

Abstract
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease with no known cure that affects at least five million people worldwide. Monozygotic twin concordance and familial aggregation studies strongly suggest that lupus results from genetic predisposition along with environmental exposures including UV light. The majority of the common risk alleles associated with genetic predisposition to SLE map to genes associated with the immune system. However, evidence is emerging that implicates a role for aberrant DNA repair in the development of lupus. Here we summarize our current knowledge of the potential association of lupus with mutations in DNA repair genes. We also discuss how defective or aberrant DNA repair could lead to the development of lupus.
Keywords: DNA repair, Systemic Lupus Erythematosus, somatic hypermutation, class switch recombination, cytoplasmic DNA, neoantigen
INTRODUCTION
Systemic lupus erythematosus (SLE or lupus) is a chronic autoimmune disease. The Lupus Foundation of America estimates that 1.5 million Americans, and at least 5 million people worldwide, suffer from a form of lupus. There are no known cures and very few novel treatments for this disease have advanced to the clinic within the last decade. SLE has 11 diagnostic criteria set forth by the American College of Rheumatology: malar rash, discoid rash, photosensitivity, oral ulcers, non-erosive arthritis, pleuritis or pericarditis, renal disorder, neurologic disorder, hematologic disorder, immunologic disorder (antibodies to native DNA, Sm nuclear antigen, or phospholipid), and the presence of high titers of antinuclear antibody [1]. Additionally, women are nine times more likely to develop SLE than men (for reviews see [2, 3]), and the disease is 2–3 times more prevalent in people of Asian, Hispanic, Native American, and African ancestry than people of European ancestry. Monozygotic twin concordance and familial aggregation studies strongly suggest that lupus results at least in part from genetic predisposition [4–6]. Numerous genome-wide association studies have identified over 50 common risk alleles associated with SLE, and the majority of them are associated with immune system function, as would be expected for an autoimmune disease (for a review see[7]).
Recent work from our laboratory shows that a single amino acid substitution in the Polb gene that alters a tyrosine to cysteine at position 265 (Y265C) in DNA polymerase beta (Pol β) leads to the development of lupus-like disease in mice. The disease is characterized by high titers of antinuclear antibodies, glomerulonephritis, and dermatitis, which are phenotypes analogous to human SLE [8]. Our previous demonstration that the Y265C variant Pol β protein is slow and unable to support base excision repair (BER) [9] suggests that defective or aberrant BER may be an underlying mechanism of lupus development. Importantly, two genome-wide association studies of individuals of Han Chinese ancestry with SLE independently replicated the association of SLE with the rs12676482 SNP, which resides in a non-coding region of the POLB gene [10, 11]. This SNP is in perfect linkage disequilibrium with rs2272733, which is highly correlated with decreased expression of the POLB gene in humans [12]. Decreased expression, as with the POLB genetic variant in Han Chinese, and low catalytic activity, as with the Polb Y265C mouse model, may play analogous roles in SLE development in humans and mice, respectively. Our work strongly implicates defective or aberrant DNA repair as a mechanism underlying lupus development.
Additional support for the possibility of DNA repair being associated with SLE comes from findings showing that cells derived from SLE patients are unable to repair DNA lesions as efficiently as control cells. An early study analyzing DNA repair and its association with autoimmunity shows that lymphocytes from SLE patients have a major defect in the removal of O6-methylguanine after treatment with N-methyl-N-nitrosourea (MNU), and cell growth of SLE lymphocytes is significantly reduced in the presence of MNU as compared to lymphocytes from controls [13]. Other studies demonstrate defective DNA double-strand break (DSB) repair of lymphoblastoid cells derived from SLE patients [14] [15]. In addition, lymphocytes derived from a subset of individuals with SLE are exquisitely sensitive to hydrogen peroxide (H2O2), perhaps implicating aberrant BER as an underlying mechanism [16]. These were among the first studies that provided evidence that defective DNA repair is potentially linked to SLE pathogenesis.
In general, there are six major pathways of DNA repair that are evolutionarily conserved including nucleotide excision repair (NER), BER, mismatch repair (MMR), DSB repair either in the form of either homology-directed repair (HDR) or non-homologous end-joining (NHEJ), crosslink repair, and various forms of direct reversal of DNA damage. DNA repair essentially functions to remove DNA lesions, breaks, and mismatched bases, eventually restoring the DNA to its original form. Importantly, DNA repair has also been co-opted by the immune system to generate antibody diversity. The manner in which aberrant DNA repair could be associated with autoimmunity is the topic of this Perspective article.
GENETICS OF DNA REPAIR AND LUPUS
Germline mutations within the DNA repair genes OGG1, NEIL3, XRCC1, POLB, and FEN1 have been suggested to be associated with predisposition to SLE or linked to lupus-like features in mice (Table 1). Interestingly, each of these genes encodes a protein that functions during BER.
Table 1.
Mutations in Base Excision Repair Genes Associated with Lupus
| Gene | Protein Function | Notes |
|---|---|---|
| OGG1 | DNA glycosylase that removes 8-oxoguanine. | rs1052133 associated with the development of lupus nephritis[20]. |
| NEIL3 | DNA glycosylase that removes Spand Gh. | Germline variant (D132V) associated with autoimmunity; nullizyogous mice treated with poly-IC develop mild nephritis [25]. |
| POLB | Fills DNA gaps during BER; functions in SHM and perhaps VDJ recombination. | rs12676482 in perfect LD1 withrs2272733, associated with SLE in Han Chinese and correlated with downregulation of Pol β PolbY265c/c mice develop lupus [8, 10, 11]. |
| XRCC1 | Scaffold protein during BER. | rs25487 associated with increased risk for lupus in a Brazilian cohort[29]. |
| FEN1 | Processes Okazaki fragments and long patch BER substrates. | Mice with mutation in catalytic residue develop high levels of ANA[31]. |
linkage disequilibrium; see text for additional details.
BER is a DNA repair pathway that recognizes and removes small non-helix distorting lesions that arise from endogenous (e.g., reactive oxygen species and spontaneous deamination of cytosine) and exogenous sources (e.g., chemotherapeutic drugs). BER occurs primarily through four steps: 1) recognition and cleavage of the nucleotide base, 2) creation of a nick 5′ of the lesion, 3) DNA synthesis, and 4) DNA ligation [17]. This process is initiated by a mono- or bifunctional DNA glycosylase that recognizes specific DNA lesions. Monofunctional glycosylases remove the nitrogenous base leaving an apyrimidinic/apurinic (AP or abasic) site, while bifunctional glycosylases have an additional DNA lyase activity that incises 5′ of the AP site by β–elimination yielding a 3′ α,β-unsaturated aldehyde and a 5′ phosphate or lead to further processing of the 3′ aldehyde to a 3′ phosphate via a δ-elimination [18]. If an AP site is present, an AP endonuclease will recognize and cleave 5′ of the lesion to create a 3′ hydroxyl and a 5′ deoxyribose phosphate. AP endonuclease can also process the 3′ α,β-unsaturated aldehyde, resulting from the bifunctional glycosylase lyase activity, leaving a 3′ hydroxyl group [18]. At this point, the process can proceed in one of two subpathways: short patch (SP) or long patch (LP) repair. SP repair is dependent on the removal of the 5′ deoxyribose phosphate moiety by Pol β to create a 1-nucleotide (nt) gap and a 5′ phosphate. After 1 nt is extended from the 3′ hydroxyl to fill in the 1-nt gap, the DNA ligase III- X-ray Cross-complimenting Complex 1 (XRCC1) complex ligates the nick. On the other hand, LP BER is initiated by Pol β, δ, and/or ε extending 2–12 nts from the 3′ hydroxyl of the cleaved strand [19] using the intact strand as a template, and displacing the 5′ deoxyribose phosphate-containing strand. This creates a “flap” of DNA that is then cleaved by Flap Endonuclease 1 (FEN1), leaving a 5′ phosphate and a 3′ hydroxyl for ligation by DNA ligase 1.
The 8-oxoguanine DNA glycosylase, OGG1, is a bifunctional glycosylase that predominantly excises 8-oxoguanine that is mainly induced by reactive oxygen species during aerobic respiration. There is a single nucleotide polymorphism (SNP), rs1052133, in the OGG1 that is associated with the development of lupus nephritis and an observed increase of 8-oxoguanine levels in plasma [20]. This SNP in OGG1 results in a serine to cysteine amino acid substitution at position 326 (S326C) for the αOGG1 isoform,[20] which is the major isoform found in the nucleus [21–23]. The Nei-like DNA glycosylase 3, NEIL3, is a bifunctional DNA glycosylase that primarily recognizes and excises the 8-oxo-dG degradation products preferentially excising spiroiminodihydantoin (Sp) and guanidinohydantoin (Gh) adducts from single-strand DNA [24]. A recent report identified three patients from a consanguineous family that presented elevated serum levels of autoantibodies to cytoplasmic, structural, and nuclear proteins. These three patients are homozygous for the D132V NEIL3 germline variant that results in a decreased glycosylase activity on single-strand DNA containing an Sp enantiomer or a Gh lesion as compared to wild type NEIL3. Interestingly, neil3−/− mice present lupus-like phenotypes upon injection with poly(IC), including high levels of autoantibodies and mild glomerulonephritis [25]. XRCC1 is a scaffold protein that recruits Pol β and other BER enzymes to damaged sites in DNA [26, 27]. The rs25487 SNP that encodes an arginine to glutamine amino acid substitution at position 399 (R399Q) of XRCC1 is associated with high titer of anti-dsDNA antibodies in a Brazilian cohort [28]. Individuals of Taiwanese Han Chinese ancestry harboring this SNP are 1.8 times more likely to develop SLE. Additionally, the rs25487 SNP is significantly associated with photosensitivity, malar rash, high titer of anti-nuclear antibodies, hematologic disorder, and arthritis [29]. Interestingly, repair of DNA strand breaks of patients harboring the R399Q variant was decreased as compared to controls in alkaline comet assays. Recent work has shown that there is reduced residency time of the R399Q variant at DNA damage sites [30], which could result in the accumulation of DNA repair intermediates. As mentioned above, specific SNPs in the POLB gene are associated with SLE in individuals of Han Chinese ancestry [10–12]. Mutations in FEN1 may also be linked to lupus predisposition as mice harboring the E160D protein variant of FEN1 that removes a catalytic residue develop high titers of anti-nuclear and anti-dsDNA [31].
