

Abstract
This paper reports a biocompatible and label-free cell separation method using ferrofluids that can separate a variety of low-concentration cancer cells from cell culture lines (~100 cancer cells/mL) from undiluted white blood cells, with a throughput of 1.2 mL/h and an average separation efficiency of 82.2%. The separation is based on size difference from cancer cells and white blood cells, and is conducted in a custom-made biocompatible ferrofluid that retains not only excellent short-term viabilities, but also normal proliferations of 7 commonly used cancer cell lines. A microfluidic device is designed and optimized specifically to shorten the exposure time of live cells in ferrofluids from hours to seconds, by eliminating time-consuming off-chip sample preparation and extraction steps and integrating them on-chip to achieve one-step process. As a proof-of-concept demonstration, a ferrofluid with 0.26% volume fraction was used in this microfluidic device to separate spiked cancer cells from cell lines at a concentration of ~100 cells/mL from white blood cells with a 1.2 mL/h throughput. The separation efficiencies were 80±3%, 81±5%, 82±5%, 82±4%, and 86±6% for A549 lung cancer, H1299 lung cancer, MCF-7 breast cancer, MDA-MB-231 breast cancer, and PC-3 prostate cancer cell lines, respectively. Separated cancer cells purity was between 25.3% and 28.8%. In addition, separated cancer cells from this strategy showed an average short-term viability of 94.4±1.3% and separated cells were cultured and demonstrated normal proliferation to the confluence even after the separation process. Owning to its excellent biocompatibility and label-free operation, and its ability to recover low concentration of cancer cells from white blood cells, this method could lead to a promising tool for rare cell separation.
Graphical abstract
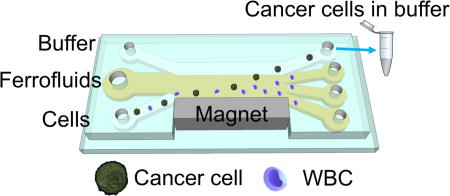
A biocompatible and label-free separation of low-concentration cancer cells of cell lines from white blood cells is developed.
Introduction
Microfluidic manipulation of cells in magnetic liquids,1 i.e., negative magnetophoresis, led to a number of recent applications in cell separation,1–4 trapping and focusing,5–8 and density measurements.9–13 Its working principle is as follows: cells without any labels placed inside a uniformly magnetic media – magnetic liquids, act as “magnetic holes”.14 A magnetic field gradient attracts the magnetic media, which causes the “magnetic holes” – cells, to be preferentially pushed away. This way, cells can be continuously manipulated in a label-free fashion. Magnetic force acting on the cells is proportional to their sizes,1, 15 very similar to buoyancy force, which allows for a size-based manipulation. Typical devices for conducting “negative magnetophoresis” assays are simple and low-cost, involving only channels and permanent magnets. Their operation does not necessitate accessories such as power supplies or function generators. Because of its label-free, low-cost and simple-to-use nature, “negative magnetophoresis” has been used recently for cell manipulation. For example, Demirci group developed a static-flow system with a form of magnetic liquids – paramagnetic salt solutions – to precisely measure subtle density differences among cell groups.9, 10 Salt solutions containing transition and lanthanide metals, such as Mn2+ or Gd3+, are weakly magnetic due to their unpaired inner-shell electrons that produce a magnetic moment. Our group demonstrated a continuous-flow ferrohydrodynamic separation of HeLa cells from whole blood in another form of magnetic liquids – ferrofluids.16 Ferrofluids are colloidal suspensions of magnetic nanoparticles with diameters of approximately 10 nm. Although both paramagnetic salt solution and ferrofluid have served as the medium in “negative magnetophoresis” assays, ferrofluids were considered to be better suited for applications such as high-throughput separation that requires a continuous flow, because of their stronger magnetic properties, while paramagnetic salt solutions exceled in static-flow applications such as density measurement.
Cell separation based on “negative magnetophoresis” in ferrofluids is facing its own challenges, especially in rare cell separation where cell integrity needs to be maintained for further analysis, while typically less than 1000 cells in one milliliter of sample are available and need to be enriched in a high-throughput and high-efficiency manner.17 The challenges associated with cell separation in ferrofluids are three-fold. First of all, ferrofluids are not natural media for cells; they need to be rendered biocompatible so that cells remain alive and their normal functions are kept intact during and after the separation for post-separation analysis. This is not trivial, although progresses were made recently through preserving viability and normal proliferation of cells in custom-made ferrofluids,3, 18 biocompatibility of ferrofluids remains to be a significant challenge for cell separation applications. For examples, although Yellen’s group developed a bovine serum albumin (BSA) coated ferrofluid in which human umbilical vein endothelia cells (HUVEC) had more than 95% viability after 2 hours of exposure and were able to maintain normal proliferation afterwards,18 this ferrofluid was only used in static-flow conditions for relatively slow cell manipulation, its colloidal stability could be an issue in high-throughput and continuous-flow cell separation applications, because of the thick BSA surfactant layer used for particle functionalization. Although Koser’s group reported a citrate-stabilized ferrofluid and demonstrated a 75% viability of blood cells in them after several hours’ exposure,3 long-term cell proliferation study in this ferrofluid was not conducted. In summary, only a very limited number of cells were studied in these custom-made ferrofluids in operation conditions that were not always compatible for continuous-flow cell separation, and data didn’t often provide both short-term and long-term impacts on them after separation. As a result, there is an urgent need for a new ferrofluid that can minimize its negative effects on mammalian cells, at the same time is colloidally stable for high-throughput and continuous-flow operation under strong magnetic fields. The second challenge comes from device design for cell separation in ferrofluids. Even with a biocompatible ferrofluid, it is still necessary to reduce the exposure time of cells in them down to an absolute minimum, because prolonged exposure time will inevitably lead to particle endocytosis and/or diffusion and affect cell viability and normal functions.19 For example, we observed in this study that long exposure time of A549 lung cancer cells in ferrofluids resulted in a higher cellular uptake of nanoparticles and slower cell growth. In previous publications, the majority of cell exposure time to ferrofluids came from sample preparation and sample extraction that could last up to hours.3, 16, 20 As a result, a new one-step device design that integrates sample preparation and extraction on chip could significantly reduce exposure time and improve overall biocompatibility of the assay. The third challenge is associated with the low concentration of target cells in rare cell separation. In order to capture a meaningful number of target cells, throughput of at least 1 mL/h and separation efficiency of at least 80% in low concentration (<1000 cell/mL) conditions are necessary.21 Although cell separation in ferrofluids was demonstrated before, they mostly focused on the separation of bacteria and yeast cells,4, 20 bacteria and red blood cells,3 and HeLa cells and mouse blood.16 The throughputs of these studies were relatively low, and the target cells were at a much higher concentration (e.g., 105–106 cells/mL) than the definition of rare cells. It is therefore necessary to systematically optimize the device and ferrofluid design so that the throughput and efficiency of separation are comparable to those needed for rare cell separation.
