMed resource. This is more than double from 2006. This is a significant advancement from the publication of Barker’s original paper noting the geographical association related to death from coronary heart disease linked to adverse influences during fetal life that was published in 1986. In the years to come, experimental studies will provide insight into mechanisms and the etiology of increased renal risk that is programmed during fetal life; translational studies will expand these findings to the bedside.
Key Issues.
CKD contributes to increased risk for cardiovascular disease, recurrent hospitalizations, and billions of dollars in health-care expenditures each year. Criteria for CKD include a reduction in glomerular filtration rate and markers of kidney damage such as albuminuria. CKD is associated with a gradual loss of renal function and progression to end-stage kidney failure can be fatal without dialysis or kidney transplant.
It is well established that birth weight is inversely associated with risk for CKD. Epidemiological studies suggest that reductions in renal function may occur as early as one year of age in LBW individuals. Confounding influences such as hypertension and diabetes, in addition to age, increase the risk for CKD in LBW individuals.
A reduction in nephron number is a common finding in LBW cohorts. Experimental models that investigate the etiology of developmental origins of kidney disease also note a consistent finding of reduced nephron number. Experimental studies indicate that compensatory glomerular hypertrophy may contribute to maintenance of absolute GFR in animal models of developmental insult, but this may occur at the expense of enhanced risk for renal injury and disease.
Preterm birth is also a risk factor for CKD. Interventions to improve renal development that occurs after birth are necessary to alleviate impaired nephrogenesis and renal risk in this population.
Epidemiological and experimental studies suggest that females in young adulthood may be protected relative to male counterparts against increased risk for renal injury and disease that has its origins in early life, but this protection is lost with age.
Birth weight is associated with heightened albuminuria in living kidney donors. Normal congenital nephron number is in excess of what is needed for normal function. However, a significant loss of renal reserve that originates during fetal life may put LBW individuals with low renal reserve at greater risk for renal injury over time, in particular following a secondary insult.
Acknowledgments
Funding
This review was supported by the National Institutes of Health (Grants: F30DK112718, T32HL105324, HL074927, HL51971, and P20GM104357), and the American Heart Association (Grant: AHA: 19900004).
Footnotes
Declaration of Interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
References
Papers of special note have been highlighted as either of interest (*) or of considerable interest (**) to readers.
- 1.United States Renal Data System. 2015 USRDS annual data report: Epidemiology of kidney disease in the United States. National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases; Bethesda, MD: 2015. [Google Scholar]
- 2.Inker LA, Astor BC, Fox CH, Isakova T, Lash JP, Peralta CA, Tamura MK, Feldman HI. KDOQI US Commentary on the 2012 KDIGO Clinical Practice Guideline for the Evaluation and Management of CKD. Am J Kidney Dis. 2014;63(5):713–735. doi: 10.1053/j.ajkd.2014.01.416. [DOI] [PubMed] [Google Scholar]
- 3.Pakula AM, Skinner RA. Acute kidney injury in the critically ill patient: A current review of the literature. J Intensive Care Med. 2016;31(5):319–324. doi: 10.1177/0885066615575699. [DOI] [PubMed] [Google Scholar]