Although not historically thought of as DNA damage, ribonucleotides accumulate in DNA are removed predominantly by ribonuclease H2 (Rnase H2) (for a review see [32]). Individuals harboring rare hypomorphic variants in the RNASEH2 gene develop SLE [33]. Compromised DNA repair has also been linked to SLE development. One study reported that SLE patients have a more condensed chromatin structure that leads to downregulation of genes that function in NER and HDR and defective DNA repair [34]. Recent work has also provided evidence for the existence of lupus autoantibodies that recognize DNA repair proteins and that are able to enter into the nucleus and inhibit DNA repair (for a review see [35]).
MECHANISMS ASSOCIATED WITH DEFECTIVE DNA REPAIR AND SLE
The association of defective or aberrant DNA repair with the development of lupus is relatively new. There are hundreds of DNA repair genes that encode proteins that participate in the repair of DNA damage (see http://sciencepark.mdanderson.org/labs/wood/DNA_Repair_Genes.html). Mutations in any one of these genes may be associated with increased risk for lupus development because aberrant or defective DNA repair, as a result of mutations in DNA repair genes, has the potential to lead to a variety of consequences including alteration of antibody diversification, cell death, increased levels and aberrant processing of cytosolic DNA, or increased levels of mutations that lead to the generation of autoantibodies.
Antibody Diversification and Defective DNA Repair
Co-opted DNA repair play a critical role in antibody diversification (Figure 1). V(D)J recombination is essentially an NHEJ DNA repair process that is important for the early stages of B- and T-cell receptor development and occurs in the primary lymphoid organs (for a review see [36]). The process is initiated by the recombination-activating gene 1 (RAG1) and RAG2 binding to recombination signal sequences that flank the variable (V), diversity (D), and joining (J) junctions. RAG1 and RAG2 then create a DSB, where the ends are sealed into hairpin loops. This structure is subsequently cleaved by the Artemis protein and the ends are joined using either classical NHEJ machinery or a form of microhomology-mediated end-joining that can lead to nucleotide additions. Together, this process provides 105–106 unique rearrangements of the V, D, J segments in both B- and T-cell receptors [37]. We recently showed that lupus-prone mice harboring the Y265C Pol β variant [9] have short complementarity determining region 3 (CDR3) junctions in the immunoglobulin heavy chain exclusively, indicating aberrant V(D)J recombination. The CDR3 region is part of the variable chain of the immunoglobulin receptor that recognizes antigens and it is known that pathogenic autoantibodies have short CDR3 regions [38]. Importantly, immature B cells that express long and positively charged antibodies are counter-selected during B cell development [39]. Perhaps autoantibodies with short CDR3 junctions escape selection and play a role in driving autoimmunity.
Figure 1. Co-opted DNA repair during antibody diversification, and its significance in generating autoantibodies.
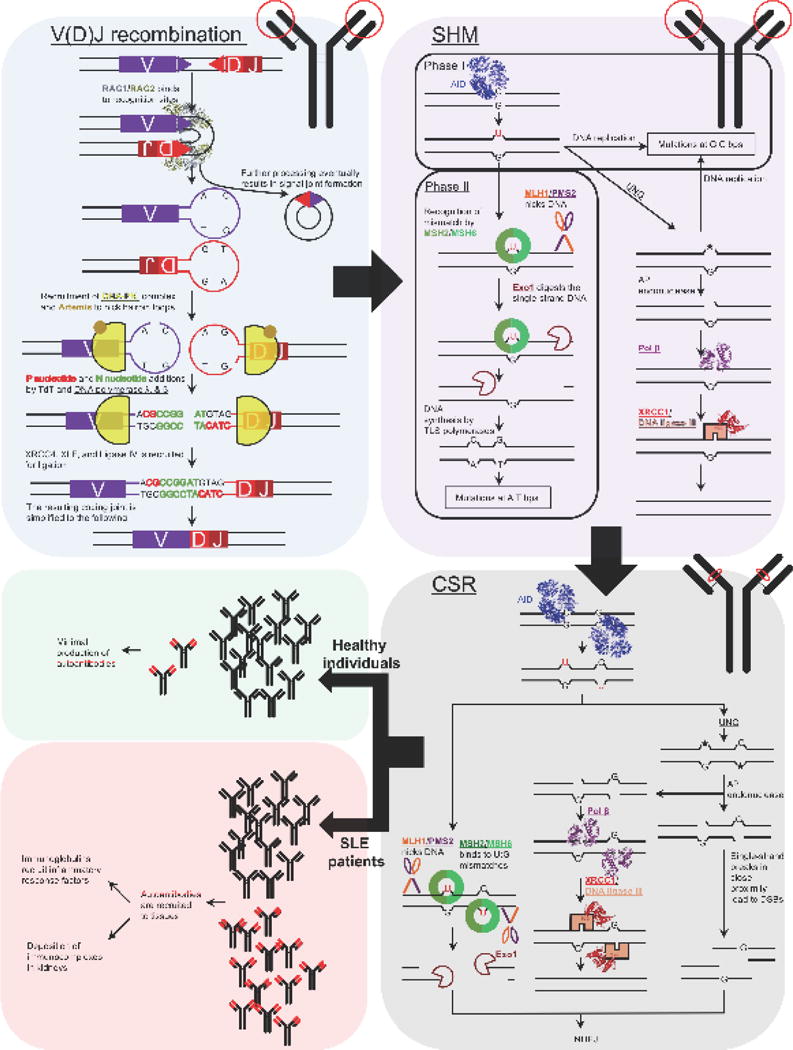
The three major processes for antibody diversification are V(D)J recombination, SHM, and CSR. The location of these processes are shown respective to the antibody (circled in red) pictured in the upper right corner of each box. Additionally, DNA repair proteins are underlined. During V(D)J recombination of the immunoglobulin heavy chain, RAG1 and RAG2 bind to recognition signal sequences (purple and red triangles) bringing the V and DJ sequences to close proximity to each other. RAG1/RAG2 create DSBs resulting in hairpin loops. The hairpin loops recruit Ku70/Ku80 and the DNA-PKcs (DNA-PK complex), and in coordination with Artemis, the hairpin DNA is nicked. This can create DNA overhangs that are filled in with P nucleotides and additional N nucleotides. Collectively, DNA synthesis at these sites is performed by the concerted activity of Polλ, Pol β, and terminal deoxynucleotidyl transferase (TdT) in the immunoglobulin heavy chain. Afterwards, the ends are ligated by XRCC4, XLF, and ligase IV. SHM occurs in the variable region of the immunoglobulin gene. AID converts deoxycytidine to deoxyuridine, which can be processed by the UNG glycosylase to create an abasic site (★). During phase I of SHM, DNA replication of the deoxyuridine- or abasic site-containing DNA results in mutations at G:C bps. Conversely, the abasic site can be repaired by BER. In phase II of SHM, MMR processes the U:G mismatches leading to DNA gap-filling by TLS polymerases, which is in contrast to canonical MMR where DNA synthesis is performed by replicative DNA polymerases. The infidelity of the TLS polymerases results in mutations in A:T bps. CSR is similar to SHM; however, this occurs only at switch regions located downstream of the recombined VDJ regions of the immunoglobulin heavy chain gene. In this case, the intermediates of MMR and BER can result in single-strand breaks, and if they are in close proximity to each other, can lead to DSBs. The DSBs are resolved by NHEJ resulting in an immunoglobulin isotype switch. Aberrant DNA repair can possibly lead to increased production of pathogenic autoantibodies and the disease phenotypes present in SLE patients as shown in the lower left panel. PDB: RAG1/RAG2 (3JBX), AID (5JJ4), Pol β (5TB8), and XRCC1 (3PC8).
