

ABSTRACT
Mutation-specific vaccines have become increasingly important in glioma immunotherapy; however, shared neoepitopes are rare. For diffuse gliomas, a driver mutation in the gene for isocitrate dehydrogenase type-1 has been shown to produce an immunogenic epitope currently targeted in clinical trials. For highly aggressive midline gliomas, a recurrent point mutation in the histone-3 gene (H3F3A) causes an amino acid change from lysine to methionine at position 27 (K27M). Here, we demonstrate that a peptide vaccine against K27M-mutant histone-3 is capable of inducing effective, mutation-specific, cytotoxic T-cell- and T-helper-1-cell-mediated immune responses in a major histocompatibility complex (MHC)-humanized mouse model. By proving an immunologically effective presentation of the driver mutation H3K27M on MHC class II in human H3K27M-mutant gliomas, our data provide a basis for the further clinical development of vaccine-based or cell-based immunotherapeutic approaches targeting H3K27M.
KEYWORDS: Glioma, K27M-mutant histone H3, neoepitope, vaccination
Introduction
Contrary to the assumption that brain tumors are hardly immunogenic, immunotherapeutic approaches have become increasingly relevant in clinical trials in malignant gliomas. As a result of promising preclinical results, more than 100 clinical trials investigating the use of antigen-specific vaccination strategies alone or in combination with chemotherapy, radiotherapy or immune checkpoint inhibition are currently performed.1 Vaccines targeting one or multiple immunogenic neoepitopes originating from oncogenic driver mutations may best meet the obvious challenge of inducing an effective anti-glioma immune response for two main reasons: (i) they represent tumor antigens, specifically expressed in tumor cells thus reducing the risk of central T-cell tolerance or autoimmune reactions and (ii) they are homogeneously expressed, therefore preventing immune escape. One example is a peptide vaccine targeting mutant isocitrate dehydrogenase-1 (IDH1R132H) that induced a CD4-driven mutation-specific immune response in a major histocompatibility complex (MHC)-humanized mouse model and reduced growth of IDH1R132H-overexpressing tumors.2 This and a similar vaccine are currently tested in two phase I clinical trials in patients with IDH1 R132H-mutated gliomas.3,4
Another example of a recently discovered oncogenic mutation occurring especially in pediatric high-grade midline and diffuse intrinsic pontine gliomas is a point mutation in the histone-3 gene (H3F3A). An amino acid exchange from lysine to methionine at position 27 (K27M) leads to the reduction of H3K27 trimethylation resulting in a distinct global methylation and gene expression pattern.5,6,7,8 Besides, gliomas harboring H3K27M lack hallmark cytogenetic aberrations usually found in high-grade gliomas and are characterized by an exceptionally aggressive tumor progression.9 From a clinical point of view, H3K27M represents—due to its significance for glioma biology—a promising target also for immunotherapeutic approaches.
Results and discussion
A widely used approach to prioritize cancer-specific genetic alterations such as somatic point mutations for their suitability as vaccine targets is the application of MHC-binding algorithms.10,11 By using NetMHC,12 a K27M containing histone-3 10mer (H3K27M p26–35) was identified as a potential binder to HLA-A*0201 (Fig. 1A). The corresponding wild type sequence as well as 8mer, 9mer, and 11mer peptides showed negligible binding affinities and epitope presentation was mainly restricted to HLA-A*02 (Fig. S1A and B). In vivo, vaccination of MHC-humanized mice expressing HLA-A*0201 HLA-DRA*0101 HLA-DRB1*0101 while lacking mouse MHC class I and II (A2.DR1 mice) with H3K27M p26–35 induced mutation-specific cytotoxic T-cell responses confirming H3K27M as a glioma neoepitope restricted to human MHC class I (Fig. 1B–D). Remarkably, the vaccine failed to induce immunogenicity in C57BL/6 mice with a murine MHC H2b haplotype (data not shown).
Figure 1.