In this study, we addressed the above-mentioned three challenges associated with rare cell separation in ferrofluids, by demonstrating a label-free separation of low-concentration cancer cells from cell culture lines at a concentration of 100 cancer cells/mL from undiluted white blood cells at a concentration of ~106 cells/mL in a newly developed biocompatible ferrofluids, with a optimized device design that achieved a throughput of 1.2 mL/h and a separation efficiency of greater than 80%. Cells were only exposed to ferrofluids for seconds in this process. We first developed a new water-based ferrofluid in which 7 commonly used cancer cell lines showed excellent short-term viability and normal proliferation to confluence after extended exposures. The ferrofluid possessed ideal properties including its pH value, tonicity, materials and surfactants of nanoparticles, as a biocompatible medium for mammalian cells, while at the same time the overall colloidal stability of this ferrofluid was well maintained to allow for high-throughput and continuous-flow separation under strong magnetic fields. We further developed a new device design that significantly reduces the exposure time of cells in ferrofluids, from hours to seconds, by taking advantage of the laminar flow nature of liquids in microchannels.22, 23 The design is explained in detail in Fig. 1. Briefly, in a frequently used setup of ferrohydrodynamic cell separation,3, 4, 16, 20 the majority of ferrofluid exposure time came from sample preparation (e.g., off-chip pre-mixing between ferrofluids and cells) and sample extraction (e.g., off-chip washing of cells after separation), as shown in Fig. 1a and 1b. However, the only time that cells needed to be exposed to ferrofluids was when they were actually being separated from each other. As a result, a device design that incorporates both on-chip sample preparation and extraction could significantly reduce exposure time and improve overall biocompatibility of the assay. In this device design, cell samples, ferrofluids, and a buffer were injected into a main channel through individual inlets, as shown in Fig. 1c. When they combined in the main channel, cell samples are mixed with the ferrofluid almost instantaneously because of strong magnetic convection,24, 25 and then separated based on their size difference. Large cells moved across the ferrofluid layer with a faster velocity than smaller ones. Towards the end of the channel, larger cells reached the ferrofluid/buffer boundary and were extracted into the buffer stream containing extremely low concentration of nanoparticles diffused from the ferrofluid stream. This way, cells were only exposed to ferrofluids when necessary (i.e., separation) and the exposure time was determined by the flow rate and channel length, which in this design was on the order of seconds. Finally, we performed a systematic parametric study of key factors influencing the performance of this separation method, and determined parameters for high-throughput and high efficiency low-concentration cancer cell separation of cell culture lines from undiluted white blood cells.
Fig. 1.

(a) Processing time of existing cell separation in ferrofluids involves time-consuming pre-mixing of cells with ferrofluids (~30 minutes) and off-chip washing steps (~30 minutes), while separation takes place within seconds. Total exposure time of cells in ferrofluids is estimated to be 1–2 hours. (b) Schematic illustration of an existing cell separation device. Cell mixtures are mixed with ferrofluids before separation and target cells are still in contact with ferrofluids after separation. (c) Schematic illustration of the proposed biocompatible cell separation in ferrofluids. Cell sample, ferrofluid, and buffer are injected into the device without pre-mixing. Cells are only in contact with ferrofluids when they are separated from each other. After separation, larger cancer cells are extracted into the buffer stream, eliminating the washing step. Total exposure time of cells to ferrofluids is estimated to be seconds. (d) Top-view of the proposed device, which consists of a microchannel and a permanent magnet, their relevant dimensions, and labeling of inlets and outlets. (e) A photo of the prototype device with a U.S. quarter for size comparison. Blue dye is used to visualize the channel.
Experimental section
Synthesis and characterization of biocompatible ferrofluids
Ammonium hydroxide solution (NH4OH, 28% w/w), iron (II) chloride tetrahydrate (FeCl2·4H2O), iron (III) chloride hexahydrate (FeCl3·6H2O), nitric acid (HNO3), iron (III) nitrate nonahydrate (Fe(NO3)3·9H2O), and sodium hydroxide (NaOH) were purchased from a commercial vendor (Sigma-Aldrich, St. Louis, MO). All reagents were used as received. Maghemite nanoparticles were synthesized by a chemical co-precipitation method.16, 26 In a typical reaction, 50 mL of ammonium hydroxide solution was quickly added to a mixture of 100 mL of 0.4 M iron (II) chloride tetrahydrate and 0.8 M iron (III) chloride hexahydrate and was followed by stirring at room temperature for 30 minutes. The suspension was then centrifuged at 2000×g for 3 minutes and the precipitate was dispersed in 200 mL of 2 M nitric acid and 0.35 M iron (III) nitrate nonahydrate. The mixture was maintained at 90 °C for 1 hour. During this time, the color of the mixture changed from black (Fe3O4) to reddish brown (Fe2O3). The maghemite nanoparticle suspension was centrifuged at 3000×g for 3 minutes and finally dispersed in 120 mL of deionized (DI) water, yielding a stable dispersion with a pH of 1.5–2. The pH of the dispersion was adjusted to 2.9 by 1 M sodium hydroxide solution. 40 mL of Atlox 4913 (Croda, Inc., Edison, NJ), a graft copolymer solution, was added to the dispersion and stirred for 5 minutes before raising pH to 7.0. The dispersion was then vigorously stirred at room temperature for 1 hour, and the resulted ferrofluid was dialyzed with a dialysis membrane (Spectrum Labs Inc., Rancho Dominguez, CA) against DI water for one week. DI water was refreshed every 24 hours. After dialysis, excess water was vaporized at 72 °C. Finally, 10% (v/v) 10× Hank’s balanced salt solution (HBSS; Life Technologies, Carlsbad, CA) was added into the ferrofluid to render it isotonic for cells followed by adjusting pH to 7.0. Sterile filtration of ferrofluid was performed with a 0.2 µm filter (VWR, Radnor, PA) and exposed to UV light for 12 hours before experimental use.
Size and morphology of maghemite nanoparticles were characterized via transmission electron microscopy (TEM; FEI Corp., Eindhoven, the Netherlands). Magnetic properties of the ferrofluid were measured at room temperature using a vibrating sample magnetometer (VSM; MicroSense, LLC, Lowell, MA) with a 2.15 T electromagnet. The magnetic moment of ferrofluid was measured over a range of applied fields from −21.5 to +21.5 kOe. The measurements were conducted in step field mode at a stepsize of 250 Oe/s. Zeta potential of the ferrofluid was measured with a Zetasizer Nano ZS (Malvern Instruments Inc., Westborough, MA).
Cell cultures and sample preparation
7 cancer cell lines (ATCC, Manassas, VA) including two lung cancer cell lines (A549 and H1299), three breast cancer cell lines (HCC1806, MDA-MD-231 and MCF-7), one cervical cancer cell line (HeLa), and one prostate cancer cell line (PC-3) were used to characterize biocompatibility of the ferrofluid. A549, H1299, HCC1806, and PC-3 cells were cultured in RPMI-1640 medium (Mediatech, Inc., Manassas, VA) supplemented with 10% (v/v) fetal bovine serum (FBS; Life Technologies, Carlsbad, CA) and 1% (v/v) penicillin/streptomycin solution (Mediatech, Inc., Manassas, VA) at 37 °C under a humidified atmosphere of 5% CO2. HeLa cells were cultured in Dulbecco’s modified eagle medium (DMEM; Life Technologies, Carlsbad, CA) supplemented with 10% (v/v) FBS and 1% (v/v) penicillin/streptomycin solution at 37 °C under a humidified atmosphere of 5% CO2. MDA-MB-231 and MCF-7 cells were cultured in DMEM supplemented with 10% (v/v) FBS, 1% (v/v) penicillin/streptomycin solution and 0.1 mM non-essential amino acid (NEAA; Life Technologies, Carlsbad, CA). All cell lines were released through incubation with 0.05% Trypsin-EDTA solution (Life Technologies, Carlsbad, CA) at 37 °C for 5–10 minutes.