Both somatic hypermutation (SHM) and class switch recombination (CSR) are also integral for the maturation of antibodies produced by B cells and occur in two similar stages in secondary lymphoid organs (e.g., lymph nodes, spleen, Peyer’s patches). Initially, both processes involve transcription-coupled deamination of deoxycytidine to deoxyuridine on single-strand DNA by the activation-induced cytidine deaminase AID in the V region. Deoxyuridine is subsequently processed by either BER or MMR enzymes in combination with translesion synthesis DNA polymerases, resulting in a mutation every 102–103 base pairs at immunoglobulin (Ig) loci, which is an ~106-fold higher mutation frequency than the rest of the genome. SHM immensely expands the specificity repertoire of antibodies allowing for the recognition of a variety of potential pathogenic epitopes; however, this can also lead to the formation of autoreactive antibodies [40]. During CSR, the concerted activity of BER, MMR, and TLS on AID-induced deoxyuridine lead to DSBs that are terminally processed by NHEJ to induce isotype switching [41, 42]. This results in the class switching of the IgM and IgD isotypes to IgG, IgE, and IgA isotypes, where their isotype-specific effector functions include recognition of pathogens (IgM), binding to allergens to trigger histamine release and provide protection against parasitic nematodes (IgE), and mounting the major immune responses (IgG). Interestingly, disease in SLE patients and lupus-prone mice is associated with increased isotype switching of autoreactive IgMs resulting in high titers of autoreactive IgGs and the deposition of immunocomplexes within the kidneys. Together, this leads to renal disease, which is one of the major clinical manifestations of SLE [43–47]. Class-switched antibodies to IgG confer transport into extravascular spaces, activation of the complement system, and binding to Fc receptors, providing the optimal potential for autoreactive antibodies to elicit proinflammatory and pathogenic responses [48]. Our Pol β Y265C lupus-prone mice also exhibit high levels of SHM compared to those of WT mice and it is known that plasma cells from patients with active SLE harbor extensively mutated V regions (for a review see [49]). Extensively mutated immunoglobulins may be more pathogenic as a result of their potential ability to recognize large numbers of epitopes, many of which are self-antigens.
Defective DNA Repair and Cell Death
Functional mutations in DNA repair genes result in defective DNA repair. An inability to repair DNA can result in genomic instability or cell death (Figure 2). Sustained activation of the DNA damage response as a result of the presence of unrepaired cellular DNA promotes apoptosis (for a review see [50]). The ATM and ATR proteins are important early sensors of DNA damage and their activation eventually leads to accumulation of p53. p53 promotes apoptosis in the presence of high levels of unrepaired DNA damage or upon accumulation of DNA repair intermediate substrates (for example see [9, 51]). ADP-ribosyltransferase diphtheria toxin-like 1 (ARTD1 or PARP1) functions in the repair of strand breaks ([52, 53]). Upon binding to strand breaks in DNA, PARP1 synthesizes poly-(ADP-ribose) (PAR) and this leads to the recruitment of additional DNA repair proteins to facilitate DNA repair. The synthesis of PAR by PARP1 also results in depletion of NAD+, leading to an energy collapse in the presence of high levels of DNA damage. This is thought to result in cellular necrosis (for a review see [54]). However, it was recently shown that activation of PARP1 in the presence of high levels of DNA damage directly suppresses glycolysis via inhibition of hexokinase 1, leading to cell death [55]. Another form of cell death termed necroptosis is also activated in cells harboring DNA damage (for a review see [56]). Cellular debris emanating from dead or dying cells is normally cleared by phagocytes and macrophages (for a review see [57]. Importantly, a major mechanism underlying lupus pathogenesis involves defective clearance of dead cells. At least seven different genetic variants in human genes that function in the clearance of cellular debris are associated with SLE (for a review see [58]). In the case of defective DNA repair, there may be an imbalance between the high levels of cell death and the ability to clear cellular debris. Persistent apoptotic debris can stimulate the immune response through activation of Toll-like Receptors (TLRs), eventually leading to autoimmune disease. Highly proliferative cells, such as activated B cells, may be especially prone to cell death as a result of defective DNA repair. For example, an inability to fill gaps in DNA after damage is excised, as observed for the Y265C Pol β variant, could result in collision of single nucleotide gaps/single-strand breaks with the replication fork and lead to cell death, and in this case, apoptosis [9]. Interestingly, high levels of broken DNA, detected with a TUNEL assay, in the germinal centers of the PolbY265C/C mice [8], suggest that high levels of cell death occur in the germinal centers. This could result in release of self-antigen in the germinal center, which may decrease selection against B cells that produce autoantibodies should it be taken up and presented by T cells present in the germinal center.
Figure 2. Molecular mechanisms contributing to the breakdown of self-tolerance.
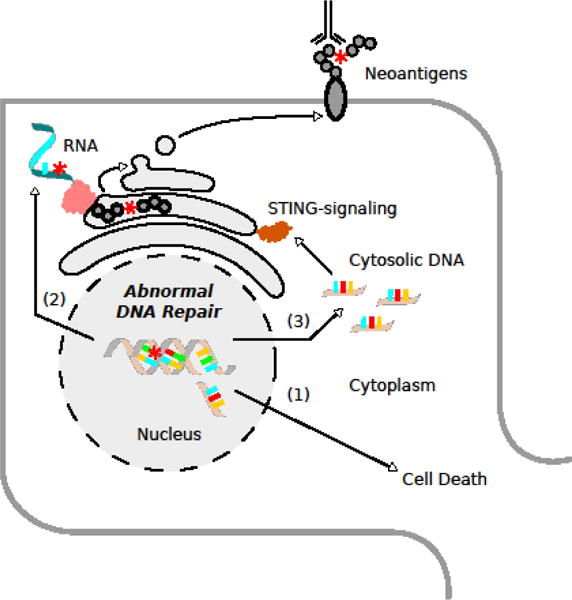
Aberrant DNA repair can lead to the accumulation of various types of DNA damage (mutations, double strand breaks, etc.), which result in (1) cell death, (2) neoantigen formation, and the (3) release of cytoplasmic DNA. Together, these factors prime the immune system and contribute to autoimmune disease.
Aberrant/Defective DNA Repair and Cytosolic DNA
When the cell is overwhelmed with DNA damage or is unable to repair it due to defective DNA repair, DNA may accumulate in the cytosol of cells (Figure 2). Treatment of cells with agents that induce DNA damage, including cytosine β-D-arabinofuranoside hydrochloride (Ara-C) and aphidicolin, leads to the presence of cytosolic DNA [59]. Cells respond to foreign DNA introduced into the cytoplasm by triggering an innate immune response. The innate immune system utilizes germline-encoded receptors, called pattern recognition receptors (PRRs), to detect non-self pathogen-associated molecular patterns (PAMPs). PAMPs stimulate an innate immune response through the expression of various interferons (IFNs), chemokines, and other cytokines. In particular, bacterial and viral DNA are potently immunostimulatory when localized in the cytoplasm or endosomes, where PRRs reside. The considerable potency of nucleic acids as triggers of the innate immune response has gained appreciation over the last decade. Distinct classes of nucleic acid sensing molecules have been uncovered that function in different cell types and subcellular compartments to coordinate innate defenses [60]. The first-described PRR for DNA, was toll-like receptor 9 (TLR9)[61], which recognizes unmethylated CpG repeat sequences in DNA that are prevalent in bacteria [61–64]. Many cells that do not express TLR9 can still robustly produce IFNs when exogenous DNA is delivered into the cytoplasm [65, 66]. This TLR9-independent cytosolic pathway, functions in a broad range of cell types including mouse embryonic fibroblasts (MEFs), pDCs, and bone marrow-derived macrophages (BMDMs) [65]. Accordingly, stimulator of IFN genes (STING) was identified as a potent inducer of IFN expression in response to cytosolic DNA [11, 67, 68]. STING itself is not a DNA PRR, but is rather a central adaptor for many cytosolic DNA sensors. Upon binding to cytosolic DNA, cyclic GMP-AMP synthase (cGAS) triggers formation of cyclic GMP-AMP (cGAMP), which binds to STING, leading to transcription of inflammatory genes. Thus high levels of DNA damage as would be expected to occur in DNA repair-defective cells can lead to an inflammatory response as a result of the accumulation of cytosolic DNA. In addition, in the presence of cytosolic DNA and an intact DNA damage response, retinoic acid early transcript 1 (RAE1) ligands, a subset of ligands recognized by the immunoreceptor NKG2D, are also induced in a STING-dependent manner, which leads to activation of natural killer (NK) cells [59].
Although the detection of pathogenic nucleic acids has a fundamental role in host defense, the inappropriate detection of self-DNA can also result in autoimmunity. Accordingly, defects in PRRs have been linked to various autoimmune diseases in vivo [69–76]. Several autoimmune diseases including SLE exhibit increased IFN, chemokine, and other cytokine expression[77–83] consistent with dysregulated innate immune signaling. Interestingly, DNA repair proteins are now known to play a critical role in the generation of innate immune signals not only in response to nuclear DNA damage but also to cytosolic DNA. However, the exact link between DNA damage response (DDR), innate immune signaling and autoimmunity remains unclear. It is possible that defective DNA repair factors functioning as PRRs may also contribute to autoimmune disease pathogenesis.
Three Prime Repair Exonuclease 1 (TREX1) is one of the first DNA processing enzymes associated with inflammatory disease and lupus in humans. TREX1 functions to degrade cytosolic DNA [77–79, 84, 85]. Mice harboring a mutation in a key catalytic residue of TREX1 develop SLE [86]. Interestingly, TREX1 is unable to degrade cytoplasmic DNA with the oxidized base 8-hydroxyguanosine (8-oxoG) [87], resulting in activation of an inflammatory response. Recent work has shown that deletion of Trex1, specifically in the dendritic cells of mice, leads to development of systemic autoimmunity [88].