H3K27M p26–35 vaccination induces mutation-specific CD8+ immune responses in MHC-humanized mice. (A) MHC peptide binding predictions for H3K27M (red bars)-containing and wild type (black bars) 10mer peptides to HLA-A*0201 using NetMHC algorithm. Peptides with binding affinity > 0.3 are defined as potential binders. (B) ELISpots of IFNγ splenocyte responses to H3K27M p26–35 (KM, red dots) or wild type (wt, black dots) after vaccination of A2.DR1 mice with the 10mer H3K27M p26–35 in Montanide®. Numbers of spots to DMSO as negative control were subtracted. Individual values and mean ± SEM of four mice per group and representative ELISpots are shown. (C, D) Intracellular flow cytometry of splenocyte granzyme B (GRANB (C) or Ki-67 (D) responses to H3K27M p26–35 (KM, red dots) or wild type control (wt, black dots) after vaccination of A2.DR1 mice with H3K27M p26–35 in Montanide®. Re-stimulation with the vehicle DMSO served as control, and gate frequencies were subtracted. Gated on living CD3+CD8+ cells. Individual values and mean ± SEM of three mice per group and representative dot plots are shown.
In contrast to the assumption of a predominantly HLA class I-restricted tumor mutanome, recent studies underline the importance of HLA class II neoepitopes as cancer vaccine targets.13 To investigate class II-restricted H3K27M-specific immune responses, A2.DR1 mice were vaccinated with a 27mer harboring the point mutation in a central position (H3K27M p14–40). Peptide cleavage and epitope predictions suggested the formation of several H3K27M p14–40 peptide fragments enabling class I as well as class II immune responses (Fig. S2A and B). Indeed, vaccination of A2.DR1 mice with H3K27M p14–40 resulted in enhanced mutation-specific IFNγ responses. Intracellular flow cytometry after application of HLA-ABC as well as HLA-DR blocking antibodies during splenocyte re-stimulation substantiated both, H3K27M-specific class I-dependent CD8+-driven as well as class II-dependent CD4+-mediated immune responses (Figs. 2 and S2C).
Figure 2.
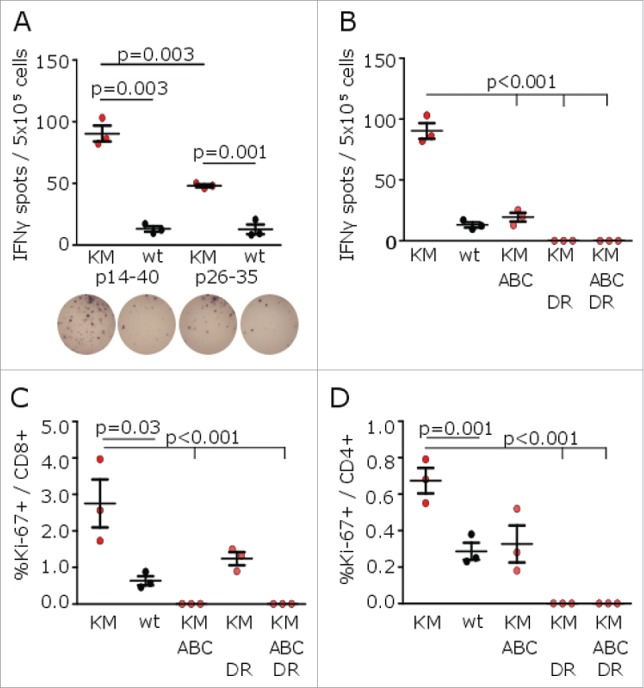
H3K27M p14–40 vaccination induces mutation-specific CD4+ and CD8+ IFNγ immune responses in MHC-humanized mice. (A) ELISpots of IFNγ splenocyte responses to H3K27M p14–40 (KM p14–40, red dots), the wild type control (wt p14–40, black dots), H3K27M p26–35 (KM p26–35, red dots) or the wild type control (wt p26–35, black dots) after vaccination of A2.DR1 mice with the 27mer H3K27M p14–40. (B) ELISpots of IFNγ splenocyte responses to H3K27M p14–40 (KM) in the presence of a pan-HLA class I blocking antibody (ABC), an HLA-DR blocking antibody (DR) or both antibodies after vaccination of A2.DR1 mice with H3K27M p14–40. (C, D) Intracellular flow cytometry of splenocyte CD8+ (C) or CD4+ (D) Ki-67 responses to H3K27M p14–40 (KM) in presence of a pan-HLA class I blocking antibody (ABC), an HLA-DR blocking antibody (DR) or both antibodies after vaccination of A2.DR1 mice with H3K27M p14–40. For ELISpot analyses, the numbers of spots to DMSO as negative control were subtracted. For flow cytometry, re-stimulation with the vehicle DMSO served as control, and gate frequencies were subtracted. Gated on living CD3+CD8+ or living CD3+CD4+ cells, respectively. Individual values and mean ± SEM of three mice per group are shown.