- 4.Palevsky PM, Liu KD, Brophy PD, Chawla LS, Parikh CR, Thakar CV, Tolwani AJ, Waikar SS, Weisbord SD. KDOQI US Commentary on the 2012 KDIGO Clinical Practice Guideline for Acute Kidney Injury. Am J Kidney Dis. 2013;61(5):649–672. doi: 10.1053/j.ajkd.2013.02.349. [DOI] [PubMed] [Google Scholar]
- 5**.Cobo G, Hecking M, Port FK, Exner I, Lindholm B, Stenvinkel P, Carrero JJ. Sex and gender differences in chronic kidney disease: progression to end-stage renal disease and haemodialysis. Clin Sci (Lond) 2016;130(14):1147–1163. doi: 10.1042/CS20160047. An expert review highlighting sex differences in CKD and potential hypotheses. [DOI] [PubMed] [Google Scholar]
- 6**.Carrero JJ. Gender differences in chronic kidney disease: underpinnings and therapeutic implications. Kidney Blood Press Res. 2010;33(5):383–392. doi: 10.1159/000320389. An expert review highlighting sex differences in CKD and lack of women in clinical studies. [DOI] [PubMed] [Google Scholar]
- 7.Coggins CH, Lewis JB, Caggiula AW, Castaldo LS, Klahr S, Wang SR. Differences between woman and men with chronic renal disease. Nephrol Dial Transplant. 1998;13(6):1430–1437. doi: 10.1093/ndt/13.6.1430. [DOI] [PubMed] [Google Scholar]
- 8**.Neugarten J, Acharya A, Silbiger SR. Effect of Gender on the Progression of Nondiabetic Renal Disease: A Meta-Analysis. J Am Soc Nephrol. 2000;11(2):319–329. doi: 10.1681/ASN.V112319. An extensive meta-analysis of the literature examining the effect of sex on chronic renal disease. [DOI] [PubMed] [Google Scholar]
- 9.Levey AS, Astor BC, Stevens LA, Coresh J. Chronic kidney disease, diabetes, and hypertension: what’s in a name? Kidney Int. 2010;78(1):19–22. doi: 10.1038/ki.2010.115. [DOI] [PubMed] [Google Scholar]
- 10**.Brenner BM, Mackenzie HS. Nephron mass as a risk factor for progression of renal disease. Kidney Int Suppl. 1997;63:S124–S127. An expert review highlighting the link between nephron number and renal disease. [PubMed] [Google Scholar]
- 11**.Barker DJP, Osmond C. Infant mortality, childhood nutrition, and ischaemic heart disease in England and Wales. Lancet. 1986;1(8489):1077–1081. doi: 10.1016/s0140-6736(86)91340-1. One of the first papers to formally propose a link between fetal environment and adult health with a study population over a large geographical distribution. [DOI] [PubMed] [Google Scholar]
- 12**.Barker DJP. In utero programming of chronic disease. Clin Sci (Lond) 1998;95(2):115–128. An expert review highlighting the developmental origins of health and disease. [PubMed] [Google Scholar]
- 13.Barker DJP. Fetal origins of coronary heart disease. BMJ. 1995;311(6998):171–174. doi: 10.1136/bmj.311.6998.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Romo A, Carceller R, Tobajas J. Intrauterine growth retardation (IUGR): epidemiology and etiology. Pediatr Endocrinol Rev. 2009;6(Suppl 3):332–336. [PubMed] [Google Scholar]
- 15.Abitbol CL, Rodriguez MM. The long-term renal and cardiovascular consequences of prematurity. Nat Rev Nephrol. 2012;8(5):265–274. doi: 10.1038/nrneph.2012.38. [DOI] [PubMed] [Google Scholar]
- 16.World Health Organization. International statistical classification of diseases and related health problems, tenth revision. World Health Organization; Geneva: 1992. [PubMed] [Google Scholar]
- 17.Hughes MM, Black RE, Katz J. 2500-g Low birth weight cutoff: history and implications for future research and policy. Matern Child Health J. 2016;21(2):283–289. doi: 10.1007/s10995-016-2131-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kramer MS. The epidemiology of adverse pregnancy outcomes: An overview. J Nutr. 2003;133(5 Suppl 2):1592S–1596S. doi: 10.1093/jn/133.5.1592S. [DOI] [PubMed] [Google Scholar]
- 19.Valero de Bernabe J, Soriano T, Albaladejo R, Juarranz M, Calle ME, Martinez D, Dominguez-Rojas V. Risk factors for low birth weight: a review. Eur J Obstet Gynecol Reprod Biol. 2004;116(1):3–15. doi: 10.1016/j.ejogrb.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 20.Wollmann HA. Intrauterine growth restriction: definition and etiology. Horm Res. 1998;49(Suppl 2):1–6. [PubMed] [Google Scholar]