The DNA-dependent protein kinase (DNA-PK) complex is a heterotrimer consisting of Ku70/80 and DNA-PK catalytic subunit (DNA-PKcs). Ku70 and Ku80 are the essential sensors for free ends in DSB repair [89]. Accordingly, the DNA-PK complex has a well-established role in the NHEJ of DSBs[90] and is thus indispensable for V(D)J recombination [91]. Interestingly, DNA-PK also functions as a PRR. The complex is capable of binding cytosolic DNA and stimulating IFN, chemokine, and other cytokine expression in a STING-dependent manner. Moreover, MEFs and mice lacking DNA-PKcs show attenuated cytokine responses to DNA. However, in contrast to its role in V(D)J recombination, DNA-PKcs activity is not required for the innate immune sensing of DNA [92], suggesting that the protein complex has multifaceted role in immunity. The MRE11-Rad50-NBS1 (MRN) HDR complex is another major sensor of DSBs [93, 94] that has been implicated as a PRR in MEFs. Similar to DNA-PK, MRN-mediated signaling occurs through STING [95].
In addition to co-opting cytosolic DNA sensing pathways, DDR proteins are also capable of modulating innate immune signaling by other means. Ataxia telangiectasia mutated (ATM) kinase is recruited by the MRN complex and plays a central signaling role in HDR [96]. However, in contrast to MRN, ATM appears to operate as a negative regulator of the STING pathway [97]. Unrepaired DNA damage in ATM-deficient cells promotes an IFN response [97, 98], and inappropriately primes the innate immune system in response to genomic stress. In humans, loss-of-function mutations in ATM results in Ataxia Telangiectasia (AT). Interestingly, AT is a complex neurodegenerative disease that results in a variety of inflammatory and autoimmune syndromes [99–103].
Specific examples of the links between defective DNA repair and the presence of cytosolic DNA are beginning to emerge, especially with regard to collapsed replication forks. Depletion of RAD51 or RPA in cells leads to the accumulation of cytosolic DNA [104] and a type 1 IFN response in a TREX1- and STING-dependent manner. In TREX1-deficient cells, RAD51 and RPA bind to ssDNA localized to the cytoplasm, which eventually results in replication stress and also leads to an IFN response. Importantly, patients with mutations in RAD51C exhibit an IFN signature and other autoimmune features. BRCA1 haploinsufficiency is associated with the presence of collapsed replication forks and replication stress [105]. The MUS81 structure-specific nuclease cuts stalled replication forks and cleavage of DNA in prostate cancer cells by MUS81 leads to the accumulation of cytosolic DNA, which stimulates STING and leads to a type I and II IFN response. Interestingly, the presence of MUS81 foci and levels of cytoplasmic DNA increase as the severity of prostate cancer increases [106]. Thus, mutated HDR genes may underlie autoimmunity either as monogenic mutations or in combination with other germline variants.
Defective DNA Repair Generates Mutations That Can Function as Neoantigens
A driver of autoimmunity is the production of self-antigens that lead to the development of autoreactive antibodies. In fact, there are ~180 known autoantibodies associated with SLE as of 2015; 90 of these antibodies are common to ≥20% of SLE patients, and 20 of these antibodies are common to ≥50% of SLE patients [107]. During lupus development, self-antigens are thought to arise as a result of cell death, as described above. However, self-antigens, or neoantigens, could also arise as a result of a high mutational burden that may occur in the absence of efficient DNA repair in people harboring one or more mutations that inactivate or alter the function of a critical DNA repair gene (Figure 2). These neoantigens are likely to arise in a tissue- and cell type-specific manner under specific conditions, as demonstrated to be the case for the finding of high levels of myositis autoantigen specifically in regenerating muscle (for a review see [108]). There is also a relationship between cancer and autoimmunity. A well-characterized example of this link centers on the autoimmune disease scleroderma. Patients with scleroderma and autoantibodies against RNA polymerase 3 subunit 1 (RPC1) are at increased risk for cancer. Moreover, patients with cancer were shown to harbor somatically acquired mutations in the POLR3 locus, which encodes RPC1, in their incipient cancer, suggesting that acquired immunity is a cancer control mechanism [109]. There are numerous examples of the link between defective DNA repair and cancer including defective NER and Xeroderma pigmentosum, defective HDR and breast and ovarian cancer, and defective MMR and colorectal and numerous other cancers (see [110]). The cancers that develop as a result of specific types of DNA repair defects are generally organ- and tissue-specific. Therefore, the mutations or neoantigens that originate from defective DNA repair could originate within a specific tissue and cellular context, leading to cancer but also to the emergence of a specific autoimmune response. Furthermore, tumors with high mutational burden are more likely to harbor neoantigens and the presence of neoantigens is correlated with improved response to immunotherapies, such as antibodies against CTLA-4 and PD-L1, that reactivate the immune system (see for example [111–113]). Although cancer immunotherapy shows great promise, there are several adverse events associated with this type of therapy, and several of them are associated with autoimmunity (for reviews see [114, 115]).
CONCLUDING REMARKS
DNA repair is typically thought of as a genomic stability maintenance system, even though it has been co-opted by the immune system as a critical player in antibody diversification. Inadequate repair may result in cell death, the accumulation of cytosolic DNA, or the generation of neoantigens, eventually triggering an inappropriate immune response, resulting in autoimmune disease. Recent studies point to an important role for DNA damage sensors in the triggering of innate immune signals not only in response to nuclear DNA damage but also to cytosolic DNA. There is much crosstalk between DNA repair and immunity. Therefore it is likely that defective DNA repair will emerge as an important underlying mechanism of the development of autoimmune disease.
Acknowledgments
This work was supported by ES019179 from the National Institute of Environmental Health Sciences.
Abbreviations
- 8-oxoG
8-hydroxyguanosine
- OGG1
8-oxoguanine DNA glycosylase
- ARTD1
ADP-ribosyltransferase diphtheria toxin-like 1
- ATM
Ataxia telangiectasia mutated
- BER
Base excision repair
- BMDMs
Bone marrow-derived macrophages
- CSR
Class switch recombination
- CDR3
Complementarity determining region 3
- cGAMP
Cyclic GMP-AMP
- cGAS
Cyclic GMP-AMP synthase
- DDR
DNA damage response
- Pol β
DNA polymerase beta
- DNA-PK
DNA-dependent protein kinase
- DNA-PKcs
DNA-PK catalytic subunit
- DSB
Double strand break
- FEN1
Flap endonuclease I
- HDR
Homology-directed repair
- H2O2
Hydrogen peroxide
- Ig
Immunoglobulin
- IFNs
Interferons
- LP
Long patch
- MMR
Mismatch repair
- MEFs
Mouse embryonic fibroblasts
- MRN
MRE11-Rad50-NBS1
- MNU
N-methyl-N-nitrosourea
- NK
Natural killer
- NEIL3
Nei-like DNA glycosylase 3
- NHEJ
Non-homologous end-joining
- nt
Nucleotide
- NER
Nucleotide excision repair
- PAMPs
Pathogen-associated molecular patterns
- PAR
poly-(ADP-ribose)
- RAG1
Recombination-activating gene 1
- RAG2
Recombination-activating gene 2
- RAE1
Retinoic acid early transcript 1
- Rnase H2
Ribonuclease H2
- RPC1
RNA polymerase 3 subunit 1
- SP
Short patch
- SNP
Single nucleotide polymorphism
- SHM
Somatic hypermutation
- STING
Stimulator of interferon genes
- SLE
Systemic lupus erythematosus
- TREX1
Three primer repair exonuclease 1
- TLR
Toll-like receptor
- XRCC1
X-ray Cross-complimenting Complex 1
- Ara-C
β-D-arabinofuranoside hydrochloride
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement: The authors declare no conflict of interest.