Due to the limited immunogenicity of H3K27M in murine gliomas and lacking availability of an A2.DR1 intracranial tumor model, a syngeneic A2.DR1 H3K27M-expressing subcutaneous sarcoma model was established to investigate the therapeutic efficacy of H3K27M peptide vaccines2 (Figs. 3A and S3A). Here, vaccination with H3K27M p14–40 significantly reduced the growth of pre-established tumors compared with a vehicle-treated control group (Fig. 3B). The presence of H3K27M-specific IFNγ-secreting CD8+ and CD4+ tumor-infiltrating lymphocytes (TILs) in tumors of mice treated with the H3K27M peptide vaccine confirmed induction of an effective cytotoxic and T-helper-1 antitumor immune response (Figs. 3C and S3B and C).
Figure 3.
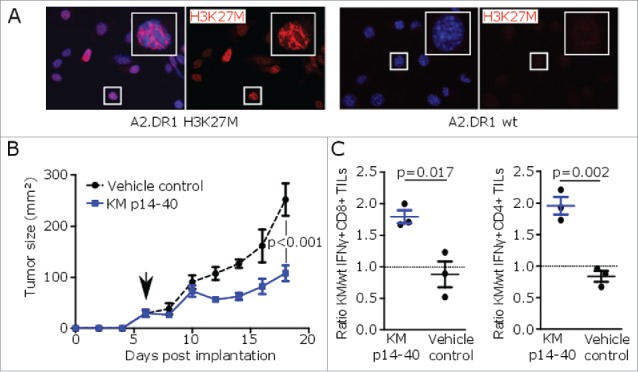
H3K27M peptide vaccination reduces growth of H3K27M+ tumors in MHC-humanized mice. (A) Nuclear H3K27M expression (red) of transduced A2.DR1 sarcoma cells (A2.DR1 H3K27M) or wild type cells (A2.DR1 wt) by immunofluorescent staining using a mutation-specific antibody. Cell nuclei are visualized with DAPI (blue). (B) Growth of pre-established H3K27M over-expressing subcutaneous syngeneic tumors in A2.DR1 mice after peptide vaccination with H3K27M p14–40 (KM p14–40, blue) or vehicle control (black) starting on day 6 (arrow). Mean ± SEM of three mice per group is shown. (C) Intracellular flow cytometry of CD8+ or CD4+ IFNγ responses of tumor-infiltrating lymphocytes (TILs) against H3K27M p14–40 relative to the corresponding wild type control of tumor-bearing A2.DR1 mice after vaccination with H3K27M p14–40 (KM p14–40, blue) or control treated mice (vehicle control, black). Re-stimulation with the vehicle DMSO served as control and gate frequencies were subtracted. Gated on living CD45+CD3+CD8+ or living CD45+CD3+CD4+ cells. Individual values and mean ± SEM of three mice per group are shown.
Finally, in situ proximity ligation assay (PLA), a method that we have previously established to prove cancer neoepitope presentation on MHC molecules,14 demonstrated specific H3K27M epitope co-localization with HLA-DR in H3K27M-mutant human glioma tissue (Figs. 4A and S3D). Using a mutation-specific HLA class I multimer, H3K27M-specific cytotoxic T cells were exclusively detected in peripheral blood mononuclear cell (PBMC) samples of 3/3 patients with H3K27M-mutant gliomas after ex vivo expansion, indicating that H3K27M is an immunogenic epitope in patients with H3K27M-mutated gliomas capable of inducing spontaneous mutation-specific T-cell responses (Figs. 4B and S3E).
Figure 4.