A549, H1299, MCF-7, MDA-MB-231, and PC-3 cells were not only used in ferrofluid’s biocompatibility characterization but also used in cell separation experiments. Therefore, these five cell lines were stained with 2 µm CellTracker Green (Life Technologies, Carlsbad, CA) at 37 °C for 30 minutes before separation. The resulting cell suspensions were then centrifuged at 200 ×g for 5 minutes and suspended in phosphate buffered saline (PBS; Life Technologies, Carlsbad, CA) with 2% (v/v) FBS before use. For a validation experiment of the simulation on cancer cells from cell culture lines and red blood cells (RBCs) separation, human whole blood (Streck, Omaha, NE) was diluted 1000 times by PBS to achieve the concentration of 2×106 cells/mL. For low-concentration cancer cells of cell culture lines separation from undiluted white blood cells (WBCs), WBCs were obtained from undiluted human whole blood (Zen-Bio, Research Triangle Park, NC) with its RBCs lysed by RBC lysis buffer (eBioscience, San Diego, CA). The concentration of WBCs was on the order of 106 cells/mL. 100 CellTracker Green pre-stained cancer cells were spiked into 1 mL of either diluted whole blood or undiluted WBCs. Cancer cells were first counted with a hemacytometer (Hausser Scientific, Horsham, PA) and serially diluted in culture medium to achieve a solution with approximately 1×104 cells/mL. Cells were then counted with a Nageotte counting chamber (Hausser Scientific, Horsham, PA) to determine the number of cells per µL. 100 cells (~10 µL) were spiked into 1 mL of WBCs. The number of cells spiked was determined by the average of two counts, with less than 5% difference between the counts.
Characterizations of cell biocompatibility after exposures to ferrofluids
Cell viability was evaluated by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) assay. A549 cells were first incubated in each well of a 96-well plate (Corning Inc., Corning, NY) for a total of 24 hours. Ferrofluids of varying concentrations (0.05%, 0.19%, 0.22%, and 0.26% v/v) were added to the plate. After incubation for 12 hours, ferrofluid and medium were removed and cells were washed three times with PBS. MTT (ATCC, Manassas, MA) assay was then performed to determine the cell viability following the manufacture’s recommended protocol. Cell viability of the other 6 cell lines was investigated by the same MTT assay with a 0.26% (v/v) ferrofluid after 2 hours’ incubation.
Cell proliferation rate was assessed by MTT assay, too. A549 cells were first incubated with ferrofluids (0.26% v/v) for 1 minute and 2 hours, respectively at 37 °C under a humidified atmosphere of 5% CO2. Cells were then washed three times with PBS and released through incubation with 0.05% Trypsin-EDTA solution. 4000 cells were seeded in each well of a 96-well plate. MTT assay was performed every 24 hours to determine the growth rate following the manufacture’s recommended protocol. The medium was changed on the third day. The proliferation of other 6 cancer cell lines was investigated by attempting to culture cells to confluence after exposing them to ferrofluids for 2 hours.
Characterizations of cell biocompatibility after cell separation experiments
Short-term viability after separation was examined using a Live/Dead assay (Life Technologies, Carlsbad, CA). 1×106 A549 cells were injected through inlet A at a flow rate of 20 µL/min. After separation, cells from outlet 4 were collected and incubated with a working solution (2 µM calcein-AM and 4 µM propidium iodide (PI)) for 30 minutes at room temperature. After the solution was removed and washed with PBS, the labeled cells were observed under a fluorescence microscope (Carl Zeiss, Inc., Germany) for counting. For long-term proliferation, the separated A549 cells were collected into a centrifuge tube and spun down to remove the buffer, and then the cells were suspended in complete culture medium and seeded into a 24-well plate (Corning Inc., Corning, NY). Cells were then cultured at 37 °C under a humidified atmosphere of 5% CO2, the medium was refreshed every 24 h during the first 3 days. Cellular morphology was inspected every 24 hours.
Cellular nanoparticle uptake
Nanoparticle uptake study was conducted with A549 lung cancer cells. 1×105 A549 cells were seeded in each well of a 4-well chamber slide (Thermo Fisher Scientific, Waltham, MA). After 24-hour incubation, ferrofluids were added and incubated with cells at 37 °C for 1 minute and 2 hours, respectively. The ferrofluids were then removed and cells were washed three times with PBS. Next, cells were fixed with ice-cold 95% ethanol (Thermo Fisher Scientific, Waltham, MA) for 15 minutes. Subsequently, cells were incubated with Prussian blue staining solution (a mixture of equal volume of 1.2 mM/L hydrochloric acid and 4% w/v potassium ferrocyanide solution; Sigma-Aldrich, St. Louis, MO) for 15 minutes at room temperature. Cells were then rinsed with DI water and counterstained with pararosaniline solution (Sigma-Aldrich, St. Louis, MO) for 10 minutes. After consecutive dehydrations with 70%, 90%, and 100% ethanol, the chamber was removed and the slide was mounted. The slide was then examined using a light microscope (Carl Zeiss, Inc., Germany).
Device fabrication and experimental setup
Microfluidic devices were made of polydimethylsiloxane (PDMS) using standard soft lithography techniques.27 The thickness of the microfluidic channel was measured to be 52 µm by a profilometer (Veeco Instruments, Inc., Chadds Ford, PA). A NdFeB permanent magnet (K&J Magnetics, Inc., Pipersville, PA) was embedded into the PDMS channel with their magnetization direction vertical to the channel during the curing stage. The magnet is 5.08 cm in length, 0.635 cm in both width and thickness. Device and magnet dimensions are depicted in Fig. 1d, and a photo of the system is shown in Fig. 1e. Flux density at the center of magnet’s surface was measured to be 390 mT by a Gauss meter (Sypris, Orlando, FL) and an axial probe with 0.381 mm diameter of circular active area. The fabricated devices were flushed by 70% ethanol (Decon Labs, Inc., King of Prussia, PA) for 10 minutes before use. During a typical experiment, a microfluidic device was placed on the stage of an inverted microscope (Carl Zeiss, Inc., Germany) for observation and recording. Three fluids were controlled by individual syringe pumps (Chemyx, Inc., Stafford, TX) with tunable flow rates. Cell samples, ferrofluids, and PBS containing 2% (v/v) FBS were injected into the device through different inlets. Images and videos of microparticles and cells were recorded with a high-resolution CCD camera (Carl Zeiss, Inc., Germany).
Polystyrene microparticles (Polysciences, Inc., Warminster, PA) with diameters of 15.7 µm and 5.8 µm were prepared in PBS at the concentration of 2×106 particles/mL for device calibration. Microparticle mixtures were injected into inlet A with a flow rate of 0.5–8 µL/min. The flow rate of inlet B was fixed at 5 µL/min for all experiments, and flow rate of inlet C (3.5–7 µL/min) was adjusted accordingly to make the ferrofluid/buffer boundary just right below the outlet 4, to allow for particle and cell extraction. The magnet was placed 1, 4 and 7 mm away from the channel, which corresponded to magnetic field strengths 300, 134, 72 mT, and magnetic field gradients 83.4, 32.2, 12.9 T/m (ESI, Fig. S1†). Ferrofluid concentrations of 0.13, 0.26 and 0.39% (v/v) were used.
For cancer cells of cell lines/RBCs and cancer cells of cell lines /WBCs separation experiments, cell mixtures were injected into inlet A at the flow rate of 20 µL/min. The magnet was placed 4 mm away from the channel and ferrofluids with a concentration of 0.26% (v/v) was used. After separation, cells from outlet 4 were collected into a 96-well plate for counting under a fluorescence microscope.