- 21.Ananth CV, Keyes KM, Wapner RJ. Pre-eclampsia rates in the United States, 1980–2010: age-period-cohort analysis. BMJ. 2013;347(f6564):1–9. doi: 10.1136/bmj.f6564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bezek S, Ujházy E, Mach M, Navarová J, Dubovický M. Developmental origin of chronic diseases: toxicological implication. Interdiscip Toxicol. 2008;1(1):29–31. doi: 10.2478/v10102-010-0029-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rondó PH, Ferreira RF, Nogueira F, Ribeiro MC, Lobert H, Artes R. Maternal psychological stress and distress as predictors of low birth weight, prematurity and intrauterine growth retardation. Eur J Clin Nutr. 2003;57(2):266–72. doi: 10.1038/sj.ejcn.1601526. [DOI] [PubMed] [Google Scholar]
- 24*.Das SK, Mannan M, Golam Faruque AS, Ahmed T, Mcintyre HD, Al Mamun A. Effect of birth weight on adulthood renal function: A bias-adjusted meta-analytic approach. Nephrology (Carlton) 2016;21:547–565. doi: 10.1111/nep.12732. A systemic review and meta-analysis linking low birth weight and chronic kidney disease. [DOI] [PubMed] [Google Scholar]
- 25.Lackland DT, Bendall HE, Osmond C, Egan BM, Barker DJP. Low birth weights contribute to higher of early-onset chronic renal failure in the southeastern United States. Arch Intern Med. 2000;160(10):1472–1476. doi: 10.1001/archinte.160.10.1472. [DOI] [PubMed] [Google Scholar]
- 26*.White SL, Perkovic V, Cass A, Chang CL, Poulter NR, Spector T, Haysom L, Craig JC, Salmi IA, Chadban SJ, Huxley RR. Is low birth weight an antecedent of CKD in later life? A systematic review of observational studies. Am J Kidney Dis. 2009;54(2):248–261. doi: 10.1053/j.ajkd.2008.12.042. A systemic review and meta-analysis linking low birth weight and chronic kidney disease. [DOI] [PubMed] [Google Scholar]
- 27.Hughson M, Farris AB, III, Douglas-Denton R, Hoy WE, Bertram JF. Glomerular number and size in autopsy kidneys: The relationship to birth weight. Kidney Int. 2003;63(6):2113–2122. doi: 10.1046/j.1523-1755.2003.00018.x. [DOI] [PubMed] [Google Scholar]
- 28.Hughson M, Gobe GC, Hoy WE, Manning RD, Douglas-Denton R, Bertram JF. Associations of glomerular number and birth weight with clinicopathological features of African Americans and whites. Am J Kidney Dis. 2008;52(1):18–28. doi: 10.1053/j.ajkd.2008.03.023. [DOI] [PubMed] [Google Scholar]
- 29.Manalich R, Reyes L, Herrera M, Melendi C, Fundora I. Relationship between weight at birth and the number and size of renal glomeruli in humans: A histomorphometric study. Kidney Int. 2000;58(2):770–773. doi: 10.1046/j.1523-1755.2000.00225.x. [DOI] [PubMed] [Google Scholar]
- 30.Denic A, Lieske JC, Chakkera HA, Poggio ED, Alexander MP, Singh P, Kremers WK, Lerman LO, Rule AD. The substantial loss of nephrons in healthy human kidneys with aging. J Am Soc Nephrol. 2016;28(1):313–320. doi: 10.1681/ASN.2016020154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luyckx VA, Brenner BM. The clinical importance of nephron mass. J Am Soc Nephrol. 2010;21(6):898–910. doi: 10.1681/ASN.2009121248. [DOI] [PubMed] [Google Scholar]
- 32.Cassidy-Bushrow AE, Wegienka G, Barone CJ2nd, Valentini RP, Yee J, Havstad S, Johnson CC. Race-specific relationship of birth weight and renal function among healthy young children. Pediatr Nephrol. 2012;27(8):1317–1323. doi: 10.1007/s00467-012-2136-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frankfurt JA, Duncan AF, Heyne RJ, Rosenfeld CR. Renal function and systolic blood pressure in very-low-birth-weight infants 1–3 years of age. Pediatr Nephrol. 2012;27(12):2285–2291. doi: 10.1007/s00467-012-2265-y. [DOI] [PubMed] [Google Scholar]
- 34.Khalsa DD, Beydoun HA, Carmody JB. Prevalence of chronic kidney disease risk factors among low birth weight adolescents. Pediatr Nephrol. 2016;31(9):1509–1516. doi: 10.1007/s00467-016-3384-7. [DOI] [PubMed] [Google Scholar]