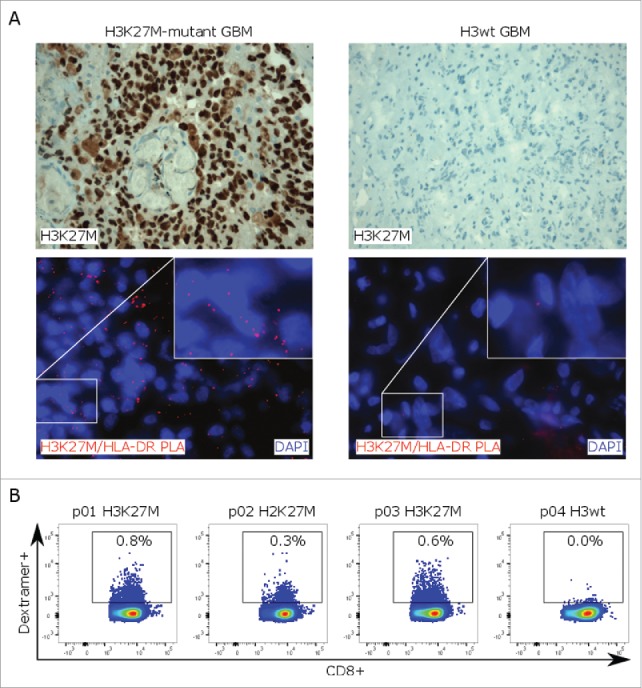
H3K27M-specific T-cell responses in glioma patients. (A)H3K27M staining (upper row) and in situ proximity ligation assay (PLA, lower row) with H3K27M and HLA-DR antibodies (red signal) on H3K27M-mutant or H3 wild type glioma tissue. Cell nuclei are visualized with DAPI (blue). (B) Detection of mutation-specific CD8+ T cells in PBMC samples of glioma patients using a H3K27M p26–35-specific HLA-A*0201 dextramer. PBMCs were analyzed after ex vivo expansion in presence of H3K27M p26–35 and hIL-2 for 9 d. Individual dot plots are shown.
In summary, we identified H3K27M as an immunogenic neoepitope targetable by a mutation-specific peptide vaccine. In an MHC-humanized mouse model, vaccination with a H3K27M 27mer induced both cytotoxic T-cell-driven as well as T-helper-1-cell-driven IFNγ immune responses suggesting in vivo processing of the peptide vaccine resulting in presentation of the histone point mutation H3K27M on both HLA class I and HLA class II. Further analyses will identify the core epitope residues, and the potential to enhance immunogenicity for various HLA types through directed amino acid substitution. For most cancer vaccines, a combination of several peptides targeting various epitopes is thought to be favorable to induce effective antitumor immune responses.13 For H3K27M, a single 27mer vaccine simultaneously targeted multiple epitopes without inducing immunity against the wild type histone-3 peptides. Mutation-specific immune responses were effective in inhibiting growth of pre-established H3K27M-expressing syngeneic tumors. One limitation is the lacking availability of an MHC-humanized orthotopic glioma model to prove vaccine efficacy against intracranial tumors. However, proximity ligation assay verified presentation of H3K27M on MHC class II in human gliomas and detection of spontaneous mutation-specific CD8+ T cells in patients with H3K27M-mutant gliomas substantiated an immunologically effective recognition by the human immune system. These endogenous immune responses might be enhanced by vaccination to gain antitumor efficacy. While most somatic point mutations are unique to the individual patient requiring extensive molecular analyses to manufacture individually tailored vaccines, H3K27M is an early genetic event occurring in essentially all tumor cells of each individual patient and can easily be detected by immunohistochemistry of glioma tissue during the routine diagnostic process. Taken together, our results provide evidence for a further clinical translation of a vaccine targeting H3K27M as part of a personalized (combinatorial) immunotherapeutic approach for HLA-A*02-positive patients with H3K27M-mutant gliomas.
Materials and methods
Mice
HLA-A*0201, HLA-DRA*0101 and HLA-DRB1*0101 transgenic mice devoid of mouse MHC (A2.DR1 mice), C57BL/6-Tg (HLA-DRA*0101, HLA-DRB1*0101), 1Dmz-Tg (HLA-A2.1-β2M), 1Bpe-IAβbtm1Doi-β2mtm1Doi-H-2Dbtm1Bpe-IAαtm1Bpe-IEβtm1Bpe were provided by M. Berard.15 All animal procedures followed the institutional laboratory animal research guidelines and were approved by the governmental authorities. 8–14-weeks old mice were assigned to age- and sex-matched experimental groups.