References
- 1.Petri M, Orbai AM, Alarcon GS, Gordon C, Merrill JT, Fortin PR, Bruce IN, Isenberg D, Wallace DJ, Nived O, Sturfelt G, Ramsey-Goldman R, Bae SC, Hanly JG, Sanchez-Guerrero J, Clarke A, Aranow C, Manzi S, Urowitz M, Gladman D, Kalunian K, Costner M, Werth VP, Zoma A, Bernatsky S, Ruiz-Irastorza G, Khamashta MA, Jacobsen S, Buyon JP, Maddison P, Dooley MA, van Vollenhoven RF, Ginzler E, Stoll T, Peschken C, Jorizzo JL, Callen JP, Lim SS, Fessler BJ, Inanc M, Kamen DL, Rahman A, Steinsson K, Franks AG, Jr, Sigler L, Hameed S, Fang H, Pham N, Brey R, Weisman MH, McGwin G, Jr, Magder LS. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. 2012;64:2677–2686. doi: 10.1002/art.34473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Petri M. Sex hormones and systemic lupus erythematosus. Lupus. 2008;17:412–415. doi: 10.1177/0961203308090026. [DOI] [PubMed] [Google Scholar]
- 3.Tedeschi SK, Bermas B, Costenbader KH. Sexual disparities in the incidence and course of SLE and RA. Clin Immunol. 2013;149:211–218. doi: 10.1016/j.clim.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 4.Alarcon-Segovia D, Alarcon-Riquelme ME, Cardiel MH, Caeiro F, Massardo L, Villa AR, Pons-Estel BA, E. Grupo Latinoamericano de Estudio del Lupus Familial aggregation of systemic lupus erythematosus, rheumatoid arthritis, and other autoimmune diseases in 1,177 lupus patients from the GLADEL cohort. Arthritis Rheum. 2005;52:1138–1147. doi: 10.1002/art.20999. [DOI] [PubMed] [Google Scholar]
- 5.Deapen D, Escalante A, Weinrib L, Horwitz D, Bachman B, Roy-Burman P, Walker A, Mack TM. A revised estimate of twin concordance in systemic lupus erythematosus. Arthritis Rheum. 1992;35:311–318. doi: 10.1002/art.1780350310. [DOI] [PubMed] [Google Scholar]
- 6.Block SR, Winfield JB, Lockshin MD, D’Angelo WA, Christian CL. Studies of twins with systemic lupus erythematosus. A review of the literature and presentation of 12 additional sets. The American journal of medicine. 1975;59:533–552. doi: 10.1016/0002-9343(75)90261-2. [DOI] [PubMed] [Google Scholar]
- 7.Ghodke-Puranik Y, Niewold TB. Immunogenetics of systemic lupus erythematosus: A comprehensive review. J Autoimmun. 2015;64:125–136. doi: 10.1016/j.jaut.2015.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Senejani AG, Liu Y, Kidane D, Maher SE, Zeiss CJ, Park HJ, Kashgarian M, McNiff JM, Zelterman D, Bothwell AL, Sweasy JB. Mutation of POLB causes lupus in mice. Cell Rep. 2014;6:1–8. doi: 10.1016/j.celrep.2013.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Senejani AG, Dalal S, Liu Y, Nottoli TP, McGrath JM, Clairmont CS, Sweasy JB. Y265C DNA polymerase beta knockin mice survive past birth and accumulate base excision repair intermediate substrates. Proc Natl Acad Sci U S A. 2012;109:6632–6637. doi: 10.1073/pnas.1200800109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sheng YJ, Gao JP, Li J, Han JW, Xu Q, Hu WL, Pan TM, Cheng YL, Yu ZY, Ni C, Yao S, He CF, Liu YS, Li Y, Ge HM, Xiao FL, Sun LD, Yang S, Zhang XJ. Follow-up study identifies two novel susceptibility loci PRKCB and 8p11.21 for systemic lupus erythematosus. Rheumatology (Oxford) 2011;50:682–688. doi: 10.1093/rheumatology/keq313. [DOI] [PubMed] [Google Scholar]
- 11.Han JW, Zheng HF, Cui Y, Sun LD, Ye DQ, Hu Z, Xu JH, Cai ZM, Huang W, Zhao GP, Xie HF, Fang H, Lu QJ, Xu JH, Li XP, Pan YF, Deng DQ, Zeng FQ, Ye ZZ, Zhang XY, Wang QW, Hao F, Ma L, Zuo XB, Zhou FS, Du WH, Cheng YL, Yang JQ, Shen SK, Li J, Sheng YJ, Zuo XX, Zhu WF, Gao F, Zhang PL, Guo Q, Li B, Gao M, Xiao FL, Quan C, Zhang C, Zhang Z, Zhu KJ, Li Y, Hu DY, Lu WS, Huang JL, Liu SX, Li H, Ren YQ, Wang ZX, Yang CJ, Wang PG, Zhou WM, Lv YM, Zhang AP, Zhang SQ, Lin D, Li Y, Low HQ, Shen M, Zhai ZF, Wang Y, Zhang FY, Yang S, Liu JJ, Zhang XJ. Genome-wide association study in a Chinese Han population identifies nine new susceptibility loci for systemic lupus erythematosus. Nat Genet. 2009;41:1234–1237. doi: 10.1038/ng.472. [DOI] [PubMed] [Google Scholar]
- 12.Zeller T, Wild P, Szymczak S, Rotival M, Schillert A, Castagne R, Maouche S, Germain M, Lackner K, Rossmann H, Eleftheriadis M, Sinning CR, Schnabel RB, Lubos E, Mennerich D, Rust W, Perret C, Proust C, Nicaud V, Loscalzo J, Hubner N, Tregouet D, Munzel T, Ziegler A, Tiret L, Blankenberg S, Cambien F. Genetics and beyond–the transcriptome of human monocytes and disease susceptibility. PLoS One. 2010;5:e10693. doi: 10.1371/journal.pone.0010693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harris G, Asbery L, Lawley PD, Denman AM, Hylton W. Defective repair of 0(6)-methylguanine in autoimmune diseases. Lancet. 1982;2:952–956. doi: 10.1016/s0140-6736(82)90159-3. [DOI] [PubMed] [Google Scholar]
- 14.Davies RC, Pettijohn K, Fike F, Wang J, Nahas SA, Tunuguntla R, Hu H, Gatti RA, McCurdy D. Defective DNA double-strand break repair in pediatric systemic lupus erythematosus. Arthritis Rheum. 2012;64:568–578. doi: 10.1002/art.33334. [DOI] [PubMed] [Google Scholar]
- 15.McCurdy D, Tai LQ, Frias S, Wang Z. Delayed repair of DNA damage by ionizing radiation in cells from patients with juvenile systemic lupus erythematosus and rheumatoid arthritis. Radiat Res. 1997;147:48–54. [PubMed] [Google Scholar]
- 16.Bashir S, Harris G, Denman MA, Blake DR, Winyard PG. Oxidative DNA damage and cellular sensitivity to oxidative stress in human autoimmune diseases. Ann Rheum Dis. 1993;52:659–666. doi: 10.1136/ard.52.9.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Y, Wilson SH. DNA base excision repair: a mechanism of trinucleotide repeat expansion. Trends Biochem Sci. 2012;37:162–172. doi: 10.1016/j.tibs.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baute J, Depicker A. Base excision repair and its role in maintaining genome stability. Crit Rev Biochem Mol Biol. 2008;43:239–276. doi: 10.1080/10409230802309905. [DOI] [PubMed] [Google Scholar]
- 19.Sattler U, Frit P, Salles B, Calsou P. Long-patch DNA repair synthesis during base excision repair in mammalian cells. EMBO Rep. 2003;4:363–367. doi: 10.1038/sj.embor.embor796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee HT, Lin CS, Lee CS, Tsai CY, Wei YH. The role of hOGG1 C1245G polymorphism in the susceptibility to lupus nephritis and modulation of the plasma 8-OHdG in patients with systemic lupus erythematosus. Int J Mol Sci. 2015;16:3757–3768. doi: 10.3390/ijms16023757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boiteux S, Radicella JP. The human OGG1 gene: structure, functions, and its implication in the process of carcinogenesis. Arch Biochem Biophys. 2000;377:1–8. doi: 10.1006/abbi.2000.1773. [DOI] [PubMed] [Google Scholar]
- 22.Furihata C. An active alternative splicing isoform of human mitochondrial 8-oxoguanine DNA glycosylase (OGG1) Genes Environ. 2015;37:21. doi: 10.1186/s41021-015-0021-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kohno T, Shinmura K, Tosaka M, Tani M, Kim SR, Sugimura H, Nohmi T, Kasai H, Yokota J. Genetic polymorphisms and alternative splicing of the hOGG1 gene, that is involved in the repair of 8-hydroxyguanine in damaged DNA. Oncogene. 1998;16:3219–3225. doi: 10.1038/sj.onc.1201872. [DOI] [PubMed] [Google Scholar]
- 24.Liu M, Bandaru V, Bond JP, Jaruga P, Zhao X, Christov PP, Burrows CJ, Rizzo CJ, Dizdaroglu M, Wallace SS. The mouse ortholog of NEIL3 is a functional DNA glycosylase in vitro and in vivo. Proc Natl Acad Sci U S A. 2010;107:4925–4930. doi: 10.1073/pnas.0908307107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Massaad MJ, Zhou J, Tsuchimoto D, Chou J, Jabara H, Janssen E, Glauzy S, Olson BG, Morbach H, Ohsumi TK, Schmitz K, Kyriacos M, Kane J, Torisu K, Nakabeppu Y, Notarangelo LD, Chouery E, Megarbane A, Kang PB, Al-Idrissi E, Aldhekri H, Meffre E, Mizui M, Tsokos GC, Manis JP, Al-Herz W, Wallace SS, Geha RS. Deficiency of base excision repair enzyme NEIL3 drives increased predisposition to autoimmunity. J Clin Invest. 2016;126:4219–4236. doi: 10.1172/JCI85647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kubota Y, Nash RA, Klungland A, Schar P, Barnes DE, Lindahl T. Reconstitution of DNA base excision-repair with purified human proteins: interaction between DNA polymerase beta and the XRCC1 protein. EMBO J. 1996;15:6662–6670. [PMC free article] [PubMed] [Google Scholar]