Simulation
Cell trajectories were simulated in three-dimensional (3D) manner by modifying previously developed models with a concentration profile of ferrofluids across the width of the microchannel.28, 29 Briefly, we used an analytical model that could predict the 3D transport of diamagnetic cells in ferrofluids inside a microfluidic channel coupled with permanent magnets (see ESI†). The magnets produced a spatially non-uniform magnetic field that led to a magnetic buoyancy force on the cells. Resulting trajectories of the cells were obtained by (1) calculating the 3D magnetic buoyancy force via an experimentally verified and analytical distribution of magnetic fields as well as their gradients, together with a nonlinear magnetization model of the ferrofluid, (2) deriving the hydrodynamic viscous drag force with an analytical velocity profile in the channel including “wall effect”, (3) solving governing equations of motion using analytical expressions of magnetic buoyancy force and hydrodynamic viscous drag force. The parameters of simulation (device dimension and geometry, fluid and cell properties, and magnetic fields) reflected exact experimental conditions.
Results and discussion
Ferrofluid properties
Fig. 2a shows size distribution and a sample TEM image of maghemite nanoparticles of the custom-made ferrofluid. The particles had a mean diameter of 11.24 nm with a standard deviation of 2.52 nm. Although nanoparticles with larger diameters were considered to be more biocompatible because they may inhibit direct diffusion across cell membrane,30–33 we chose this diameter for the nanoparticles to preserve the colloidal stability of ferrofluids against agglomeration due to gravitational settling and magnetic dipole-dipole attraction.15 Particles with a large diameter are prone to settling and agglomeration, and can disrupt continuous-flow separation. However, at a diameter of ~10 nm, thermal agitation at room temperature is sufficient to keep particles separated. As a result, our ferrofluids remained colloidally stable after at least 10 months’ storage. The nanoparticles were also functionalized with a graft copolymer as surfactants to prevent them from coming too close to one another when there was a magnetic field. In all of the cell manipulation experiments conducted here, our ferrofluids did not show any sign of nanoparticle agglomeration under magnetic fields. We measured the saturation magnetization of the as-synthesized ferrofluid to be 0.96 kA/m, as shown in Fig. 2b. Considering the bulk magnetization of maghemite is about 370 kA/m,34 we estimated the volume fraction of the magnetic content of the ferrofluid to be 0.26%. The low volume fraction of the ferrofluid not only leaded to good biocompatibility for live cells, but also enabled us to observe cell motion in microchannel directly with bright-field microscopy, which was difficult with opaque ferrofluids of high solid volume fractions. The surface charge of the particles was negative, measured by zeta potential of −27.2±11.4 mV (ESI, Fig. S2†). The ferrofluid was made to be isotonic and its pH was adjusted to 7.0 for biocompatible cell manipulation.
Fig. 2.

(a) Size distribution of the maghemite nanoparticles within the ferrofluid (11.24±2.52 nm). Inset: a TEM image of the maghemite nanoparticles in the ferrofluid. Scale bar: 20 nm. (b) Magnetization curve of the ferrofluid. The saturation magnetization of this ferrofluid is 0.96 kA/m, corresponding to a 0.26% volume fraction of magnetic materials.
Ferrofluid biocompatibility
We investigated the biocompatibility of this ferrofluid by exposing a total of 7 cancer cell lines to it, and studying their short-term viability, long-term cell proliferation, and cellular nanoparticle uptake after the exposure. These cell lines included two lung cancer cell lines (A549 and H1299), three breast cancer cell lines (HCC1806, MDA-MB-231 and MCF-7), one prostate cancer cell line (PC3), and one cervical cancer cell line (HeLa). We chose these cell lines because they were frequently used to validate new microfluidic separation technologies for cancer cells.
Short-term cell viability was examined using MTT assay. Here we use the A549 lung cancer cell line as an example to describe the results. Fig. 3a compares A549 cell viabilities after 12-hour exposures to a control medium and 4 different concentrations (0.05%, 0.19%, 0.22%, and 0.26% v/v) of custom-made ferrofluids. A549 cells showed 100±3% viability in the control medium, and gradually decreasing viabilities in ferrofluids (91±3% viability for 0.05% ferrofluid, 86±4% viability for 0.19% ferrofluid, 83±4% viability for 0.22% ferrofluid, and 83±3% viability for 0.26% ferrofluid). This is expected as the nanoparticle concentration does affect the short-term cell viability, due to either particle diffusion across cellular membrane or endocytosis of particles by cells. Still, A549 cells retained 83±3% viability in a 0.26% concentration ferrofluid after a 12-hour period of exposure, which was at least 6 times longer than current cell viability studies in custom-made ferrofluids.3, 16, 18 Such a long period of exposure is typically not necessary for high-throughput cell separation; and a more reasonable estimated time of ferrofluid exposure for current cell separation schemes is 1–2 hours. Within such a time frame, Table 1 summarizes the results of all 7 cell lines, which showed consistently over 90% viability after 2-hour exposure in a 0.26% ferrofluid. They confirmed that this ferrofluid possessed minimal detrimental effect on 7 cancer cell lines in the short term.
Fig. 3.

(a) Cell viability of A549 cells was evaluated by MTT assay. Different concentrations of ferrofluids (0.05%, 0.19%, 0.22%, and 0.26% v/v) were added in the incubation medium. Average cell viabilities were 100% in the control, 91% in 0.05% ferrofluid, 86% in 0.19% ferrofluid, 84% in 0.22% ferrofluid, and 83% in 0.26% ferrofluid, after a 12-hour incubation with ferrofluids. (b) Growth curves of A549 cells with different exposure times to ferrofluids were determined by MTT assay. Cells incubated with ferrofluids for 2 h grew more slowly than the control group. No significant difference was found between cells incubated with ferrofluid for 1 min and the control group. (c–e) Cellular nanoparticle uptake of A549 cells. The cells were incubated with ferrofluid for 0, 1 min, and 2 h and then subjected to Prussian blue staining. Positive staining was visible in the majority of the cells that were incubated with ferrofluid for 2 h (e). 1 min incubation with ferrofluid showed little cellular uptake of nanoparticles (d). (c) Control. Scale bars: 50 µm.
Table 1.
Short-term viability and long-term proliferation of 7 cancer cell lines after exposures to the custom-made biocompatible ferrofluid. Short-term viability was determined by a Live/Dead assay after 2-hour exposure to the ferrofluid with a 0.26% volume fraction ferrofluid. Long-term proliferation was determined by culturing cells after the same exposure to the ferrofluid.
| Cell line | A549 (Lung cancer) |
H1299 (Lung cancer) |
HeLa (Cervical cancer) |
MDA- MB-231 (Breast cancer) |
HCC1806 (Breast cancer) |
MCF-7 (Breast cancer) |
PC3 (Prostate cancer) |
|---|---|---|---|---|---|---|---|
| Viability after 2-hour exposure to ferrofluids | 94% | 95% | 92% | 95% | 94% | 95% | 96% |
| Proliferation to confluence after 2-hour exposure to ferrofluids | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
In addition to short-term viability, we also examined whether all cell lines were able to proliferate normally after ferrofluid exposures. As shown in Table 1, all 7 cell lines were capable of normal proliferation to confluence after 2-hour incubation in the ferrofluid. This is the first time that long-term effects of a colloidally stable ferrofluid were studied on several cancer cell lines. To the best of our knowledge, Yellen’s group conducted the only proliferation study using HUVEC after exposures to a bovine serum albumin coated ferrofluid.18
Our ferrofluids showed excellent short-term and long-term biocompatibility for 7 types of cancer cells. As discussed earlier, even with such a ferrofluid, it is still better to minimize cells’ exposure time to it, as prolonged exposure time will inevitably lead to particle endocytosis and/or diffusion, which may affect cells’ normal functions. To investigate the effect of exposure time on cell proliferation, we examined A549 cells again using both MTT assay for proliferation and Prussian blue assay for nanoparticle uptake. This time, A549 cells were either seeded directly into a 96-well plate as a control or incubated in ferrofluids for 1-minute and 2-hour. Their proliferation measurements (absorbance at 570 nm of MTT assay) was evaluated and recorded every 24-hour, and their nanoparticle uptake (iron distribution) were imaged after incubation with ferrofluids using Prussian blue assay. Fig. 3b compares cell proliferations between control, 1-minute exposure, and 2-hour exposure to ferrofluids. No significant change was found between the control and 1-minute exposure; cells incubated in ferrofluids for 1-minute were able to proliferate normally and resulted in nearly the same growth rate as the control. This was also confirmed by nanoparticle uptake comparison in Fig. 3c and 3d, which showed almost identical and little iron presence. On the other hand, 2-hour exposure to ferrofluids did affect A549 cell proliferation in a noticeable and negative way, evidenced by a lower growth rate in Fig. 3b, and a significant iron presence in Fig. 3e. The longer exposure time of A549 cells in ferrofluids led to the higher cellular uptake of nanoparticles and slower cell growth. It is therefore beneficial to minimize the exposure time of cells to ferrofluids.