Peptides
For vaccination and splenocyte or PBMC stimulation, the human histone-3.3 (H3F3A) wild type and H3K27M 27mer peptides were H3 wild type p14–40 KAPRKQLATKAARKSAPSTGGVKKPHR or H3K27M p14–40 KAPRKQLATKAARMSAPSTGGVKKPHR. Additionally, 10mer peptides H3 wild type p26–35 RKSAPSTGGV and H3K27M p26–35 RMSAPSTGGV were used. Negative control was mouse myelin oligodendrocyte glycoprotein (MOG) p35–55 MEVGWYRSPFSRVVHLYRNGK (Genscript). All other peptides were synthesized in-house. Peptides were diluted in PBS 10% DMSO at 2.0 mg/mL.
MHC binding and peptide cleavage prediction
To estimate peptide binding of H3K27M containing peptide sequences to HLA-A*0201 or HLA-DRB*0101, NetMHC or NetMHCII algorithms12 were applied for 8mers, 9mers, 10mers and 11mers (class I) or 15mers (class II) and compared with corresponding wild type peptides. Additionally, binding of H3K27M or wild type p26–35 to HLA class I was assessed. Peptides with logscore affinity >0.3 for class I or >0.8 for class II were defined as potential binders. To assess cleavage of H3K27M p14–40, the ExPASy PeptideCutter tool16 was used and peptide positions with a cleavage score >0.8 were defined as potential cleavage sites.
Tumor cells and overexpression of H3K27M
A2.DR1 sarcoma cells generated as described previously2 were transduced with a H3K27M-overexpressing vector. In brief, the full length sequence of murine H3F3A (NCBI gene ID 3020) containing a lysine to methionine exchange at position 27 generated by side-directed mutagenesis was cloned into the vector backbone of pCCL.PPT.SFFV.MCS.IRES.eGFP.WPRE.17 Lentiviral particles for the transduction of A2.DR1 sarcoma cells were produced by co-transfecting the H3K27M-containing vector in HEK293T cells using FuGENE_ HD (Promega) and the corresponding packaging plasmids (pMDLg/pRRE, pRSV-Rev, pMD2.VSVG). Infection of A2.DR1 sarcoma cells was performed in the presence of 8 mg/mL polybrene (Merck Millipore). Transduced cells were sorted for high GFP expression on FACS Aria II (BD Biosciences) using BD FACS Diva software.
Peptide vaccination and tumor inoculation
A2.DR1 mice were immunized with 100 μg H3K27M p14–40 or p26–35 in PBS emulsified with equal volume of Montanide-ISA51 (Seppic) to 1 mg/mL. Mice received two subcutaneous injections of 50 μL each into the lateral pectoral regions. 300 ng rmGM-CSF (Preprotech) in PBS was subcutaneously injected between shaved injection sites; Aldara cream containing 5% imiquimod (Meda Pharma) was applied and mice were boosted after 10 d without rmGM-CSF. Experiments were terminated after 21 d.
For tumor vaccination, 5 × 105 syngeneic H3K27M-expressing sarcoma cells were embedded in Matrigel (BD Biosciences) and 200 µL tumor cell suspension was subcutaneously injected into the shaved right flank of A2.DR1 mice. On day 6, mice were grouped according to tumor size and vaccinated with 1mg/mL H3K27M p14–40 in PBS or PBS 5% DMSO as vehicle control. Mice received two subcutaneous injections of 50 μL each into the lateral pectoral regions. 50 µg anti-mouse CD40 InVivo Mab (BE0016-2, BioXCell) in 100 µL PBS was subcutaneously injected between shaved injection sites. Additionally, Aldara cream was applied and mice received 2 × 105 IU rhIL2 (Novartis) in 200 µL PBS intraperitoneally on days 6–8. The experiment was terminated on day 19 when tumor tissue and spleens were collected.
Isolation of splenocytes
Spleens of vaccinated A2.DR1 mice were excised and mashed through a 40-μm cell strainer. Erythrocytes were lysed with ACK buffer, containing 150 mM NH4Cl, 10 mM KHCO3 and 100 μM Na2EDTA, and cells were cultured in RPMI1640 containing 10% FBS and 100 U/mL penicillin, 100 μg/mL streptomycin, 1 mM sodium-pyruvate, 2 mM glutamine (all from PAA Laboratories), 100 μM non-essential amino acids (Lonza) and 50 μM β-mercaptoethanol (Sigma-Aldrich).