- 35.Ruggajo P, Skrunes R, Svarstad E, Skjærven R, Reisæther AV, Vikse BE. Familial factors, low birth weight, and development of ESRD: A Nationwide Registry Study. Am J Kidney Dis. 2016;67(4):601–608. doi: 10.1053/j.ajkd.2015.11.015. [DOI] [PubMed] [Google Scholar]
- 36.Hirano D, Ishikura K, Uemura O, Ito S, Wada N, Hattori M, Ohashi Y, Hamasaki Y, Tanaka R, Nakanishi K, Kaneko T, Honda M. Pediatric CKD Study Group in Japan in conjunction with the Committee of Measures for Pediatric CKD of the Japanese Society of Pediatric Nephrology. Association between low birth weight and childhood-onset chronic kidney disease in Japan: a combined analysis of a nationwide survey for paediatric chronic kidney disease and the National Vital Statistics Report. Nephrol Dial Transplant. 2016;31(11):1895–1900. doi: 10.1093/ndt/gfv425. [DOI] [PubMed] [Google Scholar]
- 37.Faa G, Gerosa C, Fanni D, Monga G, Zaffanello M, Van Eyken P, Fanos V. Morphogenesis and molecular mechanisms involved in human kidney development. J Cell Physiol. 2012;227(3):1257–1268. doi: 10.1002/jcp.22985. [DOI] [PubMed] [Google Scholar]
- 38.Black MJ, Sutherland MR, Gubhaju L, Kent AL, Dahlstrom JE, Moore L. When birth comes early: effects on nephrogenesis. Nephrology (Carlton) 2013;18(3):180–182. doi: 10.1111/nep.12028. [DOI] [PubMed] [Google Scholar]
- 39.Abitbol CL, DeFreitas MJ, Strauss J. Assessment of kidney function in preterm infants: lifelong implications. Pediatr Nephrol. 2016;31(12):2213–2222. doi: 10.1007/s00467-016-3320-x. [DOI] [PubMed] [Google Scholar]
- 40.Sutherland MR, Gubhaju L, Moore L, Kent AL, Dahlstrom JE, Horne RS, Hoy WE, Bertram JF, Black MJ. Accelerated maturation and abnormal morphology in the preterm neonatal kidney. J Am Soc Nephrol. 2011;22(7):1365–1374. doi: 10.1681/ASN.2010121266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li S, Chen SC, Shlipak M, Bakris G, McCullough PA, Sowers J, Stevens L, Jurkovitz C, McFarlane S, Norris K, Vassalotti J, Klag MJ, Brown WW, Narva A, Calhoun D, Johnson B, Obialo C, Whaley-Connell Becker B, Collins AJ. Low birth weight is associated with chronic kidney disease only in men. Kidney Int. 2008;73(5):637–642. doi: 10.1038/sj.ki.5002747. [DOI] [PubMed] [Google Scholar]
- 42.Hallen SI, Euser AM, Irgens LM, Finken MJJ, Holmen J, Dekker FW. Effect of intrauterine growth restriction on kidney function at young adult age: the Nord Trǿndelag Health (HUNT 2) Study. Am J Kidney Dis. 2008;51(1):10–20. doi: 10.1053/j.ajkd.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 43.Arsenault MG, Miao Y, Jones K, Sims D, Spears J, Wright GM, Hartwig S. Estimation of total glomerular number using an integrated dissector method in embryonic and postnatal kidneys. Can J Kidney Health Dis. 2014;1(12):1–7. doi: 10.1186/2054-3581-1-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bertram JF, Cullen-McEwen LA, Egan GF, Gretz N, Baldelomar E, Beeman SC, Bennett KM. Why and how we determine nephron number. Pediatr Nephrol. 2014;29(4):575–580. doi: 10.1007/s00467-013-2600-y. [DOI] [PubMed] [Google Scholar]
- 45.Bennett KM, Bertram JF, Beeman SC, Gretz N. The emerging role of MRI in quantitative renal glomerular morphology. Am J Physiol Renal Physiol. 2013;304(10):F1252–F1257. doi: 10.1152/ajprenal.00714.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sterio DC. The unbiased estimation of number and sizes of arbitrary particles using the disector. J Microsc. 1984;134(Pt 2):127–136. doi: 10.1111/j.1365-2818.1984.tb02501.x. [DOI] [PubMed] [Google Scholar]
- 47.Kaufman JM, Hardy R, Hayslett JP. Age-dependent characteristics of compensatory renal growth. Kidney Int. 1975;8(1):21–26. doi: 10.1038/ki.1975.72. [DOI] [PubMed] [Google Scholar]