- 27.Dianova, Sleeth KM, Allinson SL, Parsons JL, Breslin C, Caldecott KW, Dianov GL. XRCC1-DNA polymerase beta interaction is required for efficient base excision repair. Nucleic Acids Res. 2004;32:2550–2555. doi: 10.1093/nar/gkh567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bassi C, Xavier D, Palomino G, Nicolucci P, Soares C, Sakamoto-Hojo E, Donadi E. Efficiency of the DNA repair and polymorphisms of the XRCC1, XRCC3 and XRCC4 DNA repair genes in systemic lupus erythematosus. Lupus. 2008;17:988–995. doi: 10.1177/0961203308093461. [DOI] [PubMed] [Google Scholar]
- 29.Lin YJ, Wan L, Huang CM, Chen SY, Huang YC, Lai CH, Lin WY, Liu HP, Wu YS, Chen CM, Tsai YH, Tsai CH, Sheu JJ, Tsai FJ. Polymorphisms in the DNA repair gene XRCC1 and associations with systemic lupus erythematosus risk in the Taiwanese Han Chinese population. Lupus. 2009;18:1246–1251. doi: 10.1177/0961203309345777. [DOI] [PubMed] [Google Scholar]
- 30.Hanssen-Bauer A, Solvang-Garten K, Gilljam KM, Torseth K, Wilson DM, 3rd, Akbari M, Otterlei M. The region of XRCC1 which harbours the three most common nonsynonymous polymorphic variants, is essential for the scaffolding function of XRCC1. DNA Repair (Amst) 2012;11:357–366. doi: 10.1016/j.dnarep.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zheng L, Dai H, Zhou M, Li M, Singh P, Qiu J, Tsark W, Huang Q, Kernstine K, Zhang X, Lin D, Shen B. Fen1 mutations result in autoimmunity, chronic inflammation and cancers. Nat Med. 2007;13:812–819. doi: 10.1038/nm1599. [DOI] [PubMed] [Google Scholar]
- 32.Williams JS, Lujan SA, Kunkel TA. Processing ribonucleotides incorporated during eukaryotic DNA replication. Nat Rev Mol Cell Biol. 2016;17:350–363. doi: 10.1038/nrm.2016.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gunther C, Kind B, Reijns MA, Berndt N, Martinez-Bueno M, Wolf C, Tungler V, Chara O, Lee YA, Hubner N, Bicknell L, Blum S, Krug C, Schmidt F, Kretschmer S, Koss S, Astell KR, Ramantani G, Bauerfeind A, Morris DL, Cunninghame Graham DS, Bubeck D, Leitch A, Ralston SH, Blackburn EA, Gahr M, Witte T, Vyse TJ, Melchers I, Mangold E, Nothen MM, Aringer M, Kuhn A, Luthke K, Unger L, Bley A, Lorenzi A, Isaacs JD, Alexopoulou D, Conrad K, Dahl A, Roers A, Alarcon-Riquelme ME, Jackson AP, Lee-Kirsch MA. Defective removal of ribonucleotides from DNA promotes systemic autoimmunity. The Journal of clinical investigation. 2015;125:413–424. doi: 10.1172/JCI78001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Souliotis VL, Vougas K, Gorgoulis VG, Sfikakis PP. Defective DNA repair and chromatin organization in patients with quiescent systemic lupus erythematosus. Arthritis Res Ther. 2016;18:182. doi: 10.1186/s13075-016-1081-3. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 35.Noble PW, Bernatsky S, Clarke AE, Isenberg DA, Ramsey-Goldman R, Hansen JE. DNA-damaging autoantibodies and cancer: the lupus butterfly theory. Nat Rev Rheumatol. 2016;12:429–434. doi: 10.1038/nrrheum.2016.23. [DOI] [PubMed] [Google Scholar]
- 36.Malu S, Malshetty V, Francis D, Cortes P. Role of non-homologous end joining in V(D)J recombination. Immunol Res. 2012;54:233–246. doi: 10.1007/s12026-012-8329-z. [DOI] [PubMed] [Google Scholar]
- 37.Di Noia JM, Neuberger MS. Molecular mechanisms of antibody somatic hypermutation. Annu Rev Biochem. 2007;76:1–22. doi: 10.1146/annurev.biochem.76.061705.090740. [DOI] [PubMed] [Google Scholar]
- 38.Dorner T, Giesecke C, Lipsky PE. Mechanisms of B cell autoimmunity in SLE. Arthritis research & therapy. 2011;13:243. doi: 10.1186/ar3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC. Predominant autoantibody production by early human B cell precursors. Science. 2003;301:1374–1377. doi: 10.1126/science.1086907. [DOI] [PubMed] [Google Scholar]
- 40.Guo Y, Orme J, Mohan C. A genopedia of lupus genes - lessons from gene knockouts. Curr Rheumatol Rev. 2013;9:90–99. doi: 10.2174/1573397111309020003. [DOI] [PubMed] [Google Scholar]
- 41.Keim C, Kazadi D, Rothschild G, Basu U. Regulation of AID, the B-cell genome mutator. Genes Dev. 2013;27:1–17. doi: 10.1101/gad.200014.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saribasak H, Gearhart PJ. Does DNA repair occur during somatic hypermutation? Semin Immunol. 2012;24:287–292. doi: 10.1016/j.smim.2012.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Devey ME, Lee SR, Le Page S, Feldman R, Isenberg DA. Serial studies of the IgG subclass and functional affinity of DNA antibodies in systemic lupus erythematosus. J Autoimmun. 1988;1:483–494. doi: 10.1016/0896-8411(88)90069-8. [DOI] [PubMed] [Google Scholar]
- 44.Ward MM, Dawson DV, Pisetsky DS. Serum immunoglobulin levels in systemic lupus erythematosus: the effects of age, sex, race and disease duration. J Rheumatol. 1991;18:540–544. [PubMed] [Google Scholar]
- 45.Tsuzaka K, Ogasawara T, Tojo T, Mimori T, Satoh M, Homma M. Nonprecipitating IgG or IgM anti-Sm antibody: clinical significance and changes in immunoglobulin class. J Rheumatol. 1993;20:822–830. [PubMed] [Google Scholar]
- 46.Saiki O, Saeki Y, Tanaka T, Doi S, Hara H, Negoro S, Igarashi T, Kishimoto S. Development of selective IgM deficiency in systemic lupus erythematosus patients with disease of long duration. Arthritis Rheum. 1987;30:1289–1292. doi: 10.1002/art.1780301112. [DOI] [PubMed] [Google Scholar]
- 47.Steward MW, Hay FC. Changes in immunoglobulin class and subclass of anti-DNA antibodies with increasing age in N/ZBW F1 hybrid mice. Clin Exp Immunol. 1976;26:363–370. [PMC free article] [PubMed] [Google Scholar]
- 48.Aschermann S, Lux A, Baerenwaldt A, Biburger M, Nimmerjahn F. The other side of immunoglobulin G: suppressor of inflammation. Clin Exp Immunol. 2010;160:161–167. doi: 10.1111/j.1365-2249.2009.04081.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dorner T, Jacobi AM, Lee J, Lipsky PE. Abnormalities of B cell subsets in patients with systemic lupus erythematosus. Journal of immunological methods. 2011;363:187–197. doi: 10.1016/j.jim.2010.06.009. [DOI] [PubMed] [Google Scholar]
- 50.Roos WP, Kaina B. DNA damage-induced cell death: from specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013;332:237–248. doi: 10.1016/j.canlet.2012.01.007. [DOI] [PubMed] [Google Scholar]
- 51.Ochs K, Sobol RW, Wilson SH, Kaina B. Cells deficient in DNA polymerase beta are hypersensitive to alkylating agent-induced apoptosis and chromosomal breakage. Cancer Res. 1999;59:1544–1551. [PubMed] [Google Scholar]
- 52.Hassa PO, Hottiger MO. The diverse biological roles of mammalian PARPS, a small but powerful family of poly-ADP-ribose polymerases. Front Biosci. 2008;13:3046–3082. doi: 10.2741/2909. [DOI] [PubMed] [Google Scholar]
- 53.Fouquerel E, Sobol RW. ARTD1 (PARP1) activation and NAD(+)in DNA repair and cell death. DNA Repair (Amst) 2014;23:27–32. doi: 10.1016/j.dnarep.2014.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Berger NA. Poly(ADP-ribose) in the cellular response to DNA damage. Radiat Res. 1985;101:4–15. [PubMed] [Google Scholar]
- 55.Fouquerel E, Goellner EM, Yu Z, Gagne JP, Barbide Moura M, Feinstein T, Wheeler D, Redpath P, Li J, Romero G, Migaud M, Van Houten B, Poirier GG, Sobol RW. ARTD1/PARP1 negatively regulates glycolysis by inhibiting hexokinase 1 independent of NAD+ depletion. Cell Rep. 2014;8:1819–1831. doi: 10.1016/j.celrep.2014.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hanson B. Necroptosis: A new way of dying? Cancer Biol Ther. 2016;17:899–910. doi: 10.1080/15384047.2016.1210732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mahajan A, Herrmann M, Munoz LE. Clearance Deficiency and Cell Death Pathways: A Model for the Pathogenesis of SLE. Front Immunol. 2016;7:35. doi: 10.3389/fimmu.2016.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tsokos GC, Lo MS, Reis PC, Sullivan KE. New insights into the immunopathogenesis of systemic lupus erythematosus. Nat Rev Rheumatol. 2016;12:716–730. doi: 10.1038/nrrheum.2016.186. [DOI] [PubMed] [Google Scholar]