IFNγ ELISpot
ELISpot white bottom multiwell plates (MAIPSWU10, Millipore) were coated with anti-mouse IFNγ (AN18, Mabtech) and blocked with RPMI1640 containing 10% FBS and 100 U/mL penicillin and 100 μg/mL streptomycin. Mouse splenocytes were seeded at 5 × 105 per well and stimulated with 10 µg/mL peptide or PBS 10% DMSO or 10 ng/mL phorbol myristate acetate (PMA, Sigma-Aldrich) with 0.5 µg/mL ionomycin (Sigma-Aldrich) as controls. Peptide presentation was blocked with an antibody (1 μg per well) against HLA-ABC (W6/32) or HLA-DR (L243, both from BioLegend). After 36h, IFNγ-producing cells were detected with biotinylated anti-mouse IFNγ (R4-6A2), streptavidin-ALP (all from Mabtech) and ALP color development buffer (Bio-Rad) and quantified using an ImmunoSpot Analyzer (Cellular Technology Ltd.).
Isolation of peripheral blood mononuclear cells
PBMC were isolated from heparin blood of five glioma patients and one healthy volunteer after obtaining a written informed consent by density-gradient centrifugation using lymphocyte separation medium (LSM) 1077 (PAA Laboratories). For ex vivo expansion of H3K27M-specific cytotoxic T cells, PBMCs were cultured in RPMI-1640 with glutamax, 1% L-glutamine, 1% sodium pyruvate, 100 U/mL penicillin, 100 μg/mL streptomycin (all from PAA Laboratories), 1% non-essential amino acids (Lonza), 0.1% β-mercaptoethanol (Sigma-Aldrich) and 8% human serum in six-well plates in the presence of 10 µg/mL H3K27M p26–35. After 2 d, 1,000 U/mL hIL-2 (Proleukine) was added to the cultures and fresh medium was supplemented on day 5. Cells were harvested on day 9.
Flow cytometry
For characterization of H3K27M-specific CD8+ T cells, splenocytes of vaccinated A2.DR1 mice were stimulated ex vivo with 10 µg/mL H3K27M p26–35 or wild type p26–35 in a U-bottom 96-well-plate. After 3 d, 10 μg/mL brefeldin-A (Sigma-Aldrich) was added for 5 h. Cells were blocked with rat anti-mouse CD16/32 (eBioscience) and stained with anti-mouse CD45-PO (BioLegend), CD3-APC/Cy7 (BioLegend) and CD8A-PE/Cy7 (eBioscience) antibodies. After permeabilization using the BD cytofix/cytoperm kit, granzyme-B-PerCP/eFluor710 and Ki-67-FITC (both from eBioscience) antibodies were added.
For identification of H3K27M p14–40-reactive T cells, splenocytes of vaccinated A2.DR1 mice were stimulated ex vivo with 10 µg/mL H3K27M p14–40, H3K27M p26–35, wild type p14–40 or wild type p26–35 After 3 d, 10 μg/mL brefeldin-A was added for 5 h. Cells were blocked with rat anti-mouse CD16/32 and stained with Fixable Viability Dye-eFluor®506 (eBioscience), anti-mouse CD3-FITC (BioLegend), CD4-PB (BioLegend) and CD8A-PerCP/Cy5.5 (eBioscience) antibodies. After permeabilization, Ki-67-eFluor660 antibody (eBioscience) was added.
For characterization of mutation-specific TILs, H3K27M-expressing flank tumors of vaccinated A2.DR1 mice were cut into small pieces, digested in HBSS containing 50 µg/mL Liberase-DL (both from Sigma) and mashed, and the lymphocytes were isolated by density gradient centrifugation using Lympholyte-M (Cedarlane). TILs were re-stimulated ex vivo with 10 µg/mL H3K27M p14–40 or wild type p14-40 in T-cell medium. After 18h, 10 μg/mL brefeldin-A was added for 5 h. Cells were blocked with rat anti-mouse CD16/32 and stained with Fixable Viability Dye-eFluor®520 (eBioscience), anti-mouse CD3-APC/Cy7 (BioLegend), CD8A-BV510™ (BioLegend) and CD4-PB antibodies (BioLegend). After permeabilization with the eBioscience Foxp3/Transcription Factor Fixation/Permeabilization kit, IFNγ-PE/Cy7 antibody (eBioscience) was added.