Device optimization and calibration
We described previously the general idea behind the device design to significantly decrease the exposure time of cells to ferrofluids. Briefly, we aimed to eliminate unnecessary exposure time including sample preparation and sample extraction and allow cells to be in contact with ferrofluids when it was absolutely necessary (e.g., separation). The flow rate and channel length determined the exposure time in a typical cell separation protocol, which was estimated to be on the order of seconds in our devices. Here we described the results of device optimization and calibration using analytical models and microparticles. This optimization was also verified by a separation experiment of cancer cells of cell culture lines and red blood cells.
We used the 3D analytical model to optimize our device (Fig. 1d) for a potential cell separation application. In this case, we allowed two groups of cells with different sizes to enter the channel and simulated their trajectories. Sample simulated trajectories of two types of cells (cancer cells with a presumed 15.7 µm diameter, and red blood cells with a presumed 5.8 µm diameter) are shown in Fig. 4a. Location of the simulation window is depicted in Fig. 4b. We chose these two cells’ sizes for simulation because microparticles with exact sizes were available for calibration purposes. From these trajectories, we calculated two outputs – a deflection in the y-direction for the larger cancer cells, denoted as Y2, and a separation distance between the two types of cells, denoted as ΔY. Both outputs were optimized using parameters including channel length (4–8 cm), magnetic fields and gradients (field: 72–300 mT; gradient: 12.9–83.4 T/m), flow rates of cell inlet (inlet A in Fig. 1c, 0.5–20 µL/min), and ferrofluid concentrations (0.13–0.39% v/v). The goal was to achieve separation of larger cancer cells from smaller blood cells, which translated to maximizing both Y2 and ΔY simultaneously. Calibration of the device used two types of microparticles with diameters of 5.8 µm and 15.7 µm. Experimental conditions for the calibration including magnetic fields, flow rates, and ferrofluid concentrations were the same as in simulation. A sample image of the microparticles’ trajectories at the outlets is shown in Fig. 4c. We extracted both outputs (Y2 and ΔY) from the images and used them to compare simulation and calibration results. Location of the experimental observation window is depicted in Fig. 4b. In this device design, cell samples from inlet A were quickly mixed with ferrofluids from inlet B because of a strong magnetic convection resulted from interactions between the ferrofluids and permanent magnet.24, 25, 35 With typical device and flow parameters used in cell separation, we estimated that a homogeneous mixing could be achieved at a channel length of a few millimeters away from the inlets (ESI, Fig. S3†), which was confirmed by experimental observations in Fig. 4d and 4e. Given that the total channel length was ~5 cm, we considered the effects from mixing on cell separation to be minimal and neglected them in the following optimization.
Fig. 4.

Device optimization via simulations and calibrations. (a) Simulated concentration profile of ferrofluids and the cells’ trajectories across ferrofluids and the buffer stream. The ferrofluid concentration is represented by gray scale. The trajectories of a cancer cell with 15.7 µm diameter are indicated by red circles, and trajectories of a red blood cell with 5.8 µm diameter are indicated by green circles. (b) Schematic of the microchannel with various simulation and observation windows. (c) A representative image of microparticle separation observed in the window from (b). Representative images of magnetic convective mixing (without microparticles) from the observation window in (b) with the magnet (d) and without the magnet (e). The observation window is 6.7 mm away from the entrance of the main channel. The gray scale intensity profile in the red dashed boxes of (d) and (e) can be found in Supplementary Information (ESI, Fig. S3†). The flow rate of inlet A is fixed at 4 µL/min, ferrofluid concentration is fixed at 0.26%, and magnetic field gradient is fixed at 32.2 T/m for (a)-(f). Numerical simulation of separation distance ΔY and deflection distance Y2 at the end of the channel with parameters including: (f) channel length, (g) magnetic field gradient, (h) flow rate of inlet A, and (i) ferrofluid concentration. Experimental calibration of these parameters for separation distance ΔY and deflection distance Y2 using microparticles: (j) ΔY and Y2 as a function of magnetic field gradient, (k) ΔY and Y2 as a function of flow rate of inlet A, (l) ΔY and Y2 as a function of ferrofluid concentration. The flow rate of inlet A is fixed at 4 µL/min and ferrofluid concentration is fixed at 0.26% (v/v) for (g) and (j). Magnetic field gradient is fixed at 32.2 T/m and ferrofluid concentration is fixed as 0.26% (v/v) for (h) and (k). Magnetic gradient is fixed at 32.2 T/m and the flow rate of inlet A is fixed at 4 µL/min for (i) and (l). Scale bars: 200 µm.
We first optmized the channel length, as the dimensions of the permanent magnet used in the separation remained constant. Fig. 4f shows under typical device and flow parameters, both ΔY and Y2 increased with the channel length, and reached saturation when the length was around 5.8 cm. It should be noted that the optimized channel length could be affected by parameters including flow rates, magnet properties, ferrofluid properties, and cell types. The second set of parameters we optimized for the device were the magnetic field strength and its gradient, both of which changed their values as we adjusted the distance between the magnet and channel (ESI†, Fig. S1). Fig. 4g and 4j show in both simulation and calibration, when the magnetic field gradient increased, the overall deflection of 15.7 µm microparticle Y2 increased, too. This was because the driving force – magnetic buoyancy force on the microparticles, was determined in part by the gradient. The larger the gradient, the larger the magnetic force and resulted deflection of microparticles. Interestingly, the simulated separation distance between two microparticles, ΔY had a peak at a medium gradient (38.2 T/m), which was confirmed by the calibration experiments. This was due to the fact that both microparticles reached their maximum deflections very quickly under a strong gradient (83.4 T/m) in the device, resulting in a mixing rather than separation of the two types of microparticles. On the other hand, separation distance also decreased when the gradient was too weak (12.9 T/m) to deflect microparticles and distinguish them. As a result, we chose to use the medium field and gradient (134 mT and 32.2 T/m) for subsequent cell separation.
A third parameter to optimize was the flow rate of cell inlet (inlet A in Fig. 1c). Both simulation (Fig. 4h) and calibration (Fig. 4k) results show a monotonically decreasing trend for Y2 and ΔY, as the magnitude of the flow rate increased. This was consistent with the findings from existing cell separation technologies,36 where a tradeoff existed between throughput (flow rate in this case) and separation efficiency (separation distance ΔY in this case).