- 59.Lam AR, Le Bert N, Ho SS, Shen YJ, Tang ML, Xiong GM, Croxford JL, Koo CX, Ishii KJ, Akira S, Raulet DH, Gasser S. RAE1 ligands for the NKG2D receptor are regulated by STING-dependent DNA sensor pathways in lymphoma. Cancer Res. 2014;74:2193–2203. doi: 10.1158/0008-5472.CAN-13-1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 61.Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 62.Ahmad-Nejad P, Hacker H, Rutz M, Bauer S, Vabulas RM, Wagner H. Bacterial CpG-DNA and lipopolysaccharides activate Toll-like receptors at distinct cellular compartments. Eur J Immunol. 2002;32:1958–1968. doi: 10.1002/1521-4141(200207)32:7<1958::AID-IMMU1958>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 63.Ewald SE, Lee BL, Lau L, Wickliffe KE, Shi GP, Chapman HA, Barton GM. The ectodomain of Toll-like receptor 9 is cleaved to generate a functional receptor. Nature. 2008;456:658–662. doi: 10.1038/nature07405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yasuda K, Richez C, Uccellini MB, Richards RJ, Bonegio RG, Akira S, Monestier M, Corley RB, Viglianti GA, Marshak-Rothstein A, Rifkin IR. Requirement for DNA CpG content in TLR9-dependent dendritic cell activation induced by DNA-containing immune complexes. J Immunol. 2009;183:3109–3117. doi: 10.4049/jimmunol.0900399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stetson DB, Medzhitov R. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity. 2006;24:93–103. doi: 10.1016/j.immuni.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 66.Ishii KJ, Coban C, Kato H, Takahashi K, Torii Y, Takeshita F, Ludwig H, Sutter G, Suzuki K, Hemmi H, Sato S, Yamamoto M, Uematsu S, Kawai T, Takeuchi O, Akira S. A Toll-like receptor-independent antiviral response induced by double-stranded B-form DNA. Nat Immunol. 2006;7:40–48. doi: 10.1038/ni1282. [DOI] [PubMed] [Google Scholar]
- 67.Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhong B, Yang Y, Li S, Wang YY, Li Y, Diao F, Lei C, He X, Zhang L, Tien P, Shu HB. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity. 2008;29:538–550. doi: 10.1016/j.immuni.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 69.Frasnelli ME, Tarussio D, Chobaz-Peclat V, Busso N, So A. TLR2 modulates inflammation in zymosan-induced arthritis in mice. Arthritis Res Ther. 2005;7:R370–379. doi: 10.1186/ar1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Su SB, Silver PB, Grajewski RS, Agarwal RK, Tang J, Chan CC, Caspi RR. Essential role of the MyD88 pathway, but nonessential roles of TLRs 2, 4, and 9, in the adjuvant effect promoting Th1-mediated autoimmunity. J Immunol. 2005;175:6303–6310. doi: 10.4049/jimmunol.175.10.6303. [DOI] [PubMed] [Google Scholar]
- 71.Prinz M, Garbe F, Schmidt H, Mildner A, Gutcher I, Wolter K, Piesche M, Schroers R, Weiss E, Kirschning CJ, Rochford CD, Bruck W, Becher B. Innate immunity mediated by TLR9 modulates pathogenicity in an animal model of multiple sclerosis. J Clin Invest. 2006;116:456–464. doi: 10.1172/JCI26078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Abdollahi-Roodsaz S, Joosten LA, Roelofs MF, Radstake TR, Matera G, Popa C, van der Meer JW, Netea MG, van den Berg WB. Inhibition of Toll-like receptor 4 breaks the inflammatory loop in autoimmune destructive arthritis. Arthritis Rheum. 2007;56:2957–2967. doi: 10.1002/art.22848. [DOI] [PubMed] [Google Scholar]
- 73.Abdollahi-Roodsaz S, Joosten LA, Koenders MI, Devesa I, Roelofs MF, Radstake TR, Heuvelmans-Jacobs M, Akira S, Nicklin MJ, Ribeiro-Dias F, van den Berg WB. Stimulation of TLR2 and TLR4 differentially skews the balance of T cells in a mouse model of arthritis. J Clin Invest. 2008;118:205–216. doi: 10.1172/JCI32639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Marta M, Andersson A, Isaksson M, Kampe O, Lobell A. Unexpected regulatory roles of TLR4 and TLR9 in experimental autoimmune encephalomyelitis. Eur J Immunol. 2008;38:565–575. doi: 10.1002/eji.200737187. [DOI] [PubMed] [Google Scholar]
- 75.Fang J, Fang D, Silver PB, Wen F, Li B, Ren X, Lin Q, Caspi RR, Su SB. The role of TLR2, TRL3, TRL4, and TRL9 signaling in the pathogenesis of autoimmune disease in a retinal autoimmunity model. Invest Ophthalmol Vis Sci. 2010;51:3092–3099. doi: 10.1167/iovs.09-4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Reynolds JM, Pappu BP, Peng J, Martinez GJ, Zhang Y, Chung Y, Ma L, Yang XO, Nurieva RI, Tian Q, Dong C. Toll-like receptor 2 signaling in CD4(+) T lymphocytes promotes T helper 17 responses and regulates the pathogenesis of autoimmune disease. Immunity. 2010;32:692–702. doi: 10.1016/j.immuni.2010.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lee-Kirsch MA, Chowdhury D, Harvey S, Gong M, Senenko L, Engel K, Pfeiffer C, Hollis T, Gahr M, Perrino FW, Lieberman J, Hubner N. A mutation in TREX1 that impairs susceptibility to granzyme A-mediated cell death underlies familial chilblain lupus. J Mol Med (Berl) 2007;85:531–537. doi: 10.1007/s00109-007-0199-9. [DOI] [PubMed] [Google Scholar]
- 78.Lee-Kirsch MA, Gong M, Chowdhury D, Senenko L, Engel K, Lee YA, de Silva U, Bailey SL, Witte T, Vyse TJ, Kere J, Pfeiffer C, Harvey S, Wong A, Koskenmies S, Hummel O, Rohde K, Schmidt RE, Dominiczak AF, Gahr M, Hollis T, Perrino FW, Lieberman J, Hubner N. Mutations in the gene encoding the 3′–5′ DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat Genet. 2007;39:1065–1067. doi: 10.1038/ng2091. [DOI] [PubMed] [Google Scholar]
- 79.Crow YJ, Leitch A, Hayward BE, Garner A, Parmar R, Griffith E, Ali M, Semple C, Aicardi J, Babul-Hirji R, Baumann C, Baxter P, Bertini E, Chandler KE, Chitayat D, Cau D, Dery C, Fazzi E, Goizet C, King MD, Klepper J, Lacombe D, Lanzi G, Lyall H, Martinez-Frias ML, Mathieu M, McKeown C, Monier A, Oade Y, Quarrell OW, Rittey CD, Rogers RC, Sanchis A, Stephenson JB, Tacke U, Till M, Tolmie JL, Tomlin P, Voit T, Weschke B, Woods CG, Lebon P, Bonthron DT, Ponting CP, Jackson AP. Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutieres syndrome and mimic congenital viral brain infection. Nat Genet. 2006;38:910–916. doi: 10.1038/ng1842. [DOI] [PubMed] [Google Scholar]
- 80.Ravenscroft JC, Suri M, Rice GI, Szynkiewicz M, Crow YJ. Autosomal dominant inheritance of a heterozygous mutation in SAMHD1 causing familial chilblain lupus. Am J Med Genet A. 2011;155A:235–237. doi: 10.1002/ajmg.a.33778. [DOI] [PubMed] [Google Scholar]
- 81.Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, Shark KB, Grande WJ, Hughes KM, Kapur V, Gregersen PK, Behrens TW. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci U S A. 2003;100:2610–2615. doi: 10.1073/pnas.0337679100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, Pascual V. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med. 2003;197:711–723. doi: 10.1084/jem.20021553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhuang H, Narain S, Sobel E, Lee PY, Nacionales DC, Kelly KM, Richards HB, Segal M, Stewart C, Satoh M, Reeves WH. Association of anti-nucleoprotein autoantibodies with upregulation of Type I interferon-inducible gene transcripts and dendritic cell maturation in systemic lupus erythematosus. Clin Immunol. 2005;117:238–250. doi: 10.1016/j.clim.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 84.Rice G, Newman WG, Dean J, Patrick T, Parmar R, Flintoff K, Robins P, Harvey S, Hollis T, O’Hara A, Herrick AL, Bowden AP, Perrino FW, Lindahl T, Barnes DE, Crow YJ. Heterozygous mutations in TREX1 cause familial chilblain lupus and dominant Aicardi-Goutieres syndrome. American journal of human genetics. 2007;80:811–815. doi: 10.1086/513443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yang YG, Lindahl T, Barnes DE. Trex1 exonuclease degrades ssDNA to prevent chronic checkpoint activation and autoimmune disease. Cell. 2007;131:873–886. doi: 10.1016/j.cell.2007.10.017. [DOI] [PubMed] [Google Scholar]