- 48.Beeman SC, Cullen-McEwen LA, Puelles VG, Zhang M, Wu T, Baldelomar EJ, Dowling J, Charlton JR, Forbes MS, Ng A, Wu Q, Armitage JA, Egan GF, Bertram JF, Bennett KM. MRI-based glomerular morphology and pathology in whole human kidneys. Am J Physiol Renal Physiol. 2014;306(11):F1381–F1390. doi: 10.1152/ajprenal.00092.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Martin H. Laboratory measurement of urine albumin and urine total protein in screening for proteinuria in Chronic Kidney Disease. Clin Biochem Rev. 2011;32(2):97–102. [PMC free article] [PubMed] [Google Scholar]
- 50.Hall JE, Guyton AC, Farr BM. A single-injection method for measuring glomerular filtration rate. Am J Physiol. 1977;232(1):F72–F76. doi: 10.1152/ajprenal.1977.232.1.F72. [DOI] [PubMed] [Google Scholar]
- 51.van Veldhuisen DJ, Ruilope LM, Maisel AS, Damman K. Biomarkers of renal injury and function: diagnostic, prognostic and therapeutic implications in heart failure. Eur Heart J. 2016;37(33):2577–2585. doi: 10.1093/eurheartj/ehv588. [DOI] [PubMed] [Google Scholar]
- 52.Cowley AW, Ryan RP, Kurth T, Skelton MM, Schock-Kusch D, Gretz N. Progression of GFR reduction determined in conscious Dahl S hypertensive rats. Hypertension. 2013;62(1):85–90. doi: 10.1161/HYPERTENSIONAHA.113.01194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schock-Kusch D, Geraci S, Ermeling E, Shulhevich Y, Sticht C, Hesser J, Stsepankou D, Neudecker S, Pill J, Schmitt R, Melk A. Reliability of transcutaneous measurement of renal function in various strains of conscious mice. PLoS One. 2013;8(8):e71519. doi: 10.1371/journal.pone.0071519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Carrara F, Azzollini N, Nattino G, Corna D, Villa S, Cerullo D, Zoja C, Abrante B, Luis-Lima S, Porrini E, Cannata A, Ferrari S, Fois M, Stucchi N, Gaspari F. Simplified method to measure glomerular filtration rate by iohexol plasma clearance in conscious rats. Nephron. 2016;133(1):62–70. doi: 10.1159/000445843. [DOI] [PubMed] [Google Scholar]
- 55.Grenier N, Mendichovszky I, de Senneville BD, Roujol S, Desbarats P, Pedersen M, Wells K, Frokiaer J, Gordon I. Measurement of glomerular filtration rate with magnetic resonance imaging: principles, limitations, and expectations. Semin Nucl Med. 2008;38(1):47–55. doi: 10.1053/j.semnuclmed.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 56*.Traynor J, Mactier R, Geddes CC, Fox JG. How to measure renal function in clinical practice. BMJ. 2006;333(7571):733–737. doi: 10.1136/bmj.38975.390370.7C. An overview of approaches used to measure renal excretory function. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Delanghe JR. How to estimate GFR in children. Nephrol Dial. Transplant. 2009;24:714–716. doi: 10.1093/ndt/gfn306. [DOI] [PubMed] [Google Scholar]
- 58.Schwartz GJ, Work DF. Measurement and estimation of GFR in children and adolescents. Clin J Am Soc Nephrol. 2009;4(11):1832–1843. doi: 10.2215/CJN.01640309. [DOI] [PubMed] [Google Scholar]
- 59.Langley SC, Jackson AA. Increased systolic blood pressure in adult rats induced by fetal exposure to maternal low protein diets. Clin Sci (Lond) 1994;86(2):217–222. doi: 10.1042/cs0860217. [DOI] [PubMed] [Google Scholar]
- 60.Woodall SM, Johnston BM, Breier BH, Gluckman PD. Chronic maternal undernutrition in the rat leads to delayed postnatal growth and elevated blood pressure of offspring. Pediatr Res. 1996;40(3):438–443. doi: 10.1203/00006450-199609000-00012. [DOI] [PubMed] [Google Scholar]
- 61.Henriksen T, Clausen T. The fetal origins hypothesis: placental insufficiency and inheritance versus maternal malnutrition in well-nourished populations. Acta Obstet Gynecol Scand. 2002;81(12):112–114. doi: 10.1034/j.1600-0412.2002.810204.x. [DOI] [PubMed] [Google Scholar]