The last parameter we chose to optimize was the ferrofluid concentration. Generally speaking, a higher concentration of ferrofluid resulted in a higher magnitude of magnetic force on the microparticles, leading to a larger deflection, which was observed in both simulation and calibration (Fig. 4i and 4l) of Y2. However, a high ferrofluid concentration was not necessarily beneficial for achieving a larger separation distance ΔY. Figures 4f shows there was an optimal ferrofluid concentration close to 0.17% (v/v) for both ΔY and Y2. Concentrations higher than 0.17% (v/v) resulted in larger Y2 but smaller ΔY. This was because both microparticles achieved sufficient deflections in a strongly magnetized ferrofluid, resulting in mixing rather than separation of the two. Microparticle calibration experiments in Fig. 4l did not capture this optimal concentration, as there were only three concentrations of ferrofluids used.
While the simulation and calibration results matched each other quite well qualitatively, we noticed quantitate differences between the two for the separation distance ΔY. The simulation results consistently yielded larger ΔY than the calibration. This might be due to the fact that the simulation did not take into account the widening of cell streams, which effectively reduced the separation distance, as shown in Fig. 4c.
Because of the device design and the diffusion between ferrofluids and buffer stream, cell separation efficiency and the amount of ferrofluids in collection outlet could affect each other. In this study, our goal was to not only achieve biocompatible and label-free cell separation with best possible separation efficiency, but also maintain cell integrity. As a result, we optimized the flow rates of ferrofluids and buffer stream so that their diffusion boundary was at exactly the boundary of outlets 3 and outlet 4 (collection outlet). This way, the majority of spiked cancer cells could be extracted, while the amount of ferrofluids was minimized in collection outlet. We estimated the concentration of magnetic nanoparticles that diffused into the collection outlet to be ~0.002% (v/v) via a simulation using typical flow rate parameters. A magnetization measurement from one experiment revealed a 0.00128% (v/v) concentration (ESI, Fig. S4†) of the liquid collected from the same outlet, which was on the same order of magnitude as the simulation. This measured concentration of nanoparticles in the collection outlet was 203-fold more dilute than the original ferrofluid, and unlikely produced detrimental effects to cells. We also estimated via a simulation the overall exposure time of cells in ferrofluids to be 4–53 seconds depending on the cell input flow rates (0.5–20 µL/min) in current devices.
Finally, we verified these optimized and calibrated parameters (magnetic field and gradient: 134 mT, 32.2 T/m, ferrofluid concentration: 0.26% v/v, channel length: 5.8 cm) with a separation of spiked cancer cells (A549 lung cancer and MCF-7 breast cancer, 100 cells/mL) from diluted human whole blood (RBC concentration: 2×106 cells/mL) at 0.9 mL/h throughput. Detailed results are summarized in supplementary information (ESI, Fig. S5 and Table S1†). Briefly, separation efficiency (defined as the ratio of captured cancer cells to spiked cancer cells) for A549 cell line was 77±6%, and the purity of cancer cells recovered (defined as the ratio of cancer cells to all cell types in collection outlet) was 62.1±0.9%. Separation efficiency for MCF-7 cell line was 84±4%, and its purity was 59.2±0.8%. We concluded that these optimized parameters could be used to enable a high-throughput and high efficiency low-concentration cell separation in ferrofluids.
Cell separation
We chose to validate the biocompatible cell separation strategy using spiked cancer cells from cell culture lines in undiluted white blood cells (WBCs). Separating spiked cancer cells from WBCs is potentially the first step to render ferrofluid-based “negative magnetophoresis” useful in rare cell separation applications such as enriching circulating tumor cells (CTCs) from peripheral blood.1 Since CTCs occur at an extremely low concentration of 1–10 cells every 1 billion RBCs and 1 million of WBCs,37–39 its enrichment requires the development of a highly efficient and high-throughput separation.21 For that purpose, we scaled up the device by increasing the depth of the device from 52 µm to 150 µm in Fig. 1d to accommodate high cell flow rates (20 µL/min, i.e., 1.2 mL/h, see ESI, Fig. S6† for device calibration), and chose the optimized magnetic field and gradient (134 mT, 32.2 T/m), ferrofluid concentration (0.26% v/v), and channel length of 5.8 cm based on previous optimization and calibration results. Mean diameters of all cells used here were measured to be: 15.5 µm for A549, 16.9 µm for H1299, 18.7 µm for MCF-7, 18.1 µm for MDA-MB-231, 18.9 µm for PC-3, and 11.1 µm for WBCs.
We validated the separation of spiked cancer cells from undiluted human blood with only WBCs. This is more challenging than separating cancer cells and RBCs, as the size differences between cancer cells and WBCs are much more subtle. We used A549, H1299, MCF-7, MDA-MB-231, and PC-3 cell lines, with a spike ratio of 100 cells/mL in 1 mL of undiluted WBCs. Cell flow rate was 1.2 mL/h. Experimental results are summarized in Fig. 5a–c and Table 2. Fig. 5a shows A549 cancer cells and WBCs were flowing near the bottom of the channel and exiting through the outlet 1, resulting in no separation of the two when there was no magnetic field. Fig. 5b shows larger A549 cancer cells deflected from the ferrofluid stream into the PBS buffer stream toward outlet 4 when there was a magnetic field. WBCs remained in the ferrofluid stream and exited through outlets 2 and 3. Fluorescence image of A549 cells confirmed such separation in Figure 5c. From Table 2, the separation efficiency for A549 cells was 80±3%. The purity of cancer cells was 25.3±0.1% from outlet 4. Similar experiments were carried out to separate multiple cancer cell lines from WBCs. The separation efficiencies were 81±5%, 82±5%, 82±4%, and 86±6% for H1299, MCF-7, MDA-MB-231, and PC-3 cells. Even with the subtle size difference between cancer cells and WBCs, we were able to achieve high separation efficiency (80–86%) using this strategy. The purity of cancer cells was on the order of 20% for all cases. This size-based separation strategy performed well in separating cancer cells from WBCs. As the diameter of cancer cells increased from 15.5 µm (A549) to 18.9 µm (PC-3), we observed a slight increase in separation efficiency, which is expected as the separation is based on size difference of cell types.
Fig. 5.

Micrographs of spiked cancer cells of cell culture lines and white blood cells (WBCs) separation processes. (a) In absence of magnetic fields, cell mixtures exited the channel through outlet 1. (b) When magnetic fields were present, larger A549 cancer cells were deflected and reached the ferrofluid/buffer boundary, exited through outlet 4 (collection outlet), while smaller WBCs exited through other outlets. (c) Fluorescence images of A549 cancer cells during cell separation. A549 cells were stained with CellTracker Green. Dashed white lines depict the microchannel boundaries. Scale bars: 200 µm.
Table 2.
Summary of cancer cell separation performance
| Cell line | No. of cells spiked |
No. of cells captured |
Efficiency | No. of WBCs | Purity |
|---|---|---|---|---|---|
| A549 | 100 | 80±3 | 80±3% | 236±22 | 25.3±0.1% |
| H1299 | 100 | 81±5 | 81±5% | 218±15 | 27.1±0.1% |
| MCF-7 | 100 | 82±5 | 82±5% | 208±29 | 28.3±0.1% |
| MDA-MB-231 | 100 | 82±4 | 82±4% | 233±48 | 26.0±0.2% |
| PC-3 | 100 | 86±6 | 86±6% | 212±32 | 28.8±0.1% |
100 CellTracker Green stained cancer cells were spiked into 1 mL of WBCs. Data are expressed as Mean±S.D., n=3.
We investigated the short-term viability and the long-term proliferation of separated A549 cancer cells collected from the device. After running the cell mixture through the device for separation, A549 cells were collected from the outlet 4 and studied for their viability using a Live/Dead assay. Fig. 6a shows the viability of A549 cells before and after separation were 95.2±2.0% and 93.8±1.5%, respectively. Fig. 6b shows representative fluorescence images of A549 cells before and after separation using a Live/Dead stain. They indicate no significant impact on cell viability from the ferrofluid exposure and cell processing. We also examined the long-term proliferation of A549 cancer cells after separation. Fig. 6c shows the images of A549 cells over a 4-day period and Live/Dead staining of the cultured cells on day 4. We concluded that A549 cells were able to proliferate to confluence.