For identification of human H3K27M-specific cytotoxic T cells, PBMCs were stained with anti-human CD3-APC/efluor780 (eBioscience) and CD8A-APC (eBioscience) antibodies after incubation with PE-conjugated HLA-A*0201 MHC class I dextramers bound to H3K27M p26–35 (Immudex, 4 µL/well). Just before acquisition, 4′,6-Diamidino-2-Phenylindole (DAPI, ThermoFisher Scientific) was added. Living single cells were gated on CD3+CD8+ cells and analyzed for dextramer binding.
In all mouse experiments, splenocytes or TILs treated with MOG or the vehicle solution DMSO were used as negative controls, and for intracellular markers, gate frequencies of DMSO were subtracted. Cells were measured using FACS Canto II (BD Biosciences) and data analysis was done using FlowJo software.
Immunocytochemistry and immunohistochemistry
H3K27M-expressing or wild type A2.DR1 sarcoma cells were seeded on glass coverslips, grown until 70–90% confluent and fixed and permeabilized with Cytofixx Pump Spray (Cell Path) at −20 °C and subsequent 4% PFA at room temperature. Primary antibody was rabbit anti-human K27M-mutant histone-H3 antibody (ABE419, Merck Millipore), and secondary antibody was goat anti-rabbit Alexa Fluor 546 (Molecular Probes, Invitrogen, A-11010). Vectashield HardSet Mounting Medium with DAPI (Vector Laboratories) was used for mounting and nuclear staining. Images were taken on DM IRB microscope (Leica).
Formalin-fixed paraffin-embedded H3K27M-mutant and H3 wild type glioma tissue was obtained from the archives of the Department of Neuropathology, Institute of Pathology, Heidelberg. H3K27M-expressing A2.DR1 mouse tumor tissue was fixed with Roti-Histofix 4.5% and paraffin embedded. Sections cut to 3 µm were incubated with ABE419 and processed using a Ventana BenchMark XT immunostainer. For visualization, ultraView™Universal DAB Detection Kit (Ventana Medical Systems) was used.
Proximity ligation assay
Glioma tissue was deparaffinized with HistoClear II (National Diagnostics) and rehydrated. Antigen retrieval was performed using Cell Conditioning Solution CC2 (Ventana Medical Systems, Inc.) for 30 min. PLA was performed as described previously14 with primary antibodies ABE419 and monoclonal mouse anti-human HLA-DR (TAL1B5, Abcam). Ligation and amplification were performed using the Detection Reagents Red. Vectashield HardSet Mounting Medium with DAPI (Vector Laboratories) was used for mounting and nuclear staining.
Statistics
Data are expressed as mean ± SEM. Experiments were repeated at least three times with similar results. Analysis of significance was performed using unpaired t-tests except for growth of flank tumors where a two-way ANOVA test was used (GraphPad Prism). p values < 0.05 were considered statistically significant.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgment
We acknowledge support by the DKFZ Light Microscopy Facility.
Funding
K.O. was supported by a physician-scientist fellowship of the Medical Faculty, University of Heidelberg, L.B. was supported by the Hartmut Hoffmann-Berling International Graduate School of Molecular and Cellular Biology MD/PhD program, University Heidelberg., K.D. was supported by the German-Israeli Helmholtz Research School in Cancer Biology, and J.K.S. was supported by the Helmholtz International Graduate School for Cancer Research at DKFZ. This study was supported by funds of the "Stiftung für Krebs-und Scharlachforschung" and the NCT 3.0 program.