- 62.Chaiworapongsa T, Chaemsaithong P, Yeo L, Romero R. Pre-eclampsia part 1: current understanding of its pathophysiology. Nat Rev Nephrol. 2014;10(8):466–480. doi: 10.1038/nrneph.2014.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Davis EF, Lazdam M, Lewandowski AH, Worton SA, Kelly B, Kenworthy Y, Adwani S, Wilkinson AR, McCormick K, Sargent I, Redman C, Leeson P. Cardiovascular risk factors in children and young adults born to preeclamptic pregnancies: a systematic review. Pediatrics. 2012;129(6):e1552–61. doi: 10.1542/peds.2011-3093. [DOI] [PubMed] [Google Scholar]
- 64.Wlodek ME, Mibus A, Tan A, Siebel AL, Owens JA, Moritz KM. Normal lactational environment restores nephron endowment and prevents hypertension after placental restriction in the rat. J Am Soc Nephrol. 2007;18(6):1688–1696. doi: 10.1681/ASN.2007010015. [DOI] [PubMed] [Google Scholar]
- 65.Alexander BT. Placental insufficiency leads to development of hypertension in growth-restricted offspring. Hypertension. 2003;41(3):457–462. doi: 10.1161/01.HYP.0000053448.95913.3D. [DOI] [PubMed] [Google Scholar]
- 66.Xiao D, Xu Z, Huang X, Longo LD, Yang S, Zhang L. Prenatal gender-related nicotine exposure increases blood pressure response to angiotensin II in adult offspring. Hypertension. 2008;51(4):1239–1247. doi: 10.1161/HYPERTENSIONAHA.107.106203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dodic M, May CN, Wintour EM, Coghlan JP. An early prenatal exposure to excess glucocorticoid leads to hypertensive offspring in sheep. Clin Sci (Lond) 1998;94(2):149–155. doi: 10.1042/cs0940149. [DOI] [PubMed] [Google Scholar]
- 68.Liu NQ, Ouyang Y, Bulut Y, Lagishetty V, Chan SY, Hollis BW, Wagner C, Equils O, Hewison M. Dietary vitamin D restriction in pregnant female mice is associated with maternal hypertension and altered placental and fetal development. Endocrinology. 2013l;154(7):2270–2280. doi: 10.1210/en.2012-2270. [DOI] [PubMed] [Google Scholar]
- 69.Weaver K. Pregnancy-induced hypertension and low birth weight in magnesium-deficient ewes. Magnesium. 1986;5(3–4):191–200. [PubMed] [Google Scholar]
- 70.Gatford KL, Mohammad SN, Harland ML, De Blasio MJ, Fowden AL, Robinson JS, Owens JA. Impaired beta-cell function and inadequate compensatory increases in beta-cell mass after intrauterine growth restriction in sheep. Endocrinology. 2008;149(10):5118–5127. doi: 10.1210/en.2008-0233. [DOI] [PubMed] [Google Scholar]
- 71.Green AS, Rozance PJ, Limesand SW. Consequences of a compromised intrauterine environment on islet function. J Endocrinol. 2010;205(3):211–224. doi: 10.1677/JOE-09-0399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Simmons RA, Templeton LJ, Gertz SJ. Intrauterine growth retardation leads to the development of type 2 diabetes in the rat. Diabetes. 2001;50(10):2279–2286. doi: 10.2337/diabetes.50.10.2279. [DOI] [PubMed] [Google Scholar]
- 73.Ortiz LA, Quan A, Zarzar F, Weinberg A, Baum M. Prenatal dexamethasone programs hypertension and renal injury in the rat. Hypertension. 2003;41(2):328–334. doi: 10.1161/01.hyp.0000049763.51269.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74*.Woods LL, Ingelfinger JR, Nyengaard JR, Rasch R. Maternal protein restriction suppresses the newborn renin-angiotensin system and program adult hypertension in rats. Pediatr Res. 2001;49(4):460–467. doi: 10.1203/00006450-200104000-00005. Study demonstrating in a rat model of developmental origins that GFR at the level of the nephron may be impaired following a developmental insult. [DOI] [PubMed] [Google Scholar]
- 75.Xie Z, Dong Q, Ge J, Chen P, Li W, Hu J. Effect of low birth weight on impaired renal development and function and hypertension in rat model. Ren Fail. 2012;34(6):754–759. doi: 10.3109/0886022X.2012.676526. [DOI] [PubMed] [Google Scholar]