Fig. 6.

(a) Short-term cell viability comparison between before vs. after separation groups. No significant difference was found between the two. (b) Representative images of Live/Dead staining of the before (top) and after separation (bottom) groups. Calcein-AM (green) and PI (red) channels were merged in these images. Scale bars: 100 µm. (c) Bright field images of cultured A549 cells collected after separation from day 1 to day 4. A Live/Dead staining of the cultured cells on day 4 showed excellent viability. A control group of cell culture was used for comparison. No significant difference was found in cell proliferation between cells in the control group and cells collected after device separation. Scale bars: 50 µm.
In summary, we developed a biocompatible and label-free cell separation method using ferrofluids, to differentiate between low-concentration cancer cells of cell culture lines and WBCs with subtle size differences in a high-throughput and high-efficiency manner. Separated cancer cells showed excellent average viability (94.4±1.3%) and normal proliferation. This could be useful in preserving cell integrity for further analysis after enrichment. We achieved on average 82.2% separation efficiency in separating a variety of cancer cells from cell culture lines from WBCs at an extremely low concentration of 100 cells/mL with a throughput of 1.2 mL/h. The efficiency obtained here is comparable the average reported efficiency of 82% from recent label-free microfluidic separation of cultured cancer cells in blood.21 For examples, the separation efficiency reported here is close to the efficiencies of methods based on standing surface acoustic wave,40 dielectrophoresis,41–43 slanted spiral channel,44 and is higher than the efficiency of vortex technology.45–47 The purity of recovered cancer cells from this method was between 25.3% and 28.8% depending on specific cell lines. It is also comparable to the reported purity values from existing label-free methods when they were used to separate spiked cancer cells from blood. These reported purities varied dramatically from 0.1% to 90%,40–47 as most of the label-free methods focused on improving separation efficiency of low-concentration cells, rather than their purity. For examples, the recovered cancer cell purity of this method is higher than purities reported from the standing surface acoustic wave method (0.1%, calculated from ~90% WBC removal rate after separation),40 and a few dielectrophoretic methods (10%42 and 16.24%43), but lower than purities from the slanted spiral channel method (50%),44 and the vortex technology (57–94%).45–47
Although the throughput of current devices (1.2 mL/h) is comparable to the throughput from methods based on standing surface acoustic wave40 and dielectrophoresis,41–43 it needs improvement in order to handle the clinically relevant amount of human blood (e.g., 7.5 mL/h for CTCs enrichment), which was demonstrated by methods such as slanted spiral channel44 and vortex technology.45–47. Further scale-up of a single device and/or potential multiplexing of several devices together could improve the throughput. As a proof-of-concept demonstration, this method was used to separate low-concentration spiked cancer cells from WBCs, with RBCs removed beforehand by a RBC lysis buffer. In the future, it is beneficial and necessary to design a two-step separation device, which can first remove the bulk of RBCs, and then further enrich cancer cells from mostly WBCs to automate whole blood processing on-chip. While other methods have been demonstrated to be able to handle clinical samples such as whole blood,40, 43–47 this method was still at its early stage of development and was limited to cultured cancer cell. Future studies using whole blood needs to be conducted to further evaluate the potential of this method in rare cell separation.
Conclusions
In this study, we reported a biocompatible and label-free separation method of low-concentration cancer cells of cell culture lines from undiluted white blood cells based on their size difference, by using a custom-made ferrofluid and integrating on-chip sample preparation, separation and extraction into a microfluidic device. The ferrofluid possessed not only ideal biocompatible properties for live cell manipulation, including its low magnetic content concentration (e.g., 0.26% volume fraction), neutral pH, isotonicity, maghemite nanoparticles and their surfactant but also excellent colloidal stability that enables high-throughput and high-efficiency continuous separation. A biocompatibility study of 7 commonly used cancer cell lines showed consistently over 90% of short-term viabilities and abilities to proliferate to confluence for all cells, even after extended exposure to this ferrofluid. Additionally, an optimized device design eliminated time-consuming off-chip sample preparation and extraction steps, which reduced overall exposure time of cells to ferrofluids from hours to seconds. To demonstrate the potential of this method in rare cell separation, a variety of cancer cells from cell culture lines in white blood cells were separated with an average efficiency of 82.2%, at a throughput of 1.2 mL/h with an extremely low concentration of ~100 cancer cells/mL. Separated cancer cells showed excellent viability and normal proliferation. This method addressed the challenges associated with cell separation in ferrofluids, including excellent biocompatibility of not only the custom-made ferrofluid, but also the assay itself, as well as device design and optimization specifically for the low concentration of target cells. While still at its early stage of development, this method could be a promising tool for rare cell separations because of its excellent biocompatibility, label-free operation, performances with culture cancer cells, along with potentials for device scale-up, multiplexing, and further optimization.
Supplementary Material
Acknowledgments
This material is based upon work supported by the National Science Foundation under Grant Nos. 1150042, 1242030, and 1359095; and by the National Institute of General Medical Sciences of the National Institutes of Health under Award No. R21GM104528. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Electronic supplementary information (EST) available.