References
- 1.NCT Clinical Trial Registration Number NCT02017717 (ClinicalTrials.gov); A Study of the Effectiveness and Safety of Nivolumab Compared to Bevacizumab and of Nivolumab With or Without Ipilimumab in Glioblastoma Patients (CheckMate 143). [Google Scholar]
- 2.Schumacher T, Bunse L, Pusch S, Sahm F, Wiestler B, Quandt J, Menn O, Osswald M, Oezen I, Ott M et al.. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature 2014; 512:324-7; PMID:25043048; https://doi.org/ 10.1038/nature13387 [DOI] [PubMed] [Google Scholar]
- 3.NCT Clinical Trial Registration Number NCT02193347; IDH1 Peptide Vaccine for Recurrent Grade II Glioma (RESIST). [Google Scholar]
- 4.NCT Clinical Trial Registration Number NCT02454634; Phase I Trial of IDH1 Peptide Vaccine in IDH1R132H-mutated Grade III-IV Gliomas (NOA-16). [Google Scholar]
- 5.Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang DA, Tönjes M et al.. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012; 482:226-31; PMID:22286061; https://doi.org/ 10.1038/nature10833 [DOI] [PubMed] [Google Scholar]
- 6.Bender S, Tang Y, Lindroth AM, Hovestadt V, Jones DT, Kool M, Zapatka M, Northcott PA, Sturm D, Wang W et al.. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell 2013; 24:660-72; PMID:24183680; https://doi.org/ 10.1016/j.ccr.2013.10.006 [DOI] [PubMed] [Google Scholar]
- 7.Lewis PW, Muller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA, Garcia BA, Muir TW, Becher OJ, Allis CD. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 2013; 340:857-61; PMID:23539183; https://doi.org/ 10.1126/science.1232245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, Qu C, Ding L, Huether R, Parker M et al.. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet 2012; 44:251-3; PMID:22286216; https://doi.org/ 10.1038/ng.1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DTW, Konermann C, Pfaff E, Tönjes M, Sill M, Bender S et al.. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 2012; 22:425-37; PMID:23079654; https://doi.org/ 10.1016/j.ccr.2012.08.024 [DOI] [PubMed] [Google Scholar]
- 10.Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, Arthur CD, White JM, Chen YS, Shea LK et al.. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 2012; 482:400-4; PMID:22318521; https://doi.org/ 10.1038/nature10755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Robbins PF, Lu YC, El-Gamil M, Li YF, Gross C, Gartner J, Lin JC, Teer JK, Cliften P, Tycksen E et al.. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med 2013; 19:747-52; PMID:23644516; https://doi.org/ 10.1038/nm.3161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lundegaard C, Lund O, Nielsen M. Accurate approximation method for prediction of class I MHC affinities for peptides of length 8, 10 and 11 using prediction tools trained on 9mers. Bioinformatics 2008; 24:1397-8; PMID:18413329; https://doi.org/ 10.1093/bioinformatics/btn128 [DOI] [PubMed] [Google Scholar]
- 13.Kreiter S, Vormehr M, van de Roemer N, Diken M, Lower M, Diekmann J, Boegel S, Schrörs B, Vascotto F, Castle JC et al.. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 2015; 520:692-6; PMID:25901682; https://doi.org/ 10.1038/nature14426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bunse L, Schumacher T, Sahm F, Pusch S, Oezen I, Rauschenbach K, Gonzalez M, Solecki G, Osswald M, Capper D et al.. Proximity ligation assay evaluates IDH1R132H presentation in gliomas. J Clin Invest 2015; 125:593-606; PMID:25555220; https://doi.org/ 10.1172/JCI77780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pajot A, Michel ML, Fazilleau N, Pancre V, Auriault C, Ojcius DM, Lemonnier FA, Lone YC. A mouse model of human adaptive immune functions: HLA-A2.1-/HLA-DR1-transgenic H-2 class I-/class II-knockout mice. Eur J Immunol 2004; 34:3060-9; PMID:15468058; https://doi.org/ 10.1002/eji.200425463 [DOI] [PubMed] [Google Scholar]
- 16.Wilkins MR, Gasteiger E, Bairoch A, Sanchez JC, Williams KL, Appel RD, Hochstrasser DF. Protein identification and analysis tools in the ExPASy server. Methods Mol Biol 1999; 112:531-52; PMID:10027275 [DOI] [PubMed] [Google Scholar]
- 17.Weiler M, Blaes J, Pusch S, Sahm F, Czabanka M, Luger S, Bunse L, Solecki G, Eichwald V, Jugold M et al.. mTOR target NDRG1 confers MGMT-dependent resistance to alkylating chemotherapy. Proc Natl Acad SciUSA 2014; 111:409-14; PMID:24367102; https://doi.org/ 10.1073/pnas.1314469111 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.