- 86.Grieves JL, Fye JM, Harvey S, Grayson JM, Hollis T, Perrino FW. Exonuclease TREX1 degrades double-stranded DNA to prevent spontaneous lupus-like inflammatory disease. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:5117–5122. doi: 10.1073/pnas.1423804112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gehrke N, Mertens C, Zillinger T, Wenzel J, Bald T, Zahn S, Tuting T, Hartmann G, Barchet W. Oxidative damage of DNA confers resistance to cytosolic nuclease TREX1 degradation and potentiates STING-dependent immune sensing. Immunity. 2013;39:482–495. doi: 10.1016/j.immuni.2013.08.004. [DOI] [PubMed] [Google Scholar]
- 88.Peschke K, Achleitner M, Frenzel K, Gerbaulet A, Ada SR, Zeller N, Lienenklaus S, Lesche M, Poulet C, Naumann R, Dahl A, Ravens U, Gunther C, Muller W, Knobeloch KP, Prinz M, Roers A, Behrendt R. Loss of Trex1 in Dendritic Cells Is Sufficient To Trigger Systemic Autoimmunity. Journal of immunology. 2016;197:2157–2166. doi: 10.4049/jimmunol.1600722. [DOI] [PubMed] [Google Scholar]
- 89.Merkle D, Douglas P, Moorhead GB, Leonenko Z, Yu Y, Cramb D, Bazett-Jones DP, Lees-Miller SP. The DNA-dependent protein kinase interacts with DNA to form a protein-DNA complex that is disrupted by phosphorylation. Biochemistry. 2002;41:12706–12714. doi: 10.1021/bi0263558. [DOI] [PubMed] [Google Scholar]
- 90.Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem. 2010;79:181–211. doi: 10.1146/annurev.biochem.052308.093131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bassing CH, Swat W, Alt FW. The mechanism and regulation of chromosomal V(D)J recombination. Cell. 2002;109(Suppl):S45–55. doi: 10.1016/s0092-8674(02)00675-x. [DOI] [PubMed] [Google Scholar]
- 92.Ferguson BJ, Mansur DS, Peters NE, Ren H, Smith GL. DNA-PK is a DNA sensor for IRF-3-dependent innate immunity. Elife. 2012;1:e00047. doi: 10.7554/eLife.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.D’Amours D, Jackson SP. The Mre11 complex: at the crossroads of dna repair and checkpoint signalling. Nat Rev Mol Cell Biol. 2002;3:317–327. doi: 10.1038/nrm805. [DOI] [PubMed] [Google Scholar]
- 94.van den Bosch M, Bree RT, Lowndes NF. The MRN complex: coordinating and mediating the response to broken chromosomes. EMBO Rep. 2003;4:844–849. doi: 10.1038/sj.embor.embor925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kondo T, Kobayashi J, Saitoh T, Maruyama K, Ishii KJ, Barber GN, Komatsu K, Akira S, Kawai T. DNA damage sensor MRE11 recognizes cytosolic double-stranded DNA and induces type I interferon by regulating STING trafficking. Proc Natl Acad Sci U S A. 2013;110:2969–2974. doi: 10.1073/pnas.1222694110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Paull TT. Mechanisms of ATM Activation. Annu Rev Biochem. 2015;84:711–738. doi: 10.1146/annurev-biochem-060614-034335. [DOI] [PubMed] [Google Scholar]
- 97.Hartlova A, Erttmann SF, Raffi FA, Schmalz AM, Resch U, Anugula S, Lienenklaus S, Nilsson LM, Kroger A, Nilsson JA, Ek T, Weiss S, Gekara NO. DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity. 2015;42:332–343. doi: 10.1016/j.immuni.2015.01.012. [DOI] [PubMed] [Google Scholar]
- 98.Yu Q, Katlinskaya YV, Carbone CJ, Zhao B, Katlinski KV, Zheng H, Guha M, Li N, Chen Q, Yang T, Lengner CJ, Greenberg RA, Johnson FB, Fuchs SY. DNA-damage-induced type I interferon promotes senescence and inhibits stem cell function. Cell Rep. 2015;11:785–797. doi: 10.1016/j.celrep.2015.03.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ammann AJ, Hong R. Autoimmune phenomena in ataxia telangiectasia. J Pediatr. 1971;78:821–826. doi: 10.1016/s0022-3476(71)80353-0. [DOI] [PubMed] [Google Scholar]
- 100.Deng X, Ljunggren-Rose A, Maas K, Sriram S. Defective ATM-p53-mediated apoptotic pathway in multiple sclerosis. Ann Neurol. 2005;58:577–584. doi: 10.1002/ana.20600. [DOI] [PubMed] [Google Scholar]
- 101.Shao L, Fujii H, Colmegna I, Oishi H, Goronzy JJ, Weyand CM. Deficiency of the DNA repair enzyme ATM in rheumatoid arthritis. J Exp Med. 2009;206:1435–1449. doi: 10.1084/jem.20082251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.McGrath-Morrow SA, Collaco JM, Crawford TO, Carson KA, Lefton-Greif MA, Zeitlin P, Lederman HM. Elevated serum IL-8 levels in ataxia telangiectasia. J Pediatr. 2010;156:682–684 e681. doi: 10.1016/j.jpeds.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 103.Westbrook AM, Schiestl RH. Atm-deficient mice exhibit increased sensitivity to dextran sulfate sodium-induced colitis characterized by elevated DNA damage and persistent immune activation. Cancer Res. 2010;70:1875–1884. doi: 10.1158/0008-5472.CAN-09-2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wolf C, Rapp A, Berndt N, Staroske W, Schuster M, Dobrick-Mattheuer M, Kretschmer S, Konig N, Kurth T, Wieczorek D, Kast K, Cardoso MC, Gunther C, Lee-Kirsch MA. RPA and Rad51 constitute a cell intrinsic mechanism to protect the cytosol from self DNA. Nat Commun. 2016;7:11752. doi: 10.1038/ncomms11752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pathania S, Bade S, Le Guillou M, Burke K, Reed R, Bowman-Colin C, Su Y, Ting DT, Polyak K, Richardson AL, Feunteun J, Garber JE, Livingston DM. BRCA1 haploinsufficiency for replication stress suppression in primary cells. Nat Commun. 2014;5:5496. doi: 10.1038/ncomms6496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ho SS, Zhang WY, Tan NY, Khatoo M, Suter MA, Tripathi S, Cheung FS, Lim WK, Tan PH, Ngeow J, Gasser S. The DNA Structure-Specific Endonuclease MUS81 Mediates DNA Sensor STING-Dependent Host Rejection of Prostate Cancer Cells. Immunity. 2016;44:1177–1189. doi: 10.1016/j.immuni.2016.04.010. [DOI] [PubMed] [Google Scholar]
- 107.Yaniv G, Twig G, Shor DB, Furer A, Sherer Y, Mozes O, Komisar O, Slonimsky E, Klang E, Lotan E, Welt M, Marai I, Shina A, Amital H, Shoenfeld Y. A volcanic explosion of autoantibodies in systemic lupus erythematosus: a diversity of 180 different antibodies found in SLE patients. Autoimmun Rev. 2015;14:75–79. doi: 10.1016/j.autrev.2014.10.003. [DOI] [PubMed] [Google Scholar]
- 108.Rosen A, Casciola-Rosen L. Autoantigens in systemic autoimmunity: critical partner in pathogenesis. J Intern Med. 2009;265:625–631. doi: 10.1111/j.1365-2796.2009.02102.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Joseph CG, Darrah E, Shah AA, Skora AD, Casciola-Rosen LA, Wigley FM, Boin F, Fava A, Thoburn C, Kinde I, Jiao Y, Papadopoulos N, Kinzler KW, Vogelstein B, Rosen A. Association of the autoimmune disease scleroderma with an immunologic response to cancer. Science. 2014;343:152–157. doi: 10.1126/science.1246886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Freidberg EC, Wood RD, Walker GC, Siede W. DNA Repair and Mutagenesis, Second ed., ASM Press, Washington, DC. 2006 [Google Scholar]
- 111.Snyder A, Makarov V, Hellmann M, Rizvi N, Merghoub T, Wolchok JD, Chan TA. Genetics and immunology: reinvigorated. Oncoimmunology. 2015;4:e1029705. doi: 10.1080/2162402X.2015.1029705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, Miller ML, Rekhtman N, Moreira AL, Ibrahim F, Bruggeman C, Gasmi B, Zappasodi R, Maeda Y, Sander C, Garon EB, Merghoub T, Wolchok JD, Schumacher TN, Chan TA. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–128. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, Biedrzycki B, Donehower RC, Zaheer A, Fisher GA, Crocenzi TS, Lee JJ, Duffy SM, Goldberg RM, de la Chapelle A, Koshiji M, Bhaijee F, Huebner T, Hruban RH, Wood LD, Cuka N, Pardoll DM, Papadopoulos N, Kinzler KW, Zhou S, Cornish TC, Taube JM, Anders RA, Eshleman JR, Vogelstein B, Diaz LA., Jr PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med. 2015;372:2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Weber JS, Levit LA, Adamson PC, Bruinooge SS, Burris HA, 3rd, Carducci MA, Dicker AP, Gonen M, Keefe SM, Postow MA, Thompson MA, Waterhouse DM, Weiner SL, Schuchter LM. Reaffirming and Clarifying the American Society of Clinical Oncology’s Policy Statement on the Critical Role of Phase I Trials in Cancer Research and Treatment. J Clin Oncol. 2017;35:139–140. doi: 10.1200/JCO.2016.70.4692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ramelyte E, Schindler SA, Dummer R. The safety of anti PD-1 therapeutics for the treatment of melanoma. Expert Opin Drug Saf. 2017;16:41–53. doi: 10.1080/14740338.2016.1248402. [DOI] [PubMed] [Google Scholar]