- 76.Gilbert JS, Lang AL, Grant AR, Nijland MJ. Maternal nutrient restriction in sheep: hypertension and decreased nephron number in offspring at 9 months of age. J Physiol. 2005;565(Pt 1):137–147. doi: 10.1113/jphysiol.2005.084202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zimanyi MA, Bertram JF, Black JM. Nephron number in the offspring of rats fed a low Pprotein diet during pregnancy. Image Anal Sterol. 2000;19(3):219–222. [Google Scholar]
- 78.Zimanyi MA, Denton KM, Forbes JM, Thallas-Bonke V, Thomas MC, Poon F, Black MJ. A developmental nephron deficit in rats is associated with increased susceptibility to a secondary renal injury due to advanced glycation end-products. Diabetologia. 2006;49(4):801–810. doi: 10.1007/s00125-006-0175-x. [DOI] [PubMed] [Google Scholar]
- 79.Ojeda NB. Low birth weight increases susceptibility to renal injury in a rat model of mild ischemia-reperfusion. Am J Physiol Renal Physiol. 2011;301(2):F420–F426. doi: 10.1152/ajprenal.00045.2011. [DOI] [PubMed] [Google Scholar]
- 80.Boubred F, Daniel L, Buffat C, Feuerstein JM, Tsimaratos M, Oliver C, Dignat-George F, Lelievre-Pegorier M, Simconi U. Early postnatal overfeeding induces chronic renal dysfunction in adult male rats. Am J Physiol Renal Physiol. 2009;297(4):F943–F951. doi: 10.1152/ajprenal.90704.2008. [DOI] [PubMed] [Google Scholar]
- 81.Gurusinghe S, Brown RD, Cai X, Samuel CS, Ricardo SD, Thomas MC, Kett MM. Does a nephron deficit exacerbate the renal and cardiovascular effects of obesity? PLoS One. 2013;8(9):e73095. doi: 10.1371/journal.pone.0073095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Boubred F, Daniel L, Buffat C, Tsimaratos M, Oliver C, Lelièvre-Pégorier M, Simeoni U. The magnitude of nephron number reduction mediates intrauterine growth-restriction-induced long term chronic renal disease in the rat. A comparative study in two experimental models. J Transl Med. 2016;14(1):331–339. doi: 10.1186/s12967-016-1086-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Van Vliet BN, Chafe LL, Antic V, Schnyder-Candrian S, Montani JP. Direct and indirect methods used to study arterial blood pressure. J Pharmacol Toxicol Methods. 2000;44(2):361–373. doi: 10.1016/s1056-8719(00)00126-x. [DOI] [PubMed] [Google Scholar]
- 84.Woods LL. Neonatal uninephrectomy causes hypertension in adult rats. Am J Physiol. 1999;276(4 Pt 2):R974–R978. doi: 10.1152/ajpregu.1999.276.4.R974. [DOI] [PubMed] [Google Scholar]
- 85.Singh RR, Denton KM, Bertram JF, Jefferies AJ, Moritz KM. Reduced nephron endowment due to fetal uninephrectomy impairs renal sodium handling in male sheep. Clin Sci (Lond) 2010;118(11):669–680. doi: 10.1042/CS20090479. [DOI] [PubMed] [Google Scholar]
- 86*.Ruta LM, Dickinson H, Thomas MC, Denton KM, Anderson WP, Kett MM. High-salt diet reveals the hypertensive and renal effects of reduced nephron endowment. Am J Physiol Renal Physiol. 2010;298(6):F1384–F1392. doi: 10.1152/ajprenal.00049.2010. This experimental study provides evidence that a reduction in nephron number is not sufficient to induce renal injury in the absence of a second hit such as hypertension. [DOI] [PubMed] [Google Scholar]
- 87.Woods LL, Ingelfinger JR, Rasch R. Modest maternal protein restriction fails to program adult hypertension in female rats. Am J Physiol Regul Integr Comp Physiol. 2005;289(4):R1131–R1136. doi: 10.1152/ajpregu.00037.2003. [DOI] [PubMed] [Google Scholar]
- 88.Black MJ, Lim K, Zimanyi MA, Sampson AK, Bubb KJ, Flower RL, Parkington HC, Tare M, Denton KM. Accelerated age-related decline in renal and vascular function in female rats following early-life growth restriction. Am J Physiol Regul Integr Comp Physiol. 2015;309(9):R1153–R1161. doi: 10.1152/ajpregu.00403.2014. [DOI] [PubMed] [Google Scholar]
- 89.Saez F, Castells MT, Zuasti A, Salazar F, Reverte V, Loria A, Salazar FJ. Sex differences in the renal changes elicited by angiotensin II blockade during the nephrogenic period. Hypertension. 2007;49(6):1429–1435. doi: 10.1161/HYPERTENSIONAHA.107.087957. [DOI] [PubMed] [Google Scholar]