References
- 1.Zhao W, Cheng R, Miller JR, Mao L. Advanced Functional Materials. 2016;26:3916–3932. doi: 10.1002/adfm.201504178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shen F, Hwang H, Hahn YK, Park JK. Anal Chem. 2012;84:3075–3081. doi: 10.1021/ac201505j. [DOI] [PubMed] [Google Scholar]
- 3.Kose AR, Fischer B, Mao L, Koser H. P Natl Acad Sci USA. 2009;106:21478–21483. doi: 10.1073/pnas.0912138106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zeng J, Deng YX, Vedantam P, Tzeng TR, Xuan XC. J Magn Magn Mater. 2013;346:118–123. [Google Scholar]
- 5.Winkleman A, Gudiksen KL, Ryan D, Whitesides GM, Greenfield D, Prentiss M. Appl Phys Lett. 2004;85:2411–2413. [Google Scholar]
- 6.Rodriguez-Villarreal AI, Tarn MD, Madden LA, Lutz JB, Greenman J, Samitier J, Pamme N. Lab Chip. 2011;11:1240–1248. doi: 10.1039/c0lc00464b. [DOI] [PubMed] [Google Scholar]
- 7.Zeng J, Chen C, Vedantam P, Tzeng TR, Xuan XC. Microfluid Nanofluid. 2013;15:49–55. [Google Scholar]
- 8.Kauffmann P, Ith A, O’Brien D, Gaude V, Boue F, Combe S, Bruckert F, Schaack B, Dempsey NM, Haguet V, Reyne G. Lab Chip. 2011;11:3153–3161. doi: 10.1039/c1lc20232d. [DOI] [PubMed] [Google Scholar]
- 9.Durmus NG, Tekin C, Guven S, Sridhar K, Yildiz AA, Calibasi G, Ghiran I, Davis RW, Steinmetz LM, Demirci U. P Natl Acad Sci USA. 2015;112:3661–3668. doi: 10.1073/pnas.1509250112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tasoglu S, Khoory JA, Tekin HC, Thomas C, Karnoub AE, Ghiran IC, Demirci U. Adv Mater. 2015;27:3901–3908. doi: 10.1002/adma.201405660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Knowlton S, Yu CH, Jain N, Ghiran IC, Tasoglu S. Plos One. 2015;10 doi: 10.1371/journal.pone.0134400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yenilmez B, Knowlton S, Tasoglu S. Advanced Materials Technologies. 2016;1:1600144–1600153. [Google Scholar]
- 13.Knowlton SM, Sencan I, Aytar Y, Khoory J, Heeney MM, Ghiran IC, Tasoglu S. Sci Rep. 2015;5 doi: 10.1038/srep15022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Skjeltorp AT. Phys Rev Lett. 1983;51:2306–2309. [Google Scholar]
- 15.Rosensweig RE. Ferrohydrodynamics. Cambridge University Press; Cambridge, UK: 1985. [Google Scholar]
- 16.Zhao W, Zhu T, Cheng R, Liu Y, He J, Qiu H, Wang L, Nagy T, Querec TD, Unger ER, Mao L. Advanced Functional Materials. 2016;26:3990–3998. doi: 10.1002/adfm.201503838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dharmasiri U, Witek MA, Adams AA, Soper SA. Annu Rev Anal Chem. 2010;3:409–431. doi: 10.1146/annurev.anchem.111808.073610. [DOI] [PubMed] [Google Scholar]
- 18.Krebs MD, Erb RM, Yellen BB, Samanta B, Bajaj A, Rotello VM, Alsberg E. Nano Lett. 2009;9:1812–1817. doi: 10.1021/nl803757u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Conner SD, Schmid SL. Nature. 2003;422:37–44. doi: 10.1038/nature01451. [DOI] [PubMed] [Google Scholar]
- 20.Zhu TT, Cheng R, Lee SA, Rajaraman E, Eiteman MA, Querec TD, Unger ER, Mao LD. Microfluid Nanofluid. 2012;13:645–654. doi: 10.1007/s10404-012-1004-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen YC, Li P, Huang PH, Xie YL, Mai JD, Wang L, Nguyen NT, Huang TJ. Lab Chip. 2014;14:626–645. doi: 10.1039/c3lc90136j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu GP, Hejiazan M, Huang XY, Nguyen NT. Lab Chip. 2014;14:4609–4615. doi: 10.1039/c4lc00885e. [DOI] [PubMed] [Google Scholar]
- 23.Tarn MD, Lopez-Martinez MJ, Pamme N. Anal Bioanal Chem. 2014;406:139–161. doi: 10.1007/s00216-013-7363-6. [DOI] [PubMed] [Google Scholar]
- 24.Hejazian M, Phan DT, Nguyen NT. Rsc Adv. 2016;6:62439–62444. [Google Scholar]
- 25.Hejazian M, Nguyen NT. Micromachines-Basel. 2017;8 [Google Scholar]
- 26.Massart R. Ieee T Magn. 1981;17:1247–1248. [Google Scholar]
- 27.Xia YN, Whitesides GM. Annu Rev Mater Sci. 1998;28:153–184. [Google Scholar]
- 28.Cheng R, Zhu TT, Mao LD. Microfluid Nanofluid. 2014;16:1143–1154. [Google Scholar]
- 29.Zhu TT, Lichlyter DJ, Haidekker MA, Mao LD. Microfluid Nanofluid. 2011;10:1233–1245. [Google Scholar]
- 30.Huang J, Bu LH, Xie J, Chen K, Cheng Z, Li XG, Chen XY. Acs Nano. 2010;4:7151–7160. doi: 10.1021/nn101643u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luciani N, Gazeau F, Wilhelm C. J Mater Chem. 2009;19:6373–6380. [Google Scholar]
- 32.Thorek DLJ, Tsourkas A. Biomaterials. 2008;29:3583–3590. doi: 10.1016/j.biomaterials.2008.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jiang W, Kim BYS, Rutka JT, Chan WCW. Nat Nanotechnol. 2008;3:145–150. doi: 10.1038/nnano.2008.30. [DOI] [PubMed] [Google Scholar]
- 34.Hwang DK, Dendukuri D, Doyle PS. Lab Chip. 2008;8:1640–1647. doi: 10.1039/b805176c. [DOI] [PubMed] [Google Scholar]
- 35.Mao L, Koser H. OVERCOMING THE DIFFUSION BARRIER: ULTRA-FAST MICRO-SCALE MIXING VIA FERROFLUIDS. The 14th International Conference on Solid-State Sensors, Actuators and Microsystems; Lyon, France. [2007 June 10–14]. 2007. [Google Scholar]
- 36.Gossett DR, Weaver WM, Mach AJ, Hur SC, Tse HTK, Lee W, Amini H, Di Carlo D. Anal Bioanal Chem. 2010;397:3249–3267. doi: 10.1007/s00216-010-3721-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alix-Panabieres C, Pantel K. Clin Chem. 2013;59:110–118. doi: 10.1373/clinchem.2012.194258. [DOI] [PubMed] [Google Scholar]
- 38.Alix-Panabieres C, Pantel K. Nat Rev Cancer. 2014;14:623–631. doi: 10.1038/nrc3820. [DOI] [PubMed] [Google Scholar]
- 39.Yu M, Stott S, Toner M, Maheswaran S, Haber DA. J Cell Biol. 2011;192:373–382. doi: 10.1083/jcb.201010021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li P, Mao ZM, Peng ZL, Zhou LL, Chen YC, Huang PH, Truica CI, Drabick JJ, El-Deiry WS, Dao M, Suresh S, Huang TJ. P Natl Acad Sci USA. 2015;112:4970–4975. doi: 10.1073/pnas.1504484112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang SB, Wu MH, Lin YH, Hsieh CH, Yang CL, Lin HC, Tseng CP, Lee GB. Lab Chip. 2013;13:1371–1383. doi: 10.1039/c3lc41256c. [DOI] [PubMed] [Google Scholar]
- 42.Gascoyne PRC, Noshari J, Anderson TJ, Becker FF. Electrophoresis. 2009;30:1388–1398. doi: 10.1002/elps.200800373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moon HS, Kwon K, Kim SI, Han H, Sohn J, Lee S, Jung HI. Lab Chip. 2011;11:1118–1125. doi: 10.1039/c0lc00345j. [DOI] [PubMed] [Google Scholar]
- 44.Warkiani ME, Guan GF, Luan KB, Lee WC, Bhagat AAS, Chaudhuri PK, Tan DSW, Lim WT, Lee SC, Chen PCY, Lim CT, Han J. Lab Chip. 2014;14:128–137. doi: 10.1039/c3lc50617g. [DOI] [PubMed] [Google Scholar]
- 45.Che J, Yu V, Dhar M, Renier C, Matsumoto M, Heirich K, Garon EB, Goldman J, Rao JY, Sledge GW, Pegram MD, Sheth S, Jeffrey SS, Kulkarni RP, Sollier E, Di Carlo D. Oncotarget. 2016;7:12748–12760. doi: 10.18632/oncotarget.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dhar M, Pao E, Renier C, Go DE, Che J, Montoya R, Conrad R, Matsumoto M, Heirich K, Triboulet M, Rao JY, Jeffrey SS, Garon EB, Goldman J, Rao NP, Kulkarni R, Sollier-Christen E, Di Carlo D. Sci Rep. 2016;6 doi: 10.1038/srep35474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sollier E, Go DE, Che J, Gossett DR, O’Byrne S, Weaver WM, Kummer N, Rettig M, Goldman J, Nickols N, McCloskey S, Kulkarni RP, Di Carlo D. Lab Chip. 2014;14:63–77. doi: 10.1039/c3lc50689d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.