- 90.Intapad S, Tull FL, Brown AD, Dasinger JH, Ojeda NB, Fahling JM, Alexander BT. Renal denervation abolishes the age-dependent increase in blood pressure in female intrauterine growth-restricted rats at 12 months of age. Hypertension. 2013;61(4):828–834. doi: 10.1161/HYPERTENSIONAHA.111.00645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tran M, Young ME, Jefferies AJ, Hryciw DH, Ward MM, Fletcher EL, Wlodek ME, Wadley GD. Uteroplacental insufficiency leads to hypertension, but not glucose intolerance or impaired skeletal muscle mitochondrial biogenesis, in 12-month-old rats. Physiol Rep. 2015;3(9):pii: e12556. doi: 10.14814/phy2.12556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Andersson SW, Lapidus L, Niklasson A, Hallberg L, Bengtsson C, Hulthén L. Blood pressure and hypertension in middle-aged women in relation to weight and length at birth: a follow-up study. J Hypertens. 2000;18(12):1753–1761. doi: 10.1097/00004872-200018120-00008. [DOI] [PubMed] [Google Scholar]
- 93.Lam NN, Lentine KL, Levey AS, Kasiske BL, Garg AX. Long-term medical risks to the living kidney donor. Nat Rev Nephrol. 2015;11(7):411–419. doi: 10.1038/nrneph.2015.58. [DOI] [PubMed] [Google Scholar]
- 94.Rogers NM, Lawton PD, Jose MD. Indigenous Australians and living kidney donation. N Engl J Med. 2009;361:1513–1516. doi: 10.1056/NEJMc0905777. [DOI] [PubMed] [Google Scholar]
- 95.Young RJ, Hoy WE, Kincaid-Smith P, Seymour AE, Bertram JF. Glomerular size and glomerulosclerosis in Australian aborigines. Am J Kidney Dis. 2000;36(3):481–489. doi: 10.1053/ajkd.2000.9788. [DOI] [PubMed] [Google Scholar]
- 96**.Berglund D, MacDonald D, Jackson S, Spong R, Issa N, Kukla A, Reule S, Weber M, Matas AJ, Ibrahim HN. Low birthweight and risk of albuminuria in living kidney donors. Clin Transplant. 2014;28(3):361–367. doi: 10.1111/ctr.12321. Clinical study highlighting potential increased renal risk in low birth weight living kidney donors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nicholson ML, Windmill DC, Horsburgh T, Harris KP. Influence of allograft size to recipient body-weight ratio on the long-term outcome of renal transplantation. Br J Surg. 2000;87:314–319. doi: 10.1046/j.1365-2168.2000.01390.x. [DOI] [PubMed] [Google Scholar]
- 98.Kim YS, Kim MS, Han DS, Kim DK, Myoung SM, Kim SI, Park K. Evidence that the ration of donor kidney weight to recipient body weight, donor age, and episodes of acute rejection correlate independently with live-donor graft function. Transplantation. 2002;72:280–283. doi: 10.1097/00007890-200207270-00021. [DOI] [PubMed] [Google Scholar]
- 99.Silverwood RJ, Pierce M, Hardy R, Sattar N, Whincup P, Ferro C, Savage C, Kuh D, Nitsch D. Low birth weight, later renal function, and the roles of adulthood blood pressure, diabetes, and obesity in a British birth cohort. Kidney Int. 2013;84(6):1262–1270. doi: 10.1038/ki.2013.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Vikse BE, Irgens LM, Leivestad T, Hallan S, Iversen BM. Low birth weight increases risk for end-stage renal disease. J Am Soc Nephrol. 2008;19(1):151–157. doi: 10.1681/ASN.2007020252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Raaijmakers A, Petit T, Gu Y, Zhang Z, Wei F, Cools B, Jacobs L, Thijs L, Thewissen L, Levtchenko E, Staessen JA, Allegaert K. Design and feasibility of "PREMATurity as predictor of children"s Cardiovascular-renal Health” (PREMATCH): A pilot study. Blood Press. 2015;24(5):275–283. doi: 10.3109/08037051.2015.1053220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wong C, Gerson A, Hooper SR, Matheson M, Lande M, Kupferman J, Furth S, Warady B, Flynn J. Chronic Kidney Disease in Children (CKiD) Study. Effect of elevated blood pressure on quality of life in children with chronic kidney disease. Pediatr Nephrol. 2016;31(7):1129–1136. doi: 10.1007/s00467-015-3262-8. [DOI] [PMC free article] [PubMed] [Google Scholar]